1. LA REGOLAZIONE DELL’ESPRESSIONE GENICA
Il GENOMA è il magazzino dell’informazione biologica in cui sono codificate tutte le informazioni relative alla caratterizzazione morfologica e funzionale di un organismo. L’unità di informazione è rappresentato dai geni che codificano proteine, rRNA, tRNA o sRNA, ma è solo l’interazione tra di essi e i rigidi meccanismi che ne regolano l’espressione fa si che sebbene in un organismo tutte le cellule contengano lo stesso corredo genetico, non tutte esprimo gli stessi geni ed allo stesso momento. I meccanismi che modulano l’espressione genica in senso spazio-temporale possono essere genetici e/o epigenetici e si esplicano a molteplici livelli (Fig. 1.1). Con ESPRESSIONE GENICA, quindi, si intende quella serie di eventi che dall'attivazione della trascrizione di un gene, conducono ad es. alla produzione della proteina corrispondente. La regolazione di questi processi è molto fine e la sua complessità aumenta salendo la scala evolutiva. Studiare la regolazione dell'espressione di un gene significa accertare in quali tessuti viene espresso, in quali condizioni e qual è l'effetto di tale espressione.
Fig. 1.1: Differenti livelli della regolazione genica.
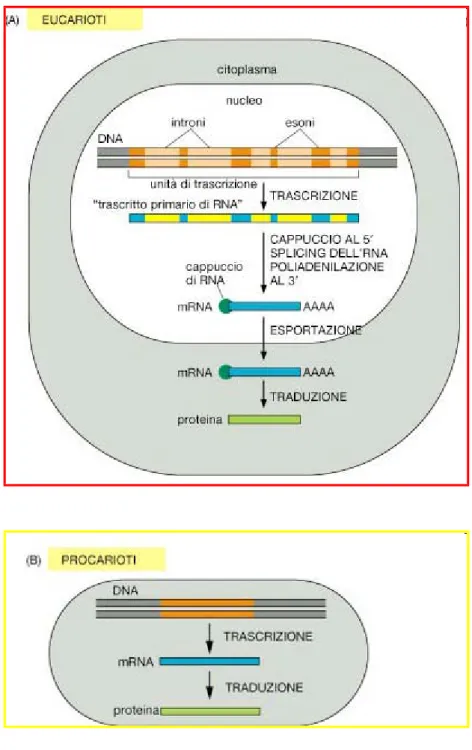
Il primo prodotto dell’espressione genica è il trascrittoma, ovvero l’insieme di copie di RNA; il processo che porta alla sua formazione è la trascrizione. In particolare le molecole di mRNA che codificano le
proteine, dirigono, poi, la sintesi del prodotto finale dell’espressione genica: il proteoma, cioè l’insieme delle proteine cellulari. Il processo che porta alla formazione del proteoma è la traduzione. L’RNA messaggero (mRNA), che raramente supera il 4% dell’RNA totale, ha una vita breve in quanto viene degradato poco dopo la sintesi. Gli mRNA batterici hanno un’emivita di non più di pochi minuti; gli mRNA eucariotici vengono degradati poche ore dopo la sintesi(Fig. 1.2). Questo continuo ricambio sta ad indicare che la composizione del trascrittoma non è fissa, e può essere rinnovata rapidamente cambiando la velocità di sintesi di ogni mRNA. La restante percentuale di RNA totale è costituita dal tipo non codificante, quale: RNA ribosomiale (rRNA), parte integrante dei ribosomi e RNA transfer (tRNA) coinvolto nella sintesi proteica e responsabile del trasferimento degli amminoacidi ai ribosomi. Gli eucariotici contengono altri RNA non codificanti, classificati in base alla localizzazione cellulare: i piccoli RNA nucleari (snRNA), i piccoli RNA nucleolari (snoRNA) e i piccoli RNA citoplasmatici (scRNA).
Per specificare come una sequenza di mRNA è traducibile in un polipeptide è utilizzato il CODICE GENETICO a triplette; nel quale ciascuna parola, o meglio codone, comprende tre nucleotidi per poter specificare i 20 amminoacidi che si trovano nelle proteine. Il codice genetico comprende anche quattro codoni di punteggiatura:
• un codone di inizio, solitamente 5’-AUG-3’
• tre codoni di terminazione 5’-UAG-3’, 5’-UAA-3’, 5’-UGA-3’.
Il codice genetico può essere definito come il linguaggio molecolare basato su l’ordine delle quattro basi azotate che si susseguono nella molecola di DNA, il codice ha una valenza universale, motivo per cui è stato indicato come la chiave per conoscere l’intima struttura degli organismi.
Fig. 1.2: Dal gene alla proteina: principali differenze della regolazione genica tra eucarioti e procarioti.
1.1 Promotore e sequenze regolatrici
La trascrizione è il processo che media il trasferimento dell’informazione genica, contenuta nel DNA, in una molecola complementare di RNA che sarà poi tradotto in un prodotto proteico, richiede l’intervento di enzimi. L’enzima responsabile della trascrizione del DNA in RNA è l’RNA polimerasi DNA-dipendente. L’RNA polimerasi è il componente centrale
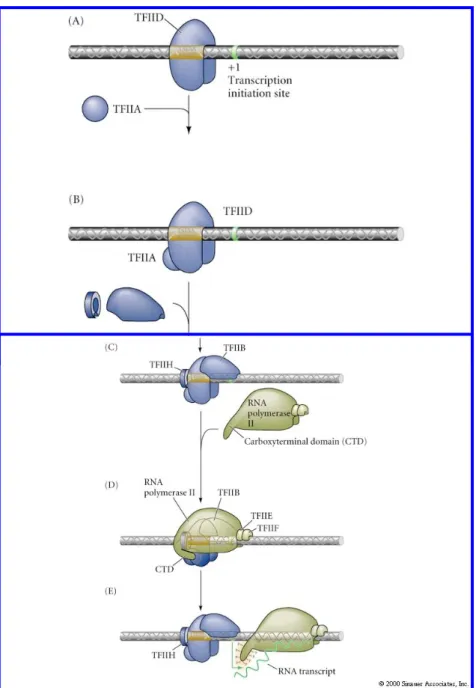
del complesso di inizio della trascrizione. Ogni volta che un gene viene trascritto, un nuovo complesso deve essere assemblato a monte del gene. I complessi di inizio della trascrizione si formano in posizioni ben precise del genoma e non in punti casuali, in quanto i loro siti di riconoscimento sono identificati da specifiche sequenze nucleotidiche, i PROMOTORI, presenti a monte del gene. Nei batteri i promotori vengono riconosciuti direttamente dall’RNA polimerasi, negli eucarioti, invece, è necessaria la presenza di una proteina intermedia che faccia da piattaforma tra il DNA e l’enzima. Il contatto viene stabilito per primo dal fattore generale di trascrizione TFIID (GTF) (Fig. 1.3) che ne riconosce il “core”, questo dà l’avvio alla costituzione del complesso nucleo-proteico necessario per l’avvio della trascrizione. Negli organismi eucarioti la regolazione del momento in cui la trascrizione deve avvenire è esercitata principalmente attraverso l’azione di fattori di trascrizione che possono rappresentare elementi attivatori e repressori, ma anche con meccanismi diversi che influenzano l’accesso degli stessi fattori di trascrizione ai geni. Come già detto, i fattori di trascrizione sono in grado di incrementare il tasso di trascrizione di un gene grazie al legame con particolari sequenze del DNA, dette siti di regolazione, che possono trovarsi anche molto distanti dal sito di inizio della trascrizione (TSS) sia a monte che a valle del gene.
Possiamo distinguere 3 classi fondamentali di fattori di trascrizione:
- Fattori di trascrizione basali, coinvolti nel processo di preiniziazione. Sono ubiquitari e interagiscono in modo aspecifico con la regione del promotore prossimale di tutti i geni di classe II (Tab. 1.1).
- Fattori di trascrizione che legano specifici siti a monte o a valle del sito di inizio della trascrizione e che sono in grado di stimolare o reprimere la trascrizione stessa.
- Fattori di trascrizione inducibili, simili ai precedenti, ma che sono a loro volta attivati o inibiti in particolari fasi o in seguito a particolari stimoli (es. stress).
Fig. 1.3: Assemblaggio del complesso di inizio della trascrizione. Il primo passo è il riconoscimento della regione TATA e della sequenza Inr da parte di TFIID. Gli altri componenti del complesso si assemblano secondo l’ordine: TFIIA, TFIIB, TFIIF/RNA polimerasi, TFIIE, TFIIH.
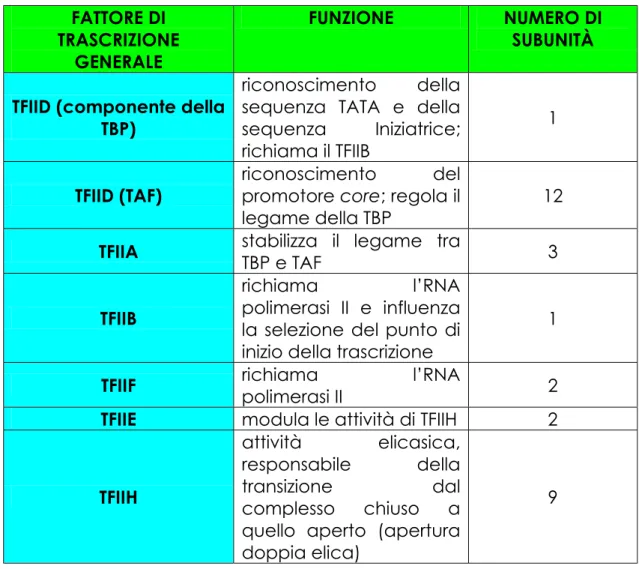
Tab. 1.1: Principali fattori di trascrizione e relative funzioni. FATTORE DI TRASCRIZIONE GENERALE FUNZIONE NUMERO DI SUBUNITÀ
TFIID (componente della TBP)
riconoscimento della sequenza TATA e della sequenza Iniziatrice; richiama il TFIIB
1
TFIID (TAF) riconoscimento del promotore core; regola il
legame della TBP 12
TFIIA stabilizza il legame tra TBP e TAF 3
TFIIB
richiama l’RNA polimerasi II e influenza
la selezione del punto di inizio della trascrizione
1
TFIIF richiama l’RNA polimerasi II 2
TFIIE modula le attività di TFIIH 2
TFIIH
attività elicasica, responsabile della transizione dal complesso chiuso a
quello aperto (apertura doppia elica)
9
1.1.a IL PROMOTORE
Il termine promotore, negli eucariotici, viene usato per descrivere tutte le sequenze importanti per l’inizio e la regolazione della trascrizione di un gene. Queste sequenze comprendono:
• Il promotore prossimale o basale situato nella regione a cavallo del sito di inizio della trascrizione, entro il quale avviene il legame con l’RNA polimerasi e i fattori di trascrizione basali. Il core del promotore prossimale è la minima regione del promotore che è capace di iniziare la trascrizione basale, contiene il sito d’inizio della trascrizione (TSS) e si estende tipicamente nel tratto da -60 a +40 relativamente al TSS.
• Il promotore distale, costituito da uno o più elementi, che come indica il nome sono situati ancora più a monte del sito di inizio, solitamente intorno a 1Kb, che controlla l’attività del promotore stesso attraverso l’interazione con fattori di trascrizione specifici
(Shahmuradov et al., 2003, 2005; Molina et al., 2005; Yamamoto et al., 2007)
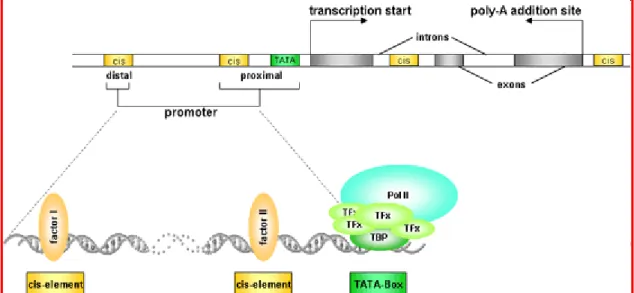
Negli organismi animali sono state descritte altre sequenze, in aggiunta a quelle dei promotore prossimale e distale, che contengono elementi ENHANCER e/o SILENCING (Barton et al., 1997; Rombauts et al., 2003). Questi elementi possono essere ritrovati sia nelle regioni a monte del TSS che negli introni, ma anche a valle dei geni che essi regolano (Fig. 1.4)
(Larkin et al., 1993; Wasserman et al., 2000).
Fig. 1.4: Elementi regolatori della trascrizione. A seconda della localizzazione rispetto al TSS, nel promotore troviamo elementi distali e prossimali. A livello del sito di inizio della trascrizione, troviamo il TATA-box sito di legame del complesso dei fattori di trascrizione e dell’RNA polimerasi. In tutta la sequenza sono interposti elementi cis riconosciuti da fattori di trascrizione che attivano o reprimono l’attività genica. All’estremità 3’ del gene è presente il sito di poli-Adenilazione.
- Caratteristiche conservate del promotore prossimale
Il promotore è caratterizzato da un’architettura costituita da “motivi” conservati sia nella sequenza sia nella localizzazione (Molina et al., 2005,
Yamamoto et al., 2007). La conservazione è sia in senso intra-specifico,
tra promotori di geni di uno stesso organismo, che inter-specifico, tra mono e dicotiledoni, ma anche tra piante e metazoi. La regione del promotore prossimale, fino a -100 pb a monte del sito di inizio della trascrizione (TSS), è caratterizzata da elementi sensibili all’orientamento e determinanti per la direzione della trascrizione che includono le sequenze Y-Patch, o Striscie di pirimidine, e i TATA-box (consenso 5’ – TATAWAW – 3’ dove W è A o T). Una regione più distale invece, oltre -100 pb dal TSS, è ricca in sequenze insensibili all’orientamento e contiene il gruppo REG (Regulatory Elements Group).
- Caratteristiche del sito di inizio della trascrizione (TSS)
Il sito di inizio della trascrizione (TSS) è caratterizzato da una sequenza conosciuta come motivo Initiator (Inr) (consenso 5’ -YYCARR - 3’). È un particolare sito di riconoscimento per la TFIID; è stato evidenziato in studi condotti su promotori di mammiferi e si è dimostrato funzionale anche nelle piante. Indagini su i TSS di Arabidopsis hanno rivelato che solo un limitato numero di promotori (meno del 10%) possiede il motivo Inr intorno al TSS, e successive mappature sulla frequenza delle basi presenti nella posizione -1/+1 tra i TSS di Arabidopsis hanno evidenziato che: la posizione -1 è preferibilmente occupata da una C o una T, mentre la posizione +1 è occupata da una A o una G. Questa “YR-Rule” (Y = C o T, R = A o G) è applicabile ad oltre il 77% dei promotori di Arabidopsis ed è da considerarsi una forma meno stringente di Inr (Fig. 1.2.3).
Un altro elemento caratterizzante il core promoter dei geni di organismi animali è l’elemento di riconoscimento della polimerasi (TFIIB Recognition
Element -BRE), solitamente localizzato appena a monte del TATA box
con una sequenza ricca in GC-(G/C)(G/C)(G/C)CGCC. Questo elemento BRE non è stato ritrovato da Yamamoto et al. (2007) come LDSS-positivo (LDSS: Local Distribution of Short Sequences) nelle piante; piuttosto nelle sequenze ai lati del TATA box viene preferito il dimero CC sia nei promotori di Arabidopsis che di riso.
- Striscie di pirimidine (Y-Patch)
Sono sequenze composte da sole C e T, ritrovate nella gran parte dei promotori di Arabidopsis. Sono localizzate da -100 a -13 pb e risultano conservate sia in monocotiledoni che dicotiledoni. Presentano tipicamente un picco nella regione a monte del promotore, distribuita tra -13 e -60 pb. In realtà il ruolo biochimico delle Y-Patch non è ancora conosciuto, ma la loro posizione, sensibile alla direzione e la loro grande abbondanza in natura suggeriscono che sono una componente del core del promotore. L’analisi LDSS (Local Distribution of Short Sequences) di Yamamoto et al. (2007) suggerisce che uomo e topo non condividono con le piante questo elemento che risulta quindi specifico per esse anche se con funzione ignota.
- TATA box
Di tutti i promotori conosciuti in A. thaliana circa il 30-50% contiene un TATA-box localizzato tra 25 e 35 bp a monte del TSS. È una sequenza ricca in T/A (Burley et al., 1996), ed è apparentemente il più conservato segnale funzionale nei promotori eucariotici. In alcuni casi può dirigere un’accurata trascrizione da parte della Polimerasi II anche in assenza di altri elementi di controllo. Molti geni fortemente espressi contengono un potente TATA-box nel loro promotore prossimale, ma grandi gruppi di
geni housekeeping e della fotosintesi mancano del TATA-box e sono indicati come promotori TATA-less. In questi promotori, l’esatta posizione del sito di inizio della trascrizione può essere controllata dalla sequenza nucleotidica della regione nell’intorno del TSS stesso, o da un elemento del promotore (DPE), che è tipicamente osservato 30 bp a valle del TSS
(Smale, 1997; Burke e Kadonaga, 1997; Shahmuradov et al., 2003, 2005).
- REG (Regulatory Elements Group)
Studi compiuti da Yamamoto et al. (2007) hanno evidenziato che la regione che si estende da -20 a -400 contiene solitamente numerosi siti di legame con fattori responsabili di una regolazione specifica della trascrizione. In questa regione si localizzano i siti del gruppo REG che comprendono più di 200 sequenze, metà dei quali corrispondono a conosciuti cis element. La loro sequenza complementare mostra lo stesso picco di distribuzione suggerendo un’indifferente sensibilità all’orientamento. Il principale REG trovato in Arabidopsis e riso, necessario nell’espressione del ciclo cellulare correlato all’attività meristematica è la sequenza GGCCCA, con una posizione intorno a -80 pb. Questa sequenza, conosciuta come Elemento II PCNA-2 di Arabidopsis, indica un buon grado di conservazione tra le specie; una mutazione nella sequenza TGAACC ne cancella l’espressione. Anche a valle del TSS, da +1 a + 50, in posizioni variabili o conservate, si ritrovano motivi ricorrenti che corrispondono a brevi sequenze di 6 - 10 bp e che possono rappresentare siti di legame per proteine. Nella Fig. 1.5 sono riportate alcune caratteristiche dei motivi più ricorrenti sia a monte che a valle del TSS.
Fig. 1.5: Analisi dei motivi più ricorrenti presenti nelle regioni [-50 , -1] e [+1 , +50] del TSS di diversi geni di Arabidopsis. I motivi sono numerati da 1 a 13 e ordinati secondo il grado di maggior rilevanza. Il primo ed il secondo numero corrisondono al numero di hits generati utilizzando il database MEME o il database AlignACE. La seconda colonna mostra la frequenza nucleotidica di distribuzione rappresentata graficamente con WebLogo, la grandezza rappresenta la frequenza. La terza colonna è il grafico delle frequenze di distribuzione (modificato da Molina e Grotewold, 2005).
Motivi 1, 2, 4, 6, 9
Comunemente trovati in Arabidopsis, mostrano una simile e frequente distribuzione nelle regioni [-50, -1] e [+1, +50]; i motivi corrispondono al microsatellite (CA)n (con n = 5). La potenziale partecipazione dei microsatelliti nel controllo della regolazione non è ancora chiaro ma recenti studi in riso e Arabidopsis, mostrano che la loro distribuzione potrebbe seguire un gradiente crescente all’avvicinarsi del sito di inizio della trascrizione.
Motivo 5
E’ il revertito complementare del Motivo 12, con la sequenza consenso AAACCCTA ed è similmente presente nelle regioni [-50, -1] e [+1,+50]; il motivo non è conforme alle sequenze tipiche dei microsatelliti.
Motivi 3, 7
Mostrano una chiara sovraespressione nell’intervallo [-50, -1] con un incremento intorno ai bordi a sinistra dell’intervallo: il Motivo 3 ha tutte le caratteristiche del TATA box ed è stato trovato in 1899 geni, rappresentando circa il 15% di tutti i geni investigati mentre il Motivo 7 è stato trovato in un piccolo numero di geni, 153, con la sequenza consenso A A/G GCCCAT/A. Il Motivo 7 è presente in geni che controllano il ciclo circadiano e nelle regioni a monte della sequenza codificante di geni di Arabidopsis indotti dal buio.
Motivo 8
Conformi ai microsatelliti (CG)n, frequenti in monocotiledoni come il riso e meno frequenti in dicotiledoni come Arabidopsis, nella quale è consistente con una bassa ma comparabile frequenza in tutte e tre le
regioni esaminate. L’apparente alta frequenza del motivo nella regione [+1,+50], comparata alla regione [-50,-1] (421 contro 282), sembra corrispondere ad un incremento del contenuto in G/C del 5' UTR, come riflesso si ha un aumento della distribuzione delle sequenze casuali con la stessa composizione nucleotidica nella corrispondente regione [+1,+50].
Motivo 10
Microsatellite (GAA)n trovato in 340 sequenze è presente nelle regioni [+1,+50], a prescindere che sia codificante o 5' UTR, ma non è ancora associato ad un ruolo funzionale in Arabidopsis.
Motivo 11
Presente in un significante numero di sequenze. È posto nel Kozak consenso (ACCATGG) per la traduzione del codone ATG di inizio (Kozak,
1987). Questo motivo è presente in sole 213 sequenze del 5' UTR [+1, +50],
ma, considerando che delle 1.352 sequenze prese in considerazione in cui è stato trovato 1.139 hanno un breve 5' UTR, questo dato rappresenta un buon controllo interno sulla validità della ricerca tra i motivi nelle regioni [- 50, -1] e [+1, +50].
Motivo 12
Sequenza presente nei telomeri di Arabidopsis, sito di legame per le proteine di tipo MYB collegate ai telomeri. Questo elemento è presente nella regione 5' UTR o nella regione promotrice di molti geni codificanti prodotti associati con l’apparato traduzionale (fattori di trascrizione basali), o coinvolti nell’espressione di geni dei meristemi radicali.
Motivo 13
Con il consenso T/A CCGGCGA, è stato trovato solo nella regione [+1, +50]. Non identifica nessun sito di legame con nessun fattore di trascrizione conosciuto.
- Isole CpG
Le isole CpG sono regioni particolarmente ricche in C e G (oltre il 50% ) della lunghezza media di circa 200 bp. Sono una caratteristica strutturale ricorrente nei promotori dei geni umani (Rombauts et al., 2003). Rappresentano regioni di regolazione della trascrizione esercitata attraverso la possibilità dei residui di citosina di essere metilati: infatti uno stato di ipermetilazione è generalmente associato a DNA inattivo trascrizionalmente (Rombauts et al., 2003). Sono stati sviluppati molti programmi per la ricerca delle isole CpG nelle regioni al 5’ dei geni
(Rombauts et al., 2003). Tuttavia, i parametri usati per individuare le isole
CpG nell’uomo e in altri vertebrati non possono essere usati per le piante; l’applicazione in quest’ultime è molto limitata a causa delle differenze nella distribuzione delle basi. Solo abbassando la percentuale di contenuto in CpG e variando il valore del rapporto CpG/CpNpG è possibile individuare nelle piante sequenze CpG nella regione del promotore che si differenzino dalla generale distribuzione delle due basi nelle regioni degli esoni e degli introni. In realtà in accordo con l’osservazione che nelle piante anche regioni a valle del TSS, per esempio il primo introne, possono essere sede di elementi di regolazione,
(Zhang et al., 1994; Gidekel et al., 1996; de Boer et al., 1999), la
distribuzione delle isole CpG e CpNpG differisce tra i promotori e gli introni da una parte e gli esoni dall’altra, indipendentemente se codificanti o no. Quindi una maggiore accuratezza è richiesta per l’identificazione di “putative” isole CpG e CpNpG nelle piante.
1.2 Sequenze regolatrici dell’estremità 3’ del gene: segnali di
poli adenilazione
Nelle cellule eucariotiche molti RNA vengono inizialmente sintetizzati come precursori o pre-RNA e prima di essere traslocati nel citoplasma e tradotti in proteina devono essere maturati (Fig. 1.8) ovvero andare incontro a tre principali modificazioni post-trascrizionali che comprendono:
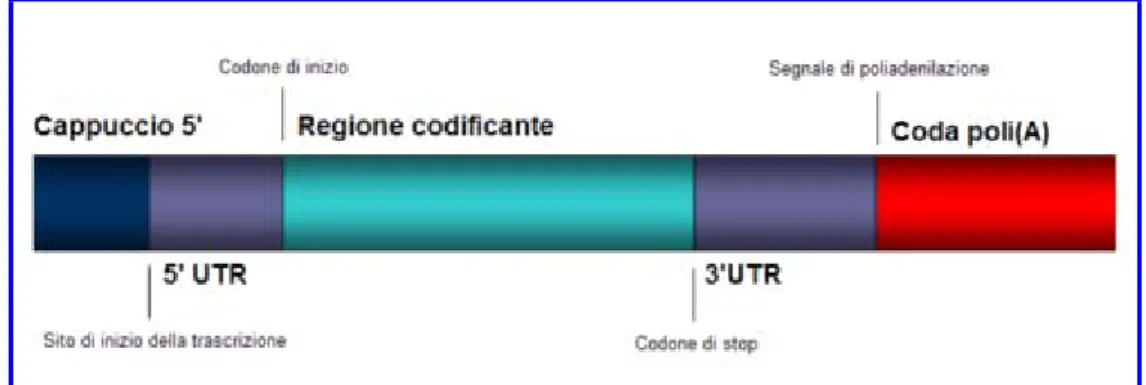
• modificazioni delle estremità, che prevede l’aggiunta di un gruppo 7- metil-guanina all’estremità 5’ (5’capping), e la poliadenilazione all’estremità 3’ che consiste nell’aggiunta di una coda poli(A) (Fig. 1.6).
Fig. 1.6: La struttura di un mRNA eucariotico maturo. Un mRNA completamente modificato include un cappuccio 5', un 5' UTR, una regione codificante, un 3' UTR ed una coda poli(A).
• lo splicing, che consiste nella rimozione degli introni dal pre-mRNA mediante reazioni di taglio e riunione (Fig. 1.7).
Fig. 1.7: Schema dello splicing. Il taglio del sito di splicing al 5’ viene promosso da un idrossile (OH) legato al carbonio-2’ di un nucleotide adenosinico presente all’interno della sequenza intronica (in azzurro). Ciò dà luogo alla formazione della struttura a “lariat”. Successivamente, il gruppo 3’-OH dell’esone a monte (in giallo) induce il taglio del sito di splicing al 3’, consentendo così che gli introni vengano ligati mentre l’introne viene rilascito e degradato.
• Modificazioni chimiche mediante l’aggiunta di gruppi chimici a nucleotidi specifici, chiamata editing dell’RNA.
Ognuno di questi passaggi è altamente regolato da particolari fattori proteici e utilizza i segnali codificati nel 3’UTR dei precursori dell’mRNA.
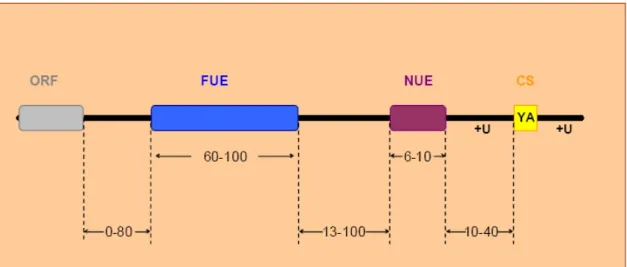
La formazione della coda poli(A) richiede una prima rimozione di un frammento 3’ UTR, effettuata attraverso il taglio endonucleolitico ATP-dipendente di un punto preciso del pre-mRNA, seguito da una immediata polimerizzazione dei residui di adenosina all’estremità 3’-OH al prodotto tagliato al 5’. Un processo apparentemente così semplice coinvolge numerose interazioni tra cis-acting factors e una moltitudine di proteine trans-acting in modo piuttosto differente tra i vari organismi eucariotici. I segnali che permettono la poliadenilazione sono dunque sequenze cis-acting particolari, raggruppabili in tre elementi comuni a mammiferi, piante e lieviti: il sito di taglio (CS), una sequenza upstream vicina al sito di taglio detta NUE (Near Upstream Element) nelle piante e AAUAAA negli animali, ed un altro elemento localizzato maggiormente a monte del CS suddetto, nelle piante, FUE (Far Upstream Element) (Fig. 1.9).Nei mammiferi troviamo un segnale addizionale localizzato a circa 20 pb a valle del CS non comunemente osservato né in piante né in lieviti. Negli animali le sequenze NUE e FUE sono state ben caratterizzate tramite analisi comparative in quanto risultano piuttosto conservate mentre nelle piante e nei lieviti il grado di conservazione tra le specie è molto basso ed è possibile comparare, in termine di sequenze, solo gli elementi segnali del sito CS (Guoli et al., 2007). Recentemente diversi studi compiuti sull’analisi delle sequenze caratterizzanti le regioni 3’UTR dei geni di Arabidopsis, hanno evidenziato delle caratteristiche ricorrenti nella composizione, ma soprattutto nella distanza delle sequenze che intervengono nella formazione della coda poli(A) (Loke et al., 2005;
Guoli et al., 2007). La regione NUE, che risulta avere una lunghezza
compresa tra 6 e 10 nucleotidi ed è localizzata a circa 10-30 pb a monte del sito di taglio, sembra essere principalmente coinvolta nella regolazione del sito di taglio. Tra le sequenze più significative ritrovate nei geni di Arabidopsis spicca il pattern AAUAAA, coincidente con il sito dei mammiferi, anche se questo è stato ritrovato solamente nel 10% dei casi seguito da molte altre meno evidenti (Loke et al., 2005). L’elemento FUE,
localizzato a circa 40-200 pb a monte del sito CS, di lunghezza intorno alle 100 pb, sembrerebbe piuttosto un elemento di controllo o di stimolazione composto da combinazioni di motivi UG piuttosto ambigui e decisamente differenti tra loro, come la sequenza UUGUAA. L’unico segnale decisamente conservato nelle piante rimane la composizione del segnale di taglio CS, formato da un dinucleotide YA (dove Y= C o U) fiancheggiato da due regioni ricche in U. A complicare la rete dei segnali di poliadenilazione delle piante, si aggiunge la constatazione che, i geni di questi organismi, contengono comunemente siti multipli di poliadenilazione controllati non da un unico elemento NUE ma da diverse regioni regolative; ad esempio nel gene rbcS-E9 del pisello ogni sito di poli(A) è controllato dai propri NUE e CS, mentre un singolo FUE controlla tre dei quattro siti e un altro FUE (che si sovrappone con uno dei NUE) controlla il sito rimanente (Li e Hunt, 1997).
Fig. 1.9: Modello dei segnali di poliadenilazione per gli mRNA di piante, i numeri ne indicano la grandezza e la distanza tra essi in pb. ORF: Open Reading Frame; FUE: Far Upstream Element; NUE: Near Upstream Element; CS: Cleavage Siteo sito di poli-adenilazione; +U: regione ricca in U.
La lunghezza della coda poli(A) aggiunta all’mRNA maturo non ha solo un’importanza funzionale di stabilizzazione del trascritto, ma svolge anche un importante ruolo di percezione delle variazione metaboliche che richiedono una rapida alterazione dei livelli di mRNA, sia per accelerarne la degradazione dei trascritti, sia per aumentarne la disponibilità in caso di rapida sintesi proteica.
- Degradazione rapida del mRNA
Subito dopo la trascrizione gli mRNA vengono degradati con un meccanismo molto efficiente per prevenire la sintesi di proteine tronche o eccessive. Questo meccanismo è particolarmente importante per quei geni che possiedono dei codoni di terminazione prematuri (ad esempio geni del TCR o gli “early-response-genes” il cui mRNA è soggetto ad un “turn-over” rapido, in quanto traduce per dei potenti regolatori cellulari come, ad esempio, i fattori di trascrizione). Studi su funghi e metazoi hanno definito 4 vie principali di degradazione:
1. Deadenilazione-dipendente e rimozione del capping seguita da una
degradazione 5’-3’.
2. Deadenilazione-dipendente e degradazione 3’-5’.
3. Degradazione non-senso mediata, che ha come bersaglio gli mRNA
con codoni prematuri terminativi.
4. Degradazione non-stop, che ha come bersaglio gli mRNA mancanti
del codone di terminazione.
Le vie 1, 2 e 4 prevedono un preliminare accorciamento o rimozione completa della coda poli(A) determinante per l’intero processo di degradazione. La rapida cinetica di deadenilazione del mRNA può essere modulato specificatamente da elementi caratterizzanti la sequenze al 3’ UTR come, ad esempio, elementi ricchi in AU, detti ARE, in grado di agire come siti di legame per specifici trans-acting factor, il cui reclutamento potrebbe influire sia direttamente sulla deadenilazione che indirettamente in diversi modi: richiamando altri enzimi deadenilasi, stimolandone la loro attività, alterando le strutture secondarie del mRNA, o infine proteggendolo da RNA-binding protein (Reverdatto et al., 2004). Vi sono tre tipi di mRNA deadenilasi: il complesso Ccr4–Pop2–Not; il complesso Pan2–Pan3; e l’enzima PARN, tutti ritrovati omologhi nei geni di Arabidopsis. AtPARN sembra essere il solo gene codificante l’enzima PARN in Arabidopsis (Reverdatto et al., 2004).
L’enzima PARN viene considerato la principale mRNA deadenilasi negli animali, anche se l’assenza di geni codificanti in lieviti e Drosophila
(Chiba et al., 2004; Reverdatto et al., 2004), fa supporre che questo
enzima non sia così indispensabile negli eucarioti e che venga compensato dalle altre classi di deadenilasi. Mutazioni in AtPARN causano difetti nella risposta a stress ambientali, a stimoli ormonali e nell’accrescimento, suggerendo che questi enzimi possiedano distinte funzioni o bersagli differenti di mRNA nelle piante. I complessi di rimozione del Poli(A) interagiscono con altre proteine, come poly(A)-binding proteins e AU-rich element-poly(A)-binding proteins. In Arabidopsis l’enzima AtPARN svolge una funzione essenziale di regolazione dei trascritti genici durante le fasi embrionali di piante ed animali, analisi su mutanti difettivi dell’enzima presentano trascritti embrione specifici iperadenilati che vanno incontro a ritardi nello sviluppo, culminanti nel blocco dello stadio cotiledonare, questo suggerisce che l’enzima sia essenziale per lo sviluppo embrionale, anche se il suo ruolo non è ben chiaro (Chiba et al., 2004). Altri studi suggeriscono il suo coinvolgimento nella degradazione sistemica dell’mRNA legati a processi di risposta a stress mediati correlati ad ormoni come l’acido abscissico (ABA). I mutanti ipersensibili all’ABA ahg2-1, in grado di mantenersi sensibili anche in stadi di sviluppo avanzato, mostrano un maggior accumulo di ABA endogeno nei semi rispetto al selvatico. Quando i mutanti venivano sottoposti a stress ambientali mostravano una maggiore espressione dei geni indotti da ABA, acido salicilico e da stress rispetto al selvatico. Studi ulteriori hanno rivelato che il gene AHG2 codifica per una ribonucleasi poli(A)-specifica identificabile come AtPARN. Analisi dettagliate dimostrano inoltre che la mutazione ahg2-1 riducendo la produzione di AtPARN, altera il tempo di degradazione dei trascritti. Questi risultati suggeriscono una regolazione di alcuni geni in risposta agli stress grazie all’attività di destabilizzazione degli mRNA effettuato da AtPARN, anche se non è ancora chiaro quale siano esattamente i geni corrispondenti ai trascritti regolati (Nishimura et al., 2005).
- Meccanismi di poliadenilazione citoplasmatica
In particolari condizioni e in determinati distretti cellulari gli RNA messaggeri possono essere immagazzinati “dormienti” per poi essere attivati, tramite un processo di estensione (fino a 1000 adenosine) della coda poli(A) che avviene nel citoplasma. Questo meccanismo di controllo post-trascrizionale consente, in presenza di un preciso segnale, un rapido incremento della sintesi proteica non più legata ai sistemi trascrizionali che necessiterebbero di tempi più lunghi. (Richter, 1999; Kim
e Richter, 2006). La poliadenilazione citoplasmatica è stata, infatti,
caratterizzata in contesti in cui è necessario un rigoroso controllo temporale o spaziale della produzione proteica, o un incremento molto rapido dei livelli proteici come è richiesto ad es. dall’oogenesi. Questi meccanismi sono stati ampiamente studiati in oociti di Xenopus, topo e Drosophila mentre per il regno vegetale non vi sono che studi preliminari. Studi condotti su Xenopus hanno evidenziato che i messaggeri interessati nella poliadenilazione citoplasmatica, sono caratterizzati da una precisa localizzazione subcellulare e da una estesa regione 3’UTR che ospita necessariamente, nella sua porzione terminale, due particolari elementi ARE (Mendez e Richter, 2001). Uno di questi è l’esanucleotide AAUAAA,
noto come elemento CPS/PAE (Cleavage Polyadenylation
Signal/Polyadenylation Element), già necessario nel processo nucleare di taglio e addizione della coda corta di poliadenilazione sul pre-mRNA. L’altro, denominato CPE (Cytoplasmic Polyadenylation Element), si trova generalmente a breve distanza dal primo ed è costituito da una sequenza ricca di uridine con il consensus UUUUUAU (Piccioni et al.,
2005).
In Xenopus l’elemento CPE sul trascritto viene riconosciuto dalla proteina di legame CPEB che, previa fosforilazione del residuo serina 174 compiuto dalla chinasi Eg2, riconosce un’altra proteina: la CPSF. A questo punto il complesso ribonucleoproteico (messaggero-CPEB-CPSF) richiama sul 3’ dello stesso messaggero l’enzima poli(A)-polimerasi
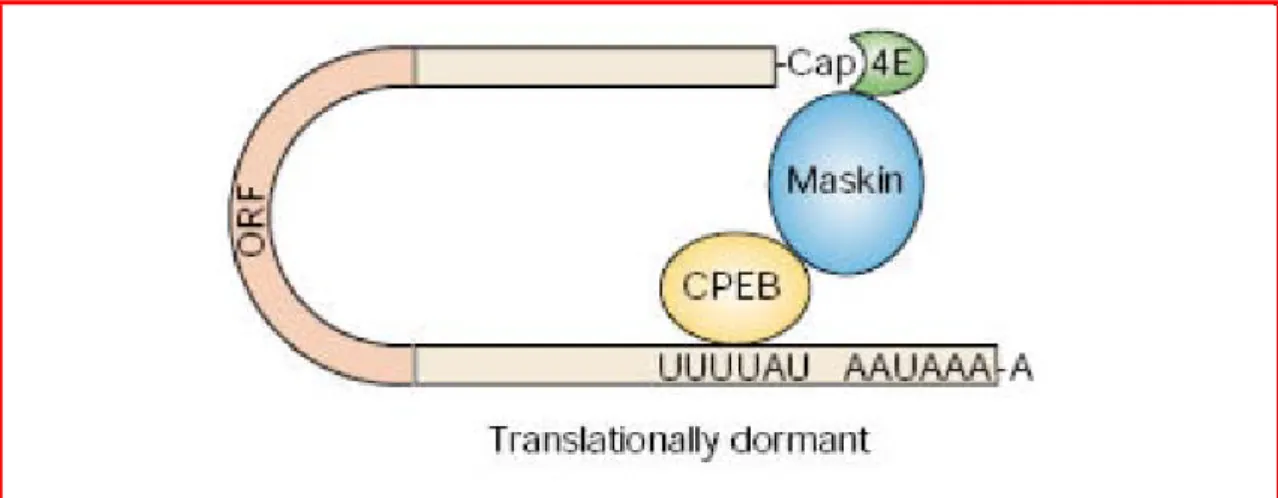
citoplasmatica (PAP) che catalizza l’allungamento della coda di poliadenilazione con un meccanismo del tutto simile a quello nucleare (Fig.1.10). L’allungamento è generalmente molto cospicuo, determinando un incremento anche di diverse centinaia di residui di adenosina (Kim e Richter, 2006). Una estesa coda di poliadenilazione consente il legame di molte unità di proteine leganti la coda poli(A) (PABP) citoplasmatiche; queste hanno la proprietà di reclutare il fattore di iniziazione eIF4G che porta ad una torsione del messaggero in un anello chiuso agli estremi 5’ e 3’. Tale evento è stato messo in relazione con una notevole facilitazione della traduzione e del processo di riciclo del ribosoma (Mendez e Richter, 2001). Sembra inoltre che le PAP siano interessate nella facilitazione della formazione di una struttura di 5’ cap-I o 5’ cap-II, eventi che conferiscono una migliore competenza traduzionale al trascritto (Mangus et al., 2003). Se il processo di poliadenilazione citoplasmatica non viene attivato, i trascritti contenenti CPE e CPS/PAE vengono rigidamente inibiti, sia rimanendo immagazzinati in comparti sub-cellulari protetti dall’azione esonucleasica (Richter, 1999), sia grazie alla proteina CPEB che nella forma non fosforilata lega un membro della famiglia delle eIF4G-BP (Eucariotic Initiation Factor 4G-Binding Protein, in Xenopus: Maskin). Questi fattori contengono un dominio di legame per eIF4G, omologo a quello presente nel fattore di iniziazione eIF4E ed agiscono da competitori, determinando una inibizione della traduzione (Fig. 1.11)
Fig. 1.10: Attivazione della traduzione in Xenopus tramite poliadenilazione citoplasmatica. La chinasi Eg2 opera la fosforilazione del CPEB, richiama i fattori di taglio e poliadenilazione specifici (CPSF) e la PAP (poli(A) polimerasi) che allunga la coda di poli A. Maskin si dissocia da eIF4E e delle proteine leganti il poli A (PAPB) si associano alla coda e al fattore IF4G, causando il ripiegamento della molecola. (Modificato da Mendez e Richter, 2001)
Fig. 1.11: Blocco della traduzione. Gli mRNA contenenti il CPE sono trascrizionalmente dormienti e residui nel complesso contenente il CPEB, Maskin e il fattore eIF4E.
CPE: elemento di poliadenilazione citoplasmatica CPEB: CPE-binding protein

![Fig. 1.5: Analisi dei motivi più ricorrenti presenti nelle regioni [-50 , -1] e [+1 , +50] del TSS di diversi geni di Arabidopsis](https://thumb-eu.123doks.com/thumbv2/123dokorg/7344416.92359/11.892.172.811.118.960/fig-analisi-motivi-ricorrenti-presenti-regioni-diversi-arabidopsis.webp)