1. INTRODUZIONE
1.1. Mieloma Multiplo: la patologia
Il Mieloma Multiplo (MM), anche detto Mieloma
plasmacellulare o Plasmocitoma, è una patologia tumorale maligna caratterizzata dalla proliferazione e dall’accumulo di un singolo clone di plasmacellule nel midollo osseo.
La definizione “multiplo” associata al mieloma deriva dalla localizzazione dei plasmociti neoplastici in attiva proliferazione, che interessano zone multiple del midollo osseo (plasmociti midollari) ma che possono (essere confinati all’osso) anche interessare i tessuti molli (plasmocitoma extramidollari)[1]. La progressione e lo sviluppo della malattia porta ad una serie di conseguenze fisiopatologiche le quali risultano in un complesso insieme di sintomi e disfunzioni gravi, tra cui si evidenziano dolore osseo associato a lesioni osteolitiche (80% dei casi), anemia,
ipercalcemia, la compromissione della produzione di
immunoglobuline normali (IgG, IgA, IgD o IgE) con conseguente insufficienza renale (60% dei casi) e suscettibilità ad infezioni. In questo quadro clinico, la prognosi dei pazienti è generalmente infausta ed il progressivo declino della qualità della vita è associato ad una aspettativa di sopravvivenza che oscilla tra i 20 e i 60 mesi.
Il mieloma multiplo è una patologia relativamente rara, rappresenta circa l’1% di tutte le malattie maligne ed in particolare il 10% tra le neoplasie ematologiche[2]. A livello mondiale
l’incidenza annua del mieloma multiplo è di circa 3 nuovi casi ogni 100.000 individui (superiore negli anziani) ed è maggiore nei maschi, mentre l’età media alla diagnosi si attesta intorno ai 68 anni. I tassi più elevati, inoltre, si osservano tra gli Afro-Americani degli Stati Uniti (33% di tutti i cancri ematologici) mentre i tassi più bassi si registrano tra le popolazioni dell’Asia e dell’Europa Orientale[3].
In Italia, l’incidenza registrata è di circa 2-4 nuovi casi ogni 100.000 abitanti all’anno, con un’età media di insorgenza che va oltre i 50 anni. Anche in Italia, l’incidenza è leggermente maggiore nei maschi rispetto alle femmine. I tassi di mortalità, relativi a tutti i gruppi di età registrati nel 1997, sono pari alla media dell’Unione Europea negli uomini (2,15) e superiore alla media (1,52) nelle donne (1,55).
Le caratteristiche del tumore corrispondono a quelle della plasmacellula mielomatosa, che è interessata da alterazioni genetiche, biochimiche e metaboliche che ne alterano la corretta funzionalità, conferendole carattere neoplastico. Le plasmacellule superiori al 20% della cellularità globale, possono arrivare a localizzarsi in sedi specifiche, con un infiltrazione massiva dello stesso spazio midollare e conseguente alterazione della crassi ematica (Figura 1.1).
Dal punto di vista funzionale, le cellule mielomatose vanno incontro ad un forte aumento dell’attività cellulare, in particolare con una produzione di elevate quantità di un immunoglobulina monoclonale (detta proteina M), solitamente di tipo IgG o IgA, oppure di un suo componente/frammento, caratteristica della patologia.
Infatti, il mieloma multiplo ha origine della trasformazione neoplastica di una cellula della fase differenziata antigene-dipendente, quali in particolare il plasmoblasto o la cellula B memoria, ovvero elementi cellulari che sono appena passati per la fase di selezione antigenica a livello del centro germinativo del midollo osseo (Figura 1.2).
Nella fase del ciclo in cui le cellule B immature diventano plasmacellule mature, avviene un processo fisiologico denominato “ricombinazione sito-specifica” dei geni delle immunoglobuline (V, D e J), che consiste nell’associazione combinatoria di differenti segmenti genici, con la conseguente generazione dell’ampio spettro di anticorpi caratteristico di ciascun individuo. In altre parole, segmenti diversi di geni diversi vengono riarrangiati da un complesso apparato biochimico cellulare, noto come recombinasi VDJ, a formare un gene che codificherà le componenti necessarie a formare l’immunoglobulina specifica di ciascun linfocita B.
A causa di questo delicato passaggio di ricombinazioni, le cellule B sono caratterizzate da una instabilità genetica intrinseca che è alla base, a sua volta, della maggior parte degli eventi oncogenici che contribuiscono alla patogenesi del mieloma multiplo. Infatti, nel cariotipo delle plasmacellule mielomatose si osservano numerose alterazioni come delezioni o traslocazioni, localizzate nelle regioni cromosomiche contenenti i geni per le immunoglobuline.
L’alterazione più frequente, che colpisce appunto la regione che contiene i geni codificanti per le catene pesanti delle immunoglobuline, è la traslocazione della banda 14q32, anormale in circa il 75% dei pazienti affetti da mieloma[4]. Il fatto che nella maggior parte delle traslocazioni a carico della banda 14q32 il punto di rottura cada all’interno della regione di “switching” delle catene pesanti fornisce una indicazione indiretta del fatto che l’evento iniziale dello sviluppo della malattia abbia effettivamente luogo nel centro germinativo, con un probabile coinvolgimento di progenitori plasmacellulare precoci.
L’80-90% dei pazienti affetti da mieloma multiplo può comunque presentare anomalie cromosomiche numeriche o strutturali (del 13q, del 17q, t(4,14)), anche diverse dalla traslocazione a carico della banda 14q32, indipendentemente dallo stadio della malattia[5].
Dal punto di vista della patogenesi del mieloma, le alterazioni e i riarrangiamenti cromosomici che si osservano nelle plasmacellule sono considerati l’evento primario nell’evoluzione della malattia, in quanto garantiscono l’immortalità della cellula, senza tuttavia essere da soli sufficienti a determinare l’effettiva trasformazione neoplastica della cellula.
Figura 1.2: La patogenesi del mieloma multiplo.
Per quel che riguarda la traslocazione della banda 14q32, ad esempio, sono finora identificati alcuni geni che agiscono come partner primari della traslocazione, quali bcl1, prad1, cicline D1 e D3 ed altri che codificano per proteine coinvolte nella regolazione del ciclo cellulare, contribuendo nell’evoluzione del carattere neoplastico. Un’altra alterazione comunemente osservata nel cariotipo di pazienti affetti da mieloma multiplo è la delezione a carico del braccio lungo del cromosoma 13 (del 13q), che si verifica precocemente nel corso della malattia ed è stata osservata nel 43% dei casi alla diagnosi e nel 70% dei pazienti con malattia avanzata o evoluta in leucemia plasmacellulare.
Dall’analisi dei pazienti che presentavano riarrangiamenti a livello della banda 14q32 e la delezione 13q sono state recentemente ipotizzate le tappe fondamentali nella patogenesi del mieloma multiplo. L’evento più precoce nello sviluppo della malattia sarebbe la ricombinazioni illegittima dei geni codificanti per la catena pesante dell’immunoglobuline con la conseguente
traslocazione a carico della banda 14q32. Tale ipotesi è confermata dalla correlazione esistente tra i riarrangiamenti a livello della banda 14q32 e il tipo di immunoglobulina prodotta dalle cellule mielomatose. Inoltre, è stato osservato che le traslocazioni della banda 14q32 sono quasi sempre associate (nel 95% dei pazienti) alla delezione del braccio lungo del cromosoma 13. In questi pazienti il riarrangiamento della banda q32 del cromosoma 14 rappresenterebbe l’evento oncogenico primario, mentre la delezione 13q l’evento oncogenico secondario.
Nel 25% dei pazienti, tuttavia, non si evidenziano traslocazioni a carico della banda 14q32, ed in questi soggetti la malattia presenta un decorso clinico indolore sino al momento in cui non si presenti la delezione 13q[6]. Studi clinici evidenziano che le anomalie a carico del cromosoma 13 sono associate ad una bassa sopravvivenza libera da eventi (EFS) e globale (OS) in seguito alle chemioterapie (convenzionale e ad alte dosi). In particolare, le delezioni, come la delezione 13q, sono associate sia ad una malattia più aggressiva che alla farmaco-resistenza del tumore[7]. Inoltre, come facilmente prevedibile, durante la progressione della malattia aumenta l’instabilità del cariotipo. Questo porta ad un accumulo, nelle cellule mielomatose, di mutazioni ed alterazioni nell’espressione di altri geni importanti per la sopravvivenza ed il controllo cellulare, quali c-myc, Nras, Kras e p53, con un conseguente decorso clinico generalmente sfavorevole (Fig. 1.2).
Un’altra importante caratteristica del mieloma multiplo è la completa dipendenza delle plasmacellule neoplastiche dal microambiente del midollo osseo, caratterizzato dalla presenza di proteine della matrice extracellulare, cellule stromali, osteoclasti, osteoblasti ed altre cellule accessorie.
L’interazione della plasmacellule con questi elementi è alla base dell’attivazione e secrezione di fattori di crescita, di fattori
anti-apoptotici e di diverse citochine (in particolare IL-6) che, attraverso una complessa rete di segnali intra- ed extra- cellulari, promuove la proliferazione, la sopravvivenza, la farmaco-resistenza e la progressione della malattia.[8].
L’interleuchina 6 (IL-6) è il principale segnale per la crescita e la sopravvivenza delle plasmacellule neoplastiche che, come le cellule stromali del midollo osseo, la producono e la secernono. In pazienti nei quali la malattia è in fase attiva, i livelli sierici di IL-6 sono aumentati e risultano, senza dubbio, associati ad una prognosi infausta. Le citochine, ed in particolare IL-6 e IL-10, oltre a essere determinanti per la crescita e la proliferazione delle cellule mielomatose, mediano anche i fenomeni di distruzione dell’osso, conseguenza principale del processo di riassorbimento da parte degli osteoclasti attivati.
La comprensione dei meccanismi molecolari della patogenesi e dell’evoluzione del mieloma multiplo è un punto cruciale nella ricerca delle cure efficaci, in quanto permetterebbe di individuare potenziali bersagli terapeutici per l’identificazione di nuove terapie in grado di superare la farmaco-resistenza e di indurre l’apoptosi delle cellule neoplastiche.
1.2. Fattori prognostici tradizionali
Sono stati individuati alcuni fattori che, valutati alla diagnosi, hanno un valore prognostico. La componente senza dubbio principale nella valutazione della condizione neoplastica è la presenza nel siero di una immunoglobulina monoclonale, o di un suo frammento, nota come proteina M. In condizioni patologiche, la proteina M è presente nel siero e/o nell’urina in quantità ingenti e viene normalmente identificata, all’elettroforesi proteica, da un picco in corrispondenza della regione delle gammaglobuline.
Oltre alla proteina M sono stati caratterizzati numerosi altri fattori prognostici, sierici e non. I fattori prognostici tradizionali, come i livelli di β2 microglobulina, della proteina C reattiva (CRP) e di albumina spiegano soltanto il 15-20% dell’eterogeneità della risposta alla terapia. Inoltre anomalie cariotipiche, presenti in un terzo dei pazienti di nuova diagnosi, sono associate ad un esito fatale più rapido e meno del 10% dei pazienti che presentano tali anomalie sopravvive oltre i 5 anni.
Mielomi iperdiploidi e positivi per le traslocazioni t(11,14)(q13q32) sono associati con traslocazioni diverse dalla t(11,14) e con delezione di parte del cromosoma 13, che predicono una prognosi estremamente peggiore[9].
Un importante limite dei fattori prognostici tradizionali individuati fino a questo momento rimane quello di essere valutabili unicamente alla diagnosi, in quanto sono sensibilmente influenzati dai regimi chemioterapici adottati nella cura della patologia.
1.3. Fattori di suscettibilità genetica
Considerando l’importante limite dei fattori prognostici tradizionali, si ipotizza che lo studio dei polimorfismi a singolo nucleotide (SNPs) in geni bersaglio coinvolti in tutti i processi cellulari possano permettere di identificare nuovi fattori prognostici.
Sulla base dei meccanismi molecolari sopra discussi, rappresentati da alterazioni genetiche, biochimiche e metaboliche che alterano la corretta funzionalità della plasmacellula conferendole carattere neoplastico, esistono diverse categorie di geni polimorfici che potrebbero modulare il rischio di sviluppare il Mieloma Multiplo.
Gli SNPs (Single Nucleotide Polymorphisms) sono molto diffusi in tutto il genoma umano: si stima che esista una variazione nucleotidica, circa, ogni mille paia di basi. L’analisi degli SNPs ha cominciato a dare un contributo importante nella ricerca di geni coinvolti nelle malattie multifattoriali attraverso studi di associazione e nel futuro darà un forte impulso alla identificazione di fattori predisponenti allo sviluppo di patologie tumorali.
Oltre a permettere lo studio della suscettibilità alla malattie complesse, lo studio dei polimorfismi a singolo nucleotide sarà in futuro sempre più importante per la progettazione di nuovi farmaci sempre più efficaci nei confronti di individui che possiedono un certo genotipo piuttosto che un altro[10 ].
I metodi di studio per l’analisi degli SNPs sono basati sull’amplificazione di DNA bersaglio mediante PCR e ibridazione con sonde fatte da oligonucleotidi allele specifiche (ASO), utilizzando enzimi che tagliano gli acidi nucleici in determinati siti, come gli enzimi di
restrizione (da cui il nome di “restriction lenght fregment polymorphisms”, RFLP); oppure metodiche basate su sistemi di ibridazione specifiche (tipo il TaqMan), altre invece basate su l’utilizzo di substrati solidi, come le metodiche basate sui micro-array. Tra queste la tecnica TaqMan sarà illustrata in dettaglio successivamente.
1.4. Suscettibilità genetica
L’esistenza della variabilità genetica può influenzare il rischio individuale di contrarre una malattia. Per questo, lo studio degli SNPs è oggi uno dei campi di maggior rilievo nella genetica molecolare umana, in quanto le varianti puntiformi contribuiscono in maniera determinante alla sostanziale diversità che caratterizza l’unicità di ciascun individuo.
Questa variabilità genetica governa i processi di assorbimento, traduzione del segnale, trasporto di soluti, metabolismo di sostanze esogene come pure la riparazione del DNA, il ciclo cellulare, l’apoptosi e altri processi che sono probabilmente responsabili delle differenze di suscettibilità tra un individuo ed un altro[11].
Molti geni coinvolti nel trasporto degli agenti esogeni e degli agenti endogeni presentano un numero considerevole di polimorfismi; sono, cioè, presenti nella popolazione forme differenti di uno stesso gene che corrispondono a trasportatori con un’attività funzionale diversa. Infatti variazioni all’interno delle sequenze nucleotidiche possono riflettersi sull’espressione, con un aumento o una diminuzione dei trascritti, provocando un alterazione della funzionalità e/o dell’espressione dei trasportatori; producendo alterazioni nell’omeostasi cellulare anche con conseguenze gravi.
Nello studio delle cause delle malattie multifattoriali, come il Mieloma Multiplo, è importante considerare ogni singolo evento/fattore che può essere concausa dello sviluppo della patologia.
Un metodo di indagine idoneo, è quello di effettuare uno studio di associazione del tipo caso-controllo. Questo si basa sul confronto di due gruppi di soggetti: gli affetti dalla malattia che costituiscono i casi, ed i controlli che costituiscono i soggetti sani. I controlli sono scelti in modo
che siano rappresentativi della popolazione generale e che abbiano caratteristiche simili per età, per sesso e/o altri parametri.
Tramite il confronto delle frequenze genotipiche osservate tra i casi e i controlli è possibile identificare SNPs che hanno un ruolo come fattori di suscettibilità alla malattia.
Nell’approccio utilizzato, il confronto delle frequenze genotipiche viene eseguito sulla base di un’analisi di regressione logistica, che permette di stabilire se sia presente una differenza significativa tra le distribuzioni genotipiche all’interno delle due popolazioni prese in esame. Infine l’analisi statistica permette di attribuire, un valore che esprime se e quanto ogni allele sia un fattore di rischio associato alla malattia oppure un fattore protettivo associato alla malattia.
ƒ(A)casi > ƒ(A)controlli = FATTORE DI RISCHIO
1.5. I Trasportatori di membrana
I trasportatori di membrana sono polipeptidi transmembrana in grado di mediare il trasporto di ioni, di molecole polari di media grandezza (amminoacidi, zuccheri, nucleotidi), di farmaci e di sostanze esogene all’esterno della cellula.
La presenza di trasportatori di membrana è indispensabile: solamente alcuni composti sono in grado di diffondere liberamente attraverso la membrana plasmatica; tutti gli altri hanno bisogno di metodi alternativi per entrare nel citoplasma. Sebbene ne esista un gran numero, alcuni di questi possiedono alcune caratteristiche in comune:
• il trasporto è selettivo per il substrato che deve essere trasportato, e basato su interazioni steriche tra il trasportatore e la molecola trasportata;
• la traslocazione attraverso la membrana è legata ad un
cambiamento conformazionale della proteina
trasportatrice, e più specificamente
all'apertura/chiusura di un "canale" all'interno del polipeptide;
• il trasporto può richiedere o meno dispendio energetico, dando vita rispettivamente ad un trasporto attivo o passivo;
• tutte le molecole trasportatrici conosciute sono proteine transmembrana, che attraversano cioè integralmente la membrana plasmatica.
I trasportatori di membrana possono essere suddivisi in tre classi principali:
o Proteine canale;
o Cotrasportatori;
o ATPasi di trasporto.
Variazioni nella sequenza nucleotidica che codifica per un trasportatore possono alterarne la funzionalità e/o l'espressione. Questo comporta differenze di efficienza dei trasportatori che possono riflettersi sull'equilibrio cellulare anche con conseguenze gravi.
1.6. MDR1 e P-glicoproteina 1
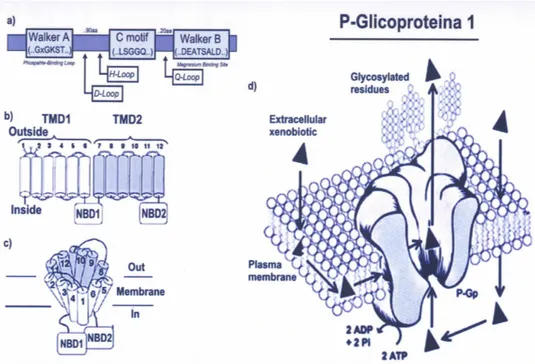
Il gene MDR1 è situato sul cromosoma 7q21.1, è lungo 209 kb ed è costituito da 29 esoni, anche se nella caratterizzazione di questo gene sono state descritte varie forme di splicing alternativo[12]. La sequenza codificante costituisce circa il 2,5% del gene, con un RNA messaggero lungo circa 4,7 kb[13]. Il gene MDR1, o ABCB1, codifica per la P-glicoproteina 1, una proteina integrale di membrana di circa 170 kDa, costituita da 1280 amminoacidi, appartenente alla superfamiglia dei trasportatori ABC (ATP-Bilnding Cassette). Per questo gene sono stati descritti numerosi polimorfismi a singolo nucleotide disseminati lungo l’intera sequenza, sia nella regione codificante che non codificante (Figura 1.3).
La P-glicoproteina 1 (P-gp1) è una potente pompa di efflusso cellulare ATP-dipendente, in grado di regolare l’uscita dalla cellula di una vasta gamma di substrati lipofilici differenti (xenobiotici), tra cui molti farmaci antitumorali, ormoni steroidei ed antibiotici.
La proteina è codificata sotto forma di un unico polipeptide, il quale è costituito da due regioni amminoacidiche ripetute, risultato di eventi di duplicazione genica, separate da una regione idrofila di collegamento (Figura 1.3b). Ognuna di queste due regioni è costituita da sei segmenti amminoacidici che attraversano la membrana cellulare, formando complessivamente un dominio transmembrana (TBD - Transmembrane Bilnding
Domain). Nella parte terminale di ciascun dominio transmembrana si trova una regione amminoacidica idrofila che costituisce il sito di legame per l’ATP (NBD – Nucleotide Bilding Domain)[14].
La P-glicoproteina 1, che è stata isolata per la prima volta in cellule di criceto resistenti alla colchicina[15], è stata descritta come un largo alloggiamento sul lato extracellulare, che assume una forma ad imbuto sul versante citoplasmatico. Recentemente si sono registrati progressi nella comprensione della struttura e del meccanismo di azione di questa proteina. Le due metà della molecola si dispongono specularmene attorno ad un asse centrale, delimitando un canale centrale la cui forma è quella di un tronco di cono rovesciato. Dal punto di vista della sua sequenza amminoacidica primaria, la P-glicoproteina 1, come tutti i membri della famiglia dei trasportatori ABC, è caratterizzata da domini conservati. In particolare, si ritrovano i motivi Walker A e B, presenti in moltissimi domini di legame per l’ATP, ed il motivo C, detto anche “signature”, che è peculiare dei trasportatori ABC. Altri motivi proteici, come i loop H, D e Q sono importanti nella formazione del dominio di legame per il nucleotidi (ATP) (Figura 1.3a). La struttura dei NBDs mostra che i due domini formano un “nucleotide-sandwich dimer”, nel quale l’ATP si lega all’interfaccia dei due domini, circondato dai domini Walker A e B di una subunità e dal motivo C e dal loop D dell’altra. L’anello dell’Adenina dell’ATP interagisce con il loop Q e, quando possibile, con il loop H[16].
Figura 1.3. a)Struttura e organizzazione della P-gp1. b) Unica sequenze
amminoacidica organizzata in 2 domini transmembrana (TMD1 e TMD2). c) Organizzazione spaziale dei domini transmembrana. d) Rappresentazione della P-gp1 nella membrana e suo funzionamento
La struttura della P-glicoproteina 1, è stata determinata mediante microscopia elettronica. È stato ricostruito il primo modello tridimensionale della proteina, nel quale si evidenziano le relazioni tra le α-eliche dei domini transmembrana e i due domini di legame per l’ATP. Con questi studi, si sono ottenute importanti indicazioni circa le modalità di azione della proteina. Nella sua conformazione nativa la regione TMD della proteina ricorda un cilindro di 5/6 nm di diametro e circa 5 nm di lunghezza. Questo cilindro circonda un poro centrale che sembra essere aperto verso il lato extracellulare e chiuso alla base[17,18,19]. Tale struttura evidenzia la disposizione speculare delle due metà della proteina a determinare il canale centrale, il cui diametro varia da un minimo di 9Å ad un massimo di 50Å [16].
Il meccanismo di azione di questa pompa ATP-asica non è stato ancora determinato con precisione. Studi recenti di
mutagenesi sito-specifica hanno evidenziato che il sito di legame del substrato è collocato all’interfaccia di contatto tra i due domini transmembrana [14] e che il legame tra substrato e trasportatore è indipendente dall’azione ATP-asica della proteina. Sono stati proposti differenti meccanismi per l’azione della pompa.
L’idrolisi dell’ATP sembra, invece, in grado di determinare cambiamenti conformazionali nei segmenti transmembrana che costituiscono il dominio di legame per il substrato, portando a profondi sconvolgimenti nella struttura della proteina, con la rotazione di una α-elica (TMD 6) e l’apertura di lacune laterali che mettono in contatto il canale centrale con l’ambiente della membrana cellulare[18]. Questa osservazione confermerebbe l’ipotesi secondo la quale la P-glicoproteina 1 interagisce anche con quei substrati ancora sospesi nella fase lipidica della membrana, escludendoli prima ancora che entrino nella cellula. L’azione dell’ATP risulta cruciale nella regolazione dei cambiamenti conformazionali. Il legame della molecola è infatti l’evento che fornisce l’energia necessaria per il riarrangiamento della struttura della proteina. Tali cambiamenti permangono fintanto che l’ADP resta legato alla proteina, mentre il suo rilascio porta la struttura a ritornare alla configurazione nativa.
Uno dei meccanismi di azione proposti per l’azione della proteina è riportato nello schema in Figura 1.4.
Secondo questa ipotesi, il legame del substrato sarebbe indipendente dal legame dell’ATP, mentre quest’ultimo determinerebbe i cambiamenti conformazionali necessari all’efflusso del substrato. Il rilascio dell’ADP determinerebbe il ripristino della conformazione nativa[18].
Sebbene permangano ancora molte lacune sul meccanismo di azione della P-Glicoproteina 1 e non esistano ancora dati precisi circa la localizzazione e la struttura del sito di legame del substrato, è ben dimostrato che la P-Glicoproteina 1 ha un’attività estremamente aspecifica, per cui è in grado di modulare il trasporto di una vastissima gamma di sostanze differenti.
Studi di mutagenesi sito-specifica hanno chiarito che il substrato interagisce con alcuni residui amminoacidici situati all’interfaccia delle α-eliche 4,5,6 del TMD1 e 9,10,11 del TMD2[14], mentre i cambiamenti conformazionale indotti dal legame dell’ATP interessano in particolare l’α-elica 6 del TMD1[18]. L’ipotesi del “substrate-induced fit” prevede che il sito di legame del substrato sia variabile, ed in particolare che dipenda dal substrato. Ciascuna molecola che interagisce con la P-Glicoproteina 1, all’interno dell’alloggiamento proteico, stabilirebbe relazioni con residui amminoacidici differenti delle sei α-eliche coinvolte, secondo la sua natura chimica. In questo modo, substrati diversi interagirebbero con residui diversi e soprattutto con forze differenti[19].
Per molti anni si è pensato che la P-Glicoproteina 1 fosse espressa unicamente nelle cellule tumorali, dove si riteneva che fosse in larga misura responsabile del fenomeno della farmaco-resistenza multipla. Tuttavia, è stato ampiamente dimostrato come questa proteina sia comunemente espressa in diversi organi e tessuti, dove è presumibilmente associata ad altre funzioni fisiologiche.
Infatti, P-gp1 è espressa a livello del fegato, dei reni, del sistema nervoso centrale (plessi corioidei e barriera emato-encefalica), della placenta, delle ovaie e dei testicoli. Ancora, la P-Glicoproteina 1 è stata individuata nelle cellule staminali ematopoietiche, nelle cellule PBM (Peripheral Blood Mononuclear Cells), nei macrofagi, nelle cellule APC (Antigen-Presenting Cells) e nei linfociti T e B; sia la funzione che la localizzazione, prevalentemente in zona di scambio, suggeriscono che la proteina abbia un ruolo determinante come barriera in grado di eliminare tossine e molecole dannose, provvedendo alla loro escrezione nella bile, nelle urine e nel lume intestinale[20].
1.7. Il polimorfismo C3435T
Il primo lavoro sui polimorfismi del gene MDR1 risale al 1989, quando Kioka et al.[21] individuarono due sostituzioni amminoacidiche, Gly185Val e Ala893Ser, nella sequenza della proteina, rilevando l’esistenza di due probabili polimorfismi genetici. Nel 1998, 11 anni dopo, Mickley et al.[22] individuarono due polimorfismi genetici, gli SNPs G2677T, situato nell’esone 21, e G2995A, situato nell’esone 24.
Ad oggi sono stati identificati più di 40 SNPs, di cui più di 30 ampiamente caratterizzati e descritti, 19 nelle sequenze esoniche e di cui solo 11 causano una sostituzione amminoacidica[13].
Il ruolo della P-gp1 sull’azione dei farmaci ha reso i polimorfismi di questo gene oggetto di un gran numero di ricerche volte a stabilire una eventuale correlazione tra l’espressione e/o l’attività della P-Glicoproteina ed una specifica configurazione allelica. Nel 2000 Hoffmeyer e colleghi osservano una alterazione dell’espressione della P-Glicoproteina 1 in relazione al polimorfismo C3435T; in particolare la quantità della proteina, rilevata mediante metodi immunoistologici, decresce in funzione del genotipo ed è maggiore negli omozigoti C/C, intermedia negli eterozigoti C/T e ridotta (fino a due volte inferiore) negli omozigoti T/T[23]. L’impatto di questa osservazione è stato notevole, tanto che le ricerche in ambito clinico degli ultimi cinque anni si sono focalizzate principalmente proprio sul polimorfismo C3435T. Altri ricercatori, a distanza di poco tempo, hanno confermato le osservazioni di Hoffmeyer.
Tuttavia, con l’aumentare delle osservazioni sono emersi anche risultati in controtendenza; infatti, in uno studio del 2002, Nakamura e colleghi[24] hanno osservato una maggiore espressione della P-gp1 in relazione al genotipo T/T, anziché al genotipo C/C[22]. Un risultato simile è stato messo in evidenza da Illmer e colleghi (2002), i quali hanno evidenziato come i livelli di mRNA per MDR1 sono minori in pazienti affetti da leucemia mieloide acuta di genotipo C/C[25].
Ancora oggi non esiste una visione comunemente accettata circa gli effetti del polimorfismo C3435T sull’espressione o sulla funzionalità della proteina.
In tempi recenti è stato proposto un ulteriore meccanismo attraverso il quale il polimorfismo silente C3435T potrebbe influenzare l’espressione delle P-Glicoproteina 1; sebbene non comporti sostituzione amminoacidica, il polimorfismo determina il cambiamento del codone, da ATC a ATT, codone sempre specifico per l’isoleucina. Questa sostituzione determina un calo nella frequenza del “codon usage” che passa dal 47% (ATC) al 35% (ATT). L’utilizzo di codoni rari potrebbe alterare il tasso di traduzione, e quindi l’espressione della proteina. Di conseguenza, la presenza dell’allele 3435T potrebbe determinare una diminuzione del tasso di traduzione per la presenza di un codone più “raro”[26].
Lo studio del polimorfismo C3435T è un settore di investigazione vasto ed importante nel campo della farmaco-resistenza e della suscettibilità genetica, volta a comprendere e a ricercare gli effetti che esso produce sull’espressione e sulla funzionalità della P-Glicoproteina 1.
1.8. SLC19A1 e RFC1
Il gene SLC19A1 è situato sul cromosoma 21q22.3, è lungo circa 28 kb. La sequenza codificante è costituita da 5 esoni (2-6), con almeno due esoni 1 alternativi, che produce un RNA messaggero lungo circa 1,7-2,8 kb.
Il gene SLC19A1, codifica per una proteina integrale di membrana di circa 65 kDa, costituita da 591 amminoacidi, appartenente alla famiglia SLC19, composta da tre proteine di trasporto con notevoli similarità strutturali e funzionali [27,28,29].
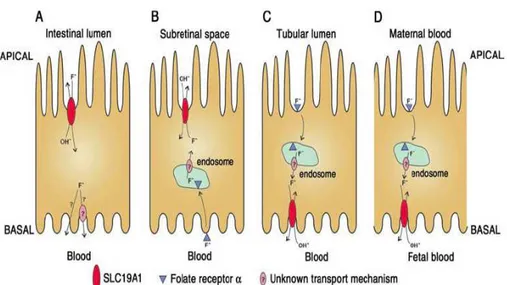
La proteina, conosciuta con i nomi di RFC1 (reduced folate carrier), RFT1 (reduced folate transporter) oppure FLOT (folate transporter) è fondamentale per l’assorbimento del folato e dei suoi derivati, insieme ad altri due componenti, il recettore del folato e la pompa H+ di tipo V.
La RFC1 è costituita da 12 domini transmembrana con almeno un sito N-glicosilato, la cui presenza è confermata sperimentalmente dall’analisi di epitopi.[30]. Comunque, sono stati proposti diversi modelli strutturali alternativi [31]. Studi di relazione struttura-funzione mostrano che il C-terminale citoplasmatico della proteina svolge un ruolo importante nella stabilità della proteina e nel meccanismo di trasporto del substrato[32,33], e che il loop intracellulare tra i domini 6 e 7 è importante per la funzione del trasportatore[34]. Studi di mutagenesi sito-specifica hanno confermato l’effettiva glicosilazione del putativo sito N-glicosilato
di SLC19A1, ed inoltre hanno confermato che il gruppo carbossilico non è implicato nella funzione di trasporto[35].
La RFC1 è una pompa di flusso, in grado di regolare il trasporto intracellulare di folato e dei suoi derivati, presentando una affinità maggiore per tetraidrofolato, N5-metiltetraidrofolato, N5-formetiltetraidrofolato e metatrexato[36]. Una caratteristica che accomuna tutti questi substrati è che sono anioni a pH fisiologico, confermando che RFC1 è attivata grazie al gradiente di pH transmembrana (pHfuori<pHdentro)[37]. Questo suggerisce che il
meccanismo di trasporto coinvolga il flusso dei substrati anionici all’interno accoppiati a H+ mediante un trasporto in sinporto, oppure coinvolga il flusso dei substrati anionici all’interno accoppiati a OH- mediante un trasporto in antiporto[38], confermando che le forme anioniche monovalenti del folato e dei suoi derivati sono i substrati preferiti da questo trasportatore.
I modelli proposti per questo trasportatore, però, sono contestati da altri studi al riguardo perché in talune tipi di cellule il meccanismo del trasportatore non sembra che sia gradiente pH-dipendente[39].
Il meccanismo di azione più attendibile proposto per la proteina è riportato nello schema in figura 1.5.
L’RNA messaggero del gene SCL19A1 è rilevabile in tutte le cellule del corpo umano. Tuttavia, l’espressione è nettamente più elevata in tessuti di assorbimento come intestino, reni e placenta rispetto ai tessuti come ad esempio il muscolo scheletrico ed il cuore.
In tutte le cellule il trasportatore è localizzato sulla membrana plasmatica[40](Figura 1.6). Inoltre la proteina RFC1 è stata anche individuata sulla membrana mitocondriale, dove può svolgere un ruolo nel traffico dei derivati del folato tra citoplasma e matrice mitocondriale[41].
1.9.
Il polimorfismo -233 G>T
Per questo gene sono stati descritti 12 polimorfismi a singolo nucleotide disseminati lungo l’intera sequenza, nelle regioni codificanti e nelle regione UTR 5’ e 3’. Ad oggi in letteratura non vi sono studi che prendano in esame il polimorfismo -233 G>T come possibile fattore di suscettibilità genetica, e non perché il gene SLC19A1 non rivesta un ruolo importante nei processi di trasporto, ma credo che sia dovuto al fatto che questo SNPs è localizzato nella sequenza UTR-3’, ed in passato queste sequenze non codificanti sono state poco considerate.
Attualmente, invece, l’interesse della comunità scientifica si è focalizzato anche sugli SNPs in queste regioni, perché si ritiene che variazioni nelle sequenze UTR possano influenzare la stabilità o l’efficienza di traduzione dell’RNA messaggero, alterandone l’espressione[38].
Considerato che il gene SLC19A1 è implicato nel trasporto del folato e dei suoi derivati, che sono substrati essenziali per la sintesi delle basi puriniche, la sua espressione alterata potrebbe avere conseguenze sul processo di replicazione del DNA e di altre componenti cellulari, favorendo l’induzione di processi degenerativi ed apoptotici.
Per questi motivi lo SNPs -233 G>T si pone come ottimo candidato per uno studio caso-controllo finalizzato a verificare il suo ruolo come fattore di suscettibilità genetica associato allo sviluppo del Mieloma Multiplo.
2. SCOPO DELLA TESI
Obiettivo della presente tesi è quello di verificare, mediante uno studio di caso-controllo, se i geni, MDR1 e SLC19A1, possano essere considerati fattori di suscettibilità per lo sviluppo del Mieloma Multiplo.
Questi geni sono entrambi polimorfici: MDR1 è implicato nel trasporto di efflusso di sostanze tossiche, e SCL19A1 implicato nel trasporto del folato, il cui metabolismo alterato può essere causa di riarrangiamenti cromosomici e induzione di processi di apoptosi. I due geni sono stati scelti per lo studio caso-controllo per la valutazione della presenza o meno di fattori di suscettibilità genetica associati allo sviluppo del Mieloma Multiplo.
Le varianti alleliche studiate per questo lavoro sono due SNPs: il polimorfismo denominato C3435T, situato nell’esone 26 del gene MDR1, ed il polimorfismo -233 G>T, situato nella regione 3’UTR del gene SLC19A1.
3) METODI DI ANALISI
3.1. Estrazione del DNA
Da un’aliquota del campione di sangue intero raccolto per ciascun paziente è stato estratto il DNA, al fine di poter effettuare l’analisi genotipica.
L’estrazione del DNA è stata fatta utilizzando il kit QIAamp DNA Blood della Qiagen, seguendo le operazioni del protocollo standard. Il processo di estrazione prevede diverse fasi di purificazione del materiale estratto, volte ad eliminare tutte le componenti cellulari e proteiche che possono inquinare il campione. In un primo passaggio, ad un volume standard di 200 µl di sangue vengono aggiunti 20 µl di proteasi K, la cui azione serve a degradare il materiale proteico, e 200 µl del tampone di lisi AL, necessario per la degradazione delle membrane biologiche. Il tutto viene agitato per pochi secondi e poi incubato in bagnetto termostatico a 58° per 10 minuti. La temperatura è importante per l’azione della proteasi K e per favorire la lisi cellulare. Al termine dei 10 minuti, la provetta contenente la miscela di reazione viene centrifugata ad 8000 rpm per 20 secondi. Si aggiungono a questo punto 200 µl di etanolo puro, che servono alla precipitazione del DNA, si agita per pochi secondi ed infine si centrifuga a 8000 rpm per 20 secondi. La soluzione così ottenuta si applica ad una colonnina QIAamp, costituita da una resina silicea che interagisce col DNA, bloccandolo nella colonna.
La colonnina viene posta all’interno di uno specifico tubo di raccolta, mentre il materiale eluito viene eliminato. Cominciano a questo punto una serie di lavaggi successivi volti a purificare il DNA estratto. Alla colonnina vengono applicati 500 µl di tampone di lavaggio AW1, ed il tutto viene sottoposto a nuova centrifugazione a 8000 rpm per 1 minuto. La colonnina viene nuovamente recuperata e posta su un nuovo tubo di raccolta da 2 ml, mentre il materiale eluito viene ancora una volta eliminato. Si applicano alla colonna 500 µl di un altro tampone di lavaggio, AW2, e si centrifuga a 14000 rpm per 3 minuti. Per il recupero del DNA, la colonnina viene posta su una eppendorf con chiusura di sicurezza da 1,5 ml e su di essa vengono applicati 200 µl di tampone AE, un tampone di eluizione necessario per il recupero del DNA. La colonna viene lasciata a temperatura ambiente per circa 5 minuti e successivamente centrifugata a 8000 rpm per 1 minuto. A questo punto, il DNA estratto si trova in soluzione nella eppendorf da 1,5 ml.
Il DNA viene tenuto alla temperatura di -20°C, mentre una piccola aliquota necessaria alle operazioni di genotipizzazione quotidiane viene conservata a 4°C per non più di un mese.
3.2. Quantizzazione e normalizzazione del DNA estratto
Una volta estratto, per poter standardizzare le analisi genotipiche successive, il DNA deve essere normalizzato, ovvero portato alla medesima concentrazione di partenza per tutti i campioni estratti. Per poter procedere alla normalizzazione del DNA, è necessaria prima la sua quantizzazione, ovvero una procedura che permette di valutarne la concentrazione.
Questo protocollo si avvale dell’uso di una sostanza fluorescente, denominata PicoGreen, che si coniuga al DNA e che emette a lunghezze d’onda comprese tra 480 nm e 520 nm ed attraverso la quantizzazione della sua luce emessa si risale alla concentrazione di DNA nel campione.
Il kit di reazione utilizzato per la quantizzazione del DNA è riportato nella tabella 3.1.
PICOGREEN 1 ml soluzione in DMSO (dimetilsulfossido)
20 X TE 25 ml Tris-HCL 200 mM, EDTA 20 mM, pH 7.5
DNA standard fago Lambda 1 ml 100 µg/ml in TE
Tabella 3.1. Kit per la quantizzazione del DNA
La quantizzazione del DNA avviene mediante la costruzione di una curva di taratura, effettuata attraverso l’utilizzo di uno standard di DNA a concentrazione nota. Lo standard, costituito solitamente da DNA del batteriofago Lambda, è fornito ad una concentrazione iniziale di 100 µg/ml, che può essere diluito 50 volte in TE per ottenere una concentrazione di 2 µg/ml di soluzione da lavoro. Una curva di calibrazione può essere disegnata, ad esempio, utilizzando 30 µl di DNA standard, mescolati a 1.47 ml di TE. In tabella 3.2 è riportato il protocollo per una curva standard di DNA.
Volume (µl) di 2 µg/ml di DNA stock Volume (µl) di TE Volume (µl) di PicoGreen diluito Concentrazio ne finale di DNA 1000 0 1000 1 µg/ml 100 900 1000 100 µg/ml 10 990 1000 10 µg/ml 1 999 1000 1 ng/ml 0 1000 1000 Bianco
Tabella 3.2. Protocollo per la preparazione di una curva standard di taratura.
I campioni ugualmente diluiti in TE e PicoGreen vengono posti su piastre a 96 pozzetti insieme al DNA standard e al controllo negativo (soluzione priva di DNA). La piastra viene inserita nello spettrofluorimetro, uno strumento in grado di rilevare e quantificare l’emissione del PicoGreen, che, con una elaborazione computerizzata, calcola la concentrazione di ogni singolo campione. Infine, i campioni vengono opportunamente diluiti al fine di portarli tutti alla stessa concentrazione finale.
3.3. PCR ed Elettroforesi di controllo
Prima di procedere alla genotipizzazione, è sempre buona norma sincerarsi che il DNA estratto sia amplificabile (integro e purificato): il DNA viene testato mediante PCR ed il risultato è controllato con elettroforesi su gel di agarosio.
La PCR (Polimerase Chain Reaction) è una tecnica che permette di ottenere copie multiple di un certo segmento di DNA. In questa reazione sono necessari i primers, o inneschi (Fw Rw), che delimitano la regione da amplificare, i nucleotidi necessari per l’estensione e l’enzima DNA polimerasi per la sintesi dei nuovi filamenti di DNA. Questi reagenti vengono addizionati ad una soluzione contenente anche MgCl2 ed un tampone ottimale per
l’attività dell’enzima ed, ovviamente, il DNA genomico da amplificare. Il volume finale della miscela di amplificazione è di 25 µl. Nella tabella 3.3 è riportata la composizione della miscela di amplificazione. REAGENTI VOLUME PCR Buffer 10X 2,5 µl MgCl2 [50 mM] 0,5 µl dNTPs [1 mM] 0,2 µl Primer Fw [100 pmol/µl] 1 µl Primer Rw [100 pmol/µl] 1 µl Taq Polimerasi [5 U/µl] 0,2 µl DNA campione [10 ng/µl] 5 µl
Acqua 14,5 µl
Totale 25 µl
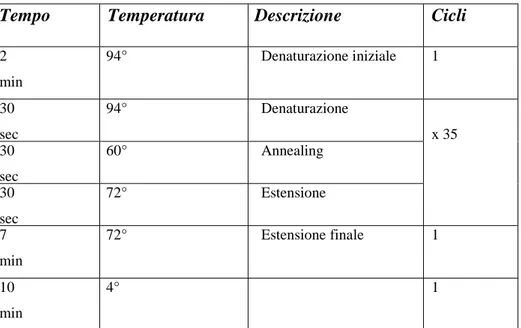
La reazione di PCR viene condotta in termociclizzatori, in grado di variare ciclicamente, secondo un programma prestabilito, la temperatura della reazione. Generalmente, la reazione di PCR prevede una fase di denaturazione, necessaria alla separazione dei due filamenti, una fase di appaiamento, per permettere ai primers di appaiarsi in maniera complementare al DNA stampo, ed una fase di estensione, in cui la DNA polimerasi amplifica la sequenza di interesse. Ciascun ciclo di denaturazione-appaiamento-estensione viene ripetuto più volte, al fine di ottenere un elevato numero di copie. Il protocollo di reazione, effettuato in termociclizzatori Perkin Elmer, viene riportato di seguito (Tabella 3.4).
Il risultato dell’amplificazione viene poi controllato mediante elettroforesi su gel di agarosio al 2%. Il gel viene preparato utilizzando una soluzione di 200 ml di tampone TBE, diluito 1X, al quale vengono aggiunti 4 g di agarosio in polvere. La beuta viene agitata manualmente per favorire l’iniziale dissoluzione dell’agarosio nel tampone e successivamente riscaldata fino a quando la soluzione non assume un aspetto limpido e trasparente e la beuta viene posta a raffreddare.
Tempo Temperatura Descrizione Cicli
2 min 94° Denaturazione iniziale 1 30 sec 94° Denaturazione 30 sec 60° Annealing 30 sec 72° Estensione x 35 7 min 72° Estensione finale 1 10 min 4° 1
Da questo momento in poi le operazioni vengono eseguite sotto cappa chimica, in quanto si utilizzano sostanze tossiche come il bromuro d’etidio e gli stessi vapori del gel. Il gel viene fatto raffreddare per circa 15 minuti e solo quando è sufficientemente freddo viene aggiunto il bromuro d’etidio ad una concentrazione di 0,5 µg/ml. Il bromuro d’etidio è un intercalante del DNA, che se illuminato con luce ultravioletta, permette la visualizzazione delle bande sul gel a corsa avvenuta. Il gel viene quindi versato nell’apposita vaschetta, precedentemente preparata con un pettine alle estremità, che verrà tolto quando il gel sarà solidificato e lascerà liberi i pozzetti di caricamento del campione. La vaschetta viene quindi coperta per evitare il decadimento alla luce del bromuro d’etidio ed il gel viene lasciato circa un’ora a solidificare.
In seguito, la vaschetta viene posta nella cella di corsa, riempita con tampone (TBE) e collegata ad un alimentatore a corrente continua, in grado di applicare una differenza di potenziale alla vasca. Si procede quindi al caricamento dei campioni amplificati per poi eseguire l’elettroforesi. Solitamente, vengono caricati 10 µl di prodotto di PCR, ai quali viene addizionato 1 µl di loading buffer necessario alla visualizzazione della corsa. Il loading buffer è costituito da un colorante, spesso il blu di bromofenolo, glicerolo e saccarosio, necessari a favorire la discesa del DNA nel pozzetto di caricamento. Comunemente, il primo pozzetto viene caricato con un ladder, una soluzione di frammenti di DNA a peso molecolare noto, utile per stabilire le dimensioni dei frammenti di DNA visualizzati sul gel. Al termine della procedura di caricamento, che deve comunque avvenire in tempi brevi per evitare che il _DNA diffonda attraverso il gel, si applica la corrente alla camera di corsa. I campioni vengono fatti
correre a 110 volt per circa 30 minuti. La presenza del colorante permette di visualizzare il fronte più avanzato della corsa.
Una volta spento l’alimentatore, la vaschetta contenente il gel viene rimossa dalla cella di corsa e portata al transilluminatore, uno strumento a luce UV che permette la visualizzazione della banda sul gel. Un esempio di gel di controllo per una PCR è riportato in figura 3.1.
3.4. La genotipizzazione dei polimorfismi C3435T e -233
G>T: il metodo TaqMan in Real Time PCR
La genotipizzazione dei locus C3435T di MDR1 e -233G>T di SLC19A1 è stata condotta attraverso l’utilizzo della Real Time PCR con metodo TaqMan, un sistema di rilevazione in tempo reale che consente di determinare il genotipo in base all’emissione di fluorofori legati a sonde allele-specifiche. Il sistema di amplificazione e rilevazione utilizzato è il sistema iCiclerTMiQ della Biorad.
Attraverso l’uso di sonde allele-specifiche per gli alleli, ciascuna delle quali legata ad un differente fluoroforo, è possibile monitorare la reazione di PCR in real time, grazie all’ausilio di un computer. Il metodo TaqMan in particolare, è stato ideato per la discriminazione allelica di polimorfismi a singolo nucleotide. Nella tabella 3.5 sono riportate le sequenze oligonuotidiche dei primers utilizzati per la reazione di PCR.
PRIMERS SEQUENZE
MDR1 (forward) 5’-CTGTTTGACTGCAGCATTGCT-3’ MDR1 (reverse) 5’-ATGTATGTTGGCCTCCTTTGCT-3’ SLC19A1 (forward) 5'-TCCTTCTGAAGTGTGTCCATCCT-3' SLC19A1 (reverse) 5'-GTGACCGGGACCAGTCC-3'
Tabella 3.5. Oligonucleotidi per l’amplificazione dei geni
Le sonde oligonuotidiche utilizzate per la genotipizzazione del gene MDR1 sono marcate all’estremità 5’ con due fluorofori differenti, ed in particolare la sonda specifica per l’allele wild type (3435C) è marcata con il fluoroforo VIC, mentre la sonda specifica per la sequenza mutata (3435T) è marcata con il
fluoroforo 6-FAM. Il fluoroforo VIC emette alla lunghezza d’onda di 530 nm, mentre il fluoroforo 6-FAM emette a 490 nm. In tabella 3.6, sono riportate le sonde allele-specifiche utilizzate. Ciascuna sonda, poi, porta legato all’estremità 3’ un silenziatore (Quencher), che assorbe la luce emessa dal fluoroforo quando si trova nelle sue immediate vicinanze.
FLUOROFORI SONDE TaqMan ALLELE
VIC 5’-VIC-TGAGGCGGGTGGATCACT-Q-3’ 3435C 6-FAM 5’-6-FAM-AGGCTGAGGCAGGTGGAT-Q-3’ 3435T
VIC 5'-VIC-TCCCACACTGGTGCCA-Q-3’ 233G
6-FAM 5'-6-FAM-CTCCCACACTTGTGCCA-Q-3’ 233T
Tabella 3.6. Le sonde allele-specifico
Dal punto di vista teorico, il principio sul quale si basa il metodo TaqMan è piuttosto semplice. I primers forward e reverse sono disegnati in maniera tale da delimitare una sequenza di DNA all’interno della quale è compreso il sito di mutazione. Durante la fase di annealing, oltre il primers, anche le sonde allele-specifiche si appaiono alla loro sequenza complementare. Quando una sonda per un allele si appaia alla sequenza specifica per l’altro allele, si crea un mismatch. La taq DNA polimerasi utilizzata nella fase di estensione è dotata, oltre che dell’attività polimerasica, anche di un’attività esonucleasica 5’-3’, che permette all’enzima di degradare eventuali frammenti di DNA che incontra legati lungo il filamento stampo. Grazie all’apposito disegno dei primers, durante la fase di estensione la Taq DNA polimerasi incontra le sonde allele-specifiche che si sono ibridate alla loro sequenza complementare sul DNA stampo. Quando l’appaiamento tra le sonde e DNA stampo è perfetto, ovvero privo di mismacth, l’attività esonucleasica della Taq DNA polimerasi provvederà alla
degradazione della sonda, liberando così il fluoroforo all’estremità 5’ che, allontanandosi dal silenziatore, potrà emettere alla sua lunghezza d’onda specifica. Al contrario, quando l’appaiamento tra DNA stampo e sonda presenta mismacth, la DNA polimerasi non degrada la sonda, bensì provvede semplicemente a scalzarla intatta, senza liberare il fluoroforo dall’effetto silenziatore del Quencher. In questo modo la presenza di emissione è indice di un perfetto appaiamento della sonda, e la discriminazione tra i due fluorofori utilizzati permette di stabilire quale é la configurazione genotipica al locus considerato. E’ anche importante sottolineare che la fluorescenza registrata è proporzionale alla quantità di DNA amplificato in ogni momento della reazione, poiché viene liberata una molecola di fluoroforo per ogni copia di DNA duplicata.
Figura 3.2. Amplificazione del DNA col metodo TaqMan Real-Time
La figura 3.2 riassume gli eventi che si verificano in un amplificazione con metodo TaqMan. Il protocollo sperimentale utilizzato per la reazione di amplificazione in Real Time, come
quello di una comune PCR, prevede più cicli di denaturazione, annealing ed estensione (Tabella 3.7).
Tempo Temp Descrizione Cicli
10 min 95 °C Denaturazione iniziale 1 15 sec 95 °C Denaturazione
1 min 60 °C Annealing+Estenzione
x 50
10 min 4 °C 1
Tabella 3.7. Protocollo sperimentale di Real Time PCR
I valori di fluorescenza emessi vengono rilevati in tempo reale da un opportuno dispositivo fluorimetrico collegato ad un computer che provvede all’elaborazione dei dati. Nelle prime fasi della reazione di PCR, la fluorescenza si mantiene a valori piuttosto bassi.
Il primo ciclo di amplificazione in cui viene rilevato un aumento evidente della fluorescenza è definito come “ciclo soglia”. Da questo punto in poi la reazione entra nella fase detta di amplificazione esponenziale, ovvero quella fase in cui tutti i campioni, indipendentemente dalla qualità di DNA iniziale, sono amplificati con la medesima efficienza. Ciascun campione ha un proprio ciclo soglia, anche se l’utilizzo di DNA precedentemente normalizzato fa sì che, più o meno, il ciclo soglia sia lo stesso per tutti i campioni. Ciascun ciclo soglia corrisponde a punto in cui le curve di amplificazione intersecano la linea di base della fluorescenza. Nella figura 3.3 sono riportate le curve di amplificazione rispettivamente per i fluorofori VIC e FAM relative alla genotipizzazione di alcuni campioni.
La qualità d campioni analizzabili contemporaneamente dipende dallo strumento: il termociclizzatore utilizzato lavora con piastre a 96 pozzetti, per cui possono essere analizzati circa 90
campioni contemporaneamente. I restanti 6 pozzetti sono solitamente riempiti con i controlli negativi (senza DNA) e positivi (DNA a genotipo noto).
Figura 3.3. Spettro di emissione per VIC-530 (a sinistra) e per Fam-530 (a
destra) in Real Time
L’allestimento della reazione di PCR prevede la preparazione di due miscele distinte. Si utilizza una miscela 2X, contenente buffer, MgCl2, nucleotidi e Taq polimerasi, ed una miscela 20X contenente le sonde ed i primers necessari. Il volume dei pozzetti contiene 15 µl di soluzione, costituiti da 4 µl di DNA da genotipizzare e 11 µl di miscela finale.
In tabella 3.8 è riportato un esempio di miscela di reazione per un unico campione. L’identificazione dei campioni analizzati è fornita ad un software, che attribuisce il genotipo a ciascun campione in base alla fluorescenza rilevata, riportata in RFU (Relative Fluorescence Unit).
Nella figura 3.5 è visibile l’output dei genotipi per alcuni campioni analizzati.
In rosso (allele 1) sono riportati gli omozigoti wild-type, in verde scuro gli eterozigoti ed, infine, in verde chiaro (allele 2) gli omozigoti mutati. Nella figura poi compaiono anche alcuni campioni blu, che corrispondono a genotipi incerti, che
necessitano quindi di un ulteriore controllo per essere attribuiti in modo univoco. In celeste sono riportati i controlli negativi. I dati ottenuti in seguito alla genotipizzazione di tutti i campioni a disposizione sono stati poi sottoposti ad un severo controllo per garantire l’univocità del genotipo attribuito.
Miscela di reazione
BufferdNTP
Taq Mix 2X (7,5 µl)
Vic e Fam (sonde) Primers
H2O
Mix 20X (3,5 µl)
DNA Campione 4 µl
Totale 15 µl
Tabella 3.8. Miscela di reazione per Real Time PCR
3.5. Analisi statistica
L’equilibrio di Hardy-Weinberg è stato verificato per ciascun polimorfismo tramite il test χ2 nei casi e nei controlli. Dei due geni esaminati sono state calcolate le frequenze genotipiche. Le frequenze alleliche calcolate sono poi state confrontate nelle due popolazioni in esame. Le associazioni tra le varianti alleliche e la patologia sono state stimate tramite un’analisi di regressione logistica multivariata, utilizzando i programmi statistici STATA (versione 8.0) e Statgraphics; calcolando gli odds ratios (ORs) e gli intervalli di confidenza (CIs) ad essi associati.
Distribuzione dell'età nei Pazienti Età 31 41 51 61 71 81 91 0 10 20 30 40 50 60 ƒ 0% 10% 20% 30% 40% 50% 60% M as chi Fe m m ine
Distribuzione dell'età nei Controlli
Età ƒ 29 39 49 59 69 79 0 20 40 60 80 0% 10% 20% 30% 40% 50% 60% 70% Maschi Femmine
4. RISULTATI
4.1. Le caratteristiche del campione
Il campione analizzato è composto da 190 pazienti affetti da Mieloma Multiplo, di cui 104 maschi e 86 femmine, con un’età media alla diagnosi di circa 62 anni (32-86 range) (figura 4.1). Il gruppo di controlli è invece costituito da 221 soggetti sani alla data di campionamento, di cui 137 maschi e 84 femmine, con un età media alla data di campionamento di circa 47 anni (figura 4.2).
Figura 4.1. Distribuzione dell’età e tra i sessi dei pazienti
4.2. I polimorfismi C3435T di MDR1 e -233G>T di SLC19A1
Il Polimorfismo di MDR1 C3435T è uno SNP silente i cui due alleli C e T possono combinarsi nei genotipi C/C, C/T e T/T. L’allele più frequente è comunque il C, che ha una frequenza riportata in letteratura del 58,3% contro il 41,7% dell’allele T. La tabella 4.1 riassume la distribuzione dei genotipi osservati nei casi e nei controlli:
Frequenze Genotipiche Frequenze alleliche
Genotipi C/C C/T T/T n C T Casi 54 28,42% 86 45,26% 50 26,32% 190 0,5105 0,4894 Controlli 57 25,79% 115 52,03% 49 22,17% 221 0,5180 0,4819
Tabella 4.1. Frequenze alleliche del polimorfismo C3435T e distribuzione dei tre genotipi nelle due popolazioni.
La distribuzione dei genotipi sia nei casi che nei controlli è in equilibrio di Hardy-Weinberg (pvalue rispettivamente di 0,429 e 0,822). Una prima valutazione della distribuzione delle classi genotipiche nei casi rispetto ai controlli si può avere dal grafico in figura 4.3:
Frequenza
0 20 40 60 80 100 120
Distribuzione Genotipi SNP C3435T tra Casi e Controlli
CONTROLLI CASI MDR1 C3435T 1 2 3
Le distribuzioni di frequenza dei genotipi tra casi e controlli sono del tutto simili, e non lasciano intravedere alcuna differenza sostanziale. L’analisi mediante il test del χ2 conferma che non c’è alcuna differenza statisticamente significativa nella distribuzione dei genotipi all’interno delle due classi considerate (χ2=1,948; Df=2; pvalue=0,3775).
Il polimorfismo -233G>T del gene SLC19A1 è invece un polimorfismo situato nella regione 3’UTR del gene, i cui possibili genotipi sono G/G, G/T e T/T. L’allele più frequente, come riportato in letteratura, è l’allele T, con una frequenza del 51,5% contro il 48,5% dell’allele G. La tabella 4.2 mostra ancora una volta le frequenze dei possibili genotipi tra casi e controlli:
Frequenze Genotipiche Frequenze alleliche
Genotipi G/G G/T T/T n G T Casi 50 26,31% 81 42,63% 59 31,05% 190 0,5105 0,4894 Controlli 47 21,27% 99 44,78% 75 33,93% 221 0,5180 0,4819
Tabella 4.2. Frequenze alleliche del polimorfismo -233G>T e distribuzione dei tre genotipi nelle due popolazioni.
Anche in questo caso, sia nei casi che nei controlli è rispettato l’equilibrio di Hardy-Weinberg (pvalue rispettivamente di 0,134 e 0,413). La figura 4.4 mostra il grafico delle distribuzioni genotipiche a confronto tra casi e controlli:
0 20 40 60 80 100 Frequenza
Distribuzione Genotipi SNP -233G>T tra Casi e Controlli
CONTROLLI CASI SLC19A1 G/G G/T T/T
Figura 4.4. Distribuzione genotipi di SLC19A1
Il test del χ2, applicato al confronto delle distribuzioni dei genotipi nelle due popolazioni dei casi e dei controlli, anche in questo caso, non mostra alcuna differenza statisticamente significativa (χ2= 1,473, Df=2; pvalue=0,4787).
E’ stata effettuata anche un’analisi di regressione logistica aggiustata per età e sesso, al fine di verificare la presenza di un rischio relativo dei genotipi con almeno un allele mutato (C/T e T/T per C3435T di MDR1 e G/T e G/G per -233G>T di SLC19A1) rispetto al wild type di sviluppare il Mieloma Multiplo. I risultati dell’analisi di regressione logistica sono mostrati in tabella 4.3:
RISCHIO MIELOMA MULTIPLO CASI/CONTROLLI OR C.I.95% PVALUE
Età 1,2098 1,168 – 1,254 0,000 Sesso (M)F (104/86) 137/84 3,6702 1,990 – 6,768 0,000 MDR1 C3435T (C/C) C/T (54/86) 57/115 0,7389 0,379 – 1,440 0,374 MDR1 C3435T (C/C) T/T (54/50) 57/49 1,1880 0,567 – 2,486 0,648 SLC19A1 -233G>T (T/T) G/T (59/81) 75/99 1,0066 0,533 – 1,897 0,984 SLC19A1 -233G>T (T/T) G/G (59/50) 75/47 1,1262 0,537 – 2,359 0,753
Tabella 4.3: Analisi del rischio relativo di sviluppare il MM in funzione dei genotipi ai loci MDR1
L’analisi di regressione logistica mostra come nessuno dei genotipi considerati per i due polimorfismi sia fattore di rischio (o protettivo) per l’insorgenza del Mieloma Multiplo.
Si può notare che vengono indicati come fattori di rischio l’età e il sesso. Mentre per il sesso è documentata in letteratura una maggiore incidenza della malattia nei maschi, che potrebbe giustificare l’indice di rischio circa 4 volte maggiore (OR=3,6702) rispetto alle femmine, il rischio associato all’età si può imputare ad un bias di selezione dei controlli, per cui l’età media di questi ultimi è molto inferiore rispetto a quella dei casi.
Se si esclude l’età come fattore di aggiustamento dell’analisi logistica i risultati rimangono sostanzialmente invariati, ed anche il rischio associato al sesso si riduce notevolmente al limite della significatività come mostrato in tabella 4.4:
RISCHIO MIELOMA MULTIPLO CASI/CONTROLLI OR C.I.95% PVALUE Sesso (M)F (104/86) 137/84 1,3753 0,924 – 2,047 0,116 MDR1 C3435T (C/C) C/T (54/86) 57/115 0,7657 0,478 – 1,224 0,265 MDR1 C3435T (C/C) T/T (54/50) 57/49 1,0581 0,613 – 1,824 0,839 SLC19A1 -233G>T (T/T) G/T (59/81) 75/99 1,0476 0,666 – 1,647 0,840 SLC19A1 -233G>T (T/T) G/G (59/50) 75/47 1,3365 0,788 – 2,264 0,281
Tabella 4.4: Analisi del rischio relativo di sviluppare il MM in funzione dei genotipi ai loci MDR1
C3435T e SLC19A1 -233G>T mediante regressione logistica aggiustata il sesso.
Eliminando un fattore confondente come è in questo caso l’età, l’analisi del rischio relativo in funzione delle variabili considerate evidenzia che nessuno dei fattori presi in considerazione è indice di rischio o protezione nell’insorgenza del Mieloma Multiplo.
5. Discussione
In questo lavoro è stata analizzata l’eventuale associazione tra i polimorfismi genetici di MDR1 e SLC19A1 e la suscettibilità genetica al Mieloma Multiplo.
Lo studio caso-controllo tra i polimorfismi C3435T e -233G>T con il Mieloma Multiplo, condotto in 190 pazienti affetti dalla malattia e 221 controlli sani, non ha evidenziato differenze significative nella distribuzione dei due alleli (rispettivamente C/T e G/T) e dei tre genotipi (rispettivamente C/C,C/T,T/T e G/G,G/T,T/T) nella popolazione affetta da Mieloma Multiplo e nella popolazione dei controlli sani.
Questi risultati confermano l’esistenza dei due polimorfismi: il polimorfismo C3435T nell’esone 26 del gene MDR1 e il polimorfismo -233G>T nella regione UTR-3’ del gene SLC19A1; in un campione indipendente di soggetti caucasici, ma non mostrano correlazione tra la distribuzione allelica dei geni, MDR1 e SLC19A1, e il fenotipo affetto da Mieloma Multiplo.
Per quanto riguardo l’analisi dei risultati del gene MDR1 essi trovano conferma da altri studi caso-controllo come ad esempio lo studio eseguito da Urayama et al. sulla ALL (Acute Lymphoblastic Leukaemia) dove non si riscontra nessuna significativa differenza per polimorfismo C3435T di MDR1 all’interno delle frequenze alleliche dei casi e dei controlli[42]. In contrasto a questo altri studi, come quello condotto da Jamroziak e colleghi sempre sulla ALL confermano l’impatto di questo polimorfismo C3435T del gene MDR1 come fattore di suscettibilità nello sviluppo della malattia [43].
Come anche altri studi condotti su diverse malattie, come il cancro colon-rettale, il cancro al seno o la B-CLL (B-cell Chronic Lymphocytic Leukemia), confermano con esito positivo questo polimorfismo C3435T a fattore di suscettibilità[43,44,45].
Infatti nel lavoro di Siegsmond et al., si evidenzia come gli SNPs in MDR1, ed in particolare questo polimorfismo, possano alterare il ruolo fisiologico della P-gp1, e come influenzino il rischio di sviluppo di malattia[46].
Inoltre si ritiene che la P-Glicoproteina-1 giochi un ruolo importante su alcune citochine, come l’interleuchina-2 e -4 (2, IL-4), l’interferone-γ (INF-γ) e il fattore di necrosi tumorale (TNF-α). La variante polimorfica C3435T di MDR1 è stata associata al rischio delle precedenti citochine[47]. Ancora, è stato suggerito che l’alterazione dell’espressione di MDR1 è stata associata con una riduzione dell’attività delle Natural Killer (NK) e dei linfociti T citotossici (CD8+), coinvolti nella regolazione della risposta immunitaria e nell’uccisione delle cellule tumorali[48].
Detto questo però altri lavori confermano i risultati del nostro lavoro, affermando che il polimorfismo C3435T non sia significativamente associato a fattori di suscettibilità genetica nello sviluppo di patologie.
Uno studio condotto da Bae et al (2006) mostra come non ci sia alcuna differenza nella distribuzione dei genotipi al sito C3435T di MDR1 e in un campione di 111 pazienti affetti da cancro colon-rettale (CRC) e 93 controlli coreani[49]. Ancora, uno studio su 146 pazienti affetti sempre da CRC e 160 controlli sani caucasici (bulgari) eseguito da Petrova et al sottolinea l’assenza di associazione tra il rischio di sviluppare la malattia e il genotipo al sito dello SNP C3435T[50].
Questi lavori dimostrano che il polimorfismo C3435T di MDR1 può effettivamente giocare un ruolo nella suscettibilità alle patologie tumorali, anche se è palese l’esistenza di evidenze in contrasto tra loro.
Maggiore chiarezza si potrà avere soltanto quando verrà chiarito il ruolo funzionale dello SNP C3435T. Questa mutazione è infatti una mutazione silente (Ile1145Ile), e per molto tempo è prevalso lo scetticismo circa la sua reale influenza sull’attività e/o sulla funzione della P-gp1. Studi recenti hanno invece avanzato almeno due possibili spiegazioni circa l’effetto di questa variazione sulla funzionalità della proteina. In un articolo di Wang e Sadee del 2006[51] viene mostrato come la variazione silente 3435T alteri drasticamente la struttura dell’mRNA di ABCB1 rispetto alla variante wild type (3435C), tanto da indurre gli autori a supporre una differenza nella stabilità della molecola in conseguenza della quale si verifica una diminuzione nell’espressione della P-gp1[52,53](figura 5.1).
Figura 5.1. Stabilità mRNA ABCB1
Tuttavia, ancor più convincente sembra essere la spiegazione proposta da Kimchi-Sarfaty et al, secondo i quali la diminuzione dell’espressione della P-gp1 sarebbe dovuta ad un effetto di “rare codon usage”, ovvero al fatto che l’allele 3435T specifichi un codone per l’Ile più raro, e quindi più difficilmente reclutabile durante la fase di sintesi della proteina. Questo determinerebbe un “ritardo” per cui