Indice
Pagina
1. Prevenzione del Diabete Mellito tipo 2 ……… 2
2. Geni e Prevenzione ………. 8
3. Genetica del diabete tipo 2 ………. 14
- Diabete monogenico ……… 14
- Diabete neonatale ………. 15
- Maturity-onset diabetes of the young ……….. 19
- Altre forme di diabete monogenico ………. 20
- Diabete tipo 2 multifattoriale ……….. 20
4. Geni candidati ………. 23 - PPARγ ……….. 26 - KCNJ11 – KCNQ1 ……… 27 - Geni MODY ……….. 28 - WFS1 ……….…………. 29 - ENPP1 ………..…….. 30 - CAPN10 ……… 31 - Cromosomi 20 e 1 ………. 31 5. Il gene TCF7L2 ……….. 33
6. Studi di associazione in tutto il genoma (GWAS) ………. 39
7. Geni e terapia del diabete ………. 51
Bibliografia ……….. 58
Parte Sperimentale Il polimorfismo del gene TCF7L2 è associato a ridotta secrezione insulinica in soggetti a rischio di diabete ………. 79
Introduzione ………. 80
Soggetti e Metodi ……….. 82
- Genotipizzazione ………..………. 83
- Analisi statistica ……… 83
Risultati ………. 85
Caratteristiche cliniche in accordo con il genotipo TCF7L2 ….. 87
Discussione ……….. 91
1. Prevenzione del Diabete Mellito tipo 2
Il diabete mellito è una delle malattie croniche più frequenti. È la 4ª-5ª causa di morte in molti dei Paesi sviluppati e presenta diffusione epidemica in molti Paesi industrializzati e in quelli di recente sviluppo. La sua prevalenza è aumentata in tutto il mondo nel corso degli ultimi decenni; si stima, infatti, che i pazienti diabetici fossero 171 milioni nel 2000 ed è previsto un aumento del tasso globale di diabete fino a 221 milioni di pazienti nell’anno 2010 (1) e 366 milioni nel 2030 (2). Anche i costi economici e sociali del diabete mellito e delle sue complicanze sono elevati ed aumenteranno enormemente (3). Pertanto la diagnosi e la prevenzione precoci sono importanti per ridurre una delle maggiori voci di spesa sanitaria.
La prevenzione primaria del diabete mellito tipo 2 è stata proposta per la prima volta ottanta anni fa ed è stata recentemente sollecitata dall’Organizzazione Mondiale della Sanità (OMS) (4). Sebbene lo sviluppo del diabete mellito tipo 2 sia influenzato dall’interazione tra fattori genetici e ambientali (figura 1), gli interventi di prevenzione oggi adottati non riflettono il contributo dei primi. La strategia attuale di prevenzione si basa essenzialmente sull’identificazione di individui ad alto rischio ed è appropriata in comunità di rischio genetico da basso a moderato in cui la maggioranza della popolazione non svilupperà la malattia.
Gli studi di prevenzione del diabete tipo 2 recentemente svolti hanno suggerito che le strategie preventive si dovrebbero concentrare sui soggetti con ridotta tolleranza al glucosio [IGT- Impaired Glucose Tolerance: glicemia due ore dopo carico orale di glucosio ≥ 140 e < 200 mg/dl (5)] o alterata glicemia a digiuno [IFG- Impaired Fasting Glucose: glicemia a digiuno ≥
Figura 1. Interazione gene-ambiente e fenotipo del diabete.
100 e < 126 mg/dl (5)] (6) ed hanno evidenziato come un intervento efficace sullo stile di vita possa migliorare la sensibilità insulinica, ridurre la glicemia a digiuno e post-prandiale, diminuire i livelli di acidi grassi liberi, migliorare il profilo lipidico e ritardare o prevenire la progressione da IGT a diabete. In effetti, gli interventi messi in atto consistono nel miglioramento dello stile di vita e nell’impiego di farmaci; entrambi gli interventi tendono a ridurre l’insulino-resistenza e a promuovere e sostenere la funzione della β-cellula pancreatica. L’adozione di un corretto stile di vita significa ridurre il peso corporeo di almeno il 5%, eseguire un’attività fisica aerobica di moderata intensità e della durata di almeno 20-30 minuti al giorno o 150 minuti alla settimana ed acquisire abitudini alimentari corrette. La maggior parte delle evidenze disponibili indica che è più importante la qualità piuttosto che la quantità totale dei nutrienti: in particolare, gli acidi grassi saturi aumentano il rischio di diabete tipo 2, mentre la parziale sostituzione di questi con acidi grassi insaturi (poli- e monoinsaturi) lo riduce (7). Un discorso a parte meritano gli acidi grassi ω3 e/o il consumo di pesce,
F
F
e
e
n
n
o
o
t
t
i
i
p
p
o
o
d
d
i
i
a
a
b
b
e
e
t
t
i
i
c
c
o
o
G
G
e
e
n
n
i
i
Trait intermedi: massa β-cellulare, secrezione insulinica, azione dell’insulina, distribuzione dell’adipe, obesità
A
sembrerebbe, infatti, che il pesce abbia un effetto protettivo nei confronti del diabete tipo 2. Per quanto riguarda i carboidrati, la maggioranza degli studi epidemiologici osservazionali suggerisce che una dieta ricca in fibre e in alimenti a basso indice glicemico sia protettiva nei confronti del rischio di diabete tipo 2. L’intervento farmacologico si basa sull’impiego di farmaci ipoglicemizzanti orali come metformina, tiazolidinedioni, acarbosio oppure sull’impiego di altri farmaci, in particolare l’orlistat.
I due studi più recenti di prevenzione primaria del diabete tipo 2 sono il Finnish diabetes prevenction study (FDPS) ed il Diabetes Prevention Program (DPP); essi hanno mostrato che il cambiamento dello stile di vita è efficace nel prevenire lo sviluppo di diabete mellito in soggetti IGT. Lo studio FDPS ha valutato gli effetti della dieta e dell’esercizio fisico sulla progressione da IGT a diabete in 552 soggetti di mezza età, sovrappeso (BMI ≥ 25 Kg/m2) e con IGT, che sono stati seguiti
per 3,2 anni. I soggetti sono stati randomizzati in due diversi tipi d’intervento sullo stile di vita. Al gruppo di controllo sono state date istruzioni generali sulla dieta e sull’esercizio fisico mentre al gruppo d’intervento sono stati dati consigli individualizzati volti alla riduzione del peso corporeo, all’alimentazione corretta, allo svolgimento di un’adeguata attività fisica. L’incidenza cumulativa del diabete dopo 4 anni è stata dell’11% nel gruppo d’intervento e del 23% nel gruppo di controllo (58% di riduzione del rischio di diabete) (8). Nello studio DPP, 3234 individui con IGT o IFG e BMI > 24 Kg/ m2 sono stati seguiti per una media di 3 anni. I
soggetti sono stati randomizzati in tre gruppi: placebo, metformina, programma di modifica dello stile di vita (riduzione del peso corporeo ed attività fisica). In questo studio si è osservata una riduzione nel rischio relativo di sviluppare diabete
pari al 58% nel gruppo d’intervento sullo stile di vita in confronto al gruppo trattato con placebo.
Anche gli studi d’intervento farmacologico hanno dimostrato una riduzione nello sviluppo del diabete mellito tipo 2. Nello studio DPP, la metformina ha ridotto il rischio relativo di diabete del 31% in confronto al placebo (9). Più recentemente, l’uso della metformina è stato valutato anche in uno studio indiano con risultati qualitativamente simili a quelli ottenuti nel DPP (10). È interessante sottolineare che in questo studio la combinazione metformina + modifiche dello stile di vita non era più efficace dei due interventi praticati isolatamente.
Nello studio Troglitazone in the Prevention of Diabetes mellitus (TRI-POD), il troglitazone si è rivelato in grado di prevenire l’insorgenza di diabete nel 58% in confronto al placebo in donne ispaniche con precedente storia di diabete gestazionale (11), sebbene lo studio sia stato terminato precocemente a causa della tossicità epatica fatale del troglitazone. Lo studio DREAM ha valutato l’impiego del rosiglitazone in soggetti a rischio. In questo studio sono stati arruolati 5269 soggetti (età >30 anni) con IGT o con IFG. I soggetti sono stati assegnati al gruppo placebo o al gruppo rosiglitazone. Il farmaco ha ridotto il rischio di sviluppare il diabete del 60%, in maniera statisticamente significativa rispetto al placebo (12). Lo studio multicentrico STOP NIDDM (Study TO Prevent Non-Insulin Dependent Diabetes Mellitus), della durata di tre anni, è stato condotto su 1429 soggetti con IGT randomizzati in due gruppi: 715 individui sono stati trattati con acarbosio (inibitore della α-glicosidasi intestinale, che riduce l’iperglicemia post-prandiale) e 714 con placebo. Questo trial ha rivelato una riduzione del rischio relativo di sviluppare diabete del 25% nel gruppo trattato con acarbosio rispetto ai soggetti trattati con placebo. Alla fine dello studio, i
pazienti sono stati valutati dopo un periodo di sospensione del trattamento (farmaco o placebo) di circa 3 mesi, durante i quali il 15% dei pazienti trattati con acarbosio ha sviluppato diabete rispetto al 10,5% dei pazienti di controllo. Questi risultati hanno dimostrato che l’intervento farmacologico con acarbosio nei pazienti con IGT può ritardare la progressione verso il diabete mellito. Questo effetto, però, scompare alla sospensione del trattamento (13). Lo studio XENDOS (XENical in the prevention of Diabetes in Obese Subjects) ha valutato il ruolo dell’orlistat (un inibitore della lipasi gastrica e pancreatica che limita del 30% l’assorbimento dei lipidi introdotti con l’alimentazione) nella prevenzione del diabete mellito tipo 2 in soggetti obesi (BMI>30 kg/m2), dei quali il 21% con IGT. In questo studio il farmaco è
stato associato a dieta ed esercizio fisico. Dopo 4 anni di terapia, si è registrata una riduzione complessiva del 37% del rischio di diabete, che nei soggetti con IGT ha raggiunto il 45% (14).
Da questi studi appare evidente che il diabete mellito tipo 2 può essere prevenuto e che sia il cambiamento dello stile di vita, sia l’intervento farmacologico sono misure efficaci di prevenzione. Rimane da determinare se il risultato sarebbe simile in altri gruppi di rischio per il diabete mellito tipo 2, ma si ritiene ragionevole ipotizzare che gli interventi proposti siano efficaci anche in altre categorie a rischio di diabete (ad esempio in soggetti con IFG, obesità, familiarità per diabete, ecc.); tuttavia al momento non sono disponibili dati che permettano di valutare il bilancio costo-beneficio dell’implementazione di programmi di prevenzione in categorie diverse dall’IGT (5).
Nel DPP inoltre è stato effettuato un confronto fra due tipi di intervento terapeutico (farmacologico e sullo stile di vita) da cui è emerso che le modificazioni dello stile di vita hanno un’efficacia
circa doppia nel prevenire il diabete rispetto all’intervento con metformina (riduzione relativa rispettivamente del 58% e del 31%). Quest’approccio terapeutico, oltre a prevenire (o ritardare) l’insorgenza di diabete mellito tipo 2, ha anche altri effetti benefici: riduzione della pressione arteriosa, riduzione del livello di trigliceridi, aumento del colesterolo HDL plasmatico. Ciò deve quindi spingere gli operatori sanitari e lo stesso Sistema Sanitario ad incoraggiare, in ogni occasione, l’acquisizione di uno stile di vita più salutare, in particolare nei pazienti a rischio di diabete.
2. Geni e Prevenzione
Il diabete mellito tipo 2 è considerato una malattia poligenica dal momento che la suscettibilità individuale allo sviluppo del diabete dipende da numerose varianti geniche o combinazioni di varianti. Una malattia di questo tipo è, spesso, indicata anche come complessa, a causa del suo pattern di ereditarietà e, inoltre, la suscettibilità genetica al diabete tipo 2 è un requisito necessario, ma non sufficiente, allo sviluppo della malattia che richiede la compartecipazione di fattori ambientali; questi ultimi appaiono come l’elemento scatenante della malattia. A causa di queste interazioni tra fattori genetici e ambientali una malattia poligenica complessa può essere indicata anche come multifattoriale.
Esiste un’ampia evidenza che il diabete mellito tipo 2 abbia una forte componente genetica. La concordanza per il diabete mellito tipo 2 in gemelli monozigoti è approssimativamente del 70% in confronto al 20-30% dei gemelli dizigoti (15,16). Poiché la penetranza della malattia aumenta con l’aumentare dell’età, è chiaro che, più lungo è il follow-up, più è alto il tasso di concordanza (16). Il diabete mellito tipo 2 tende ad aggregarsi in gruppi familiari. Il rischio di sviluppare la malattia nel corso della vita è di circa il 40% nei figli di un genitore con diabete mellito tipo 2 (17), maggiore se ne è affetta la madre (18), il rischio si avvicina al 70% se entrambi i genitori hanno il diabete. Un parente di primo grado di un paziente con diabete mellito tipo 2 ha un rischio circa tre volte maggiore di sviluppare la malattia rispetto alla popolazione generale (19). Anche le differenze etniche circa la prevalenza del diabete tipo 2 sono state attribuite a componenti genetiche (20,21).
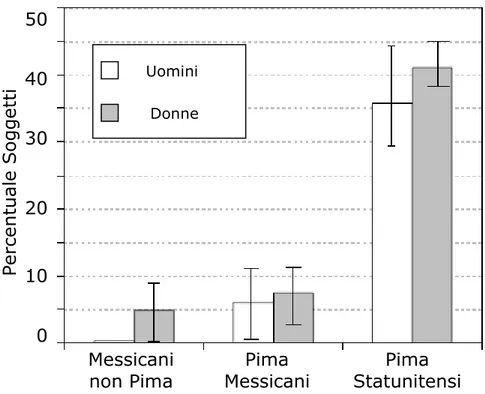
L’acquisizione di uno stile di vita occidentale gioca un ruolo chiave nell’incremento epidemico della prevalenza del diabete tipo 2 nelle varie parti del mondo (figura 2). Questo cambiamento si è verificato negli ultimi 50 anni. Chiaramente, durante questo periodo, non si è verificata una modificazione del background genetico, ma questo non esclude un ruolo importante dei geni nel rapido incremento di prevalenza della malattia, poiché i geni o le varianti di un gene sono in grado di determinare la risposta dell’individuo nei confronti di determinanti ambientali.
Figura 2. Effetti dell’ambiente tradizionale ed occidentale sulla prevalenza del diabete tipo 2 negli Indiani Pima in Messico e negli Stati Uniti.
Diabetes Care, Volume 29, August 2006, 1866-1871.
Uomini Donne
Messicani
non Pima Messicani Pima Statunitensi Pima 50 40 30 20 10 0 Pe rc en tu al e S o g g et ti
Per spiegare come si realizza l’interazione tra geni ed ambiente sono state proposte varie ipotesi, tra cui quella del gene risparmiatore. Neel (22) ha suggerito che gli individui che vivono in un ambiente con apporto di cibo incostante, come nomadi e cacciatori, avrebbero maggiori probabilità di sopravvivenza se avessero a disposizione maggiori riserve di energia. In tali ambienti, la selezione genetica favorirebbe i genotipi in grado di utilizzare substrati energetici in modo economico, conservandone più grandi quantità, preservando il glucosio per le richieste del cervello in condizioni di mancanza di cibo; tutto ciò condurrebbe ad una propensione genetica verso l’insulino-resistenza dei tessuti periferici. Inoltre sappiamo che il grasso, specialmente il grasso intra-addominale, è una forma di accumulo di energia più efficiente rispetto al glicogeno muscolare ed epatico. Questa tendenza si osserva nei fenotipi insulino-resistenti e spiega perché la prole di soggetti con diabete mellito tipo 2 abbia un accumulo più precoce di grasso centrale (18). Nel mondo occidentale, infatti, il cibo è, in generale, facilmente disponibile e abbondante, per cui i cosiddetti geni ‘risparmiatori’ sono male-adattabili alla società moderna e potrebbero contribuire alla suscettibilità per l’obesità e per il diabete tipo 2. Comunque, sebbene queste teorie evoluzionistiche, che si concentrano su potenziali vantaggi per la sopravvivenza conferiti da geni risparmiatori, divenuti adesso male-adattabili, sono di grande interesse, esse sono speculative e difficili da provare (23).
L’accumulo e il ridotto consumo di energia possono avvenire in più modi: attraverso un metabolismo efficiente nel risparmiare energia; attraverso l’adipogenesi, cioè la propensione a un rapido aumento di peso; in maniera fisiologica, mediante la capacità di far slittare processi non essenziali (come le capacità riproduttive, termogenetiche ed immunitarie); a seguito di una
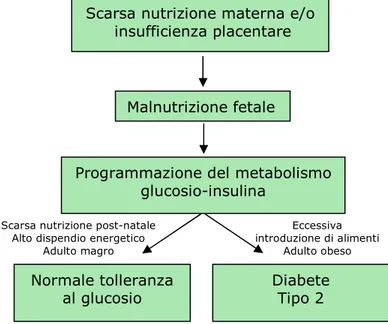
tendenza ad alimentarsi in modo eccessivo in presenza di larghe disponibilità di cibo e infine attraverso la tendenza a risparmiare energia attraverso l’inattività fisica (24). Il risparmio fisiologico probabilmente non è causa di obesità e/o diabete tipo 2, perché la capacità di inibire processi non essenziali, in condizioni di assenza di cibo, non esprime chiaramente una condizione di male-adattamento in caso di normale o eccessiva introduzione di alimenti. Le altre forme di risparmio potrebbero essere imputabili a geni che sostengono un mal adattamento nella società moderna. È stata proposta anche una spiegazione alternativa, la cosiddetta ipotesi del fenotipo risparmiatore che pone l’accento sulla programmazione intrauterina (25) (figura 3). Secondo questa ipotesi, la malnutrizione intrauterina condurrebbe a un basso peso alla nascita ed incrementerebbe il rischio di sindrome metabolica (aggregazione di diversi fattori di rischio cardiovascolare come obesità addominale, dislipidemia, ipertensione e intolleranza al glucosio) nella vita adulta.
Figura 3: Ipotesi del fenotipo risparmiatore.
Scarsa nutrizione materna e/o insufficienza placentare
Malnutrizione fetale
Programmazione del metabolismo glucosio-insulina
Normale tolleranza al glucosio
Trends in Endocrinology e Metabolism
Scarsa nutrizione post-natale Alto dispendio energetico
Adulto magro Eccessiva introduzione di alimenti Adulto obeso Diabete Tipo 2
Quest’ipotesi è stata riprodotta in parecchi studi, nei quali è stato anche dimostrato che il rischio di basso peso alla nascita è aumentato nelle famiglie con sindrome metabolica (26) suggerendo che un basso peso alla nascita possa essere la manifestazione fenotipica di un gene risparmiatore.
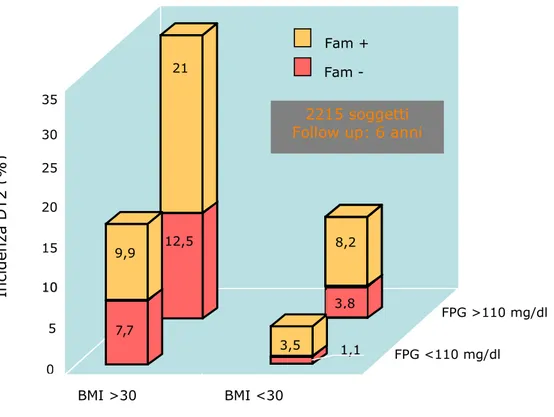
L’importanza di diversi fattori di rischio per il diabete tipo 2 differisce tra le etnie ma l’aumento di rischio associato alla storia familiare sembra l’elemento più costante (19), sebbene il suo effetto relativo tenda a diminuire con l’incremento di frequenza del diabete tipo 2 nelle popolazioni. Il valore predittivo di una storia familiare di diabete mellito tipo 2 è relativamente basso anche in soggetti giovani i cui familiari non hanno ancora sviluppato la malattia (figura 4). Altri fattori di rischio sono un basso livello di attività fisica, l’obesità addominale e la presenza di sindrome metabolica (27,28). Inoltre, un’elevata concentrazione di glucosio di per sé è un forte elemento predittore di sviluppo di diabete tipo 2 (19,29). Nello studio prospettico Botnia, 2115 individui non diabetici sono stati seguiti per sei anni. Ciò ha consentito di dimostrare che questi soggetti, con una storia familiare di diabete tipo 2, con un BMI ≥ a 30 Kg/m2 e una glicemia a digiuno ≥ 100 mg/dl, avevano un rischio
16 volte più alto di sviluppare diabete mellito tipo 2 (19).
Data l’importanza dei fattori genetici per la suscettibilità al diabete mellito tipo 2, si ritiene che la prevenzione possa essere condotta ad un livello superiore, cioè individuando tali geni di suscettibilità. La determinazione delle combinazioni genotipiche del diabete tipo 2 e delle interazioni gene-ambiente potrà, infatti, chiarire il ruolo dei fattori genetici ed ambientali nello sviluppo della malattia, potrà fornire un nuovo strumento per
l’identificazione degli individui ad alto rischio, che possono ricevere maggior beneficio dalla prevenzione, e potrà anche fornire misure preventive migliori e personalizzate, basate sulle informazioni genetiche.
Figura 4 Effetti della familiarità in relazione a Indice di Massa Corporea (BMI) ed glicemia a digiuno (FPG) sul l’incidenza di diabete tipo 2 (19).
0 5 1 100 15 20 25 30 35 FPG <110 mg/dl BMI <30 BMI >30 Fam + Fam - 21 12,5 9,9 7,7 In ci d en za D T 2 ( % ) 8,2 3,5 11,,11 3,8 2215 soggetti Follow up: 6 anni
3. Genetica del diabete tipo 2
L’identificazione e caratterizzazione delle varianti genetiche che causano o predispongono al diabete è uno dei maggiori obiettivi della ricerca biomedica. Già dall’inizio del 2007, sono state comprese le basi molecolari di molte forme monogeniche di diabete che causano una disfunzione della β-cellula e, successivamente, ci sono stati progressi nel delineare l’eziologia genetica del diabete neonatale. La ricerca dei geni che predispongo alle più frequenti forme multifattoriali di diabete tipo 2 rappresenta la sfida più grande. Nel 2006 si è assistito all’identificazione del più importante gene di suscettibilità per il diabete tipo 2 finora noto, il gene TCF7L2, e nel 2007 gli studi di associazione su tutto il genoma, condotti su larga scala, hanno fornito nuove intuizioni riguardo all’architettura genetica e alla biologia del diabete tipo 2.
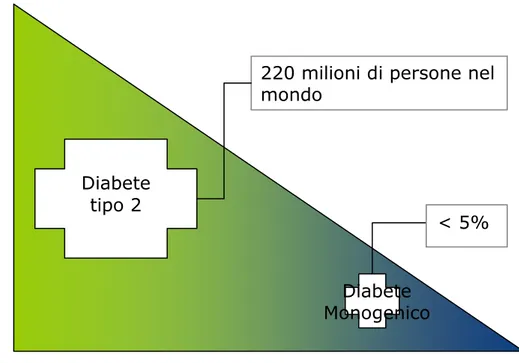
Come per altre malattie complesse, molte informazioni di rilievo per il diabete tipo 2 sono state ottenute dall’analisi delle forme di diabete monogenico. Sebbene relativamente infrequenti (cumulativamente rendono conto di circa il 5% dei casi di diabete, figura 5), potenti correlazioni tra genotipo e fenotipo hanno contribuito alla scoperta di geni, fornito intuizioni preziose sul meccanismo dell’omeostasi del glucosio e facilitato il rapido trasferimento di queste informazioni sul piano clinico.
Diabete monogenico
Nel corso degli ultimi 15 anni, le tecniche di clonaggio posizionale e gli studi sui geni candidati hanno permesso di individuare i geni implicati nello sviluppo di forme monogeniche e sindromiche di diabete. In generale, il successo è stato maggiore nei casi in cui il fenotipo predominante è la disfunzione della β-cellula
Figura 5. Prevalenza del diabete in relazione all’ambiente e al genotipo.
pancreatica, come accade nel MODY (maturity-onset diabetes of the young) e in varie forme di diabete neonatale non autoimmune, piuttosto che nei casi in cui è presente una grave insulino-resistenza (figura 6). Le caratteristiche cliniche del diabete monogenico in relazione ad altre forme di diabete sono riportate nella Tabella I.
Diabete neonatale
Senza dubbio, alcuni dei progressi più recenti e significativi sono stati quelli che hanno condotto alla classificazione molecolare del diabete che insorge nei primi sei mesi di vita (storicamente chiamato diabete neonatale, anche se l’espressione è, a rigor di termini, inaccurata). In precedenza, si parlava di “diabete mellito neonatale permanente” (permanent neonatal diabetes mellitus - PNDM) per quei bambini che avevano una forma permanente di diabete, e di “diabete mellito neonatale transitorio” (transient
Geni A m b ie n te Diabete tipo 2
c
c
o
o
m
m
u
u
n
n
220 milioni di persone nel mondo
Diabete Monogenico
Figura 6. Caratteristiche metaboliche associate con il diabete monogenico.
Tabella I. Caratteristiche fenotipiche delle varie tipologie di diabete tipo 2.
LADA Diabete tipo 2 a insorgenza precoce Diabete tipo 2 a insorgenza tardiva MODY
Malattia genetica Poligenico Poligenico Poligenico Monogenico
autosomico dominante Età d’esordio tipica
(anni) Di solito > 35 < 40 > 60 < 25
Familiari affetti 0 2 1 1
Obesità Rara Comune Comune Rara
Anticorpi Si, GAD65 No No No
Terapia insulinica Variabile,
di solito mesi o anni
Non
inizialmente Non inizialmente Non inizialmente
LADA: Latent Autoimmune Diabetes in Adults Adattato da Tooke JE, JRCP 2000; 34: 332-3350
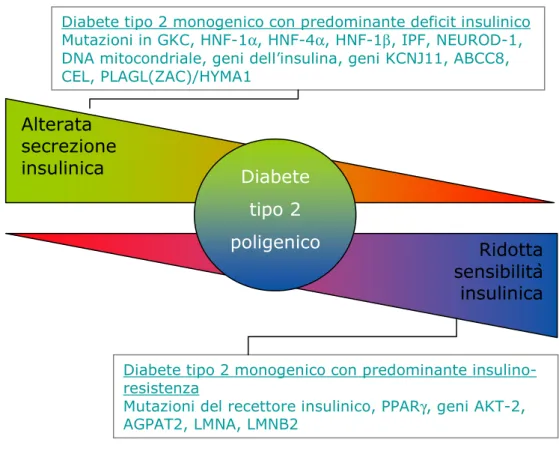
Diabete tipo 2
poligenico Ridotta
sensibilità insulinica
Diabete tipo 2 monogenico con predominante deficit insulinico Mutazioni in GKC, HNF-1α, HNF-4α, HNF-1β, IPF, NEUROD-1, DNA mitocondriale, geni dell’insulina, geni KCNJ11, ABCC8, CEL, PLAGL(ZAC)/HYMA1
Diabete tipo 2 monogenico con predominante insulino-resistenza
Mutazioni del recettore insulinico, PPARγ, geni AKT-2, AGPAT2, LMNA, LMNB2
Alterata secrezione insulinica
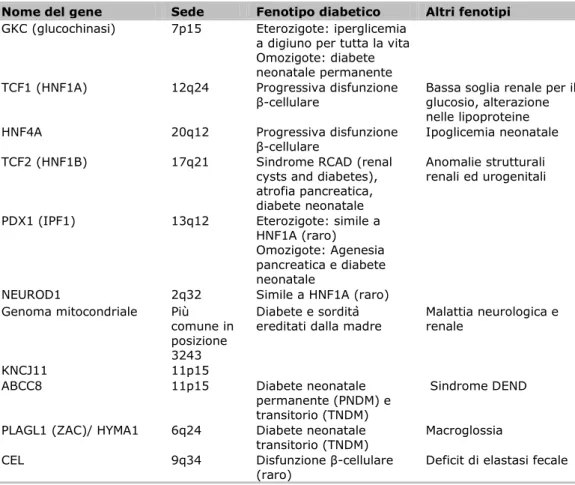
neonatal diabetes mellitus – TNDM) per quelli che avevano forme il cui decorso era caratterizzato da precoce remissione e ricomparsa del diabete tipo 2 in età adulta. Molti casi di TNDM sono dovuti a sovra espressione dei geni ZAC e HYMA1 sul cromosoma 6q (31). Anche alcuni casi di PNDM hanno un’eziologia genetica (Tabella II) mentre i rimanenti sono, spesso, forme precoci di diabete tipo 1 (autoimmune).
Data la profonda disfunzione β-cellulare che caratterizza il diabete mellito neonatale permanente, l’attenzione si è concentrata sul gene KCNJ11 poiché esso codifica per Kir6.2, un componente del canale del potassio, che svolge un ruolo centrale nella secrezione dell’insulina stimolata dal glucosio e che è il bersaglio delle sulfoniluree (farmaci ipoglicemizzanti orali). Le mutazioni in questo gene sono state inizialmente identificate in più di un terzo dei casi di PNDM (32), osservazione questa che ha ricevuto successive conferme (33-35). In alcuni individui (di solito quelli in cui gli studi funzionali mostrano un’estrema compromissione della funzione dei canali del potassio), sono presenti debolezza muscolare, epilessia e caratteristiche dismorfiche (che determinano ritardo di sviluppo, epilessia e diabete neonatale; la cosiddetta sindrome DEND - developmental delay, epilepsy and neonatal diabetes) (32,36), che riflette l’espressione extrapancreatica di Kir6.2. È stato in seguito dimostrato che le mutazioni di KCNJ11 sottendono anche molti dei casi di TNDM che non sono attribuibili ad anormalità nel cromosoma 6q (37). Molte delle mutazioni coinvolte in queste forme di diabete neonatale compromettono la normale secrezione di insulina stimolata dal glucosio, infatti, l’ATP, generato dal metabolismo intracellulare del glucosio, consente la chiusura del canale; nei soggetti in cui tali mutazioni sono presenti, ci sarebbe una mancata trasduzione del segnale dell’ATP. Questo suggerisce che le sulfoniluree, le quali
Tabella II Cause monogenetiche di disfunzione β-cellulare (30).
Nome del gene Sede Fenotipo diabetico Altri fenotipi
GKC (glucochinasi) 7p15 Eterozigote: iperglicemia
a digiuno per tutta la vita Omozigote: diabete neonatale permanente
TCF1 (HNF1A) 12q24 Progressiva disfunzione
β-cellulare Bassa soglia renale per il glucosio, alterazione nelle lipoproteine
HNF4A 20q12 Progressiva disfunzione
β-cellulare Ipoglicemia neonatale
TCF2 (HNF1B) 17q21 Sindrome RCAD (renal
cysts and diabetes), atrofia pancreatica, diabete neonatale
Anomalie strutturali renali ed urogenitali
PDX1 (IPF1) 13q12 Eterozigote: simile a
HNF1A (raro) Omozigote: Agenesia pancreatica e diabete neonatale
NEUROD1 2q32 Simile a HNF1A (raro)
Genoma mitocondriale Più
comune in posizione 3243
Diabete e sordità
ereditati dalla madre Malattia neurologica e renale
KNCJ11 11p15
ABCC8 11p15 Diabete neonatale
permanente (PNDM) e transitorio (TNDM)
Sindrome DEND
PLAGL1 (ZAC)/ HYMA1 6q24 Diabete neonatale
transitorio (TNDM) Macroglossia
CEL 9q34 Disfunzione β-cellulare
(raro) Deficit di elastasi fecale
conducono alla chiusura del canale attraverso un meccanismo ATP-indipendente, potrebbero essere efficaci in tali individui (piuttosto che l'insulina con cui venivano trattati fin dalla diagnosi nella quasi totalità dei casi). Ed infatti è stato dimostrato che molti pazienti PNDM con mutazioni di KCNJ11 possono essere trattati con successo utilizzando le sulfoniluree (38). Inoltre, sembra che il conseguente ripristino di qualche grado di reattività della β-cellula ad altri agenti metabolici (per esempio le incretine) causi un miglioramento significativo nel controllo glicemico rispetto a quanto si può raggiungere con la somministrazione di insulina esogena (38). Un lavoro più recente ha stabilito che anche le mutazioni nel gene ABCC8, che codifica per il recettore delle sulfoniluree (l’altro componente del canale
K-ATP della β-cellula) possono portare ai fenotipi TNDM e PNDM (39, 40). È stato anche dimostrato, usando markers genetici (HLA) e immunologici, che il diabete tipo 1 non contribuisce in modo significativo al diabete che compare nei primi 6 mesi di vita (41).
Maturity-onset diabetes of the young
A differenza del diabete neonatale, il MODY (diabete della maturità che insorge nel giovane) è una forma relativamente frequente di diabete monogenico che esordisce in giovani adulti. Le mutazioni dei geni GCK, che codifica per la glucochinasi, TCF1 (o HNF1A), che codifica per il fattore di trascrizione HNF-1α, TCF2 (o HNF1B) che codifica per il fattore di trascrizione HNF-1β, e HNF4A, che codifica per HNF-4α, rendono conto di quasi il 90% dei casi di MODY (Tabella II). Queste mutazioni causano manifestazioni fenotipiche, e quindi cliniche, diverse, tali che la diagnosi molecolare ha molto da offrire in termini di definizione della prognosi e della terapia (42). La disponibilità di ampie coorti di soggetti affetti dalle forme più rare di MODY ha consentito di evidenziare ulteriori differenze fenotipiche. Per esempio, gli individui con MODY attribuibile a mutazioni di HNF1A e HNF4A condividono molte caratteristiche (43), che riflettono l’interazione tra queste due molecole nello sviluppo e nella funzione delle β-cellule. È stato recentemente dimostrato, comunque, che individui con mutazioni HNF4A presentano iperinsulinemia fetale e neonatale, responsabile di aumento di peso alla nascita e ipoglicemia neonatale (44). Il meccanismo molecolare che determina queste differenze rimane da spiegare. In alcuni casi, differenze nell’espressione (sia temporale che spaziale) di fattori di trascrizione strettamente correlati potrebbero spiegare la marcata eterogeneità fenotipica. Per esempio, HNF-1α e HNF-1β (codificati da TCF1 e TCF2,
rispettivamente) sono eterodimeri che presentano lo stesso motivo che lega il DNA. In termini di mutazioni del TCF2, tuttavia, le manifestazioni predominanti sono anomalie strutturali di sviluppo (particolarmente urogenitali); tale forma di diabete è conseguenza dell’atrofia pancreatica piuttosto che di una disfunzione isolata delle insulae e gli individui affetti mostrano una significativa insulino-resistenza (45). A differenza degli altri fattori di trascrizione associati al MODY, per TCF2 spesso sono presenti delezioni dell’intero gene (46).
Altre forme di diabete monogenico
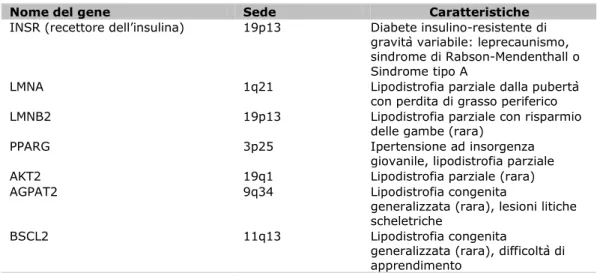
Sono costanti i progressi nell’identificazione e caratterizzazione di geni che sono causa di altri fenotipi monogenici. È stato descritto un legame tra disfunzione esocrina pancreatica e diabete in due alberi genealogici di famiglie norvegesi; questo legame è determinato da mutazioni nel gene della lipasi degli esteri carbossilici (CEL) (47). Finora, questo gene si considera una causa rara di diabete familiare. Nelle forme di insulino-resistenza monogenica, la lipodistrofia sembra essere un elemento comune ed è dovuta a mutazioni sia nei geni della lamina nucleare LMNA (48) e LMNB2 (49) sia nei geni codificanti il recettore attivato dal proliferatore dei perossisomi γ (PPARG) (50) (Tabella III).
Diabete tipo 2 multifattoriale
Di fronte ai progressi raggiunti nell’identificazione dei geni che causano il diabete monogenico, la ricerca delle varianti di suscettibilità per il diabete tipo 2 procede lentamente. Questo è dovuto all’effetto relativamente modesto di tali varianti di suscettibilità. L’ampliamento delle dimensioni dei campioni, la replicazione ampia, le analisi e interpretazioni scrupolose sono stati passi cruciali nel produrre risultati solidi e confermati. Un dato emerso dagli studi sul diabete tipo 2 è la sovrapposizione
Tabella III. Cause monogenetiche di insulino-resitenza (30).
Nome del gene Sede Caratteristiche
INSR (recettore dell’insulina) 19p13 Diabete insulino-resistente di
gravità variabile: leprecaunismo, sindrome di Rabson-Mendenthall o Sindrome tipo A
LMNA 1q21 Lipodistrofia parziale dalla pubertà
con perdita di grasso periferico
LMNB2 19p13 Lipodistrofia parziale con risparmio
delle gambe (rara)
PPARG 3p25 Ipertensione ad insorgenza
giovanile, lipodistrofia parziale
AKT2 19q1 Lipodistrofia parziale (rara)
AGPAT2 9q34 Lipodistrofia congenita
generalizzata (rara), lesioni litiche scheletriche
BSCL2 11q13 Lipodistrofia congenita
generalizzata (rara), difficoltà di apprendimento
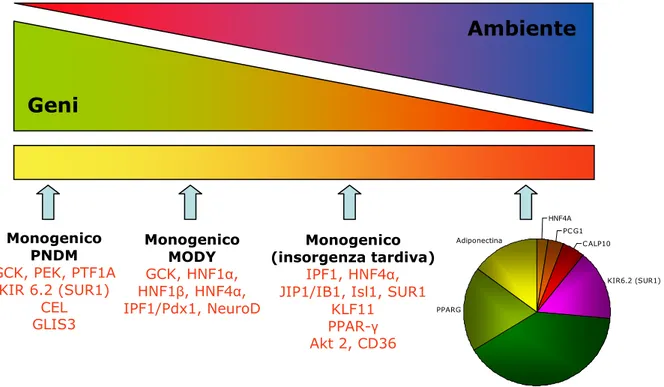
tra geni con un ruolo eziologico per le forme sia monogeniche sia multifattoriali di diabete; questo non deve sorprendere se si considerano, per esempio, i diversi fenotipi associati con la variazione di sequenza nel KNCJ11, i quali forniscono un buon esempio di un continuum che si estende dalle forme severe di diabete neonatale (DEND), attraverso il diabete neonatale transitorio, al diabete mellito tipo 2 (multifattoriale) (Figura 7). Risultati analoghi sono stati ottenuti per PPAR-γ: una rara mutazione di questo gene conduce ad una sindrome da insulino-resistenza (50), mentre un polimorfismo di un singolo nucleotide si associa ad aumento del rischio di diabete tipo 2. Mutazioni rare del gene HNF1A causano MODY 3 ed una variante comune ha effetto sul rischio di diabete negli indiani Oji-Cree (51); varianti frequenti del promoter P2 del gene HNF4A, responsabile anche del MODY 1, sono implicate nel rischio di diabete tipo 2 (52); rare mutazioni di GCK causano PNDM e MODY 2 (53) e una variante comune di un promoter altera la glicemia a digiuno negli adulti (54). Varianti comuni nel gene LMNA hanno effetto sul rischio di diabete tipo 2 e sindrome metabolica (55). Per contro, non ci sono prove che mutazioni rare nel TCF7L2, il più
importante gene di suscettibilità per il diabete tipo 2 ad oggi individuato, possano spiegare forme di diabete monogenico (56,57).
Figura 7. Effetti dei geni e dell’ambiente sui diversi tipi di diabete.
Geni
Ambiente
Monogenico PNDM GCK, PEK, PTF1A KIR 6.2 (SUR1) CEL GLIS3 Monogenico (insorgenza tardiva) IPF1, HNF4α, JIP1/IB1, Isl1, SUR1KLF11 PPAR-γ Akt 2, CD36 Monogenico MODY GCK, HNF1α, HNF1β, HNF4α, IPF1/Pdx1, NeuroD HNF4A PC G1 C ALP10 KIR6.2 (SUR1) TCF7L2 PPARG Adiponectina
4. Geni candidati
La ricerca dei geni per il diabete mellito tipo 2 è iniziata da quasi tre decenni. Il primo risultato è stato la clonazione del gene dell’insulina umana. L’identificazione e caratterizzazione delle interazioni gene-gene è stata, tuttavia, limitata dalla mancanza di potenti metodi statistici e dalla mancanza di campioni di grandi dimensioni (58), elementi necessari dal momento che lo sviluppo del diabete mellito tipo 2 dipende da parecchi geni i cui effetti sono deboli e questo rende la ricerca più complicata. In effetti, la ‘debolezza’ della componente genetica del diabete mellito tipo 2 si evidenzia anche dal confronto con altre malattie comuni, studiate anch’esse attraverso studi di associazione su tutto il genoma (vedi dopo). Il rischio relativo di malattia per un fratello di un paziente con diabete tipo 2 è, al massimo, 3-4 volte superiore a quello della popolazione generale; per l’artrite reumatoide è di 5-10 volte, 15 per il diabete tipo 1, 7-10 per i disordini bipolari, 17-35 per il morbo di Crohn, 2-7 per l’infarto miocardico precoce e 2.5-3.5 per l’ipertensione (59). Inoltre la scelta dei geni da studiare è resa difficoltosa anche per l’incompleta conoscenza dell’eziopatologia del diabete tipo 2. Infine gli studi per mappare i geni candidati, solitamente, esaminano solo una quota di varianti, o anche una singola variante, per gene (di frequente in una porzione codificante l’unità trascritta). Ecco perché, in passato, i risultati sono stati spesso deludenti.
Il metodo più semplice per individuare i geni di suscettibilità sarebbe quello di sequenziare l’intero genoma in individui diabetici e non, ma questo, per ragioni pratiche, non è ancora possibile. La situazione potrebbe cambiare rapidamente con l’introduzione di nuovi strumenti di sequenziamento su larga
scala. Inoltre sono stati descritti parecchi approcci per stimare se un’associazione osservata può giustificare un collegamento causale (60). Senza la conoscenza del dato funzionale non è sempre possibile sapere se il linkage e l’associazione rappresentano la causa genetica della malattia. Per molti disordini complessi questo può richiedere un’enorme serie di studi in vivo e in vitro.
Inizialmente sono state utilizzate le analisi di linkage familiare mentre, per la valutazione dell’azione e della secrezione dell’insulina, sono stati usati gli studi sui geni candidati (tabella IV). Gli studi di linkage consistono in analisi di regioni genomiche condivise da familiari con tassi di frequenza più alti di quanto atteso e sono attuati utilizzando markers di polimorfismo come i micro satelliti o le ripetizioni in tandem. Questo tipo d’indagine consente di determinare la posizione cromosomica di un locus, responsabile di una malattia o di un carattere genetico, rispetto a dei marcatori polimorfici, di cui è nota la posizione. Si tratta di un approccio che, analogamente alla clonazione posizionale, si è rivelato di successo nell’identificare varianti genetiche rare, responsabili di disordini monogenici (a trasmissione mendeliana) come il MODY (maturity-onset diabetes of the young) (61,62).
Tabella IV. Significato e risultati degli studi genetici nel DT2.
• Le scansioni su tutto il genoma per le analisi di linkage (LOD
scores) sono state deludenti
• Gli studi sui geni candidati esaminano proteine note, vie e
meccanismi implicati nella regolazione dei livelli ematici di glucosio (alcuni successi)
• Gli studi di associazione su tutto il genoma (GWAS) mettono in
evidenza proteine, vie metaboliche e meccanismi nuovi e imprevisti (molti lavori recenti; 19 geni potenziali)
Più difficoltosa è invece l’identificazione dei numerosi geni implicati in malattie più comuni, a ereditarietà complessa, per cui non ci sono stati risultati altrettanto incoraggianti.
Una volta divenuto chiaro che i metodi di associazione condotti in campioni più grandi erano preferibili alle analisi di linkage (31), i ricercatori, generalmente, hanno rivolto la loro attenzione ai geni candidati biologici. Negli studi sui geni candidati si tenta di correlare variazioni biologiche (il fenotipo) e variazioni nelle sequenze del DNA (il genotipo) nella forma del polimorfismo di un singolo nucleotide, cioè il cambiamento di una singola base nella sequenza del genoma umano. Queste varianti, in introni o esoni del gene, potrebbero causare un’alterazione dell’espressione e/o della funzione del prodotto genico che, a loro volta, potrebbero compromettere una funzione biologica o causare una malattia. I geni candidati, nel caso del diabete mellito tipo 2, sono, per esempio, geni che codificano per proteine che partecipano al segnale dell’insulina all’interno della cellula, codificano per enzimi coinvolti nel metabolismo del glucosio o nella regolazione della secrezione insulinica da parte della β-cellula pancreatica. Le varianti candidate sono state scoperte attraverso lavori di sequenziamento, che sono stati agevolati dal parallelo progresso del Progetto Genoma Umano (64) e dalla disponibilità dei polimorfismi di un singolo nucleotide in database pubblici (65). Alcuni dei risultati ottenuti con questo approccio hanno mantenuto la loro validità nel tempo. Questi sono: la variante P12A nel gene del recettore attivato dal proliferatore dei perossisomi γ (PPARG) (66), la variante E23K nel gene KNCJ11 (67-69), le frequenti varianti dei geni del fattore di trascrizione 2 epatico (TCF2, ora noto come HNF1B) (70, 71) e del gene della sindrome di Wolfram 1 (WFS1) (72). Tutti questi geni codificano per proteine che hanno legami
biologici forti con il diabete. Rare ma severe mutazioni in tutti e quattro questi geni causano forme monogeniche di diabete (32, 73-75) e due sono il bersaglio della terapia anti-diabetica (KCNJ11 e PPARG).
PPARγ
Il gene PPARγ, posto sul braccio corto del cromosoma 3, codifica per il recettore attivato dal proliferatore dei perossisomi γ2, un recettore nucleare presente in molti tessuti ma espresso prevalentemente negli adipociti, dove regola la trascrizione di geni coinvolti nell’adipogenesi. Questa proteina è il bersaglio molecolare dei tiazolidinedioni (glitazoni), farmaci ipoglicemizzanti orali utilizzati nella terapia del diabete mellito tipo 2 e, per questo, è un gene candidato interessante. Una variante di questo gene è caratterizzata dalla presenza di un residuo di prolina, in posizione 12, al posto dell’alanina (P12A) (76). Questa variazione è un esempio di polimorfismo di un singolo nucleotide di tipo missense, dove un cambiamento di una singola base altera la sequenza aminoacidica e, presumibilmente, anche la funzione della proteina. È stata suggerita una forte interazione tra il polimorfismo del PPARγ Pro12Ala e il rapporto tra acidi grassi polinsaturi/acidi grassi saturi della dieta nei confronti dell’indice di massa corporea e del livello di insulina a digiuno (77). È stato anche dimostrato che, i soggetti omozigoti per l’allele prolina, quando adottano uno stile di vita occidentale, tendono ad avere un peso corporeo superiore a quelli con l’allele alanina (78). Gli individui omozigoti per il più frequente allele con prolina hanno un maggior grado di insulino-resistenza (79) e circa il 20% ha una maggiore probabilità di sviluppare diabete tipo 2 (80) rispetto ai soggetti portatori dell’allele con alanina, come dimostrato definitivamente in una meta-analisi (66). Tale studio ha anche il merito di aver evidenziato la necessità di
disporre di campioni di grandi dimensioni (ottenuti, in genere, solo attraverso meta-analisi di diverse coorti) per rilevare associazioni genetiche dagli effetti modesti. Il raro allele Ala è presente in circa il 15% degli Europei e ha dimostrato di essere associato con un’incrementata attività trascrizionale, incrementata insulino-sensibilità e protezione verso il diabete tipo 2 (79). La riduzione di rischio individuale conferita dall’allele Ala è moderata, circa il 15%, ma poiché l’allele di rischio Pro è molto frequente, esso si traduce in un rischio attribuibile alla popolazione (PAR) pari al 25% (66). Il PAR esprime la frazione di malattia che potrebbe essere eliminata dalla popolazione se la variante di rischio non esistesse. Un fatto molto importante su cui porre l’accento è che questi dati dimostrano che l’identificazione delle interazioni tra fattori genetici ed ambientali è fondamentale per comprendere la patogenesi del diabete mellito tipo 2.
KCNJ11 – KCNQ1
Un altro bersaglio di farmaci impiegati in pazienti con diabete tipo 2 è il recettore della sulfonilurea SUR1. Il gene codificante per tale recettore, ABCC8, si trova poche kilobasi a monte del gene KCNJ11, che codifica per il suo partner funzionale, il canale del potassio ATP-sensibile Kir6.2. Insieme, formano una proteina ottamerica che regola il potenziale transmembrana e, in tal modo, la secrezione dell’insulina stimolata dal glucosio, nella β-cellula pancreatica. La chiusura del canale del potassio è un prerequisito perché si possa avere la secrezione di insulina. Un polimorfismo missense in KCNJ11 (per cui il glutammato è scambiato con la lisina nel codone 23, E23K) è stato associato con il diabete tipo 2 in uno studio di meta-analisi (81); questo risultato è poi stato confermato in un successivo studio di associazione su larga scala (OR 1.2; p=10-5) (67). L’allele di
rischio è associato anche con una modesta alterazione della secrezione di insulina (68, 69). Inoltre, una mutazione attivante il gene causa una grave forma di diabete neonatale (32). Mentre la mutazione neonatale provoca un’attivazione del canale del potassio ATP-dipendente dieci volte maggiore, la variante E23K causa un incremento di attività di solo due volte (82). Anche un altro canale del potassio, codificato dal gene KCNQ1, è stato recentemente implicato nel diabete tipo 2 mediante studi di associazione su tutto il genoma (83,84).
Geni MODY
Il Maturity onset diabetes of the young (MODY) è una forma monogenica di diabete mellito ad ereditarietà autosomica dominante. Esistono almeno sei forme di MODY causate da mutazioni in un distinto gene. Ad eccezione del MODY2, che è provocato da una mutazione del gene della glucochinasi, le altre forme sono causate da mutazioni in fattori di trascrizione come HNF4α (MODY1), HNF1α (MODY3), IPF1 (MODY4), HNF1β (MODY5) e neuroD (MODY6). Questi difetti genetici determinano un’alterata secrezione di insulina da parte della β-cellula e, di solito, mostrano forte eterogeneità allelica. Perciò, in numerosi studi, si è cercato di capire se, alcune varianti frequenti (e più ‘miti’) di questi geni, potessero contribuire alla suscettibilità verso il diabete tipo 2 ad esordio tardivo (70, 85-89).
In effetti, un’analisi combinata di più di 15000 campioni ha evidenziato un’associazione tra un polimorfismo di singolo nucleotide intronico (rs757210) nel gene HNF1B (responsabile del MODY 5 e chiamato TCF2, in passato) e il diabete tipo 2 (OR 1.12 e p<10-6) (70). Risultati simili sono stati ottenuti in un altro
studio su larga scala (88) e una conferma indipendente è giunta da un recente studio di associazione su tutto il genoma (71). Inoltre quest’ultimo studio, progettato per identificare varianti
genetiche associate con il cancro della prostata, ha mostrato che esiste un allele, di un SNP altamente correlato con rs757210, che protegge contro il cancro della prostata e, simultaneamente, incrementa il rischio di diabete mellito di tipo 2, un’interessante associazione che richiede ulteriori indagini (90).
Un altro gene oggetto di studio è HNF4A, responsabile di MODY1 e che si trova a livello di un picco di linkage ampiamente replicato; per esso sono state individuate due varianti genetiche nella regione promoter associate con un aumento del rischio di diabete tipo 2 (52, 85, 91-93); tuttavia le prove raccolte non raggiungono la significatività statistica (valore p) che, per tutto il genoma, è stata empiricamente fissata a ~5x10-8 (94).
Sono state analizzate anche le varianti missense A98V e I27L nel gene HNF1A (codificante per il fattore nucleare epatocitario 1α) e responsabile, se mutato, del MODY 3 (86, 87): in una meta-analisi per la variante A98V è stato calcolato un odds ratio pari a 1.17 (95% CI 1.03-1.33, p=2x10-2) (95), mentre l’allele leucina
(I27L) sembra incrementare il rischio di diabete tipo 2 in persone sovrappeso e/o anziane (88, 96-98) e sembra predire prospetticamente il diabete mellito tipo 2 (89).
Questi studi hanno anche il merito di aver enfatizzato la necessità di disporre di campioni di ampie dimensioni e la necessità di valutare il BMI ed l’età nelle analisi statistiche poiché, geni che causano difetti lievi nella secrezione insulinica, sono smascherati con maggior probabilità in individui con incrementato bisogno di insulina, per esempio in individui insulino-resistenti (96).
WFS1
Un’altra forma monogenica di diabete è la sindrome di Wolfram, caratterizzata da diabete insipido, diabete mellito ad esordio giovanile, atrofia ottica e sordità (per questo detta anche
sindrome DIDMOAD). È causata da mutazioni del gene WFS1 (73) che codifica per la wolframina; tali mutazioni rendono difettiva questa proteina. Il gene WSF1 può essere considerato il quarto gene candidato per il diabete mellito tipo 2. In effetti, recentemente, sono stati studiati 1536 SNP in 84 geni candidati per valutarne l’eventuale associazione con il diabete tipo 2 (72) e da quest’analisi sono emersi, come associati, solamente due SNP del gene WFS1 (p~10-7, OR~1.11). Il risultato è stato riprodotto
in 9533 casi e 11398 controlli. Quest’associazione ha raggiunto la significatività a livello di tutto il genoma attraverso la replicazione in coorti indipendenti (99). Sembra, inoltre, che le varianti di rischio abbiano effetto sulle funzioni della β-cellula (100, 101).
ENPP1
Infine, un altro gene candidato che è stato studiato estensivamente è ENPP1, codificante per la fosfodiesterasi alcalina/pirofosfatasi nucleotidasica, un inibitore del segnale dell’insulina. L’allele Q (glutammina) nella variante missense K121Q è stato nominalmente associato con diabete mellito tipo 2, obesità e insulino-resistenza in piccole coorti (102) e, mentre studi successivi più ampi segnalano un’associazione positiva con il diabete tipo 2 (103, 104), altri studi non hanno replicato questo risultato (105, 106). Tuttavia, in una recente meta-analisi di circa 42000 campioni, è stata documentata un’associazione di questa variante con il diabete tipo 2 in popolazioni europee secondo un modello recessivo (OR 1.38, 95% CI 1.10-1.74, p=5x10-3) (107). Sebbene non si raggiunga completamente la
significatività a livello di tutto il genoma, questa limitazione potrebbe essere dovuta al modello recessivo di trasmissione del rischio e/o all’evidente dipendenza da un concomitante background obesogenico, necessario affinché la variante di
rischio possa esercitare il suo effetto (103, 104, 107-109). Ciò nonostante, sia le persistenti segnalazioni positive di associazione riguardo a questa variante, sia le prove molecolari e fisiologiche che rilevano il ruolo di questo gene nell’insulino-resitenza, lo rendono un candidato genetico e biologico molto suggestivo.
CAPN10
Anche questo approccio d’indagine ha portato a interessanti risultati, in particolare per il gene CAPN10, codificante per la calpaina 10 (110), una cisteina proteasi con funzioni largamente sconosciute. Questo gene non era un candidato ovvio per il diabete tipo 2; tuttavia alcuni SNP nella regione promoter, gli SNP intronici 43, 44 e 63, sembrano associati con un aumento del rischio di diabete tipo 2, anche se con un basso valore p (110-113). Più recentemente, inoltre, si sono accumulate prove funzionali di un possibile ruolo della calpaina 10 nel metabolismo del glucosio (114). Non è stato facile capire come le variazioni introniche in questo gene potessero aumentare il rischio. L’allele G (glicina) del polimorfismo 43 è associato con una ridotta espressione del gene nel muscolo scheletrico e con insulino-resistenza (115). Come questo si traduca in un incrementato rischio di diabete tipo 2 non è noto e saranno necessari ulteriori studi funzionali. CAPN10 non è stato confermato come gene candidato per il diabete mellito tipo 2 negli studi di associazione su tutto il genoma.
Cromosomi 20 e 1
Un modesto picco di linkage nel cromosoma 20q è stato osservato in più popolazioni (116-122). In questa regione il più suggestivo gene candidato è il gene HNF4A, responsabile anche del MODY 1; esso è stato ampiamente genotipizzato ed
analizzato in vari studi di associazione. Analogamente, un linkage è stato osservato per il braccio lungo del cromosoma 1, con il diabete tipo 2 o trait metabolici correlati, negli Indiani Pima, nel Framingham Heart Study, in soggetti di razza bianca Statunitensi, Francesi e Inglesi e in individui Cinesi (123-128). I ricercatori dell’International Chromosome 1q Consortium hanno eseguito un mappaggio dettagliato e un’analisi di associazione di questa regione, ma non si è ancora giunti ad individuare un segnale convincente in grado di identificare un locus di predisposizione al diabete di tipo 2.
5. Il gene TCF7L2
Finora l’associazione più forte con il diabete mellito tipo 2 è quella osservata nel 2006, dai genetisti del gruppo DeCode, per il gene TCF7L2 (129). Questa scoperta ha fatto seguito all’identificazione di una regione di linkage sul cromosoma 10q. I ricercatori hanno identificato parecchi SNP associati con diabete tipo 2 in una regione di forte linkage disequilibrium nel gene TCF7L2. Quest’associazione è stata replicata in quasi ogni popolazione esaminata: dapprima in individui Islandesi, poi soggetti Statunitensi e Danesi, e successivamente in diversi campioni di individui di origine Europea, Asiatica e Africana (178-180), con un odds ratio di quasi 1.4 per l’allele di rischio (sostanzialmente più alto di tutte le altre varianti geniche, per le quali l’odds ratio oscilla tra 1.10 e 1.20) e un incontrovertibile valore p<10-80 (133, 134). L’affermazione che TCF7L2
rappresenta il più importante gene del diabete tipo 2 si riflette anche nelle dimensioni di campione necessarie per avere una potenza statistica adeguata. Sono richiesti approssimativamente 1380 casi e 1380 controlli per rivelare l’effetto del TCF7L2 con una potenza dell’80% (p=5x10-7), sulla base della frequenza
allelica nella popolazione del Regno Unito. Il segnale più forte dopo TCF7L2, il gene CDKN2A-2B, richiede 6200 casi e 6200 controlli per avere potenza statistica analoga (135). Sembra probabile che le frequenti varianti in TCF7L2 rappresenteranno il locus più importante per il diabete tipo 2 in termini di effetti sul rischio e di frequenza dell’allele di rischio.
TCF7L2 è uno dei primi geni di cui si è scoperta l’associazione con una malattia, in questo caso il diabete tipo 2, senza che ci fosse una precedente conoscenza delle sue funzioni, perciò non ‘nasce’ come gene candidato, piuttosto è da considerare un risultato inaspettato ottenuto attraverso analisi di linkage sul
Figura 8. Gene TCF7L2 nella sua posizione genomica.
Il prodotto del gene TCF7L2 è un “high mobility group (HMG) box” che contiene fattori di trascrizione implicati nell’omeostasi del glucosio ematico.
braccio lungo del cromosoma 10 (figura 8). La principale fonte del segnale di associazione nella popolazione europea sembra essere un SNP intronico (rs7903146) (129, 136, 137) che, almeno sul piano statistico, appare essere la variante con probabile significato causale (130), come dimostrato dall’estensivo risequenziamento della regione e dalle analisi di associazione. Studi prospettici hanno confermato l’impatto della variante di TCF7L2 sul rischio di diabete tipo 2 (131, 138, 139) e, tra i partecipanti del Diabetes Prevention Program, essa si è rivelata fortemente associata con la progressione da alterata tolleranza al glucosio (IGT) a diabete (con un tasso di rischio di 1.55 tra gli omozigoti) (139). Il SNP rs7903146 si trova in un introne (una regione non codificante del gene) senza un chiaro meccanismo in grado di influenzare l’attività di TCF7L2 (130). Poiché non ci sono polimorfismi codificanti correlati con rs7903146, è probabile che la variante causale agisca influenzando l’espressione di TCF7L2, piuttosto che alterando la struttura della proteina espressa.
Il gene TCF7L2, conosciuto anche come TCF-4, codifica per un fattore di trascrizione che è espresso nel pancreas fetale ed è coinvolto nel Wnt signaling, un complesso network di proteine, note per il loro ruolo nell’embriogenesi e nel cancro, ma coinvolte anche in alcuni normali processi fisiologici in animali adulti. Il WNT signaling ha una funzione critica per la proliferazione cellulare, la motilità e la normale embriogenesi, inoltre sembra
regolare la miogenesi e l’adipogenesi (140, 141). È fondamentale anche per lo sviluppo del pancreas e delle cellule insulari durante la crescita embrionale (142). L’importanza della via di signaling Wnt nell’omeostasi del glucosio è evidenziata dalla recente scoperta che anche varianti comuni di una regione del cromosoma 10q contenente HHEX (ma anche due altri geni candidati: KIF11 e IDE) si associano con il rischio di diabete tipo 2 (143-147). HHEX è un bersaglio del signaling Wnt.
TCF7L2 agisce come recettore nucleare per CTNNB1 (β-catenina) (148) formando un eterodimero; ciò conduce ad una traslocazione nucleare che provoca, a sua volta, la trascrizione di alcuni geni, compreso il pro-glugagone intestinale (figura 9). Nonostante l’incertezza riguardo alla variante causale, l’allele di rischio del TCF7L2 condurrebbe ad alterata secrezione di insulina (132, 137, 139, 103, 149, 150); ciò è compatibile con l’osservazione che l’associazione del TCF7L2 è più forte tra soggetti magri piuttosto che obesi con diabete tipo 2. Florez et al. (139) hanno mostrato, usando misure basali ottenute dal Diabetes Prevention Study, che i portatori dell’allele di rischio T rs7903146 hanno livelli significativamente più bassi di secrezione insulinica rispetto agli omozigoti CC. Saxena et al. (137) hanno scoperto che individui omozigoti per l’allele di rischio rs7903146 hanno una riduzione di circa il 50% dell’indice insulinogenico (p=3x10-3) e dell’insulin disposition index (p=4x10-3) che è il
prodotto di due misure quantitative: funzione β-cellulare x insulino-sensibilità. Altri studi hanno condotto a risultati simili, con un effetto additivo, per ogni allele di rischio in più, per le misure di secrezione insulinica ma non per quelle di insulino-resistenza (138, 151-153).
Una prova aggiuntiva del ruolo del TCF7L2 nella regolazione della secrezione insulinica deriva dallo studio che ha valutato il peso alla nascita. Freathy et al. (154) hanno genotipizzato la variante
Figura 9. Rappresentazione schematica del signaling WNT/TCF. Le proteine Wnt secrete si legano ai recettori FZD e LRP, che, a loro volta, inattivano il complesso di degradazione che comprende Axina, DVL e GSK3B. Questo consente l’ingresso della β-catenina non fosforilata nel nucleo dove viene attivata la trascrizione genica. TCF7L2 potrebbe regolare l’espressione di parecchi geni e influenzare specificamente sia la secrezione che la sensibilità insulinica.
rs 7903146 in 15709 neonati e 8344 madri. Ogni copia fetale dell’allele di rischio per il diabete tipo 2 è associata con un incremento del peso alla nascita di 18 grammi (p=1x10-3) mentre ogni copia materna è associata con un incremento del peso alla nascita del neonato pari a 30 grammi (p=2.8x10-5).
L’associazione sussiste anche quando si effettua una stratificazione in base al genotipo fetale (31g, valore p corretto=3x10-3). Questo suggerisce che l’associazione sia legata al genotipo materno, mentre l’associazione con il genotipo fetale sarebbe il risultato della forte correlazione tra genotipi di madre
WNT FZD LRP DVL GSK3B β-Catenina β-Catenina Trascrizione Genica Adipogenesi In su lin o r e si st e n za S e cr e zio n e in su lin ic a AXINA Crescita e differenziazione
e feto. Inoltre sono stati analizzati trait correlati al diabete in 10314 individui non diabetici. Da queste analisi, è emerso che l’allele di rischio avrebbe effetto sul peso alla nascita, a causa di una riduzione nella secrezione insulinica materna. Questo provocherebbe un’elevata glicemia materna durante la gravidanza e quindi un incremento di peso del neonato alla nascita.
Attraverso quali meccanismi la versione difettosa del TCF7L2 altererebbe la secrezione insulinica non è ancora chiaro. È stato suggerito che possa essere presente una riduzione della secrezione del peptide glucagone-simile 1 (GLP-1, la cui trascrizione è regolata da TCF7L2) da parte delle cellule entero-endocrine (129). Quindi ci sarebbe un diminuito effetto delle incretine (GLP-1 ma anche GIP), in grado di stimolare la secrezione di insulina dopo l’introduzione di cibo (149,155). Questa ipotesi si basa anche sul presunto ruolo di TCF7L2 nello sviluppo del cancro colon rettale. In aggiunta o in alternativa a questo meccanismo potrebbero esserci difetti nel processamento dell’insulina (151,156). Infatti, uno dei bersagli di TCF7L2 è il gene HHEX, che si trova in una regione genica che comprende anche i geni KIF11 (per il kinesin-interacting factor) e IDE (insulin degrading enzyme). Alternativamente, poiché TCF7L2 è parte del WTN signaling, si ipotizza un effetto sulla massa β-cellulare, sullo sviluppo della β-cellula pancreatica e/o sulla funzione β-cellulare. È anche possibile che il gene sia coinvolto nella proliferazione delle β-cellule in risposta ad aumentate richieste di insulina. Nei soggetti all’esordio del diabete l’espressione del TCF7L2 nelle isole di Langerhans è aumentata di 5 volte ed è tanto più alta quante più copie ci sono dell’allele di rischio. Ciò rifletterebbe un difetto di trascrizione o traslazione del TCF7L2 stesso dal momento che, la sua sovra-espressione nelle insulae umane si traduce in un’alterata secrezione di
insulina. I dati sull’indice di massa corporea ed alcune associazioni preliminari con i livelli di leptina e grelina (130), comunque, potrebbero indicare un meccanismo centrale. Tale incertezza evidenzia quanto sia affascinante quest’approccio inverso che fornisce l’opportunità di scoprire nuovi meccanismi cellulari e fisiologici alla base di una patologia.
Infine, si è visto che il cambiamento dello stile di vita può attenuare significativamente il rischio genetico (139,157). Un iniziale studio retrospettivo farmacogenetico ha indicato, inoltre, che i portatori dell’allele di rischio hanno una maggior probabilità di una inadeguata risposta alla terapia con sulfoniluree (158).
6. Studi di associazione in tutto il genoma (GWAS)
Negli ultimi anni sono stati realizzati enormi progressi che hanno avuto un ruolo chiave nell’estendere, all’intero genoma, le analisi di associazione di geni candidati. In primo luogo, grazie al completamento del Progetto Genoma Umano, milioni di SNP (inclusi la maggioranza di quelli più comuni) sono stati scoperti e depositati in database pubblici; è stato così possibile studiare quale fosse lo specifico contributo di queste varianti frequenti nei confronti della componente genetica di una malattia (94). In secondo luogo, l’International HapMap Project (HapMap) ha genotipizzato quasi quattro milioni di SNP in 270 campioni di DNA e identificato i cosiddetti tag-SNP cioè, polimorfismi di un singolo nucleotide correlati (in linkage disequilibrium) con varianti frequenti e che quindi possono essere considerati come proxies per tali varianti (sebbene la regione di DNA che le contiene non sia stata analizzata direttamente) (94,159). Più semplicemente, sono stati individuati degli aplotipi, intesi come subsets di markers. In terzo luogo, sono divenute disponibili tecnologie di genotipizzazione high-throughput che consentono l’esecuzione di studi di associazione su tutto il genoma (Genome Wide Association Studies - GWAS) con tempi ed a costi significativamente minori rispetto al passato. Inoltre, sono ora disponibili strumenti analitici che consentono di interpretare queste grandi serie di dati (160-169). E infine, attraverso collaborazioni internazionali, sono state messe insieme e condivise parecchie grandi raccolte con fenotipi ben caratterizzati.
Un’altra acquisizione che ha contribuito ai progressi fatti finora è quella relativa all’importante ruolo del DNA non codificante proteine (come mostrato, ad esempio, da regioni non codificanti
conservatesi nel corso dell’evoluzione o dai microRNA) (170, 171); ciò ha posto al centro dell’attenzione tutto il genoma e non più i singoli geni o, in particolare, le sole regioni codificanti.
Le due principali piattaforme per la genotipizzazione dei polimorfismi di singolo nucleotide in tutto il genoma disponibili in commercio sono Affymetrix 500k array e Illumina HumanHap300 array. Le varianti della piattaforma Affymetrix 500k sono state selezionate per fornire una distribuzione approssimativamente casuale di SNP nel genoma, senza prendere in considerazione i patterns di linkage disequilibrium. In contrasto, il chip Illumina HumanHap300 contiene approssimativamente 317k di SNP, specificamente selezionati per individuare comuni variazioni (minore frequenza allelica >5%), facendo uso di dati di correlazione tra polimorfismi di singolo nucleotide della fase I del progetto HapMap, in un campione di residenti dello Utah di discendenza europea. Entrambe le piattaforme consentono una buona copertura di tutto il genoma per i comuni SNP in un singolo esperimento (65% e 75% rispettivamente) (172, 173). Ogni piattaforma presenta vantaggi e limitazioni. Per esempio, il chip Affymetrix consente di coprire una piccola porzione di variazioni comuni in tutto il genoma ma non è popolazione-specifico ed è ridondante (caratteristica che dipende dal disegno agnostico per il linkage disequilibrium, sulla base del quale è stata costruita questa piattaforma). Né Affymetrix né Illumina sono esaustive (per esempio, SNP rari e variazioni strutturali sono sottorappresentati), tuttavia sono state progettate nuove generazioni di arrays per dare più ampia copertura a tutto il genoma.
Grazie a questi progressi, il 2007 ha registrato un vero passo avanti nella conoscenza della genetica del diabete tipo 2. Sono
stati pubblicati parecchi studi di associazione sull’intero genoma; questi studi hanno utilizzato frammenti di DNA con più di 500.000 SNP in un grande numero di pazienti con diabete tipo 2 e controlli; i cinque principali GWAS per il diabete mellito tipo 2 hanno coinvolto, approssimativamente, 55.000 individui (59, 143-147). Questi studi hanno molte caratteristiche in comune. In primo luogo, come detto, tutti hanno usato campioni di dimensioni relativamente grandi con un numero di casi oscillante tra 686 e 1924 e un numero di controlli da 669 a 5275 in ciascuno studio. In secondo luogo, tutti gli studi hanno usato campioni di DNA che erano stati raccolti da una ben definita area del nord Europa e tutti i partecipanti erano di discendenza nord europea. Questo ha ridotto il probabile impatto della mescolanza di popolazione, uno dei pochi possibili fattori di confondimento che può presentarsi in studi di associazione genetica e che consiste in un processo che porta ad un pool genico composito in cui, almeno alcuni individui, ‘derivano’ da più di una popolazione. In terzo luogo, tutti e cinque hanno usato estesamente studi di follow-up caso-controllo e hanno incluso uno stadio di replicazione, dove è stata testata la solidità statistica delle associazioni identificate. Un’associazione, per essere considerata statisticamente significativa, in questi studi richiede valori p inferiori a 5x10-8. Sebbene ci siano state delle differenze nella
definizione del fenotipo tra i vari studi (alcuni hanno usato pazienti affetti da diabete tipo 2 di età più giovane alla diagnosi o casi e controlli con valori di BMI analoghi) tutti gli approcci sono stati simili e i risultati osservati nelle cinque pubblicazioni sono coerenti, con una confidenza statistica che oscilla tra 1x10-12 e
1x10-19. Alcune pubblicazioni riportano come associato con
diabete tipo 2 lo stesso locus ma un diverso polimorfismo di singolo nucleotide, in tutti questi casi i polimorfismi sono sempre fortemente correlati con la malattia sulla base di alti valori r2 (si