67
MATERIALS AND METHODS
1. Materials
Luria Broth and Luria Agar, kanamycin, doxycycline, ampicillin, imidazole, ATP, IMP, inosine, 2,3-BPG, DTT, BSA, DMSO, ammonium formate, ammonium molybdate, ascorbic acid, sodium metaarsenite, sodium citrate, molecular mass standards for gel filtration experiments and for native electrophoresis and GenElute gel Extraction Kit were from Sigma Aldrich.
SDS, Magnesium Chloride, Sodium Chloride, -mercaptoethanol were from BDH Chemicals.
Ammonium persulfate, acrylamide and bis-acrylamide, TEMED, Coomassie Brilliant Blue G250, agarose were from BioRad Laboratories.
Tris (base), Glycine, Glycerol were from J.T. Baker. Dithiothreitol was from Sigma Chemical Co.
[8-14C] inosine and [8-14C] IMP were from Moravek Biochemical.
Prestained standards for SDS-PAGE/native electrophoresis were from Invitrogen. Ni-NTA Agarose resin, QIAEX II Gel Extraction Kit were from Qiagen S.p.A. DE-81 chromatographic papers were from Whatman.
Polyethyleneimine (PEI)-cellulose pre-coated thin layer plastic sheets (0.1 mm thick) were from Merck.
Rabbit anti chicken IgG horseradish peroxidase-conjugated was purchased from Chemicon International. Primary mouse anti-human -actin antibody and rabbit
anti-mouse IgG horseradish peroxidase-conjugated were kindly provided by Dr. R. Moschini.
Sephacryl HR S-300 was from Amersham.
Superdex- 200 was purchased from GE Healthcare.
ECL kit, 0.45 μm and 0.22 μm Syringe Driven Filter were from Millipore.
Hi safe II Scintillation liquid was purchased from Wallac.
T4 PNK, EcoRI, ClaI, MluI, FspI, MscI, BspEI, BstBI, T4 DNA ligase and ColorPlus Prestained Protein Marker were from New England BioLab.
68 Blue/orange Loading Dye, 1 Kb DNA ladder, DpnI, CIAP, dNTP mix, Pfu DNA polymerase, GoTaq, T4 Ligase, Ethidium bromide solution, Blue/Orange 6X Loading
Dye, TBE buffer, Wizard Plus Minipreps DNA Purification System, PureYield Plasmid Maxiprep System, PureYield Plasmid Midiprep System were from Promega.
Rapid DNA Ligation Kit was from Fermentas.
DNA mi-1kb (1 μg/μl) DNA Markers and mi-100bp (1 μg/μl) DNA Markers were from
Metabion.
pLVTHM, pLVCT-tTR-KRAB, psPAX2, pMD2.G plasmids were from Addgene.
293T, Stable2 and JM109 E. coli strains, PEI solution were kindly provided by Prof. M. Pistello.
DNA standards for Ethidium Bromide Spot Test were kindly provided by Dr. M.G. Careddu. DMEM high glucose (4.5 g/L Glucose), RPMI with L-Glutamine , FBS EU Standard - South
America Origin (Brazil), FBS tet-free, Glutamine 200 mM, Penicillin-Streptomycin Mixture 5K/5K 5000 μg/ml, Trypsin-Versene (EDTA) Mixture 1X, Trypan Blue were from
LONZA.
GFP-CertifiedApoptosis/Necrosis detection kit was from Enzo Life Science.
All other chemicals were of reagent grade.
2. Methods
2.1 Molecular biology methods
2.1.1 PET expression system and site-directed mutagenesis
The pET Expression System was used for cloning and expressing the recombinant wild-type and mutant cN-II from calf thymus in E.coli as previously reported (Allegrini et al., 1997)1. CN-II coding sequence is cloned in the bacterial pET-28c plasmid under control of strong bacteriophage T7 transcription signals; for this reason the ricombinant vector is transferred to an E. coli strain containing a chromosomal copy of the gene for T7 RNA polymerase. Although T7 lac promoter is 1
It is important to underline that the deduced aminoacid sequences of human and bovine cytosolic 5’-nucleotidase II determined from their cDNA sequences show a degree of homology of 99.5%: in fact, cN-II from calf thymus differs from human one in two conservative substitutions (Thr 2 vs Ser; Val 335 vs Ile) and in the lack of a glutamic residue at the acidic C-terminus (Allegrini et al., 1997; Bretonnet et al., 2005).
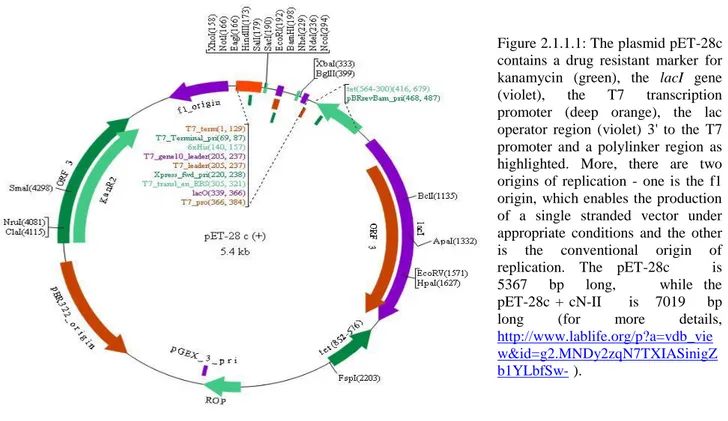
69 inducible by IPTG, there were no need to use it. To facilitate purification of the target protein, pET-28c vector carries an N-terminal His Tag-thrombin-T7 Tag configuration; more, this plasmid contains a drug resistant marker for kanamycin useful for selection (read the caption of figure 2.1.1.1 for more details).………
….. ………..
Site-directed mutagenesis experiments were performed in order to obtain non-conservative and conservative point mutants both at the effector sites (effector site 1 and 2) and at the interfaces
(interface A and interface B). To choose the residues to be mutated we proceeded from the human cN-II crystal structures described by Walldén et al., using Polyview-3D server (http://polyview.cchmc.org/polyview3d.html) and RCSB PDB Ligand Explorer 3.5 software (refer to chapter “Introduction”, paragraph 1.3.4; Walldén et al.,
2007b).
The mutants I built are as follows:
R144E, I152D, N154D for the study of effector site 1; F127E, H428D, M436W for the study of effector site 2; F36R, Y115A, D396A for the study of interface A; K311A, G319D for the study of interface B.
Figure 2.1.1.1: The plasmid pET-28c contains a drug resistant marker for kanamycin (green), the lacI gene (violet), the T7 transcription promoter (deep orange), the lac operator region (violet) 3' to the T7 promoter and a polylinker region as highlighted. More, there are two origins of replication - one is the f1 origin, which enables the production of a single stranded vector under appropriate conditions and the other is the conventional origin of
replication. The pET-28c is 5367 bp long, while the pET-28c + cN-II is 7019 bp long (for more details,
http://www.lablife.org/p?a=vdb_vie w&id=g2.MNDy2zqN7TXIASinigZ b1YLbfSw- ).
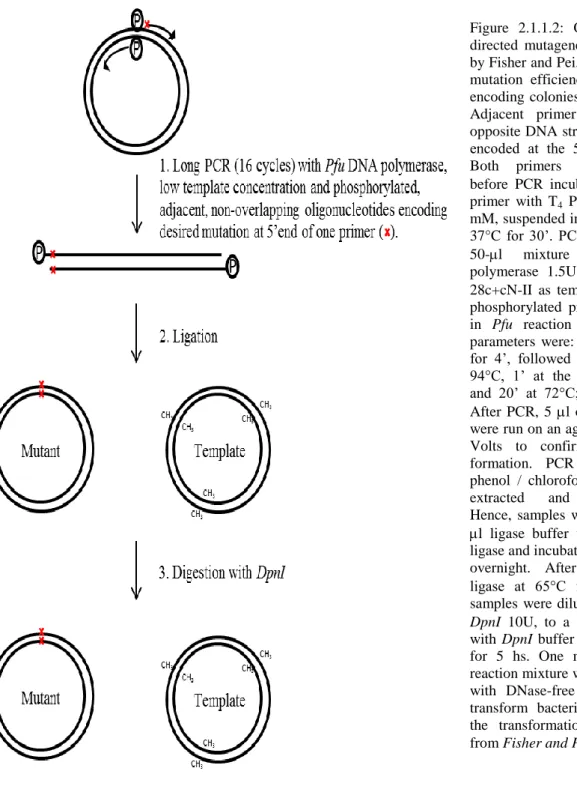
70 The point mutants F36R, Y115A, F127E, I152D, R144E, N154D, K311A, G319D, D396A and M436W were obtained following the PCR-Based Site-Directed Mutagenesis Method described by Fisher and Pei (figure 2.1.1.2 for more details; Fisher and Pei, 1997), while the point mutant
H428D was obtained by a faster and cheaper method, as described by the QuikChange® Site-Directed Mutagenesis Kit manual (figure 2.1.1.3 for more details; Stratagene).
Figure 2.1.1.2: Overview of the site-directed mutagenesis protocol described by Fisher and Pei, which has a very high mutation efficiency and yields mutant-encoding colonies in less than 48 hours. Adjacent primers were designed on opposite DNA strands with the mutation encoded at the 5’ end of one primer. Both primers were phosphorylated before PCR incubating 20 M of each primer with T4 PNK 10U and ATP 0.9 mM, suspended in the T4 PNK buffer, at 37°C for 30’. PCR was carried out in a 50-l mixture using Pfu DNA polymerase 1.5U with 10 ng of pET-28c+cN-II as template, 0.5 M of each phosphorylated primer, dNTPs 0.2 mM in Pfu reaction buffer. Amplification parameters were: an initial step at 94°C for 4’, followed by 16 cycles of 1’ at 94°C, 1’ at the annealing temperature and 20’ at 72°C; finally, 10’ at 72°C. After PCR, 5 l of the reaction mixture were run on an agarose gel in TBE at 80 Volts to confirm amplified product formation. PCR samples were then phenol / chloroform / isoaminoalcohol-extracted and ethanol-precipitated. Hence, samples were resuspended in 20 l ligase buffer with 10U of T4 DNA ligase and incubated at room temperature overnight. After heat-inactivation of ligase at 65°C for 20’, 4 l of the samples were diluted, in the presence of
DpnI 10U, to a final volume of 10 l
with DpnI buffer and incubated at 37°C for 5 hs. One microliter of the final reaction mixture was then diluted tenfold with DNase-free water and used to transform bacteria; see hereinafter for the transformation protocol (modified from Fisher and Pei, 1997).
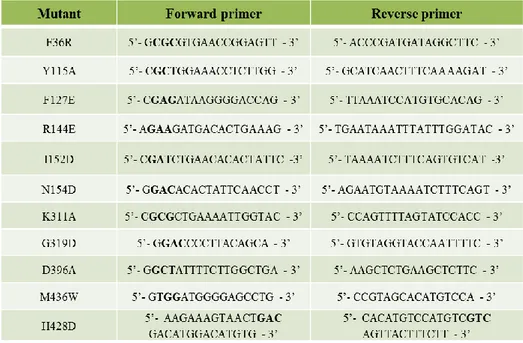
71 The primers used for site-directed mutagenesis are described in table 2.1.1.1.
Table 2.1.1.1: Primers used for site-directed mutagenesis. In bold are depicted the mutagenic codons. For the design of the primers Lasergene DNA STAR v 7.0 Software was used. The primers were synthesized by Bio-Fab Research s.r.l.
Figure 2.1.1.3: This rapid four-step procedure generates mutants with greater than 80% efficiency; besides, it requires no specialized vectors, unique restriction sites or multiple transformations.
The reaction mixture contains 0.3 mM of both primers (forward and reverse), 5 ng of
Pet-28C+cN-II, dNTPs 0.2 mM, Pfu DNApol 1.5U in
the respective buffer to a final volume of 10 l.
Amplification parameters were: an initial step at 95°C for 2’, followed by 17 cycles of 30’’ at 95°C, followed by 20’’ at 50°C and 20’ at 72°C; finally, 10’ at 72°C. Three microliters of the reaction mixture was then used to transform bacteria; see hereinafter for the transformation protocol (from
QuikChange® Site-Directed
Mutagenesis Kit manua,
Stratagene).
72
2.1.2 Preparation and transformation of competent cells
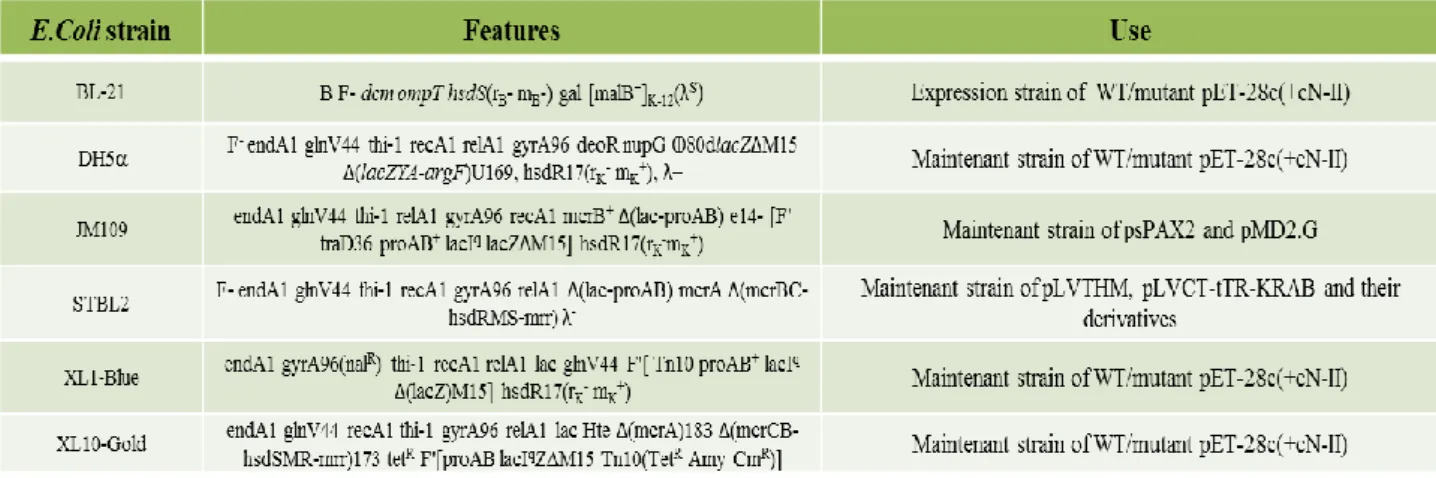
As the method both for preparing and for transforming competent cell is universal, during this paragraph I will refer simply to bacteria; read the table at the end for more details regarding the several E.coli strains used (2.1.2.1).
The bacteria strains were grown in Luria Broth medium at 37°C overnight. Starting from pre-culture, few microliters were inoculated in 100 ml of fresh LB medium and incubated with shake at 37°C. When bacterial culture reached an adsorption of 0.5 at 600 nm, the cells were harvested by centrifugation at 1100g (10 minutes, 4°C). Then, the bacteria were carefully resuspended in 12.5 ml of TFBI buffer (KAc 30 mM, MnCl2 50 mM, RbCl 100 mM), incubated on ice for 10 minutes and harvested by centrifugation at 1100g (10 min, 4°C). Hence, the pellet was resuspended in 2 ml of TFBII buffer (MOPS 10 mM, CaCl2 75 mM, RbCl 10 mM, glycerol 15%). 100 l aliquotes of the competent bacteria were stored at -80°C until transformation.
For transforming bacteria, one aliquote was transferred to an ice-cold tube containing 50 ng of DNA (which can be WT or mutant pET-28c+cN-II; psPAX2, pMD2.G, pLVTHM, pLVCT-tTR-KRAB vectors): the mixture was incubated for 1 hour on ice, then 45 seconds at 42°C and subsequently 2 minutes on ice. Afterwards, 800 l of fresh LB medium were added to the tube, which was then transferred at 37°C for 1 hour. The entire volume was plated out on plates containing Luria Broth agar and kanamycin 50 M. Plates were incubated overnight at 37°C in order to obtained monoclonal cultures. The following day, each colony was collected and incubated for 24 hours at 37°C in 5 ml of fresh Luria Broth: 1.4 ml of the culture was store at -80°C in the presence of 15% glycerol, while the rest - after DNA purification with Promega PureYield Plasmid System - was sent to Bio-Fab Research s.r.l. for sequencing analysis.
73
2.1.3 Cloning of shRNA into conditional LV
In 2008, Careddu and colleagues demonstrated that among three shRNA sequences specific for human cN-II, targeting the positions 473, 660 and 1171 respectively in the cN-II mRNA, sh660 (actually sh661, due to one base-reading-frameshift; henceforward, I will refer to it as sh661) was effective in silencing cN-II (Careddu et al., 2008). In fact, in ADF astrocytoma cells transduced with adenovirus carrying sh661 at MOI of 30, after 53 hours from infection, cN-II mRNA level decreased to about 20% of control cells (similar results were obtained also in L2, a rat lung epithelial cell line) while cN-II activity remained pretty much the same. After 72 hours from transduction, instead, enzyme activity was decreased from 0.7 mU/mg in control ADF cells to 0.45 mU/mg (note that at 68th hour from transduction, cN-II activity was still unchanged in silenced ADF cells in comparison with control cells), while cell viability decreased up to 0.59 (fold vs control) and caspase-3 activity increased from 136 pmol min−1 mg−1 in control cells to 639 pmol min−1 mg−1 in silenced cells (Careddu et al., 2008). Therefore, astrocytoma cells began to die practically immediately afterwards cN-II began to decrease, indicating that cN-II is essential for cell survival.
This observation prompted us to undertake a study aimed at assessing the physiological role of cN-II. To do this, we decided to silence cN-II both in HEK 293T and ADF cell lines in a
conditional way, allowing the control of cN-II expression in a quantitative and temporal manner: in this way, it is possible, on one hand, to silence cN-II little by little without inducing apoptosis and, so, to evaluate cN-II involvement in purine metabolism; on the other to induce apoptosis gradually and, so, to study the apoptotic pathway activated by cN-II silencing (note that ADF cell line lacks the “intrinsic apoptotic pathway” because of a point mutation in the prodomain region of caspase-9; Ceruti et al., 2005).
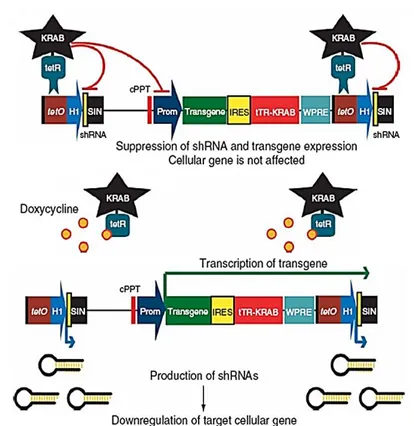
Nowadays several drug-inducible gene expression systems are available; among these, we chose the lentiviral vector-based tetracycline-inducible control system described by Szulc and colleagues, a high versatile, single vector tool that allows for conditional transgene expression from both polymerase II and III promoters with a high degree of efficacy and without significant leakiness in vitro and in vivo (Szulc et al., 2006; Szulc and Aebischer, 2008).
This is a repression-based system, in which the transcriptional activity of cellular promoters is modulated by drug-controllable epigenetic repressors, in this case containing a Krüppel-associated box (KRAB) domain. The system, in fact, takes advantage of the promiscuous repression activity of tTRKRAB, a fusion protein between the KRAB domain found in many vertebrate zinc finger transcriptional regulators and the tetracycline repressor (tetR) of E. coli. KRAB domain can silence
74 both Pol II and Pol III promoters by triggering heterochromatin formation: when tethered to specific DNA regions within the context of chimeric proteins, KRAB recruits a multimulecolar complex that leads to histone deacetylation and methylation and binding of heterochromatin protein 1 (HP-1), thus creating a local heterochromatin state extending up to 3 kb from its binding site (Margolin et al., 1994; Moosmann et al., 1997; Senatore et al., 1999; Urrutia et al., 2003; Wiznerovicz and Trono, 2003; Wiznerovicz et al., 2006). When KRAB is fused to the tetR DNA-binding domain, the resulting tTRKRAB chimeric protein allows for the doxycycline-mediated control of any promoter placed nearby tetO sequences; to do this, we chose the “Tet-On” version (figure 2.1.3.1 ).
As mentioned, this drug-inducible expression system is based on lentiviral vectors. Lentiviral vectors (LVs) are competent gene transfer vehicles, used for both research and gene therapy applications, because of their stable integration in non-dividing and dividing cells and long-term transgene expression. In these last years, researchers have been devoting significant efforts in designing LVs with improved efficacy and biosafety features. Currently, vectors combine improved safety features - decreasing the risk of generation of replication competent lentiviruses (RCLs) and vector mobilization - with increased transduction efficiency.
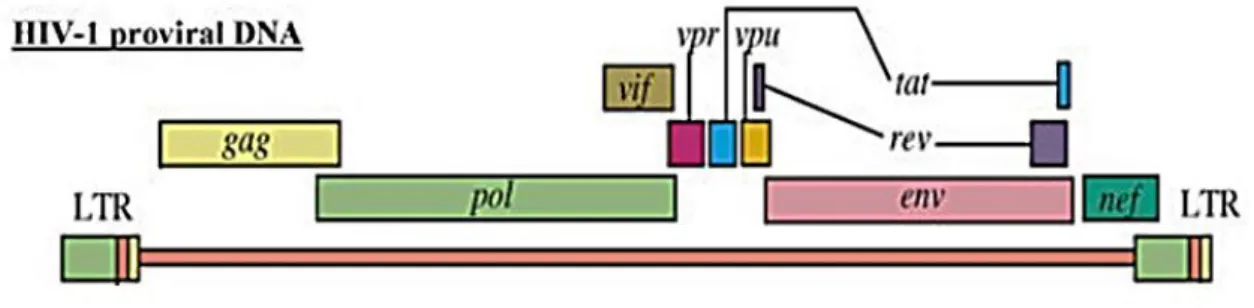
Figure 2.1.3.2 is a schematic representation of HIV provirus: each end of the proviral genome is made up of regions called long terminal repeats (LTRs) which contain the proviral U3, R and U5 regions (the rearrangement of both termini of the viral genome during reverse transcription enables appropriate expression of the viral genes). The U3 region of the 5’ LTR (copied from the U3 region Figure 2.1.3.1: The tetracycline-controllable single lentiviral vector system we used (tet-on versi(tet-on; read further for details about single-vector structure). In the absence of tetracycline, tTRKRAB chimeric protein binds to tetO and suppresses the expression of the transgene as well as its own preventing the production of small hairpin RNAs (anyway there is a low basal expression!). In the presence of tetracycline, tTRKRAB does not bind to tetO, thus allowing transgene expression and downregulation of the target gene by RNA interference. The system is a versatile tool as it is fully reversible.
Bicistronic vectors carry an internal ribosomal entry site (IRES) between the two coding sequences, id est a sequence which directly recruits ribosome. SIN, self inactivating; cPPT, central polypurine tract; Prom, promoter; IRES, internal ribosome entry site; WPRE, woodchuck hepatisis virus post-transcriptional regulatory element (from Szulc et al., 2006).
75 at the 3’ end of the RNA genome) contains the viral promoter and enhancers responsible for initiation of transcription of the viral genome at the 5’ U3/R junction. Gag, pol and env are the structural genes common to all retroviruses. The gag gene encodes viral core structural proteins (matrix, capsid and nucleocapsid); the pol gene encodes the viral replication enzymes (protease, reverse transcriptase and integrase), while the env gene encodes the envelope glycoprotein. In addition to the gag, pol and env genes, HIV-1 contains two regulatory genes, tat and rev, essential for virus replication, and four accessory genes, vif, vpr, vpu and nef involved in viral pathogenesis.
Figure 2.1.3.2: HIV-1 provirus (for explation, read the text).
The design of safe viral vectors derived from WT HIV-1 first requires that the cis-acting sequences directing viral genome transfer (provided by a transfer vector) have to be separated from the transacting sequences encoding viral structural proteins (provided by the packaging construct). This approach produces LV transfer vectors, containing a transgene expression cassette with an internal promoter that can drive transgene transcription flanked by the two viral LTRs, and packaging construct, characterized by the substitution of the viral LTRs at the 5' and 3' positions by a strong promoter and a polyadenylation signal, respectively.
Several additional strategies have been employed to further improve the biosafety of LVs, as accessory gene removal and separation of functional viral components into different expression plasmids (“third generation” packaging system; table 2.1.3.1). Besides, another efficient strategy to improve the safety of LVs involves deletion of the promoter and enhancer elements located in the transfer vector 3’LTR U3 region, giving rise to SIN (self inactivating) vectors which significantly decrease the risk of recombination between the transfer vector and packaging constructs (Zufferey et al., 1998; Pauwels et al., 2009). More, in order to design an efficient system, as WT HIV-1 infection is restricted to human cells expressing the CD4 receptor, the natural HIV envelope gene was replaced with the vesicular stomatitis virus glycoprotein (VSV-G) gene, which greatly broadened cellular tropism (to most, if not all, mammalian cell; Burns et al., 1993; Pauwels et al., 2009).
76 Table 2.1.3.1: Comparison of Lentiviral Production Systems; lentiviral expression systems described in this table all have a separate construct expressing Vesicular Stomatitis Virus G glycoprotein (VSV-G) instead of the env gene encoding by the WT HIV-1 envelope (from Pauwels et al., 2009).
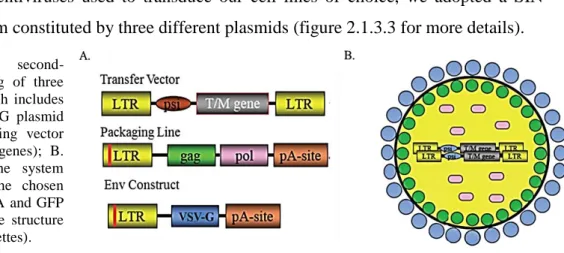
For the production of lentiviruses used to transduce our cell lines of choice, we adopted a SIN second-generation system constituted by three different plasmids (figure 2.1.3.3 for more details). Figure 2.1.3.3: A. SIN second-
generation system consisting of three vectors (transfer vector which includes the desired sequence; VSV-G plasmid as envelope vector; packaging vector consisting of gag and pol genes); B. Lentivirus derived from the system described in A carrying the chosen transgene, in our case shRNA and GFP (colour legend underlines the structure codified by the different cassettes).
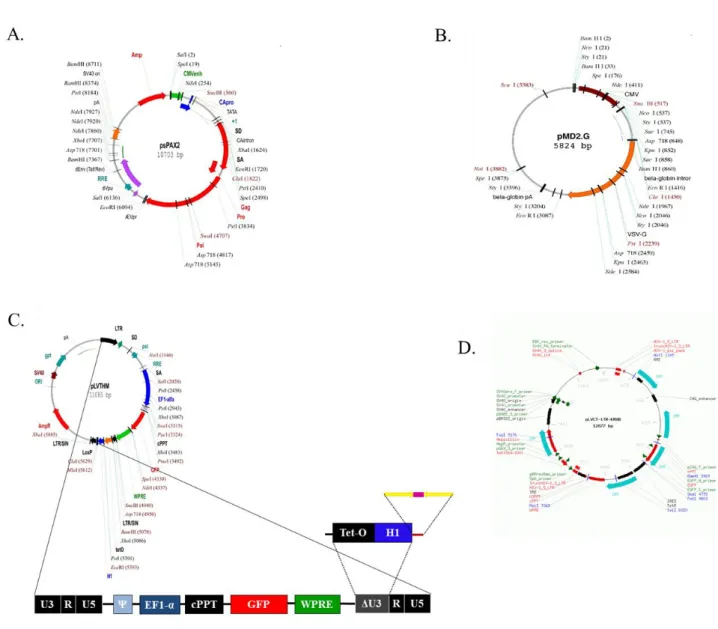
77 As packaging vector, we used psPAX2 (figure 2.1.3.4 A), which contains the structural genes gag and pol and the regulatory genes tat and rev; while, as envelope plasmid, we chose pMD2.G, which carries the cassette codifying VSV-G (figure 2.1.3.4 B). Both vectors contains heterologous promoters and polyadenylation signals instead of LTRs. Only the transfer vector has the shRNA inserted between modified LTRs (this is a SIN system!). In this way, not only the risk of production of RCLs is decreased, furthermore only the transgene can integrate in host cell genome. As this is a SIN system, the deletion in U3 region is replaced by the tetO_H1_shRNA cassette (figure 2.1.3.4 C), where tetO is the tetracycline operator specific for the binding of tetR and H1 a strong human promoter pol-III dipendent (the shRNA is depicted in yellow and fuchsia). As transgene vector we
Figure 2.1.3.4: SIN second-generation system used for the production of lentiviruses carrying the shRNA of interest. A. Packaging vector; B. Envelope vector; C. and D. transgene vectors convenient for a constitutive and a conditional silencing, respectively. pLVTHM carries the tetO_H1 cassette (see the zoom), which will be clone into pLVCT-tTRKRAB. PLVCT-tTRKRAB, instead, contains the cassette for tetR-KRAB. Ψ, encapsidation signal; EF1-, pol II elongation factor 1- promoter; cppt, central polypurine tract; GFP, green fluorescent protein; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element. For more details, read the text (from www.addgene.org).
78 used the pLVTHM, which is necessary for the cloning strategies we adopted and which guarantees a constitutive silencing, and pLVCT-tTRKRAB useful for the conditional silencing (figure 2.1.3.4 D). As you can see in the map, both these vectors are ampicillin-resistant and carry GFP as internal monitoring device; SIN LTRs; WPRE (woodchuck hepatitis virus posttranscriptional regulatory element) in order to enhance the expression of the transgene; Ψ (psi), as encapsidation signal; cppt (central polypurine tract), fundamental for initiation of second plus-strand synthesis and for virus nuclear entry (Buchschacher, 2002; Manganini et al., 2006; Szulc et al., 2006; Zufferey et al., 2009).
AS sh661 seemed to be very efficient in silencing cN-II, we decided to use it in our RNAi experiments (figure 2.1.3.5 A). As control, we utilized the same unrelated control sequence described by Careddu et al. (named shCTR; figure 2.1.3.5 B; Careddu et al., 2008). The oligomers were designed according to Szulc and Aebischer and synthesized by Bio-Fab Research s.r.l. (Szulc and Aebischer, 2008).
Figure 2.1.3.5: Oligomer sequences carrying (A) the sh661 and (B) the shCTR described by Careddu et al. (top/bottom sequences represent forward/reverse oligos; Brummelkamp et al., 2002; Careddu et al., 2008; Ge et al., 2010).
Before cloning shRNAs (sh661 and shCTR) into conditional LV, two preliminary steps were performed: the annealing and the phosphorylation of the oligomers depicted in figure 2.1.3.5 as described by Szulc and Aebischer (Szulc and Aebischer, 2008). For the annealing, both oligos (forward and reverse) were dissolved in water to the final concentration of 1 M; then 2 μl of each oligo were mixed with 46 μl of annealing buffer (potassium acetate 100 mM, HEPES 30 mM pH 7.4, magnesium acetate 2 mM) and, after a denaturation passage for 4 minutes at 95°C, incubated for 10 minutes at 70°C. Hence, 5 μl of the oligos thus annealed were incubated with 10 U of T4 PNK in a volume of 20 l in the presence of the T4 ligase buffer containing 1 mM ATP for 30 minutes at 37°C. PNK was, then, heat-inactivated incubating the mixture for 10 minutes at 70°C.
79 After these two steps, I carried out the digestion of 2 μg of pLVTHM vector, first, with ClaI (for one hour at 37°C; NEB buffer 3; BSA 0.1 mg/ml; total volume of 20 l) and, second, with MluI directly added to the mixture (for one hour at 37°C; further 5 l in NEB buffer 3) after the heat-inactivation of ClaI ( 65°C, 20 minutes).
Then 5 μl of phosphorylated oligos were incubated in the presence of 80 ng of pLVTHM vector digested with MluI/ClaI, 2 μl of T4 ligase buffer and 5 U of Rapid T4 ligase at 22°C for 20 minutes (total volume of 20 μl). Thus, Stbl 2 bacteria were transformed with 5 μl of the ligation reaction by heat-shock as described in the paragraph 2.1.2.
After the screening of plasmidic DNA1 purified from several putative positive clones with Wizard Plus Minipreps DNA Purification System, 1 μg of pLVTHM_sh661/shCTR was digested with FspI and MscI (in NEB buffer 4; final volume of 60 μl), incubating for 90 minutes at 37°C. Contemporaneously, 7.4 μg of pLVCT-tTR-KRAB were digested with FspI and MscI in the same conditions. The digested products was, then, gel-purified after gel electrophoresis using GenElute Gel Extraction Kit and quantified by Ethidium Bromide Spot Test. Thus, in order to avoid the possibility of self-ligation, linearised pLVCT-tTR-KRAB was treated with 0.06 U of CIAP (Calf Intestinal Alkaline Phosphatase) for 30 minutes at 37°C. The reaction product was, then, gel-purified and quantified by Ethidium Bromide Spot Test.
Finally, 40 ng of dephosphorylated pLVCT-tTRKRAB digested with FspI/MscI and 60 ng pLVTHM_sh661/shCTR digested with FspI/MscI were incubated with 20 U of T4 Ligase at 16°C for 16 hours (final volume of 10 μl). Thus, Stbl 2 bacteria were transformed with 5 μl of ligation reaction by heat-shock as described in the paragraph 2.1.2. The plasmidic DNA2 purified from several putative positive clones with Wizard Plus Minipreps DNA Purification System was then screened.
1
Three screening tests were performed:
- through a PCR reaction and gel electrophoresis (Forward Primer: 5’-GAATCTTATAAGTTCTGTATGA GACCACGC-3’; Reverse Primer: 5’-ATTGTCGTTAGAACGCGGCTAC-3’): 100 pg of pLVTHM or plasmidic DNA purified from putative positive clones, primers 1 μM, dNTPs 0.2 mM, GoTAQ using protocol provided by manufacturer (total volume 20 μl). Amplification parameters were an initial step at 95°C for 3’, followed by 30 cycles [30’’ a 95°C, 30’’ a 57°C, 35’’ a 72°C] and a final step at 72°C for 5’. After agarose gel-electrophoresis, real positive clones showed 448 bp-band (while in correspondence to the control a 398 bp-band was evident).
- by EcoRI/ClaI digestion (300 ng of template; NEB buffer 4; BSA 0.1 mg/ml, restriction enzymes put together; 37°C for 40’) followed by gel-electrophoresis: the real positive clones were 10849 + 286 bp, while the negative/control was 10849 + 236 bp.
- through sequencing analysis performed by Bio-Fab Research s.r.l. 2
Also in this case three screening tests were performed: - through a PCR reaction as described above;
- by MluI digestion, as described before (the real positive clones gave rise to two bands, while the negative ones/control only to one);
80 Regarding basilar molecular biology techniques, as DNA electrophoresis, DNA extraction,
Ethidium Bromide Spot Test refer to Molecular Cloning – A laboratory manual.
2.2 Biochemistry methods
2.2.1 Expression and purification of the recombinant proteins
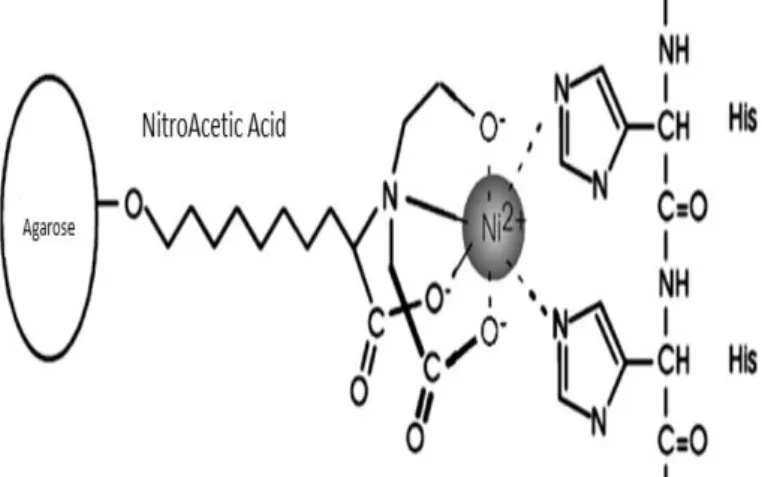
An aliquote of BL-21 bacteria transformed with wild type or mutant pET-28c grew in 200 ml of fresh Luria Broth containing kanamycin 50 M overnight (at least 16 hours) at 37°C with shake. When OD600 reached 0.5, bacteria were harvested for 20 minutes at 1000g at 4°C; then the pellet, resuspended in 4 ml of lysis buffer (Tris-HCl 50mM pH 8.0, NaCl 300mM, imidazole 10 mM) containing 1 mg/ml lysozyme, was kept shaking for one hour at room temperature. Cell lysis was completed performing 10 cycles of ice cold sonication. Lysates were, thus, centrifuged at 12000g for 40 minutes. Hence, the supernatant was loaded on a batch containing 1.2 ml of Ni-NTA agarose resin (nickel-nitroacetic acid metal-affinity chromatography matrice is specific for biomolecules which have been tagged with 6 consecutive histidine residues, as cN-II; figure 2.2.1.1; refer to paragraph 2.1.1) at 50% and maintained shaking on ice for one hour. The resin was, then, washed thrice with 5 ml of wash buffer (Tris-HCl 50mM pH 8.0, NaCl 300mM, imidazole 20 mM). Finally, the recombinant proteins were eluted from resin with 500 l of elution buffer (Tris-HCl 50 mM pH
8.0, NaCl 300 mM, imidazole 250 mM). The protein thus purified was kept at +4°C. Physical purity of the protein was evaluated by its electrophoretic homogeneity through
SDS-PAGE, which was performed essentially according to Laemmli (Laemmli, 1970). The protein concentration was determined according to Bradford, using BSA as standard (Bradford, 1976).
Figure 2.2.1.1: Interaction between Ni-NTA and a 6xHis-tagged protein. NTA is a tetradentate chelating adsorbent that occupies four of the six ligand binding sites in the coordination sphere of the nickel ion, leaving two sites free to interact with the 6xHis-tag. As reported in paragraph 2.1.1, pET-28c carries an N-terminal His Tag-thrombin-T7 Tag configuration. The 6xHis-tagged cN-II was, thus, purified using the Ni-NTA Agarose method, but without cleaving the His-tag by using thrombin, as this protease seems to cleave cN-II even if it doesn’t carry any thrombin consensus sequence. It is worthy of remarking that the presence of the tag doesn’t influence any properties of this enzyme.
81 As regard purification of cN-II from Saccharomyces cerevisiae RS112 strain (read further for more information), partially according to Invitrogen manual (Catalog no. V825-20), cell pellet provided by Dr. A. Galli was resuspended in the mininum volume of TRIS-HCl 100 mM pH 7.4 and then submitted to 3 freeze/thaw cycles; hence, an equal volume of acid-washed glass beads was added and the mixture thus formed was vortexed for 30 seconds - followed by 30 seconds on ice - at least four times. Cell lysate, then, was loaded on a batch containing 500 l of Ni-NTA agarose resin per 100 l of cell lysate. The recombinant enzyme was eluted from resin with the minimum volume of elution buffer (Tris-HCl 50 mM pH 8.0, NaCl 300 mM, imidazole 250 mM). The protein thus purified was kept at -20°C. Also in this case, protein purity was evaluated by its electrophoretic homogeneity and protein concentration was determined according to Bradford, using BSA as standard (Laemmli, 1970; Bradford, 1976).
2.2.2 Preparation of cell crude extract
Regarding mammalian cell lines (HEK 293T, ADF), cells were first trypsinized and centrifugated for 5 minutes at 1000g; then the pellet was resuspended in the minimum volume of TRIS-HCl 100 mM pH 7.4 and transferred in a 1.5 ml tube. Crude extracts were, thus, obtained by 3 freeze/thaw cycles followed by centrifugation at 10000g at 4°C for 40 minutes to pellet cell debris. Supernatants were analysed for cN-II activity, immunoblotting or stored at -80°C.
2.2.3 cN-II assays
As I have already underlined, the cytosolic 5’-nucleotidase II is a bifunctional enzyme, being able to transfer the orthophosphate (phosphatase activity) from a 6-hydroxypurine 5’-monophosphate not only to water (acting as a nucleotidase), but also to the 5’ position of a purine acceptor (acting as a phosphotransferase) by a ping-pong reaction mechanism (refer to chapter “Introduction”, paragraph 1.3.2 and figure 1.3.2.1). Thus, theoretically, it is possible to follow the nucleotidase, the phosphotransferase and the phosphatase activity measuring - respectively - the rate of formation of the nucleoside, of the nucleoside 5’-monophosphate and of orthophospate in the reaction mixture. This is true for a purified cN-II; in fact, this concept can’t be applied to a cell crude extract, which contains phosphatases and nucleotidases whose substrate specificity overlap that of cN-II. Anyway, as cN-II is the only enzyme able to phosphorylate inosine, it is possible to evaluate phosphotransferase activity even in a crude extract.
82 Each kinetic parameter (Km, K50) was measured at least thrice in the same conditions through no less than seven experimental points, using appropriate concentration ranges for the substrates and for the effectors.
One unit of enzyme activity is the amount of enzyme required to convert 1 mol of substrate to product/min under the assay conditions.
When testing a crude extract, we used 20-40 g of protein, while assaying purified enzyme we used 20-40 ng of protein.
2.2.3.1 5’-nucleotidase activity assay
cN-II was assayed as 5’-nucleotidase activity measuring the rate of the [8-14C] Inosine synthesis from [8-14C] IMP and unlabelled inosine. Incubation were performed at 37°C in a medium containing ATP 5 mM, MgCl2 20 mM, [8-14C] IMP 2mM (4500 dpm/nmol), DTT 1 mM, TRIS-HCl 100 mM pH 7.4, Inosine 1.4 mM in a total volume of 50 µl. At different time intervals (0, 10’, 20’, 30’), the reactions were stopped by rapidly drying portions of 10 µl of the incubation mixture on PEI-cellulose pre-coated thin-layer plastic sheets; then a chromatogram was developed in water to separate Inosine from IMP. In this separation, Inosine standards were used and detected as ultraviolet adsorbing areas, which were excised and counted for radioactivity with 4 ml Hi safe II Scintillation liquid (Tozzi et al., 1991).
2.2.3.2 Phosphotransferase activity assay
cN-II was assayed as phosphotransferase activity measuring the rate of [8-14C] IMP formation from [8-14C] Inosine and unlabelled IMP. Incubations were performed at 37°C in a medium containing ATP 5 mM, MgCl2 20 mM, IMP 2 mM, DTT 1 mM, TRIS-HCl 100 mM pH 7.4, [8-14C] Inosine 1.4 mM (4500 dpm/nmol) in a total volume of 50 µl. The reaction was stopped at different time intervals (0, 10’, 20’, 30’) by spotting 10 µl of the incubation medium on DE-81 paper disks, which were, then, washed once for 15 minutes in 1 mM ammonium formate and twice for 10 minutes in water. The disks were dried and placed in counting vials filled with 4 ml of Hi safe II Scintillation liquid (Tozzi et al., 1991).
2.2.3.3 Free phosphate colorimetric detection
The rate of IMP phosphorolysis was monitored according to Chifflet method, a colorimetric assay used to detect inorganic phosphate in solution; if, in the reaction mixture is present also a nucleoside acceptor as inosine, the rate of orthophosphate synthesis corresponds to the phosphatase
83 activity net of phosphotransferase one (Chifflet et al. 1988). Previously a calibration curve was constructed: colour development was detected as absorbance change at =850 nm and was function of the known Pi concentration. Blank consisted in a mixture assay without enzyme.
For this method, the following solutions are required: Solution A: 12% SDS;
Solution B: 6% Ascorbic acid in HCl 1M (prepared at the time of use); Solution C: 1% Ammonium molybdate;
Solution E: 2% Sodium citrate, 2% sodium metaarsenite, 2% acetic acid.
Enzyme incubation were performed at 37°C in a medium containing 2,3-BPG 4.5 mM, MgCl2 20 mM, IMP 2 mM, TRIS-HCl 100 mM pH 7.4 - in the presence/absence of Inosine 1.4 mM - in a total volume of 180 µl. At different time intervals (0, 15’, 30’), 50 µl of the mixture were taken off from the incubation mixture and the reaction was stopped by addition of solution A (50 µl), followed by vortexing. Then, 100 µl of solution BC (1:1) were added in order to complex Pi with ammonium molybdate and, after six minutes of incubation at room temperature, 150 µl of solution E were added to mixture in order to remove the excess of ammonium molybdate. After 10 minutes at 37°C, there was an incubation at room temperature for 5 minutes and then the colour absorbance was read at 850 nm.
2.2.3 Western and Immnoblotting
After SDS-PAGE, performed following the method of Laemmli (as far as the quantities of protein applied and the percentage of polyacrylamide used for the gel refer to the specific cases in chapter “Results”), Western blotting was performed using PVDF membrane (90 Volts for one hour or one hour and a half in the case of gel 0.75 mm- or 1.00 mm-thick, respectively; transfer buffer: [TRIS 0.025 M + Gly 0.129 M] : Methanol : MilliQ water = 1 : 1 : 3) and processed for immunodetection (Laemmli, 1970). Membrane was incubated with primary chicken anti-human cN-II antibody preparation (1:1000) overnight at 4°C, after having been washed for one hour and a half in milk 5% in PBS+Tween-20 0.05% with shake. Then, after a cycle of washes (5’ in PBS; 5’ in PBS+Tween 20 0.05%; 5’ in PBS), immunoreactive band was performed by using rabbit anti chicken IgG horseradish peroxidase-conjugated (1:100000) for one hour and a half in ice. Visualization was carried out using ECL kit reactives, after another cycle of washes.
As regards the visualization of -actin immunoreactive band, after the Western blotting, PVDF membrane - washed twice in PBS for 5 minutes - was incubated with primary mouse anti-human -
84 actin antibody preparation (1:10000), shaking, at room temperature for one hour. Then it was submitted to a cycle of washes (5’ in PBS; 5’ in PBS+Tween 20 0.05%; 5’ in PBS) and incubated with rabbit anti-mouse IgG horseradish peroxidase-conjugated (1:2,000), shaking, for one hour at room temperature. After two washes in PBS, immunoreactive band was performed with ECL kit reactives.
2.2.4 Gel filtration chromatography
Preliminary studies to evaluate the molecular mass of some recombinant mutant proteins were performed by gel filtration chromatography on Sephacryl S-300, loading 2 mg/800 l of purified protein in a column of 1.2 cmx90cm (Running Buffer: TRIS-HCl 50 mM, NaCl 200 mM, pH 7.4; flow rate of 20 ml/h). Fractions of 1.95 ml were collected. As molecular standards, we used: thyroglobulin (6.6x105 Da), apoferritin (4.4x105 Da) and alcohol deydrogenase (1.5x105 Da). V0 was evaluated using blue dextran ( 2x106 Da ). Elution profile was built by Abs280nm measurements; activity profile was measured as the rate of IMP production in the presence of inosine (phosphotransferase activity).
Then, gel-filtration chromatography was performed on a FPLC system utilizing a Superdex-200 column (1.2cmx32 cm). Purified recombinant WT or mutant cN-II (150 g) was loaded onto the column and the chromatography was performed at a flow rate of 0.3 ml/min using the same running buffer described above. Fractions of 0.1 mL were collected. As molecular standards, we used: thyroglobulin (6.6x105 Da), apoferritin (4.4x105 Da), -amylase (2.1x105 Da), alcohol deydrogenase (1.5x105 Da) and BSA (0.6x105 Da). V0 was evaluated using blue dextran ( 2x106 Da). Elution profile was built by Abs254nm measurements; activity profile was Measured as the rate of IMP production in the presence of inosine (phosphotransferase activity).
For the study of the correlation between oligomerization extent of cN-II and presence of the effectors, the column was equilibrated in Tris-HCl 50 mM, NaCl 200 mM pH 7.4 + ATP 5 mM/Pi 5 mM and the recombinant WT cN-II was incubated for 2 hours at 4°C in the same buffer.
As regard the treatment of recombinant WT cN-II in order to evaluated an enigmatic chromatographic peak, we incubated 115 g of protein with 0.5 l DNase (10 mg/ml in TRIS-HCl 50 mM, NaCl 150 mM) and 10.5 l RNase (10 mg/ml in H2O nuclease free in the presence of MgCl2 5 mM) for 2 ½ hours at 25°C.
85
2.2.5 Discontinuous native gradient electrophoresis
In order to determined, unambiguously, the relationship between the quaternary structure of WT/mutant cN-II and enzymatic activity, I tweaked a method of detection of 5’-nucleotidase activity after discontinuous native gradient electrophoresis. Hereinafter, I will report the final
protocol; then, as both the optimization of the native gradient electrophoresis and the tweak of 5’-nucleotidase detection took a rather long time, I will explain how we obtained a good protocol.
3-15% Polyacrylamide gradient gel was made in TRIS-HCl 100 mM pH 8.8. Sample volumes corresponding to 30 g of proteins were loaded (sample loading buffer 5X: Tris 500 mM pH 8.0, glycerol 50%). Run was performed for 4 hours, at 330 Volts at 4°C using two different buffers (Cathode Buffer: TRIS-His 100 mM pH 8.0; Anode Buffer: TRIS-Cl 100 mM pH 8.8.). Then, after Western blotting (read paragraph 2.2.3), the PVDF membrane was incubated, at 37°C for 90 minutes, in a reaction mixture containing IMP 2.0 mM, 2,3-BPG 4.5 mM, MgCl2 20.0 mM in TRIS-HCl 100 mM pH 7.4 to a final volume of 2.0 ml. Then the active bands were picked out incubating the membrane in 4 ml of BC solution (the same described in paragraph 2.2.3.3; that is 1:1=6% Ascorbic acid in HCl 1M:1% Ammonium molybdate) for 6 minutes, at room temperature, shaking; thus, 6 ml of solution E (2% Sodium citrate, 2% sodium metaarsenite, 2% acetic acid) were added; after 10 minutes at 37°C, the bands were evident. We have tried to detect 5’-nucleotidase on the gel adopting the method described by Wu and colleagues, but we obtained very poor results, maybe due to the fact that we use a 1.00 mm-thick gel instead of 0.50 mm-thick one as them (Wu et al., 1999).
The first hump to get over was making recombinant cN-II entry inside the gel; we tried using different percentage of polyacrylamide (starting from 6%) and different buffers (the cathode/anode buffer described above and borrowed from Niepmann and the continuous buffer system TRIS-HCl 200 mM pH 8.0; Niepmann et al., 2006; Niepmann, 2007; Current Protocol in Protein Science, 2003). To be sure to have choose the right pH for the running buffer we confirmed the theoretical pI (6.1) of cN-II by a 2-D electrophoresis performed by Dr E. B. Maserti e Dr A. Podda of the Institute of Biophysics, CNR, Pisa (figure 2.2.5.1 A). The only way to solve this step was to use a discontinuous electrophoresis using the above-mentioned cathode and anode buffers and a percentage of polyacrylamide for the lower extremity of the gel gradient not greater than 4%.
Another hang-up point was the use of Serva Blue, which confers negative charges to the protein without denaturing it. At the beginning, we decided to use it in order to promote the entrance of cN-II inside the gel; but it left a deep blue background on the gel that make difficult to read the band(s). Hence, we decide not to use it.
86 After that, two parameters remained to consider: the polyacrylamide gradient gel and the time/voltage needed for the electrofocusing. After several attempts, we were able to assess that running at 330 Volts for 4 hours on a 3-15% polyacrylamide gradient gel was the ideal condition for a good resolution and clear interpretation of the results. Figure 2.2.5.1 summarizes the main efforts which led to the tweak of this technique.
Figure 2.2.5.1: A. 2-D electrophoresis (9% polyacrylamide gel) performed by Dr E. B. Maserti e Dr A. Podda; the pI value of cN-II is 6.1; B. Experimental excursus followed to optimized the discontinuous native gradient electrophoresis protocol.
Regarding basic biochemistry techniques, as determination of protein concentration, SDS-PAGE electrophoresis, recipes of different solutions (PBS, SDS-PAGE running buffer etc.) refer to Current Protocol in Protein Science.
2.3 Cell biology methods
2.3.1 Cell culture
293T (a human kidney epithelial cell line, also named HEK) cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% Foetal Bovine Serum (FBS), 2 mM L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin. ADF (astrocytoma cell line) cells
were maintained in RPMI medium supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin. Both cell lines were grown at 37 °C in a humidified 5% CO2/95% air atmosphere.
To induce cN-II silencing, doxycycline - a semisynthetic tetracycline - was used, at the concentrations and for the times specified in chapter “Results”. All induction experiments were performed in complete medium supplemented with 10% doxycycline-free FBS. As I will describe up ahead, in some experiments FBS added to the medium was dialysed (cut-off 10000 Da) against
87 physiological solution (NaCl 0.9 M); more, when specified, cells were starved prior to induction.
Starvation was performed culturing cells in medium supplemented with 2% FBS, 2 mM L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin for 24 hours and, subsequently, in
complete medium without FBS for one hour.
Cells were routinely tested for Mycoplasma contamination.
2.3.2 Lentivirus production
At the beginning, I tried to transfect 293T cells with the three vectors described before (transgene, packaging and envelope vectors) with a cheap and efficient method, that is the calcium-phosphate-mediated transfection (Soneoka et al., 1995; Naldini et al., 1996; Sakoda et al., 1999). In our hands, this system gave very poor results, even in the presence of sodium butyrate (Gorman et al., 1993 a/b).
Thus, after having improved the quality of the plasmidic purifications and checked again the identity of the vectors used, I changed transfection method, adopting the PEI (polyethylenimine)-mediated transfection protocol.
Figure 2.3.2.1: DNA uptake inside the cell. The transfection efficiency of PEI partially relies on its ability to capture the protons which are transferred into the endosomes during their acidification; PEI, in fact, possesses substantial buffering capacity below physiological pH (every third atom of PEI is a protonable amino nitrogen atom), which makes the polymeric network an effective "proton sponge" at virtually any pH. This means extensive lysosome buffering that protects DNA from nuclease degradation and consequent lysosomal swelling and rupture that provide an escape mechanism for the PEI/DNA particles (“proton sponge hypothesis”; Boussif et al., 1995; Kichler et al., 2001; Akinc et
88 PEI is a polycationic molecule: there are no evidences demonstrating the way it promotes DNA uptake by cells; it is hypothesized that endocytosis may be the mechanism responsible for this phenomenon (read the caption of figure 2.3.2.1 for more details; Schlaeger and Christensen, 1999). The day before transfection, 3x106 293T cells per 10-cm plate were seeded in 10 ml of DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin and grown at 37 °C in a humidified 5% CO2/95% air atmosphere. After 24 hours, the medium was replaced with 5 ml of DMEM supplemented with 2 mM L-glutamine and a mixture prepared at the moment and composed by PEI and plasmids (PEI 10 M; 20 g of pLVCT-tTR-KRABsh661/shCTR; 10 g of psPAX2; 5 g of pMD2.G in NaCl 0.15 M; total volume of 1.4 ml; the mixture was incubated for 15 minutes at room temperature before use) was added drop-wise to the plate. After six hours, the medium was changed again and fresh DMEM supplemented with 10% FBS, 2mM L-glutamine, 100 U/ml Penicillin and 100 μg/ml Streptomycin was added. Two days later, the medium was collected, clarified by centrifugation at 1500g for ten minutes and 0.45
m filtered. The viral surnatant was stored at -80°C.
To evaluate the efficiency of transfection, 293T cells treated as described above were analyzed on a FACScan using a CELLQuest Version 2 (BD Biosciences, Milan, Italy) which let us to measure the percentage of GFP-positive cells.
The viral titer we obtained was very low; even centrifugation for 20 minutes at 3000g using vivaspin column with a 100000 cut-off did not improve it, on the contrary viral particles were lost during the process. Anyway, spinoculation was not necessary for transduction.
2.3.3 Cell transduction
Twenty-four hours prior to transduction, target cells (293T or ADF) were split into 24-well tissue culture plate (50000-70000 cells per well) in 1 ml of the relative complete medium. Then, medium was replaced with 1 ml of viral supernatants (adding the cross-linker molecule polybrene - 8 g/ml - did not enhance transduction efficacy, probably because it is a VSV-G-pseudotyped virus; Pluta and Kacprzak, 2009). After 72 hours from infection, cells were harvested and analyzed to evaluate the percentage of GFP-positive cells as described above.
As regards transduced 293T cells, individual clones were selected by serial dilution of cells in 96-well plates. Transduced ADF cells, instead, were submitted to fluorescence-activated cell sorter (FACS) analysis in the laboratory of Professor F. Annunziato, Laboratory of Immunology, University of Florence.
Figure 2.3.3.1 summarises the experimental pathway we carried out from virus production to cell transduction.
89 Figure 2.3.3.1: Schematic of non-replicating lentiviral vector for stable shRNA expression. Suitable host cells (in our case 293T cells) are transfected with a mixture of plasmids consisting of an shRNA expression cassette (pLVCT-tTRKRAB derivative), a packaging cassette (psPAX2) and a heterologous viral envelope expression cassette (pMD2.G). The generated lentivirus is then used to transduce the desired cell type for shRNA expression (in our case, 293T and ADF). Because only the vector containing the shRNA expression cassette (devoid of the viral structural genes) integrates into the host cell genome in the transduced cells, shRNA is continually expressed but infectious virus is not produced (from Manjunath et al., 2009).
2.3.4 MTT assay
To evaluate cell viability, we chose MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Mosmann, 1983). The method is based on the ability of metabolically active cells to reduce the MTT tetrazolium salt, water-soluble, into a purple-coloured, water-insoluble MTT formazan salt by the mitochondrial enzyme succinate dehydrogenase.
Dox-induced 293T/ADF cells were seeded in 96-multi well plate at 2.5×104 cells/5×104 per well in 100 l of complete medium in order to have 6 wells per experimental condition. Then, 10 l of MTT (5 mg/ml) was added to the culture medium of each well and cells were incubated at 37°C/5%CO2 for one hour (293T)/two hours (ADF); the reaction was stopped by adding 100 μl of 0.04 N HCl in isopropanol and the formazan salts were dissolved by gentle shaking for 10 minutes
at 37°C. Formazan salts were quantified spectrophotometrically by reading the absorbance at 570 nm.
90 Moreover, GFP-CertifiedApoptosis/Necrosis detection kit (Enzo Life science) was used for wide field fluorescence microscopy according to the manufacturer.
Regarding basilar cell biology techniques, as Mycoplasma test, freezing/thawing cells, refer to Cell and tissue: laboratory procedures in biotechnology (1999).
2.4 Methods in collaboration
As regard the experimental branches carried out in collaboration with and by other laboratories, afterwards I will report few information and the relative bibliography.
- 2-D electrophoresis was performed by Dr. E. B. Maserti e Dr. A. Podda according to Maserti et al. (Maserti et al., 2011).
- Light scattering experiments were performed by Dr. G. Strambini and Dr. M. Gonnelli as described in the article by Pesi et al. (Pesi et al., 2010).
- Bovine recombinant cN-II cloning inside pYES2 vector was carried out by Dr. S. Allegrini (figure 2.4.1). Yeast (S. cerevisiae, diploid strain RS112 genotype: MATa/MAT ura3-52/ura3-52 leu2-3,112/leu2-98 trp5-27/TRP5 ade2-40/ade 2-101 ilv1-92/ilv1-92 arg4-3/ARG4 his3 5’-pRS6-his33’/his3-200 LYS2/lys2-801) transformation, growth and induction were performed by Dr. A. Galli as described by Collavoli et al. (Collavoli et al., 2008; Caligo et al., 2009).
Figure 2.4.1: A. Primers used to clone cN-II cDNA into pYES2 vector; B. PYES2 vector (GAL1 promoter permits inducible expression of genes cloned into the vector, in green; CYC1 transcription termination signal allows efficient termination and stabilization of mRNA; pUC origin for the maintenance and high copy replication in E. coli; ampicillin resistance gene for selection of transformants in E. coli; URA3 gene for selection of yeast transformants in uracil-deficient medium, in red; 2μ origin for the maintenance and high copy replication in yeast; f1 origin the rescue of single-stranded DNA).