1. Introduction
1.1. The retina
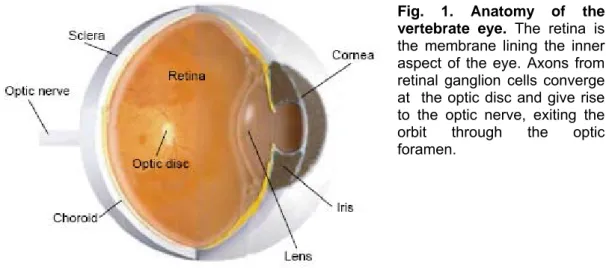
Processes leading to visual perception proceed through a set of consequential stages. The light penetrating through the cornea is first projected on the eye back (Fig.1), where it is transformed in electrical signals by the retina, a highly specialized sensory organ.
Then, these signals are brought through the optic nerve to superior visual centers where they are further elaborated.
During embryonic development, the vertebrate retina and the optic nerve originate as outgrowths of the developing brain. Hence, the retina is part of the Central Nervous System (CNS). It is the only part of the CNS that can be imaged directly and it has a very intricate and aesthetically pleasing cytoarchitecture. The combination of highly specialized cell types in well-organized networks performing complex modulatory activities results in an amazing and flexible sensory processing system and makes it a unique model for the comprehension of relationship between structure and function inside the CNS (for a review, see Wässle, 2004).
Various retinal cell types (photoreceptors, bipolar, horizontal, amacrine, glial and ganglion cells) are neatly localized in different layers based on their hierarchical relevance.
Fig. 1. Anatomy of the vertebrate eye. The retina is the membrane lining the inner aspect of the eye. Axons from retinal ganglion cells converge at the optic disc and give rise to the optic nerve, exiting the orbit through the optic foramen.
1.1.1. The mouse retina
In a vertical section of mouse retina (as in other vertebrate retinas) it is possible to recognize (fig.2):
• Photoreceptor layers: formed by the outer and inner segments of rods and cones;
• Outer limiting membrane (OLM);
• Outer nuclear layer (ONL): constituted by photoreceptor cell bodies;
• Outer plexiform layer (OPL) where processes of photoreceptors, bipolar and horizontal cells are synaptically connected;
• Inner nuclear layer (INL): formed by cell bodies of bipolar, horizontal, amacrine, and Müller glial cells;
• Inner plexiform layer (IPL) where processes of bipolar, amacrine and ganglion cells synaptically connect;
• Ganglion cell layer (GCL), containing the cell bodies of ganglion cells and displaced amacrine cells;
• Optic nerve fiber layer (OFL), constituted by axonal bundles of ganglion cells and supporting astrocytes,
• Inner limiting membrane (ILM)
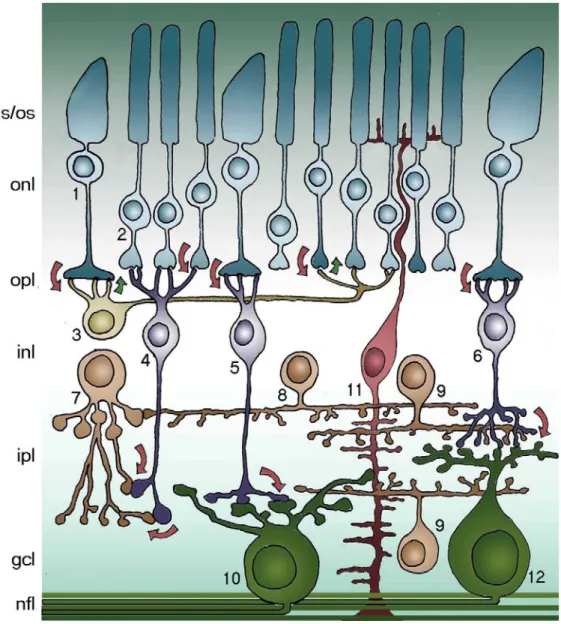
Fig. 3 summarizes the main connections among retinal neurons. Photoreceptors - rods and cones - convert light information electrical to chemical signals relayed to interneurons in the outer retina. At low light levels only rods have sufficient sensitivity to capture the few photons that are available. Cones permit day and color vision. Photoreceptors are connected to each others by electrical synapses (gap junctions) and make chemical synapses onto bipolar and horizontal cells. Two types of bipolar cells, rod and cone bipolars, make contacts with the homologous photoreceptor populations. Cone bipolar cells are further divided in two functional classes, called “ON” and “OFF” that respectively depolarize and hyperpolarize in response to the illumination of their receptive field centers. Rod Fig. 3. Schematic representation of the mouse retina. 1. cones (125,000); 2. rods (4,5*106);
3. horizontal cells (17,000); 4. rod bipolar cells (200,000); 5. cone bipolar cells ON; 6. cone bipolar cells OFF (total CB 400,000); 7. AII amacrine cells; 8. dopaminergic amacrine cells; 9. cholinergic amacrine cells; 10. ON ganglion cells; 11. Müller cells; 12. OFF ganglion cells (50,000 GCs total). is/os photoreceptor inner/outer segments; onl outer nuclear layer; opl outer plexiform layer; inl inner nuclear layer; ipl inner plexiform layer; gcl ganglion cell layer; nfl optic nerve fiber layer. Arrows indicate the flow of visual information.
bipolar cells belong to the single functional category of ON cells. Bipolar cells are then connected to RGCs to the same type (ON or OFF): this connection can be direct as in the case of cone bipolar cells, or through a chain of neurons comprising dedicated amacrine cells as for rod bipolar cells.
Differently from most mammals, mice have only one type of horizontal cells (Peichl and Gonzales-Soriano, 1994). Horizontal cells contribute to enhance contrast between adjacent light and dark regions. This is because they are constituted by two different, functionally distinct portions, by which they can control bipolar cell sensitivity: the dendritic arborization makes synapses with cones, while the axonal arborization is linked to rods. The connection horizontal cells - photoreceptors is bidirectional: not only horizontal cells are post- synaptic to rods, but also pre-synaptic to cones on which they exert a negative feed-back. Amacrine cells (more than 30 types) modulate signals from bipolar cells by providing inhibition directly onto ganglion cells; in addition, they modulate transmitter release from bipolar cells.
Light information leaves the retina and reaches other stations in the brain via axons of RGCs that collectively form the optic nerve.
Beside these above mentioned five classes of neurons, the retina contains one type of macroglial cells, and namely Müller cells. These have a radial arrangement spanning the depth of the retina and provide important, structural and functional support to retinal neurons.
Embedded within this basic organization, many specialized subcircuits are present in the vertebrate retina: they work both in sequence and in parallel to process different features of the visual image such as luminosity, contrast, chromatic composition and direction of motion (Weng et al., 2005).
RGCs represent the only exit neurons from the retina: their axons form the optic nerve that projects to the higher visual centers. The commonly employed
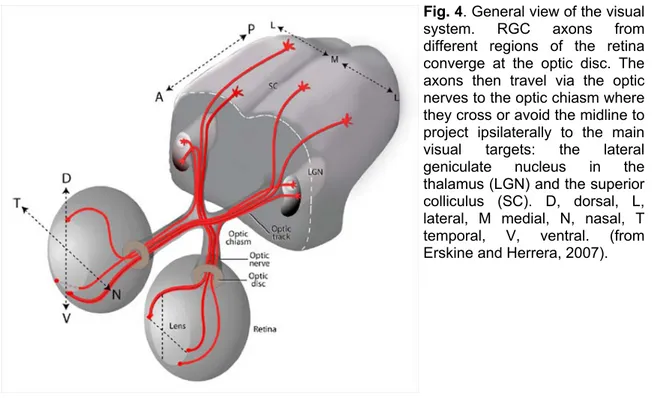
C57Bl6/J mouse retina contains approximately 50,000 ganglion cells. Most of them project contralaterally, since their axons cross at the optic chiasm; only less than 5% of the entire ganglion cell project ipsilaterally (Fig. 4). As in all vertebrates, RGCs are the largest of the mouse retinal neurons and have large diameter axons capable of generating action potentials.
The optic nerve collects all the axons of the ganglion cells and is a bundle of about 50,000 fibers in most mouse strains (Jeon et al., 1998) (more than a million in humans). Retinal fibers terminate in different visual nuclei of the thalamus, responsible for various functions of the visual system as the superior colliculus (SC), suprachiasmatic nucleus (SN) and the dorsal lateral geniculate nucleus of the thalamus (dLGN). In the thalamus, visual information is processed and then carried by LGN axons to the primary visual cortex in the occipital lobe of the brain, whereas the information going to the midbrain (SN) does not reach conscious levels but rather produces pupillary reflexes (which are controlled by the autonomic nervous system) and eye movements.
1.1.2. Mouse Retinal Ganglion Cells (RGCs)
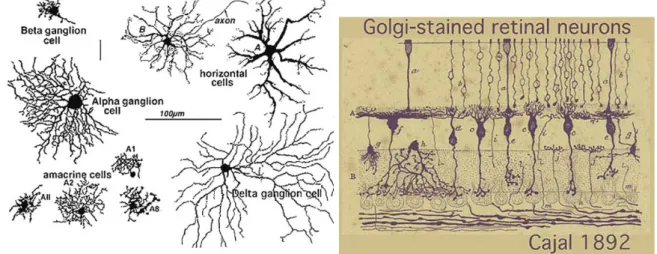
In his monumental work on Golgi staining of the vertebrate retinas, Cajal was able to classify many different varieties of ganglion cell based on their shape (dendritic morphology), extent (cell body and dendritic tree size), and number of sublayers in which they arborize (stratification levels in the IPL) (Fig.5 and 6).
Fig. 4. General view of the visual
system. RGC axons from different regions of the retina converge at the optic disc. The axons then travel via the optic nerves to the optic chiasm where they cross or avoid the midline to project ipsilaterally to the main visual targets: the lateral geniculate nucleus in the thalamus (LGN) and the superior colliculus (SC). D, dorsal, L, lateral, M medial, N, nasal, T temporal, V, ventral. (from Erskine and Herrera, 2007).
By the 70nties, it was known that α and β cells of Boycott and Wässle (1974) could be subdivided into separate subtypes depending on whether they branch in sublamina a or sublamina b of the IPL (Famiglietti and Kolb, 1976). Sublamina a contained dendrites of cells with physiologically defined OFF-center receptive fields and sublamina b dendrites of cells with ON-center receptive fields (Nelson et al., 1978) (Fig.7).
The advent of a technique to perform Golgi staining on whole mount retinas allowed a reinterpretation of many of the earlier classifications, because the entire dendritic tree of a RGC could be visualized in its third dimension. The whole mounts could also be compared with images of intracellularly injected cells after physiological recordings. These studies established a strict correspondence among dendritic morphology of RGC types and physiological function in 10 to 15 RGC types of many mammalian species (reviewed in Masland, 2001). For example, large RGCs, with open, radiate branching patterns, process fast, Fig. 6. A drawing by Cajal illustrating some
retinal neurons in vertical section (http://images.google.it/imgres?imgurl=http://w ebvision.med.utah.edu/imageswv/Catgolgi.
Fig. 5. Golgi-stained neurons of cat retina as
seen in whole mounts views (by http://images.google.it/imgres?imgurl=http://we bvision.med.utah.edu/imageswv/Catgolgi).
Fig. 7. Schematic organization of
ON- and OFF- centre ganglion cells into sublamina a and b. From:
http://webvision.med.utah.edu/G CPHYS1.HTM, after Nelson et al., (1978).
transient impulse trains and in all vertebrate retinas are concerned with motion detection, contributing to alert the animal about threatening, moving visual imagery. On the contrary, small, bushy ganglion cell types are concerned with processing small stationary, fine details in tonically activated messages. Others examples are ON and ON-OFF direction selective cells that respond to stimuli moving in a preferred direction and are inhibited by stimuli moving in the opposite or null direction; and melanopsin-containing cells that contain a photopigment and therefore directly sense light (Provencio et al., 1998 and 2002). Thus, each RGC subtype is characterized by a unique dendritic morphology and light response properties, constituting an independent and parallel channel coding a particular aspect of the visual scene; parallel information-coding channels and underlying information-coding strategies appear to have been evolutionarily conserved (Diao et al., 2004).
The number of morphologically characterized RGC types (or subtypes) in different mammalian species varies roughly between 10 and 20. Eight types of RGCs have been found in monkeys using retrograde labeling from the lateral geniculate nucleus (Dacey et al., 2003). Eleven types of RGCs have been reported in cats: α, β, γ, δ (Boycott and Wässle, 1974), epsilon (Leventhal et al., 1980), theta (Isayama et al., 2000), eta (Berson et al., 1999b), zeta (Berson et al., 1998), iota (Berson et al., 1997), kappa (Berson et al., 1999a), lambda (Berson et al., 1999b). Eleven types of RGCs have been characterized in rabbits (Rockhill et al., 2002). Thirteen types of RGCs have been reported in rats (Huxlin and Goodchild, 1997; Sun et al., 2002b), and 17 types in mice (Sun et al., 2002a). Dendritic morphology of several subtypes has been shown to be well-conserved across mammalian species; the α cell is a particularly good example of this concept (Peichl et al., 1987). Morphological equivalents of well-established ON and ON-OFF direction selective RGCs have also been found in cats (Berson et al., 1997), rats (Huxlin and Goodchild, 1997; Sun et al., 2002a), and mice (Sun et al., 2002b).
Three recent papers have defined classes of mouse RGCs on the basis of quantitative measurements (Badea and Nathans, 2004; Kong et al., 2005 and Coombs et al., 2006). Badea and Nathans (2004) provided a cluster analysis of retinal interneurons as well as of RGCs in the mouse, based on the expression of alkaline phosphatase (AP) activated by Cre-mediated recombination in a small fraction of cells.
A cluster analysis of mouse RGCs has been also provided by Kong et al. (2005), who relied on several different methods to label cells. Finally, Coombs et al. (2006) have used a line of transgenic mice expressing YFP controlled by a Thy-1 regulator in some RGCs. All these three studies assemble RGCs in clusters based on the k-means partitioning technique. Using more than 30 parametric measurements, RGCs are assigned to a specific cluster, among a number fixed a priori, utilizing a hill-climbing algorithm. This is a brilliant type of analysis because it is concentrated on avoiding errors that could derive by qualitative shape recognition of RGCs. On the other hand, it has some large limits due to the a priori fixed number of clusters and to the use of hill-climbing algorithm because when more parameters are added, further clusters may emerge in a certain group or subtype (Kong et al., 2005).
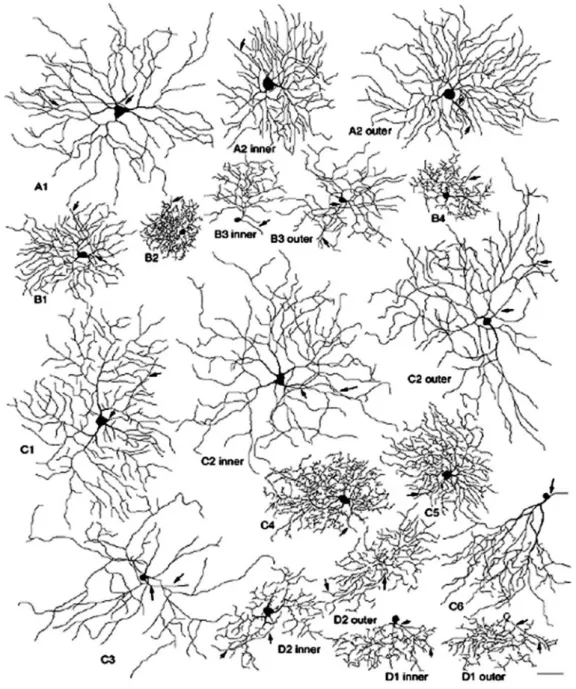
The classification of Sun et al., (2002a) (Fig. 8) adopts a more simple system of classification including the qualitative aspects of a cell characteristic morphology (shape recognition), and a few important morphometric parameters, considered fundamental in all classifications present until now. These are essentially: 1) the soma size; 2) the dendritic tree size; 3) the depth of stratification in the IPL. According to these three parameters and the shape evaluation, RGCs are classified into four principal groups. Cells with large soma and large dendritic tree are classified as A, cells with small to sized soma and small to sized dendritic tree as B, and cells with small to medium sized soma and medium-sized to large dendritic tree as C. Bistratified cells are classified as D.
A few cells, located in the peripheral retina, do not fit into this classification scheme. They are called “unclassified RGCs” and described as having big body (23 µm in diameter) and small dendritic tree (212 µm in diameter). One type stratifies at the inner IPL (68%) and the other one at the outer IPL (26%).
Thus, according to Sun classification, there are 17 ganglion cell types in the retina of the mouse (see Table.1), embedded within 4 main groups. For each given type, cells might have different stratification depths. A2 type cells consist of two subtypes, A2 inner (or ON) and A2 outer (or OFF); similarly, B3 type cells comprise B3 inner and B3 outer cells, while C2 cells comprise C2 inner and C2 outer. B2, B3, C2, C5 and D2 cell types are the most representative RGCs, in the sense that they occur more frequently in the mouse retina. In the mouse retina, as in all vertebrates, physiologically identified direction selective ganglion cells (DSGCs) are found to have bistratified morphology. The proximal dendrites of
these neurons arborize in the ON-sublamina and the distal dendrites in the OFF sublamina of the IPL, forming two independent layers with separate ON- and OFF inputs. According to Sun classification, D1 and D2 cells are bistratified RGCs.
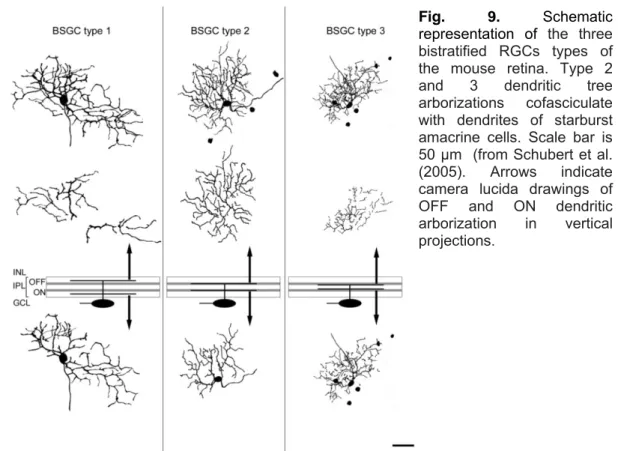
They have extremely thin, curvy and recursive dendrites distinctly stratified at two different depths (see Table.1). In particular, D2 cells exhibit a medium-density tree composed of recursive and loop-forming dendrites that make them very similar in morphology to the DSGCs of the rabbit (Sun et al., 2002a). Schubert et al. (2005) and Badea and Nathans (2004) provide a morphological classification of three different types of bistratified RGCs in the mouse retina and identify putative
Fig. 8. Schematic representation of different types of mouse RGCs from Sun et al. (2002a).
DSGCs through their co-fasciculation with the cholinergic bands of starburst amacrine cells (Fig. 9). In particular, two types (2 and 3) display homologous dye coupling and have dendrites co-fasciculating with the dendrites of starburst amacrine cells.
Table. 1. Schematic diagram of 17 types of RGCs according to Sun et al. (2002a). Each type is accompanied by three quantitative range values in which their body diameter (BD), dendritic tree diameter (DT) and stratification depth (SD) fall. Red labels in D1 and D2 types indicate the distal dendritic arborization in sublamina a (OFF) of the IPL (modified from Sun et al., 2002a). Green spots indicate RGC types analyzed in this work.
On the contrary, type 1 cells stratify independently from the cholinergic bands and show no dye no coupling. Therefore most likely they are not directional selective RGCs. The bistratified cells types 2 and 3 resemble the D2 and D1 types described by Sun et al. (2002a). The dendritic morphology of type1 cells partially resembles RGC3, but this RGC is described by Sun et al., as melanopsin-like ganglion cells because of its very large and sparse dendritic tree.
Melanopsin is an opsin-like protein, which is expressed by a subset of mouse, rat and human RGCs recently discovered. The notion that mice lacking rods and cones are capable of regulating their circadian rhythms by light has provided the conceptual framework for the discovery of an entirely new photoreceptor system within the mammalian eye (Provencio et al., 1998; Hattar et al., 2002; Provencio et al., 2002). A small fraction of RGCs (about 2% in humans) is directly photosensitive and utilizes an opsin/vitamin A-based photopigment, precisely melanopsin, maximally sensitive to the blue part of the spectrum (about 470 nm). Most recently, it has become clear that the melanopsin pigments are only distantly related to visual pigments and share biochemical features with invertebrate photopigments (Hankins et al., 2008). Melanopsin RGCs mediate a broad range of physiological responses to light, ranging from the regulation of circadian rhythms and melatonin release to the control of pupil constriction.
Melanopsin-Fig. 9. Schematic
representation of the three bistratified RGCs types of the mouse retina. Type 2 and 3 dendritic tree arborizations cofasciculate with dendrites of starburst amacrine cells. Scale bar is 50 µm (from Schubert et al. (2005). Arrows indicate camera lucida drawings of OFF and ON dendritic arborization in vertical projections.
generated information is carried by the retino-hypothalamic tract directly to the SN. Thanks to anti-melanopsin antibody generation (Hattar et al., 2002) it has became possible to reveal this peculiar population of RGCs by fluorescent immunocytochemistry. In rats, more than 95% of melanopsin labeled cell bodies (representing 2.5% of all GCs) are in the GCL, while the remainders are displaced to the INL. Within the GCL or displaced to the INL, melanopsin-expressing RGCs, extend their dendrites into the IPL, where they arborize most extensively at the border with the INL. Some arborizations invade and often terminate within the INL. Displaced RGCs have more planar and sparse dendritic arborizations. Dendrites from adjacent cells overlap extensively, forming a reticular network. The density of melanopsin-positive cells is slightly higher in the superior and temporal quadrants of the rat retina. In mice, 3 types of cells labeled by melanopsin antibodies have been described: ON, OFF, and ON/OFF (Coombs et al., 2006); there is no evidence for displaced, melanopsin-positive RGCs in mice.
Displaced RGCs are located near to the inner margin of the INL, and, in mouse, they are about 2% of all RGCs. They have mainly peripheral localizations; they are abundant and tightly clustered in the nasal quadrant, and loosely scattered in the temporal retina; they preferentially project to the ipsilateral side of the brain, representing 21% ipsilateral of all ipsilateral projections (Dräger and Olsen, 1981).
1.2. The Thy1-GFP transgenic mouse
Green fluorescent protein (GFP) isolated from the jellyfish has revolutionized our ability to view cellular details in living tissues and intact organisms. A distinct advantage of expressing fluorescent proteins compared to dye labeling is that bleaching and phototoxicity are much reduced. Also, bleaching can recover over time as more protein is synthesized.
Recently, Feng et al. (2000) have generated transgenic mice in which red, green, yellow or cyan fluorescent proteins (together termed XFPs) are selectively expressed in neurons. All four XFPs label neurons in their entirety, including axons, nerve terminals, dendrites, and dendritic spines. The Authors have chosen mice expressing XFP under the control of neuron specific element from the thy1 gene. Thy1 is an immunoglobulin superfamily member expressed by projection neurons in many parts of the nervous system, as well as by several non-neuronal cell types, including thymocytes (hence its name). Early transgenic analysis
revealed that neural and non-neural expression depends on distinct genomic elements and that deletion of a particular intron selectively abolishes expression in non-neural cells (Vidal et al., 1990). A construct lacking this intron has been successfully used to overexpress β-galactosidase and growth-promoting molecules in neurons with minimal non-neural expression (Caroni, 1997). Transgenic mice obtained by Feng et al. (2000) express various XFP under the control of these neuronal thy1 regulatory elements. Remarkably, they show that there is variability in patterns of XFP expression among mice generated from the same construct; expression is similar among offspring of each transgenic founder, but differs among lines, indicating that the variation reflects differences in integration site and/or copy number. In addition, Thy1 is a “specific” marker for RGCs in the retina, although it is present at low levels in some amacrine, bipolar and Müller cells as well (Fig.10).
Thus, GFP expression is most prominent in neurons that are Thy1 positive. Consistent with this pattern, the transgene in expressed in at least some RGCs of the 24/25 line, in amacrine cells of the 12/25 line, in bipolar cells of the 5/25 line and in Müller cells of the 4/25 line. One of these lines, called Thy1-GFP-M (Fig. 10) expresses a few GFP-positive RGCs. It has been employed successfully in recent work on retinal synaptic connections (Lin and Masland, 2005) and RGC classification (Kong et al., 2005; Coombs et al., 2006). The Thy1-GFP mouse has
Fig.10. Pattern of transgene expression of in the Thy-1GFP lines. Patterns are grouped into
large categories. “All” means a widespread expression in all the neurons of the indicated class (show in red); “Many” indicate expression in 10-80% of neurons (orange); “Few” means expression in <10% of neurons (yellow-orange). Numbers under “cortex” indicate layers in which bodies of labelled neurons are observed. In the line Thy1-GFP-M (arrow),used in this study, and in –O, some labelled non-neuronal cells (fibroblast or satellite cells) are present in the superior cervical ganglion (SGC). Dorsal root ganglion cells (DRG) and cerebellar cells express GFP in this line; A+B+M means expression in amacrine, bipolar and Müller cells. Modified from Feng et al., (2000).
also been used in the present study to exploit the advantages of single cell labeling (Fig. 11).
1.3. Retinitis Pigmentosa (RP)
RP comprises a group of heterogeneous genetic disorders that cause severe visual impairment in as many as 1.5 million patients worldwide (for a review, see Mendes et al. 2005; Kennan et al., 2005; Paskowitz et al. 2006). The first cells to become affected are generally rod photoreceptors, which mediate vision in conditions of dim light. Once rods start to die (as a result of a genetic abnormality), patients experience night-vision limitations and develop visual field
Fig.11. Some examples of RGCs labelled in the Thy1-GFP-M strain. The GFP signal is
amplified with the antibody anti-GFP conjugated to AlexaFluor-488 and then acquired at a confocal microscope. Single RGCs are easily distinguished in their parts: axons, bodies and dendritic arborizations with minimal overlapping.
constriction (or tunnel vision) (Fig. 12) and electroretinographic (ERG) abnormalities consequent to the degeneration of rods at the retinal periphery.
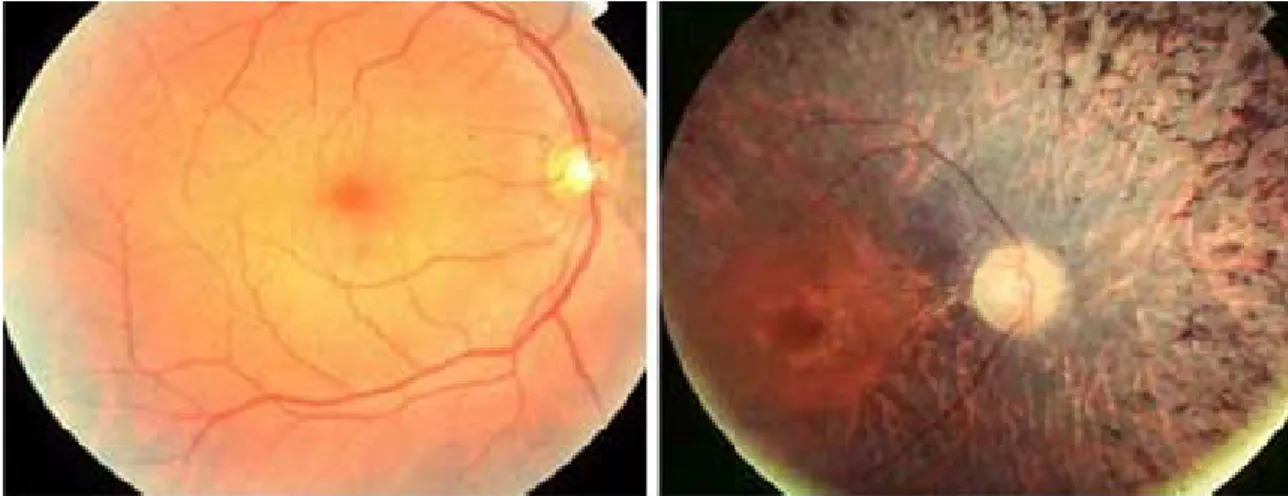
The death of rod photoreceptors has a secondary, detrimental effect on cone photoreceptors, which are responsible for vision in daylight, and these too begin to die, eventually resulting in the loss of central vision. The progressive demise of photoreceptors also precipitates other pathological symptoms in the retina, including the attenuation of the retinal vasculature and the accumulation of intra-retinal pigment deposits, from which the disease gets its name (Fig. 13).
It is unusual for patients with RP to become totally blind as most of them retain some useful vision well into old ages. Forms of RP and related diseases include,
Fig. 12. Tunnel vision (right) is a typical example of eye field degradation in RP
patients.
From http//www.pocklington- trust.org.uk/Templates/Internal.asp?NodeID=89018
Fig. 13. Fundus views of the retina of a normal human subject (left) and of a RP patient (right).
A typical brown pigmentation is present in the pathological condition. Pigment deposits, named bone spicules for their typical shape, are responsible for the name of the disease. A clear attenuation of retinal blood vessels is also visible.
among others, Usher syndrome, Leber’s congenital amaurosis, rod-cone disease, Bardet-Biedl syndrome, and Refsum disease.
1.3.1. Genetics and molecular mechanisms of RP
The genetics of RP are complex. It can be sporadic, autosomal dominant (ad), autosomal recessive (ar), or X-linked. More than 30 RP genes are known to be associated with RP; sometimes the same gene is involved in different inheritance traits. At present, about 60% of RP phenotypes have no known genetic cause. Among the large number of mutations causing RP, there are deletions, insertions, or substitutions, in turn producing missense mutations or truncations (Wang et al., 2005). Only for a small number of mutations, a clear correlation between translated protein function and retinal disease is known. Typically, this number include mutations in proteins of the phototransduction pathway, factors playing a role in the maintenance of cellular structure and in the transcriptional control of photoreceptor genes (Mendes et al. 2005; Kennan et al., 2005).
Fig. 14 shows the phototransduction cascade. Most of the genes codifying for phototransduction proteins undergo mutations eventually leading to RP and thus causing the death of photoreceptors by apoptosis. Many of these mutations have been characterized because their homologous gene is mutated in humans and in animals (mice, rats or dogs) (Bedell et al., 1997a and b).
In particular, two mutations of the phototransduction system will be taken into account here: the first (RHO) is the best studied and responsible for various forms of autosomal dominant RP (adRP). The second, (Pdeb), might cause autosomal recessive RP (arRP) and is also carried by the mouse models used in this study.
1.3.1.1. RHO mutations
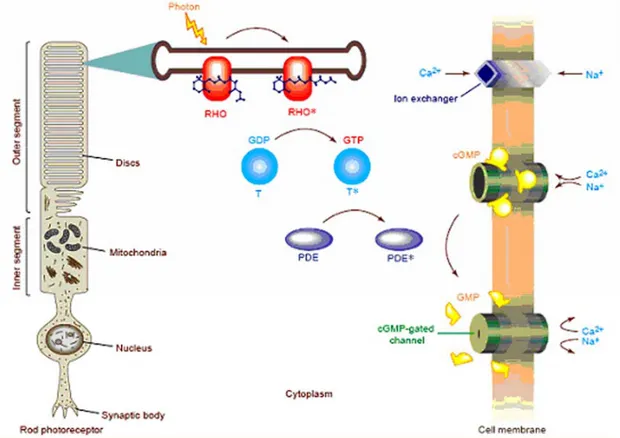
Rhodopsin (Fig. 14 above) is the light absorbing protein that initiates the visual transduction cascade. The visual pigment rhodopsin is a prototypic protein member of the G-protein coupled receptor family; it is composed of a 348 amino
Fig. 14 The rod phototransduction cascade. In the rod outer segment (ROS) membrane,
cGMP binds to a cGMP-gated cation channel in the dark, keeping it open and enabling Na+ and
Ca2+ ions to flow into the ROS. Following light absorption, the visual pigment rhodopsin, a seven transmembrane G-protein-coupled receptor, is activated by conversion of the 11-cis retinal chromophore bound form of rhodopsin to its all-trans isomer. This reaction leads to the formation of Meta II rod-opsin or Rho*, which initiates the visual cascade by binding transducin (T) and catalyzing the exchange of GDP for GTP on its a-subunit, thus converting transducin into an active form. Activated transducin, in turn, activates cGMP-phosphodiesterase (cGMP-PDE), which brings about the hydrolysis of channel bound cGMP to 5’-GMP. The resulting rapid decrease in intracellular cGMP levels causes the cGMP-gated channels to close and the rod cell becomes hyperpolarized. Following the closure of the channels, the release of glutamate at the synaptic region is inhibited and Ca2+ levels in the ROS are decreased. The hyperpolarization of the cell results in the transmission of an electrical signal, via synaptic junctions, through the different layers of the retina and on to the brain, via the optic nerve. After photoexcitation, the photoreceptor cell returns to its dark state with the shutdown of the visual cascade system and re-synthesis of cGMP, mediated by a calcium feedback mechanism. Activated rod-opsin is phosphorylated by rod-opsin kinase and is then bound by arrestin, which prevents further interaction with transducin. Transducin and phosphodiesterase (PDE) are inactivated by the hydrolysis of GTP to GDP on the transducin a-subunit by the intrinsic GTPase activity of transducin. Guanylate cyclase, the enzyme responsible for the synthesis of cGMP from GTP, is activated by the decrease in intracellular Ca2+ after photoexcitation. As the cGMP concentration increases, the cGMP-gated channels re-open, and the photoreceptor cell is returned to its depolarized state (from Kandel, ”Principles of Neural Science”, fourth edition; Part V: perception; cap. 26).
acid chain organized into three distinct domains: cytoplasmic, transmembrane and intradiscal, plus a chromophore (11-cis-retinal). At now, over than 120 point mutations in rhodopsin have been identified (Retnet http://www.sph.uth.tmc.edu/RetNet); they are subdivided in different classes depending on the involved domain; many of them eventually cause arRP but the majority lead to adRP (reviewed in Mendes et al., 2005). Some mutations in the C-terminus of the protein affect the post-Golgi trafficking and impair its normal targeting to the photoreceptor outer segment (Tam et al., 2000). Others adRP mutations are characterized by an intradiscal, transmembrane and cytoplasmic domain localization which result in a misfolding that affects the post-Golgi trafficking and impairs normal targeting of the protein to the photoreceptor outer segment. Incorrectly-folded rhodopsin might also display inability to form a functional chromophore with 11-cis-retinal. In other cases, post-translational modifications (such as in the T4R mutation that causes adRP in dogs) lead to an impairment of rhodopsin stability; finally, in others mutants a constitutive activation of opsin in dark conditions occurs. Dominance in adRP patients could be due to loss-of-function, gain-of-function or dominant negative mutations, or to any of the above in combination. At now there is strong evidence that the dominant alleles might be due to gain-of function or dominant-negative mutations. For example, from experiments on transfected cells, it has been shown that heterologous wild-type rhodopsin translocates to the plasma membrane, whereas P23H and K296E mutant opsin forms aggregates that have many of the characteristic features of an aggresome. Given the fact that these aggregates are ubiquitinated, they recruit cellular chaperones and disrupt the intermediate filament network. Thus, mutant opsin expression can disrupt the processing of normal opsin. Co-transfection showed that the wild-type protein is recruited to mutant opsin aggregates (Saliba et al., 2002).
In addition, mutant opsins, such as P23H (the most common cause of ADRP in North America), are retained within the endoplasmic reticulum (ER) causing a disturbance of Ca2+ homeostasis and a subsequent stress response of the cell, known as “unfolded-protein response” (UPR, or ER-stress response) (Rutkowski and Kaufman, 2004). This culminates in cell death by apoptosis. The P23H mutation of rhodopsin leads to photoreceptor death through this cellular mechanism (Mendes et al., 2005).
The retention of mutant opsins in the ER is followed by their retro-translocation from the ER and degradation by the ubiquitin–proteasome system (UPS) (Mendes, 2005). As is the case of other aggregation proteins, the mutant opsin not degraded by the UPS aggregates in the cytosol. Protein aggregates then coalesce into ubiquitinated proteic inclusions (Saliba et al., 2002). Many neurodegenerative diseases are characterized by the intracellular aggregation of ubiquitinated proteins and by the formation of inclusion bodies (Mendes et al., 2005). Protein aggregation can directly impair the function of the UPS (Rutkowski and Kaufman, 2004); actually, P23H rhodopsin expression inhibits UPS activity in HEK-293 cells (Illing et al., 2002). Ubiquitin-dependent proteolysis has a central role in regulating many fundamental cellular events, and such an inhibition by mutant opsins could stimulate further aggregation and initiate a cascade that would lead to apoptosis.
Several animal models of rhodopsin-associated RP have been generated and include the rhodopsin knockout mouse, which lacks any functional rhodopsin and thus rapidly undergoes retinal degeneration, and dominant transgenic models such as the P23H mouse (for a review, Kennan et al., 2005). These animals are an extremely valuable resource in the study of rhodopsin-induced degeneration mechanisms, providing in vivo evidence of the cell death pathways proposed for cell-culture systems. Although an extensive body of research is focusing on rhodopsin-induced degeneration of photoreceptors, an overall picture of how mutations in this gene induce apoptosis in photoreceptors is far from being complete.
1.3.1.2. Pdeb mutations
When a photon is absorbed by rhodopsin, cGMP-PDE and transducin are activated (Fig. 14 above). cGMP-PDE catalyzes the degradation of cGMP to guanosine-5’-monophosphate (5’-GMP), closing ionic channels gated by cGMP and eventually producing a hyperpolarization in the photoreceptor membrane. Thus, cGMP-PDE is pivotal in photoreceptor conversion of light to electric signals (Yau & Baylor, 1989). The cGMP PDE enzyme is a tetrameric molecule, composed of two large catalytic subunits, α and β, and two small inhibitory γ subunits (Baehr et al., 1979; Deterre et al., 1988).
Mutations in the Pdeb gene were first identified in rodless mice (r; Keeler, 1924), which carry a nonsense mutation in the Pdeb gene coding for the β-subunit of cGMP phosphodiesterase of rods. This spontaneous mutation was later rediscovered in the retinal degeneration mouse (rd1 or rd; actual gene symbol
Pde6brd1; Bowes et al., 1990; Pittler and Baehr, 1991a,b; Pittler et al., 1993) and
subsequently in humans with arRP (McLaughlin et al. 1993, 1995) and adRP form of night blindness (Gal et al. 1994). Linkage mapping correlates with the identification of the β subunit of cGMP-PDE as the defective candidate gene in rd mice (Bowes et al., 1989; Bowes et al., 1990).
In mice, many of the commonly used inbred strains, including C3H and its derivatives and CBA/J, are homozygous for rd1. In particular, two mutations in the
Pdeb gene of rd1 mice have been identified: besides the nonsense mutation that
causes truncation of the protein (Bowes et al. 1990), the intronic insertion of an endogenous mouse leukemia virus (Xmv28) causes an incorrect splicing. Xmv28 has also been found in strains derived from wild type mice, as well as from inbred laboratory strains that not known to have recent common origins (Bowes et al. 1993). This suggests a surprisingly high degree of evolutionary stability for a mutation that causes loss of vision in mice. Evidently, there is little or no selective pressure to maintain a functional β-PDE gene, and therefore vision, in these nocturnal animals.
Biochemical studies comparing retinas from normal and rd1 mice have show that the lack of cGMP-PDE activity causes a dramatic increase in cytoplasmic cGMP concentration (Farber and Lolley, 1974). This, in turn, results in permanent opening of the cGMP gated cation channels on the photoreceptor membrane, allowing the excessive entry of extracellular ions, particularly Ca2+. It has been suggested that this increase in intracellular Ca2+ causes a metabolic overload of the cells, eventually leading to cell death by apoptosis (Chang et al., 1993). A clear picture that describes the apoptotic pathways involved in photoreceptor degeneration in the rd1 mouse has yet to emerge. Recently Rohrer et al. (2004) evaluated gene expression over the time course of photoreceptor degeneration in the rd1 and wild type mouse retina. Their results indicate that the Pdeb gene-triggered cell death alters the expression of genes involved in diverse cellular pathways including Ca2+ homeostasis, catabolism, neuroinflammation and tissue remodelling (as blood-retina barrier breakdown).
In mouse photoreceptors, normal Ca2+ levels range between ~250 nmol/liter (in complete darkness) and ~60 nmol/liter (in light) (Woodruff et al., 2002). However, in the rd1 photoreceptors, Ca2+ levels are increased up to ~190% over wt levels (Fox et al., 1999). Consequently, messages for Ca2+-binding proteins, and in particular Ca2+ sensors such as calbindin, are up-regulated in the rd1 photoreceptors. Genes activated by Ca2+ through cAMP- binding protein sites in their promoters include genes involved in the immune response such as β2-microglobin and major histocompatibility complex class I receptors (Gobin et al., 2001). So far, the focus has been on Ca2+ as major trigger of the apoptosis cascade, while the role of high cGMP levels remains unanswered yet.
Up- and down-regulations of some proteins produced by genes involved in rod degeneration pathway have been studied with a proteomic approach on rd1. It has been shown that, in response to retinal degeneration, there is a loss of proteins involved in the rod-specific phototransduction cascade (as expected), as well as induction of proteins from the crystallin family, possibly in virtue of their protective role (Cavusoglu et al., 2003).
1.4. Examples of RP mutant mice: The rd10 mutant mouse
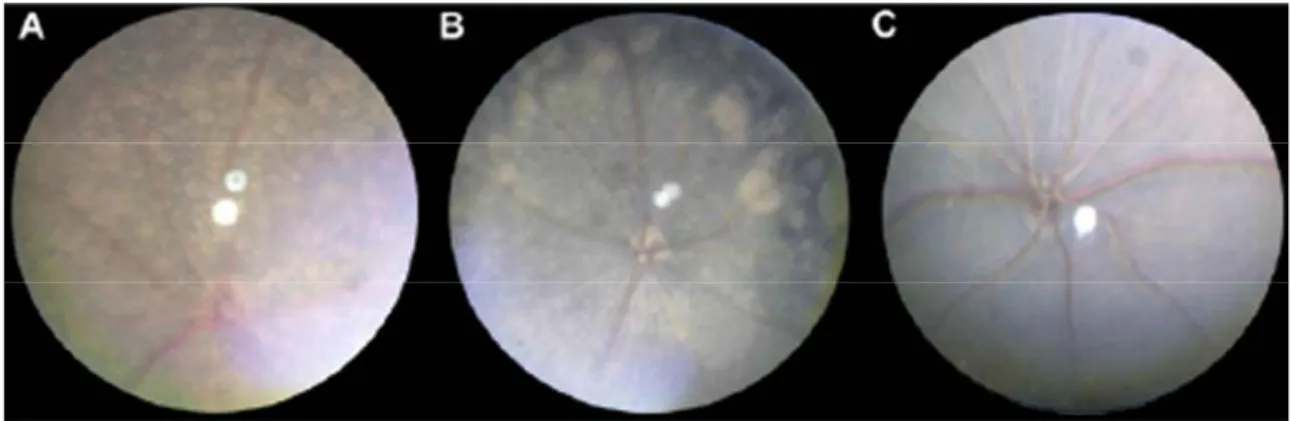
As it has been shown in the previous paragraphs, spontaneous mutations leading to retinal degeneration occur in various mammals, including mice. Like in humans, retinal alterations can be detected macroscopically by fundus examination and photography (Fig.15). Some of these mutants are invaluable tools to study the cell biology of RP.
Fig. 15. Fundus appearance of various mouse strains at two months of age: rd1 (A), rd10 (B),
and C57BL/6J wild-type (C). The retinal degeneration in the rd10 strain is easily distinguished from wild-type and rd1 retinal appearance at two months of age by fundoscopy (from Chang et al., 2002).
Besides the earliest discovered animal models of retinal degeneration, including the rd1 mutant mouse, we consider here another example of autosomic recessive RP, isolated at the Jackson Laboratories in the USA (Chang et al., 2002), and namely the rd10 (retinal degeneration 10) mouse. Genetic analysis shows that this strain carries an autosomal recessive mutation that maps to mouse chromosome (Chr) 5. Sequence analysis shows that the retinal degeneration is caused by a missense point mutation in exon 13 in the Pde6b gene of the β-subunit of the rod cGMP phosphodiesterase (β-PDE) gene (Pde6b). The mutation changes codon 560 from CGC to TGC resulting into an arginine to a cysteine change and in the loss of a CfoI site. The exon 13 missense mutation is the first known occurrence of a re-mutation in the Pde6b gene in mice and may provide a good model for studying the pathogenesis of autosomal recessive RP in humans (arRP). It may also provide a better experimental model for RP because of its later onset and milder phenotype compared to the more common rd1 mutant (Otani et al., 2004; Rex et al., 2004). In fact, in rd10 mutant mice, photoreceptor degeneration starts around P18 (about 10 days after the rd1 mutation). Rod death follows a central to periphery gradient; then a slower degeneration of cones occurs. Atrophic retinal vessels are found at four weeks of age, consistent with retinal degeneration (Chang et al., 2007; Gargini et al., 2007). Electroretinograms (ERGs) of rd10 mice are never normal, but rod and cone ERG a- and b-waves can be measured at P18 and steadily decline over 90% by two months of age. Interestingly, rearing rd10 mice in total darkness delays degeneration for at least a week, after which morphological and functional loss progresses irregularly (Chang et al., 2002).
1.5. RP experimental therapeutic strategies: Transplants and implants
1.5.1. Transplants
The possibility of retinal transplantation represents a hope for the restoration of vision through cell-replacement therapy. However, getting the transplanted cells to establish the right connections within the retina has been a major problem by far. It appears that an early developmental stage of the retina of acceptor’s represents a key feature of a successful transplant. Despite decades of experimental
attempts, transplants have yet to produce better vision in mammals with retinal degeneration because the transplanted cells do not wire up properly.
The first successful transplant of a mammalian retina dates back to 1959, when Royo and Quay transplanted fetal rat retinas into the eyes of adults of the same strain. Although the transplanted retinas did not seem to connect with the host retinas, they survived for months. Since then, intact retinal sheets from embryonic mice and rats have been transplanted to the sub-retinal space. Transplants appear to develop many characteristics of a normal retina (Seiler et al., 1990; Zhang et al., 2003) and grow to form a second retinal layer underneath the host tissue. Also, transplants of ‘micro-aggregates’ — clumps of few retinal neurons — from newborn mice, develop most characteristics of rod photoreceptors, including the expression of the rhodopsin and even the characteristic outer segments (Gouras et al., 1991 and 1994).
Unfortunately, both the intact embryonic retinal sheets and the micro-aggregates of photoreceptors remain isolated, without interacting or integrating effectively with the host retinal neurons.
Integration with the host retina is much higher when transplanting retinal progenitors, the immature cells responsible for producing all retinal cells during embryonic development. These progenitors (or retinal stem cells)are derived from fetal or newborn mice or rats, or from human fetuses. They can be maintained in cell culture and continue to proliferate and generate new neurons and specialized retinal support cells as Müller glia (Anchan et al., 1991; Reh et al., 1998; Tropepe et al., 2000). When these cells are transplanted into either normal or degenerated (dystrophic) retinas of rats and mice, they migrate into all retinal layers and develop morphological characteristics of various retinal cell types (Chacko et al., 2000; Klassen et al. 2004; Coles et al., 2004; Qiu et al., 2005), mainly differentiating into photoreceptors (Coles et al., 2004; Klassen et al., 2004).
Recently, MacLaren et al. (2006) have shown in mice that a crucial point might be the employment of cells at a particular stage of their development. The Authors have found a method to combine the migration and integration potential of the progenitor cells with the photoreceptor differentiation properties of retinal sheets. In a first set of experiments, they have tested whether retinal progenitor cells are more prone to migrate into the host retina compared to mature, differentiated neurons. Using cells taken from the retina of embryonic or newborn mice at different postnatal ages, they find, surprisingly, that postnatal cells integrate into
the ONL most effectively. This despite the fact that the embryonic cells include the highest percentage of progenitor cells. The transplanted rod cells develop morphology identical to normal host rods, making well differentiated outer segments. On further analysis, the Authors found that the best donor cells have to be from 3 to 5 days old, roughly corresponding to two or three days after the peak of rod photoreceptor production in the mouse retina. This suggests that, contrary to expectations, newly born rods, rather than the progenitor cells, are the best candidates for cellular transplantation.
In addition to the striking morphological evidence for integration of the transplanted rod photoreceptors into the normal outer retina, this study provides evidence that the transplanted photoreceptors connect up to host retinal neurons, and can partly restore the response to dim light levels in mice that are blind because they are deficient in the rod protein rhodopsin. These results represent at the moment the best example that cell-replacement therapy for blindness.
However, if this scenario had to be extended to humans, one would have to obtain newly generated rods from a developmental stage comparable to postnatal days 3–7 of the mouse. This is likely to fall in the second trimester of pregnancy and therefore access to these cells is not devoided of ethical issues. However, a recent report (Lamba et al., 2006) shows that cells expressing the protein Nrl — a marker of newly born rods — can be obtained from human embryonic stem-cell lines, given certain culture conditions. Further studies are needed to determine whether the stem-cell-derived, Nrl+ cells behave like the mouse rod photoreceptor progenitors before these can be considered for cell-replacement therapy. Anyway, at now, the message emerging from these studies is that the specific time at which the replacement cell is harvested makes all the difference in its potential for integration and functional differentiation in the host retina.
On a different rationale, promising results for RP treatment have been obtained by Otani et al. (2004), still using a cell-based approach. These Authors have shown that injections of hematopoietic stem cells (lineage-negative called Lin-HSCs) into the eye of mouse models of RP (rd1 and rd10 mice) results into a dramatic rescue of both blood vessels and photoreceptors, mainly cones. These mice show an improvement of the ERG at an age when usually it is completely extinct and the rescue effects are long lasting.
1.5.2. Implants
The development of artificial implants inserted in substitution of an entire part of the pathological retina represents an active line of investigation to treat both RP and other retinal diseases, such as macular degeneration.
Advances in microtechnology have facilitated the development of a variety of prostheses that can be connected to the brain or implanted in the eye; these implants have a greater potential for visual restoration as they are well tolerated and remain viable for several years. Some of these approaches have already improved the eyesight of patients with major visual impairments.
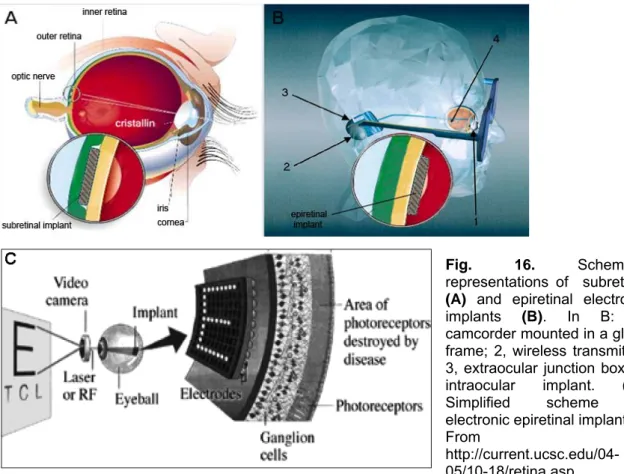
Of course, retinal prostheses are only effective if the visual pathway distal to the retinal implant is still intact and functional. Two types of prostheses are under development according to the site of retinal implant: epiretinal (on the surface of the retina) and subretinal (under the retina) prostheses (Fig. 16. A and B and C in more detailed).
Fig. 16. Schematic representations of subretinal
(A) and epiretinal electronic
implants (B). In B: 1, camcorder mounted in a glass frame; 2, wireless transmitter; 3, extraocular junction box; 4, intraocular implant. (C). Simplified scheme of electronic epiretinal implant. From
http://current.ucsc.edu/04-05/10-18/retina.asp.
The following is a brief illustration of their main features, with a focus on the advantages and limits of the prosthetic approach (for a review, see Zrenner, 2002; Chow et al., 2004; Hossain et al., 2005).
1.5.2.1. Subretinal prostheses
Subretinal prostheses contain microphotodiodes attached to microelectrodes. These implants, such as the artificial silicon retina (5000 microelectrodes), are placed in the subretinal space between the outer retina and retinal pigment epithelium (fig. 16. A). the photodiodes are stimulated by light passing trough the retina, and the resulting electric current excites adjacent retinal sensory neurons. The specifications for subretinal implants vary. For example, a typical device measures 50-100 µm wide, has a diameter of 2-3 mm, and carries microphotodiodes on a microelectrode array (Chow et al., 2004). These implants do not require an external electrical source as incident light is sufficient for stimulation (Zrenner, 2002).
The viability of subretinal devices has been assessed in animal experiments. Several species have shown tolerance to the implants, some for up to 30 months. Histological evidence has shown no relevant changes in the architecture of the retina. Visual perception is improved in six patients with RP who received an artificial silicon retina, including subjective improvement in appreciation of brightness, contrast color, movement, shape and visual field. Some patients showed an improvement in visual acuity.
Two major advantages of subretinal prostheses are the utilization of existing forces between the neural retina and retinal pigment epithelium to maintain their position and the potential of a high spatial resolution, as they are positioned close to retinal nerve cells and can stimulate neurons by means of low electrical currents. The main disadvantages of subretinal implants include impaired nourishment of the inner retina due to the creation of a mechanical barrier between the outer retina and the choroid and the occurrence of trauma to the retina during the implanting. These prostheses also show poor dissipation of heat and therefore could damage the retina. The mechanism of action of these implants may be by direct stimulation of retinal neurons. A specific neurotrophic effects elicited locally onto photoreceptors by means of the surgical procedure cannot be excluded: a study of subretinal artificial silicon implants in rats showed
a temporary protective effect on the retina, resulting in increased generation of photoreceptors.
1.5.2.2. Epiretinal prostheses
Epiretinal prostheses are composed of an array of electrodes implanted on the surface of the retina between the vitreous and inner limiting membrane. The implants receive electrical signals from a camera positioned outside the body (FIG.16.B and C in more detailed; see also Fig. 17, for in vivo application). In one such device, the camera transmits light signals to a microchip within the camera. This microchip deciphers the signal and relays it, using wireless transmission, to a microchip in the epiretinal implant which in turn stimulates the RGCs (Zrenner, 2002; Humayun et al., 1999a). Early studies in dogs and rabbits showed the flexibility of using epiretinal prostheses: a few clinical trials in humans have reported simple visual perception as phosphenes. Perception of light only is reported by three “completely blind” patients with RP who received the so-called “second sight” model: these implants have survived for up two years.
One advantage of epiretinal implants over subretinal devices is that the camera can process signals before they reach the implant: this allows optimization of the signal quality, which may lead to improved visual perception. Epiretinal implants can also use the heat dissipating properties of the vitreous and are therefore less likely than subretinal implants to damage the retina. The superficial location of the implant reduces risk of trauma during implantation and allows the replacement of
Fig. 17. Fundus photography showing an epiretinal electronic implant.
the implant. The main disadvantages of epiretinal implants are the need for complicated microtechnology and surgical techniques that ensure secure fixation of the implant on the retina. The device also requires a higher electrical current than the subretinal implant (for a review, Lakhanpal et al., 2003). Implants are being developed to generate their own electrical currents on stimulation.
Some epiretinal prostheses used to attempt restoring sight in RP patients, are designed to produce direct or near stimulation of RGCs. The success of such electronic devices is partly based upon the hope that RGCs are still viable after photoreceptors death. Yet, recent data show that some RP subject lack phosphenes in respond to epiretinal stimulation (Delbeke et al., 2001). In addition, it has been reported that perceptual thresholds for electrical activation of the retina are surprisingly high in RP patients (Rizzo et al., 2003). This is partially confirmed by the work of Stasheff with microelectrode arrays on RGCs in the rd1 mouse (Stasheff, 2008). Here, RGCs have a much higher spontaneous frequency than normal, sometimes in rhythmic bursts that are distinct from the developmental “waves”. A probable explanation could be that strong remodeling RGCs has occurred, and their membrane has become relatively unexcitable.
According to the group of Fishman (Stanford Nanofabrication Facility), all the present types of prosthetic chips (either sub or epiretinal) stimulate neurons electrically with limited spatial control and without cell specificity. For example, extracellular electrodes can excite retinal cells but cannot inhibit them (unlike physiological neurotransmitters such as GABA and glycine). An ideal chip should deliver a chemical stimulation so that different transmitters are recognized by different cells and produce different, and specific, effects. To overcome these limits, researchers are working on the ‘‘artificial synapse chip”, in which the advantages of two technologies, electronic engineering and cell biotechnology, are combined. The prototype chip should drive growth of retinal cell dendrites and axons directly into the chip, essentially mimicking a synapse, by controlled, repeatable release of neurotransmitters, as occurs in natural vision (Peterman et al., 2003 and 2004).
1.5.3. Other therapeutic strategies to treat RP
1.5.3.1. Neuroprotective factors
It is estimated that, in humans, it would be sufficient to preserve 5% of all cones to maintain a useful vision, while 50% of cone functionality would ensure normal vision acuity.
With this respect, neuroprotection, that is to say the possibility to slow down the natural RP course, or the preservation of cone function, appear as a promising and feasible approach. In addition, neuroprotection would work independently of the underlying genetic mutation causing RP, and therefore could bypass the tremendous genetic heterogeneity of this disease and the high incidence of sporadic cases. Many trophic factors have been experimented in various animal models of RP: bFGF (Faktorovich et al., 1990), GDNF, BDNF (LaVail et al., 1998) and others. So far, CTNF (ciliary neurotrophic factor) has been demonstrated as the most effective neurotrophin to protect photoreceptors from degeneration (Sieving et al., 2006). As experience with this approach is gained, other retinal degenerative disorders may be treated similarly.
Recently, a brilliant delivery system based on the endovitreal implant of special capsule (Fig. 18.bottom) containing engineered cells (Fig. 18.top) has been devised by Tao and co-workers (2002). These cells produce CTNF with interesting effects: apparently, the early supply of a cocktail of neurotrophic factors to the pathological retina can slow down photoreceptor degeneration process. Exogenous administration methods rely upon intraocular injections (Whiteley et al., 2001). A recent phase I trial of an encapsulated cell therapy delivering ciliary neurotrophic factor has been started. This treatment is presently under experimentation on humans with RP (MacDonald et al., 2007).
A very important discovery is the existence of intraretinal viability factors. It is postulated that these molecules are normally released by rods (among other cells) and sustain the survival of cones. The latter would undergo secondary degeneration when the density of rods (and therefore the supply of factor) falls below a certain threshold. The French laboratory of Sahel and collaborators has been the first to isolate one of these factors (Lèveillard et al 2004a and b).
These Authors observed that in retinal cultures from chicken embryos cones degenerated after a few days. However, the degeneration could be considerably delayed by supplementing the culture with medium derived from wilde-type mouse retinal explants. This early experiment led to the recent biochemical characterization of a secreted protein expressed in rods and necessary for the survival of cones, named RdCVF (rod derived cone viability factor). The injection of antibodies anti RdCVF in the subretinal space of wt mice causes a remarkable reduction in cone number. The administration of RdCVF in rd1 mice preserves about 40% of cones from degeneration. At present, the French group is working on the mechanism of action of this secreted factor, also devising pre-clinical trial studies for its application on humans.
1.5.3.2. Gene therapy
This therapeutic approach consists in the introduction into the retina of adeno-associated viral vectors carrying copies of the gene whose mutation causes the disease. Recently, the group of Bennett succeeded in restoring vision in blind dogs mimicking Leber’s congenital amaurosis. This is a severe form of congenital blindness caused by a point mutation in RPE65, a gene specific for the pigment
Fig.18. Top: prototype of immunoinsulating membrane encapsulating human cells producing CNTF. Bottom: capsule containing CNTF-producing cells for endovitreal implants by which the factor CTNF can be released into the vitreous body (Modified from Tao et al. 2002). The capsule has a diameter of 1mm and a length of 10mm.
epithelium cells (Acland et al., 2001; Bennett, 2004). More than three years later, Lancelot, the first dog treated with gene therapy for RPE65, continues to see well without any apparent complication.
The eye, differently from other organs, appears as a favorable site for gene therapy, possibly for its anatomic segregation allowing a restricted and long-lasting localization of the virus used to transfer the gene. Many research lines are in progress to enhance the transfection efficiency and to treat both loss and gain of function mutations. Pre-clinical safety studies also continue with gene therapy for Leber’s congenital amaurosis and human gene therapy is progressing through preclinical trials (see the web site: http://www.blindness.org/).
1.6. Aim of the project
All the reviewed experimental approaches for RP, assume, more or less explicitly, that photoreceptor degeneration is restricted to these cells and does not interfere with the structure and function of the inner retina until late stages of the disease. However, it is clear that it would be ineffective to use any transplantation approach or to implant any artificial photoreceptor-substitute if photoreceptor degeneration had triggered irreversible damage to the neurons that process the signals generated in rods and cones, i.e. horizontal, bipolar, amacrine and ganglion cells, with their glial support.
Studies that describe the effects associated with RP upon inner retinal cells are limited to few morphological and electrophysiological reports (as an example, see Li et al., 1995; Aguirre et al., 2002; Humayun et al.1999a; Tao et al., 2002). Overall, an in depth-examination of the effects of RP onto inner retinal cells is still missing, especially when compared to the effort dedicated to understanding the genetic, biochemistry and electrophysiology of photoreceptor degeneration.
During the last few years, the three research groups of A Milam, R. Marc and E. Strettoi demonstrated profound, previously unsuspected remodeling of inner retinal neurons accompanying photoreceptor death. This is mainly obtained by studying rodent models of human RP (Milam et al., 1998; Banin et al., 1999; Peng et al., 2000; Strettoi an Pignatelli, 2000; Strettoi et al., 2002; Pignatelli et al., 2004; Jones et al., 2003; Marc et al., 2003a and b; Varela et al., 2003), including the well know rd1 and the crx-null mice that mimic autosomal recessive RP and Leber’s congenital amaurosis, respectively. Remodeling invests primarily the
dendritic and axonal compartments on bipolar and horizontal cells (that are directly connected to photoreceptors). Dendrites retract progressively, when photoreceptors start to die; along with dendrites, synaptic receptors to neurotransmitters are being lost. In addition, some neuronal processes become hypertrophic and sprout. Eventually, bipolar and horizontal cells die out. Furthermore, in rd1 mice, in which photoreceptor death occurs at a time when synaptogenesis in the outer retina is still taking place, dendrites and axonal arbors of rod bipolar cells do no develop properly and remain abortive (Strettoi and Pignatelli, 2000). The rd10 mutant mouse shows similar but slower modifications of rod bipolar and horizontal cells (Chang et al. 2002, Chang et al., 2007; Gargini et al., 2007). Furthermore, important data indicate that analogous alterations of the morphology of second order neurons also occur in the retina of human RP patients (Milam et al., 1998;Strettoi, unpublished data).
These data are not too surprising after all: they highlight the fact that the retina reacts as other CNS areas when deafferentation occurs. An example of this analogy is found after experimental injury of mammalian visual cortex that causes the retrograde degeneration of the thalamus-cortical neurons projecting to the damaged cortical area. Similarly, pyramidal neurons of the rat olfactory cortex, after complete bulbectomy, undergo an apoptotic process (Capurso et al., 1997). Thus, it is reasonable that a highly integrated piece of CNS, such as the retina, reacts heavily the complete ablation of its only outer source of input represented by photoreceptors. In addition, these cells constitute almost 90% of all the retinal cells: it is expected that their massive degeneration (albeit of variable genetic origin) produces adverse effects at least on those neurons synaptically connected to the degenerating cells. Vice versa, the presence of viable photoreceptors is necessary for the development first, and later on for the maintenance, of a proper morphology of second order neurons.
In RP, residual retinal cells (i.e. surviving photoreceptors and inner retinal cells) represent the true biological platform of therapeutic treatments.
In both the rd1 and rd10 mutants, it is found that the morphology of amacrine cells resists much better to the effects of photoreceptor degeneration (Strettoi et al. 2003) as compared to that of second order neurons. So far, nothing is known about RGCs.
If we assume that remodeling proceeds from the outer to the inner retina we should expect an even higher degree of morphological preservation in the RGCs
of the same mutant mice. Yet, it has to be considered that RGCs, because of their spiking activity and the necessity of sustaining a long axon, have a high metabolic demand. As major vascular changes accompany the progression of the rd1 and rd10 degeneration from early stages (Wang et al., 2000; Otani et al., 2004), it is not unlikely that RGCs suffer from adverse remodeling (i.e. dendritic atrophy, loss of ionic channels mediating excitability etc.) independently of other cellular types of the inner retina. In analogy with what found for rod bipolar and horizontal cells after photoreceptor death (i.e. regressive remodelling and subsequent degeneration), it is possible that the same type of events also take place in RGCs. On the other hand, the literature dealing with RGC preservation in retinal degeneration addresses mostly the issue of their survival, which can be very variable (see Santos et al., 1997; Stone et al., 1992; Wang et al., 2000). Recently, evidence shows that they react profoundly to retinal detachment (Coblentz et al., 2003) and diabetes (Thanos et al., 2007). However no studies are available upon the morphology and connectivity of RGCs in diseased retinas, at the single neuron level.
To address these issues, the experimental work of this thesis aimed at providing an in-depth morphological study of RGCs in mice with moderately slow photoreceptor degeneration (rd10), at various stages of the disease progression, to find out whether and to what extent the process of inner retinal remodeling is propagated from second order neurons to the deepest retinal layers after photoreceptor degeneration.
While studying RGCs, we also tried to uncover general rules that govern inner retinal remodelling, ultimately to find appropriate time windows for rescue therapies.
1.7. Experimental design
Our approach has been largely based upon the development of a new mouse transgenic line, the rd10/Thy1-GFP mutant, in which photoreceptor degeneration is combined to the expression of the fluorescent protein GFP in a restricted number of RGCs. This allowed a systematic morphometric study of the RGC dendritic arborization complexity as a function of photoreceptor degeneration at different ages.
Retinal immunocytochemistry and confocal microscopy were the methods we used to highlight the fine dendritic structure of RGCs after photoreceptor degeneration. First, single types of RGCs were recognized and classified according to an existing database. Subsequently, parameters indicative of dendritic tree complexity were measured during the progression of the pathology. Later, we evaluated RGC survival rate at a time point advanced in the progression of the disease and in the life-span of the mouse.
Indications of cellular functions for RGCs were obtained by evaluating their capability to transport anterogradely a fluorescent marker. Finally, we evaluated parameters indicative of retinal blood vessel complexity, with the rationale that this important source of trophic supply becomes severely affected in human RP.