1. Introduzione
1.1 Regolazione redox cellulare
Per molto tempo le specie reattive dell’ossigeno (ROS) sono state studiate in relazione agli effetti citotossici da loro causati sui sistemi biologici. Negli ultimi anni, però, un sempre maggior interesse è stato dedicato alle potenziali funzioni regolatorie espletate dalle ROS: queste sembrano infatti intervenire nella regolazione di tutta una serie di meccanismi molecolari che stanno alla base di processi quali la risposta immunitaria, l’adesione e la proliferazione cellulare, l’infiammazione, il metabolismo, l’invecchiamento e la morte cellulare (Sen, 1998). Per questo motivo, nel descrivere questi processi, al termine di “stress ossidativo” si è andato sostituendo quello di “regolazione redox” (Cotgreave and Gerdes, 1998). Mentre con il primo termine si indica, infatti, una situazione patologica in cui la produzione di ROS supera le capacità difensive dell’organismo, con il secondo (regolazione redox) si intende una serie di oscillazioni fisiologiche e reversibili dell’equilibrio tra produzione di ROS e meccanismi antiossidanti in grado di regolare numerose funzioni cellulari.
Numerose evidenze sperimentali sono state ottenute al riguardo. E’ stato infatti osservato che livelli bassi, non tossici, di anione superossido e di acqua ossigenata possono stimolare la proliferazione cellulare (Nose et al., 1991; Gallagher et al., 1993; Burdon, 1995; Herbert et al., 1996) e che l’esposizione di cellule a bassi livelli
di H2O2 o di altri agenti ossidanti può svolgere un ruolo protettivo nei confronti di un successivo stimolo di natura apoptotica (Genaro et al., 1995; Clément et al., 1996; Maellaro et al., 1996). Negli ultimi anni si sono andate accumulando numerose evidenze sperimentali relative al fatto che diversi fattori di crescita, citochine o altri ligandi causano produzione di ROS quando si legano ai loro corrispettivi recettori di membrana. Ne sono esempio fattori di crescita come NGF, EGF e PDGF, citochine come TGF-β1, IL-1 e TNF-α, ormoni come l’insulina (Suzuki et al., 1997). La produzione di ROS sembrerebbe mediare un effetto a feedback positivo sulla trasduzione del segnale, dal momento che il processo è spesso favorito o dalle ROS oda un’alterazione del rapporto tioli/disolfuri intracellulari a favore del secondo (Dröge, 2002).
La regolazione redox promossa dalle ROS si espleterebbe mediante reazioni di ossido-riduzione a carico di tutta una serie di bersagli molecolari redox-sensibili, tra i quali un ruolo fondamentale sarebbe giocato dai residui di cisteina delle proteine (Lander, 1997; Suzuki et al., 1997; Monteiro and Stern, 1996). Questi ultimi, infatti, sono spesso presenti all’interno dei domini funzionali delle proteine e mostrano una particolare sensibilità alle variazioni dello stato redox della cellula. I gruppi sulfidrilici delle cisteine possono infatti andare incontro a una serie di possibili modifiche ossidative mediate dalle ROS; su queste basi è stata avanzata l’ipotesi che le cisteine possano funzionare come dei “nanotrasduttori del segnale redox”, ovvero possano modulare la funzione delle proteine e quindi la risposta cellulare ad un certo stimolo in funzione del tipo di modifica redox a cui sono andate incontro (Cooper et al., 2002; Forman et al., 2002).
In conclusione, dunque, numerose evidenze sperimentali mostrano come l’equilibrio redox svolga un ruolo critico nella regolazione di molte funzioni cellulari: in questo processo, in cui l’”informazione ossidativa” portata dalle specie reattive è trasdotta su bersagli redox sensibili, un ruolo importante nel modulare la risposta cellulare è svolto dai livelli intracellulari di antiossidanti ma in particolar modo dal glutatione e dagli enzimi ad esso connessi.
1.2 Il glutatione
Il glutatione (GSH) è un tripeptide ubiquitario presente in tutti i tessuti dei Mammiferi. Composto da acido glutammico, cisteina e glicina (γ-glutamil-cisteinil-glicina), possiede la particolare caratteristica di avere i residui di cisteina e di acido glutammico uniti da un legame γ-glutamilico invece che da un normale legame peptidico (Figura 1.1). Il legame γ-glutamilico rende il GSH resistente alle peptidasi intracellulari e permette alle cellule di conservare in questa forma la cisteina, altrimenti tossica anche a basse concentrazioni. Il GSH è presente nelle cellule di Mammifero a concentrazioni dell’ordine delle 5-10 millimoli/litro, mentre nel plasma e negli altri fluidi extracellulari non supera le 5-15 micromoli/litro. Il GSH rappresenta circa l’80% dei tioli non proteici a basso peso molecolare presenti nella cellula ed è prevalentemente dislocato a livello citosolico, ad eccezione di una piccola parte (10-15%) che si trova a livello mitocondriale e nel reticolo endoplasmico (Meredith and Reed, 1982; Meister and Anderson, 1983; Deneke and Fanburg, 1989; Hwang et al., 1992; Lu, 1999).
Figura 1.1 Struttura del glutatione ridotto (GSH). Il legame γ-glutamilico si forma tra il
gruppo γ-carbossilico del glutammato ed il gruppo amminico della cisteina (Lu et al., 1999).
1.2.1 Funzioni del GSH
Il GSH espleta molte funzioni negli organismi viventi, ma la sua funzionalità è condizionata dall’equilibrio tra la sua forma ridotta (GSH) e la sua forma ossidata (GSSG), che deriva dall’unione con un ponte disolfuro di due molecole di GSH. La forma ridotta è ampiamente predominante all’interno della cellula, con un rapporto che in condizioni normali è nell’ordine di 1:10/1:100 (Meister and Anderson, 1983; Reed et al., 1983; Lu, 1999). La presenza del gruppo sulfidrilico della cisteina conferisce al GSH proprietà antiossidanti che gli permettono di interagire con specie reattive dell’ossigeno o con altre sostanze elettrofile nell’ambito di numerosi sistemi antiossidanti intracellulari, sia enzimatici che non.
In questo senso il GSH interviene nel metabolismo dell’acqua ossigenata, degli idroperossidi e di altri perossidi organici operando come substrato degli enzimi della famiglia delle glutatione perossidasi che catalizzano le reazioni:
H2O2 + 2GSH --> GSSG + 2H2O
ROOH + 2GSH → GSSG + H2O + ROH
Il GSH è inoltre in grado di inattivare specie molecolari quali il radicale idrossile, l’acido ipocloroso, il perossinitrito, radicali perossilici, radicali alcossilici e l’ossigeno singoletto; interviene nel ripristino della forma ridotta di altri antiossidanti come la vitamina C e la vitamina E.
Il GSH è infine coinvolto nei processi di detossificazione dei farmaci xenobiotici e dei loro metaboliti mediante le reazioni catalizzate dalla glutatione S-transferasi, di cui è particolarmente ricco il fegato (Halliwell and Gutteridge, 1999).
Nell’espletare le sue funzioni antiossidanti, il GSH viene ossidato a GSSG. Questo ultimo è substrato dell’enzima glutatione reduttasi che catalizza la sua riduzione a spese del NADPH:
GSSG + NADPH + H+ → 2GSH + NADP+
In alcune circostanze lo stress ossidativo a cui è sottoposta una cellula può superare la sua capacità di ridurre il GSSG a GSH: in questo caso il GSSG può essere trasportato attivamente all’esterno della cellula oppure può reagire con i gruppi sulfidrilici delle proteine determinando la formazione di disolfuri misti in un processo noto come S-glutatiolazione proteica. E’ stato osservato che la formazione di disolfuri misti determina in vitro l’inattivazione di molti enzimi e ciò potrebbe spiegare perché le cellule mantengono livelli intracellulari di GSSG molto bassi in condizioni normali. L’elevato rapporto GSH/GSSG agirebbe dunque garantendo il mantenimento dei residui di cisteina delle proteine allo stato ridotto, prevenendo l’ossidazione dei gruppi SH oppure riducendo i disolfuri generati da agenti ossidanti (Lu, 1999).
Negli ultimi decenni, si sono accumulate molte evidenze sperimentali riguardo all’importanza dell’interazione reversibile del GSH con i gruppi cisteinici di molte proteine cellulari durante l’insorgere dello stress ossidativo. Il fatto che l’attività di molte proteine sia sensibile alle modificazioni redox dei tioli e l’esistenza di enzimi come la glutatione reduttasi, il sistema tioredoxina/tioredoxina reduttasi, la glutaredoxina e la proteina disulfuro isomerasi che sono in grado di modulare lo stato dei disolfuri misti delle proteine stesse, ha fatto ipotizzare un possibile ruolo del GSH nella regolazione di complessi processi biochimici delle cellule (Cotgreave and Gerdes, 1998). Poiché anche altri tioli non proteici a basso peso molecolare come cisteina, omocisteina e cisteamina possono partecipare a questo processo, si parla, in generale, di S-tiolazione.
Come osservato in precedenza, molti gruppi sulfidrilici delle proteine si trovano nei siti attivi degli enzimi. Se da una parte, dunque, la S-tiolazione determinata da agenti ossidanti protegge i sulfidrili delle proteine da un’ossidazione irreversibile, alterando al contempo anche la funzione di enzimi (Coan et al., 1992; Schuppe-Koistinen et al., 1994; Sahaf et al., 2005), dall’altra, il processo di riduzione dei disolfuri misti (detiolazione) ne ripristina la funzione alterata (Del Corso et al., 1993; Ravichandran et al., 1994; Cabiscol and Levine, 1996). Sono stati proposti diversi meccanismi per spiegare la S-tiolazione proteica come, ad esempio, lo scambio tiolo-disolfuro (1), la riduzione dell’acido sulfenico indotta da perossidi (2), la formazione di radicali tiile indotta da specie reattive come il radicale idrossile (3):
1) GSSG + proteina-SH → proteina-SSG + GSH
2) proteina-SH + H2O2 → proteina-SOH + OH. proteina-SOH + GSH → proteina-SSG + H2O 3) proteina-SH + OH. → proteina-S. + H
2O proteina-S. + GSH + O2 → proteina-SSG + O2-.
Si ritiene che la S-tiolazione proteica sia un processo dinamico che avviene normalmente in condizioni fisiologiche, come ad esempio durante l’oxidative burst dei neutrofili (Chai et al., 1994). Come osservato in precedenza, è stato ipotizzato che questo fenomeno possa rappresentare una forma di modificazione post-traduzionale reversibile in grado di modulare il funzionamento cellulare in presenza di sostanze ossidanti: in questo modo, una “informazione ossidativa” sarebbe trasdotta dagli agenti ossidanti intracellulari a proteine contenenti “tioli regolabili” tramite il GSH (Cotgreave and Gerdes, 1998).
Appare chiaro, quindi, che il GSH non svolge solamente funzioni antiossidanti negli organismi viventi, ma è implicato anche nella modulazione della trasduzione del segnale con importanti ripercussioni su processi come la proliferazione cellulare o la risposta immunitaria. In questo senso, quindi, i meccanismi che presiedono alla biosintesi ed all’omeostasi del GSH hanno effetti che vanno al di là del semplice mantenimento dei livelli plasmatici e cellulari di un antiossidante.
1.3 La γ-glutamiltransferasi
1.3.1 Localizzazione, struttura ed espressione dei geni della GGT
La γ-glutamiltransferasi (EC 2.3.2.2.) è un enzima localizzato sulla membrana plasmatica di molti tipi cellulari che catalizza il primo passaggio del processo di degradazione del glutatione (GSH) attraverso la scissione del legame γ-glutammilico (Tate and Meister, 1981). I primi a riscontrarne la presenza nel rene di ratto sono stati, nel 1948, Binkley e Nakamura.
In condizioni fisiologiche la GGT è presente preferenzialmente a livello di tessuti epiteliali implicati in attività secretorie e di assorbimento. La più alta attività di GGT è stata trovata nel rene, sulla superficie luminale delle cellule del tubulo convoluto prossimale, mentre le cellule del tubulo distale e dei glomeruli ne sono praticamente prive. Nel fegato la GGT è concentrata nelle cellule epiteliali delle vie biliari extraepatiche e dei canalicoli epatici. Nel pancreas la maggiore attività di GGT è presente nelle cellule acinose. Studi immunoistochimici hanno dimostrato che sono GGT positive anche le cellule endoteliali dei capillari del cervello (es. del plesso corioideo e del corpo ciliare) e del midollo spinale, le cellule delle ghiandole sudoripare, delle ghiandole sottomandibolari, dei dotti galattofori, dell’epitelio bronchiale, dell’epididimo, delle vescicole seminali, della prostata (Hanigan and Pitot, 1985; Hanigan and Frierson, 1994).
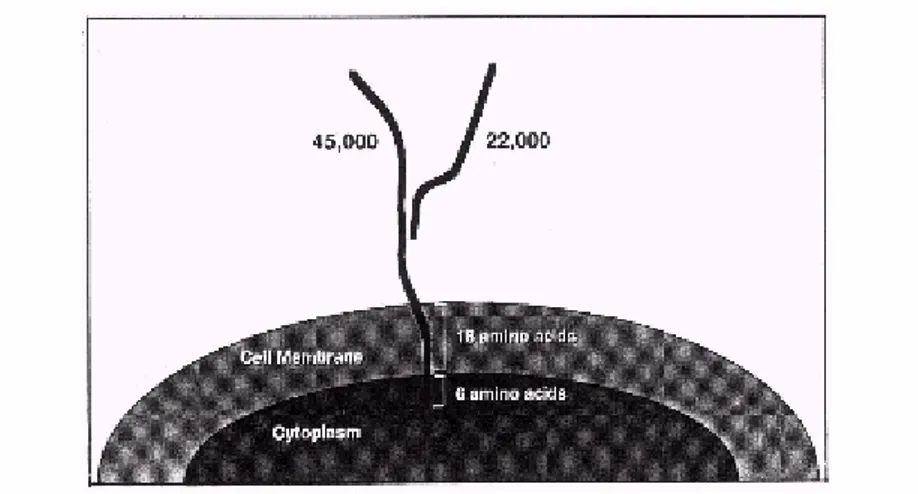
Da un punto di vista strutturale, la GGT è una glicoproteina dimerica sintetizzata da un unico propeptide (Curthoys and Hughey, 1979) che viene scissa in 2 subunità mature, una pesante (55-62 KDa) e una leggera (20-30 KDa), prima di raggiungere la membrana plasmatica (Barouki et al., 1984; Finidori et al., 1984).
La subunità leggera, che presenta un ectodominio carbossi-terminale con attività catalitica, è unita alla subunità pesante con interazioni elettrostatiche (Tate and Meister, 1981); sulla porzione ammino-terminale della catena pesante è presente un dominio idrofobico che permette l’inserzione dell’enzima sul lato esterno della membrana cellulare, in questo modo entrambe le subunità risultano in contatto con l’ambiente extracellulare (Finidori et al., 1984) (Figura 1.2).
Figura 1.2 Rappresentazione della GGT. Tutta l’attività catalitica dell’enzima è presente
all’esterno della cellula (Hanigan,1999).
Gardell e Tate (1979) hanno dimostrato che la subunità leggera della GGT, purificata dal rene di ratto, bovino e coniglio, è una proteasi, in grado di idrolizzare la stessa subunità pesante.
L’espressione dei geni della GGT riflette una complessa organizzazione tra le diverse specie e anche nell’uomo esistono differenze a seconda dell’organo d’origine e/o del tipo cellulare preso in esame (Chikki et al., 1999). Nel ratto e nel topo la GGT è
codificata da un gene presente in singola copia e può essere trascritta a partire da differenti promotori (Brouillet et al., 1994; Shi et al., 1995; Chikki et al., 1999). Ogni promotore dà origine ad uno o più mRNA che differiscono per un’unica sequenza 5’ non tradotta (Lieberman et al., 1995).
Nel genoma umano è presente una famiglia multigenica per la GGT, composta da almeno sette membri (Courtay et al., 1994), mappati sul cromosoma 22 nella regione 22q11.1-q11.2 vicino ai loci BCR (breakpoint cluster region) e IG-λ (Bulle et al., 1987; Collins et al., 1997). Sequenze correlate, probabilmente pseudogeni, sono state identificate sui cromosomi 18, 19, 20 (Figlewicz et al., 1993).
1.3.2 Attività catalitica della GGT
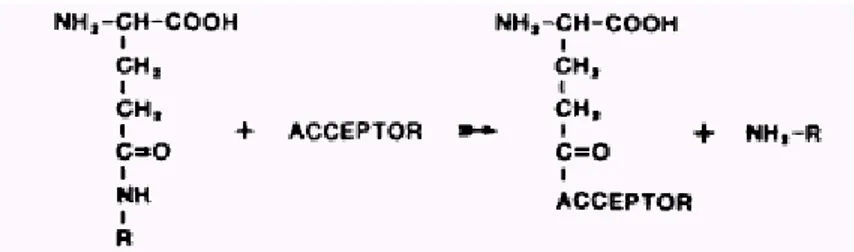
La GGT è in grado di idrolizzare il legame γ-glutamilico presente tra acido glutammico e cisteina, promuovendo il trasferimento del gruppo glutamilico su di un accettore amminoacidico o dipeptidico (Meister et al., 1981; Tate and Meister, 1981) (Figura 1.3).
I principali accettori del gruppo γ-glutammilico sono: cisteina, glutammato, L-alanilglicina, glicilglicina, L-serilglicina (Tate and Meister, 1981). Il glutatione è il più importante substrato fisiologico dell’enzima, ma la GGT può usare una più ampia varietà di γ-glutammil composti (Magnan et al., 1982).
Figura 1.3. Reazione catalizzata dalla GGT (Hanigan, 1999).
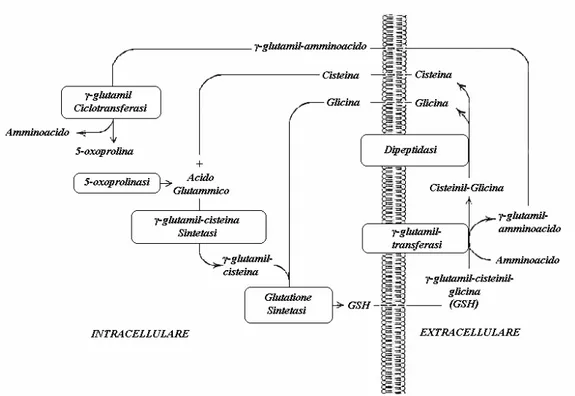
Da questa reazione si vengono a generare dei glutamil-amminoacidi o dei γ-glutamil-dipeptidi da una parte, e cisteinil-glicina dall’altra. I primi vengono trasportati all’interno della cellula e sono il substrato dell’enzima γ-glutamil-ciclotransferasi che li scinde nei corrispettivi amminoacidi ed in 5-oxoprolina: quest’ultima può essere nuovamente convertita ad acido glutammico dall’enzima 5-oxoprolinasi in una reazione energia-dipendente e essere usata nella risintesi di GSH. La cisteinil-glicina è invece idrolizzata dalle dipeptidasi di membrana a dare cisteina e glicina che sono quindi trasportate all’interno della cellula e possono partecipare alla sintesi de novo del GSH. L’insieme delle reazioni collegate all’attività di GGT e quelle che portano alla sintesi ed all’efflusso del GSH costituiscono quello che è stato definito il “ciclo del γ-glutamile” (Figura 1.4). Poiché è stato documentato che un efflusso continuo di GSH è effettuato da molti tipi cellulari mediante specifici trasportatori, è stato ipotizzato che la GGT possa favorire il recupero del GSH extracellulare che altrimenti sarebbe perso dalla cellula (Lu et al., 1996).
Figura 1.4 Ciclo del γ-glutamile (Lieberman et al., 1995).
Su queste basi, dunque, l’attività di GGT, coinvolta nel recupero degli amminoacidi costituenti e nella sintesi del GSH (Meister e Anderson, 1983; Deneke e Fanburg, 1989; Meister, 1995; Lu, 1999), è stata considerata per molto tempo un membro dei sistemi antiossidanti della cellula e, in effetti, almeno in certe condizioni, l’arricchimento delle cellule in GGT causa l’aumento della resistenza agli agenti tossici ed agli insulti proossidanti (Hiraishi et al., 1994; Prezioso et al., 1994). In questo senso è interessante osservare che elevati livelli di GGT sono espressi in numerosi tumori maligni umani, cosa che ha fatto ipotizzare che la GGT possa conferire un qualche importante vantaggio selettivo anche nell’ambito della proliferazione delle cellule tumorali.
1.3.3 Azione proossidante della GGT
Le prime evidenze sperimentali di una possibile funzione proossidante della GGT risalgono ad esperimenti condotti in vitro in cui viene descritta l’abilità della GGT di determinare la produzione di radicali liberi e specie reattive dell’ossigeno con conseguenze che portano all’ossidazione delle proteine e dei lipidi poliinsaturi della membrana plasmatica.
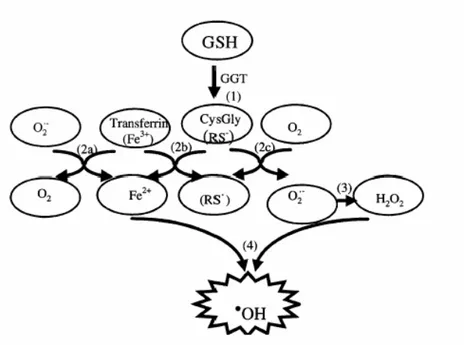
Questi eventi, che possono innescarsi esclusivamente all’esterno della membrana plasmatica, ma le cui conseguenze si ripercuotono su tutta la cellula, sono basati sulla capacità dei tioli, come il GSH, ma anche dei prodotti derivanti dalla sua idrolisi ad opera della GGT, quali la cisteinil-glicina e la cisteina, di interagire con i metalli di transizione, ed in particolare con il Fe3+ (Stark et al.,1993). Infatti, la dissociazione del gruppo tiolico –SH ad anione tiolato –S¯ consente la cessione, da parte di questo ultimo, di un elettrone al Fe3+, che viene ridotto a Fe2+, dando luogo contemporaneamente alla formazione di un radicale tiile –S˙
Il radicale tiile e il Fe2+ sono in grado di innescare una serie di eventi a cascata che porta alla produzione di anione superossido e quindi di acqua ossigenata per azione dell’enzima superossido dismutasi. L’anione superossido e l’acqua ossigenata, in presenza di Fe3+ libero o chelato all’ADP, possono generare radicali ossidrili che insieme al radicale tiile danno inizio alle reazioni a catena della perossidazione lipidica, con conseguente perdita della struttura e della stabilità della membrana cellulare, nonché delle sue importanti funzioni (Zalit et al., 1996).
Figura 1.5 Reazioni proossidanti conseguenti all’attività della GGT. 1.Formazione della
cisteinil-glicina. 2a,2b,2c.Riduzione del ferro con formazione del radicale tiile e dell’anione superossido. 3. Formazione dell’acqua ossigenata. 4. Reazione di Fenton. (Drozdz et al., 1998).
L’abilità della GGT di stimolare la perossidazione lipidica GSH-dipendente in sistemi contenenti complessi di ioni ferrici (Fe3+) come catalizzatori e acido linoleico purificato come substrato perossidabile è stata dimostrata in esperimenti condotti in vitro (Stark et al., 1993). E’ stato ipotizzato che l’effetto proossidante della GGT sia legato al catabolismo del GSH ed alla formazione di cisteinil-glicina, un tiolo molto più reattivo ed in grado di promuovere la riduzione del Fe3+ a Fe2+ molto più efficientemente del GSH. L’effetto di questa riduzione darebbe il via ad un vero e proprio ciclo redox del ferro che porterebbe alla formazione di ROS ed al verificarsi di reazioni ossidative come la perossidazione lipidica (Stark et al., 1993).
In accordo con queste ipotesi è stato osservato che il GSH sarebbe di per sé un riducente del complesso ADP-Fe3+, ma il gruppo α-carbossilico dell’acido glutammico è in grado di bloccare l’interazione tra il gruppo tiolico ed il Fe3+ (Paolicchi et al., 1999). La massima riduzione del complesso Fe3+-ADP da parte del GSH si osserva in una miscela nella quale sia presente anche la GGT che, rimuovendo l’acido glutammico dal GSH, rende libero il gruppo –SH della cisteinil-glicina di interagire con il Fe3+. Inoltre, poiché la pKa della cisteinil-glicina è più bassa di quella del glutatione (6,4 e 8,56 rispettivamente) (Stark et al., 1989), la cisteinilglicina, a pH fisiologico, sarà prevalentemente nella forma di anione tiolato (Stark et al., 1993).
In accordo con quanto detto, la cisteinil-glicina è capace di ridurre il complesso ADP-Fe3+ più efficacemente del GSH formando Fe2+ nella stessa quantità osservata nella miscela contenente GSH e GGT. Risultati simili sono stati ottenuti con la cisteina che viene prodotta dall’idrolisi della cisteinil-glicina operata dalle dipeptidasi di membrana.
L’azione proossidante della GGT è legata alla presenza di metalli redox attivi nell’ambiente extracellulare, cosa che in vivo è fortemente prevenuta grazie alla formazione di complessi con ferritina, transferrina e ceruloplasmina che non permettono ai metalli di catalizzare reazioni con radicali liberi. A tal proposito è interessante osservare che l’attività di GGT è in grado di ridurre e di promuovere il rilascio degli ioni ferro legati alla transferrina (Drozdz et al., 1998; Dominici et al., 2003a) e che l’azione proosidante è stata osservata anche in presenza di ceruloplasmina ( Glass and Stark, 1997), ovvero due sorgenti fisiologiche di metalli
di transizione. Inoltre il rilascio di ioni ferro dalle loro forme di immagazzinamento è stato osservato in molte condizioni fisiopatologiche, un processo questo che potrebbe rendere i metalli disponibili per promuovere l’azione proossidante della GGT (Paolicchi et al., 2002a).
Numerose evidenze sperimentali dimostrano le capacità proossidanti legate all’attività di GGT: è stato infatti osservato che il ciclo redox del ferro GGT-mediato è in grado di promuovere la perossidazione lipidica in lesioni preneoplastiche ricche di GGT indotte chimicamente nel fegato di ratto (Pompella et al., 1996); analoghi risultati sono stati ottenuti in epatociti isolati di ratto e in cellule di epatoma umano HepG2, in cui la perossidazione lipidica determinata dalla GGT aggiunta, nel primo caso, o espressa, nel secondo caso, è stata osservata a carico sia delle membrane cellulari che di microsomi incubati in presenza delle cellule (Paolicchi et al., 1997). E’ stato anche osservato che l’attività della GGT può promuovere l’ossidazione GSH-Fe dipendente di LDL umane isolate promuovendo un processo che si ritiene giocare un ruolo fondamentale nella formazione della placca aterosclerotica e del danno vascolare (Berliner and Heinecke, 1996; Paolicchi et al., 1999).
Gli effetti dannosi non sono però l’unica conseguenza dell’azione proossidante della GGT. Negli ultimi anni, infatti, numerose evidenze sperimentali hanno dimostrato come le specie reattive prodotte dall’attività dell’enzima giochino un ruolo “non tossico” nella modulazione redox di proteine cellulari. I bersagli diretti di queste specie ossidanti sarebbero i tioli delle proteine sul lato esterno della membrana plasmatica, anche se, mediante la formazione di acqua ossigenata che può
liberamente diffondere all’interno della cellula, la GGT sembra essere in grado di modulare anche l’attività di proteine intracellulari.
L’ossidazione dei tioli proteici della superficie cellulare non sembra essere dovuta alla sola interazione dell’H2O2, prodotta dall’attività della GGT, con i gruppi sulfidrilici delle proteine (Radi, 1991; Quesada, 1996), ma è anche legata a processi di S-tiolazione proteica promossi dalla GGT.
Il GSSG che si accumula in seguito a condizioni di elevata attività di GGT, può interagire con i residui di cisteina delle proteine: si formano in questo modo dei disolfuri misti (proteina-non proteina) tra il glutatione e i gruppi sulfidrilici delle proteine. Il processo, definito S-glutatiolazione, può essere così schematizzato:
GSSG + proteina-SH ↔ proteina-SSG + GSH
Poiché anche altri tioli non proteici a basso peso molecolare, come cisteina, omocisteina e cisteamina, possono partecipare a questo processo, si parla più in generale di S-tiolazione, come già illustrato nella sezione 1.2.1 .
Poiché molti dei gruppi sulfidrilici delle proteine si trovano nei siti attivi degli enzimi, la S-tiolazione ha quindi la duplice funzione di protegge i sulfidrili delle proteine da un’ossidazione irreversibile mediata dalle specie reattive dell’ossigeno, e di alterare, in modo reversibile, la funzione di molti enzimi (Coan et al., 1992; Schuppe-Koistinem et al., 1994; Sahaf et al., 2005). Infatti la “detiolazione”, processo attraverso cui si ha la riduzione dei disolfuri misti, ne ripristina la funzione
alterata (Del Corso et al., 1993; Ravichandran et al., 1994, Cabiscol and Levine, 1996).
In maniera del tutto analoga anche la cisteinil-glicina originatasi dal catabolismo del GSH ad opera della GGT può concorrere alla formazione di ponti disolfuro con le proteine (Pompella et al., 2006).
Un numero sempre maggiore di targets è coinvolto nel processo di S-tiolazione, sia tra le proteine di membrana che tra quelle citosoliche (Sen and Parker, 2002; Sies and Parker, 2002). La S-glutatiolazione può, infatti, interessare componenti cellulari coinvolti nella trasduzione di segnali implicati nella proliferazione cellulare, come H-ras (Mallis et al., 2001), la chinasi p59 delle cellule T (Hehner et al., 2000), la fosfatasi PTP1B (Barrett et al., 1999), c-jun (Klatt et al., 1999) NF-kB/p50 (Pineda-Molina et al., 2001) e la caspasi 3, implicata in processi apoptotici (Davis et al., 1997). I residui di cisteina, presenti sul dominio di legame al DNA di p53 possono subire modificazioni derivanti dall’ossidazione, con specifici effetti sulla capacità di legare il DNA (Parks et al., 1997).
La S-tiolazione GGT-dipendente può assumere due significati. In primo luogo, può essere interpretata come una difesa nei confronti dei danni ossidativi irreversibili (Coan et al., 1992)). In questa ottica, essendo la GGT espressa ad alti livelli sia nei tumori che nelle metastasi, potrebbe contribuire alla resistenza delle cellule cancerogene nei confronti degli effetti citotossici dello stress ossidativo prodotto dai farmaci antitumorali ad attività proossidante (Daufeub et al., 2002; Paolicchi et al., 2002b; Paolicchi et al., 2003). In secondo luogo una interessante possibilità è che la cisteinil-glicina prodotta dall’attività di GGT, inducendo la formazione di disolfuri
misti con le proteine (processo che per analogia è detto S-cisteinil-glicilazione), intervenga anch’essa nei processi regolatori come la S-glutatiolazione. Il fatto che, in alcuni esperimenti, l’aumento della S-cisteinil-glicilazione comporti una diminuzione della S-glutatiolazione potrebbe essere considerato come un meccanismo mediante il quale le cellule esprimenti attività di GGT, come le cellule tumorali (Tew et. al, 1996), riescono a modulare lo stato redox e la funzione di proteine importanti presenti nella matrice extracellulare e sulla superficie di altri tipi cellulari, quali per es. le cellule del sistema immunitario o endoteliali (Corti et al., 2005).
Gli effetti modulatori del catabolismo del GSH GGT-mediato sono ben documentati. Nella linea di istiocitoma umano U937 è stato osservato che la stimolazione dell’attività di GGT determina un incremento dell’ossidazione dei gruppi sulfidrilici delle proteine, mentre la sua inibizione lo previene. Questo effetto sembra essere dovuto sia all’H2O2 prodotta dall’attività della GGT nell’ambiente extracellulare, sia a reazioni di S-tiolazione GGT-mediate. Il coinvolgimento del perossido di idrogeno in questo processo è indicato dal fatto che l’ossidazione dei tioli proteici è impedita dalla catalasi, un enzima che catalizza la dismutazione dell’H2O2 in acqua e ossigeno molecolare; lo stesso effetto si ha, almeno in parte, per le reazioni di S-tiolazione, infatti l’inibizione della GGT con acivicina è di per sè sufficiente a produrre un aumento dei tioli proteici ridotti (Dominici et al., 1999).
Durante il catabolismo del GSH GGT-mediato vengono prodotte specie proossidanti che sono in grado di modificare lo stato redox dei gruppi –SH delle proteine della superficie cellulare (Dominici et al., 1999). Nella linea cellulare Me665/2/60, esprimente alta attività di GGT, sono stati valutati i rapporti tra l’attività dell’enzima
e lo stato di fosforilazione delle proteine ed i dati ottenuti suggeriscono che le reazioni proossidanti della GGT hanno un ruolo importante nella modulazione di questi processi (Pieri et al., 2003).
NF-kB è un fattore di trascrizione ubiquitario implicato nella regolazione di un ampio numero di geni che controllano vari aspetti delle risposte immunitarie ed infiammatorie (Baeuerle and Henkel, 1994). Diverse sono le evidenze sperimentali che hanno verificato il coinvolgimento di questo fattore di trascrizione nei cambiamenti redox conseguenti al catabolismo del GSH. Nella linea cellulare V79-GGT di fibroblasti polmonari di criceto (transfettata e stabilizzata), caratterizzata da elevata produzione di GGT umana (Visvikis et al., 1991), è stato dimostrato che la produzione di ROS, ed in particolare di H2O2, inducono l’attivazione del fattore NF-kB ed il legame col DNA (Accoui et al., 2000). Ulteriori studi su cellule di melanoma umano Me665/2/60 hanno dimostrato che la stimolazione dell’attività dell’enzima, o la sua inibizione, determinano rispettivamente la stimolazione o l’inibizione della traslocazione nucleare di NF-kB (Maellaro et al., 2000). L’aumentata traslocazione conseguente alla stimolazione dell’attivita dell’enzima, ottenuta fornendo GSH e cys-gly come substrati, è tuttavia accompagnata da un diminuito legame di NF-kB al DNA. Questo è stato interpretato come un possibile meccanismo di regolazione verso una eccessiva attivazione di NF-kB in condizioni di persistente stress ossidativo (Dominici et al., 2003b).
Altre evidenze sperimentali hanno mostrato che l’attività di GGT è in grado di promuovere il legame di AP-1 al DNA; l’acivicina, ma anche altri inibitori della
attività di GGT, sopprimono questo effetto, a conferma del coinvolgimento dell’enzima (Paolicchi et al., 2002a).
1.3.4 La GGT nei tumori
La scoperta del collegamento tra cancerogenesi e GGT è dovuta a Fiala e collaboratori nel corso di studi compiuti su tessuti epatici di ratto e di topo in cui inducevano chimicamente epatomi (Fiala et al., 1972).
L’espressione di GGT è considerata come un marker di progressione neoplastica in diversi modelli sperimentali (Warren et al., 1993; Hanigan, 1999). Elevati livelli di GGT sono stati riportati in un ampio numero di neoplasie maligne umane, come nell’ovaio (Paolicchi et al., 1997), nel colon (Murata et al., 1997), nel polmone (Blair et al., 1997), nel fegato (Tsutsumi et al., 1996), in sarcomi (Hochwald et al., 1997), nel melanoma (Supino et al., 1992), nella leucemia (Täger et al., 1995); in molti casi i livelli di GGT delle metastasi sono maggiori di quelli presenti nei corrispettivi tumori d’origine (Prezioso et al., 1993). In una serie di 60 differenti linee cellulari tumorali, la GGT è significativamente espressa nel 70% dei casi (Tew et al., 1996). Recenti ricerche hanno mostrato che l’esposizione di vari tessuti GGT-negativi (fegato di ratto, pelle di topo, epitelio tracheale di criceto) all’azione di cancerogeni, determina la comparsa di attività di GGT spesso associata all’incremento della capacità proliferativa e alla comparsa di tumori maligni (Pompella et al., 2006). I meccanismi che sono alla base dell’aumentata espressione della GGT a seguito di trattamenti con cancerogeni non sono ancora chiari, nonostante sia stato osservato che la transfezione di epatociti con l’oncogene ras permette a tali cellule di crescere
in topi nudi e induce l’espressione della GGT (Braun et al., 1987). Ciò è stato recentemente confermato in cellule CC531 di carcinoma del colon trattate con raggi γ: l’elevata espressione della GGT è correlata all’attivazione dell’oncogene ras da parte delle radiazioni (Pankiv et al., 2006).
I meccanismi cellulari e molecolari che sono alla base della correlazione tra progressione neoplastica e attività di GGT rimangono tuttora da chiarire; inizialmente è stato ipotizzato che la maggiore disponibilità di cisteina (Rajpert-De Meytes et al., 1992; Hanigan and Ricketts., 1993; Hanigan, 1995) e di GSH intracellulare (Prezioso et al., 1994) potessero conferire alle cellule tumorali importanti vantaggi sia in termini di sopravvivenza che di crescita. Infatti il frequente riscontro di elevate attività di GGT all’interno dei tumori umani e sperimentali ha fatto ipotizzare che proprio come conseguenza dell’elevata attività di GGT, le cellule tumorali possano godere di un miglior approvvigionamento di precursori per la sintesi intracellulare del GSH approfittando della disponibilità di GSH extracellulare di provenienza plasmatica (Hanigan and Pitot, 1985).
Il GSH ha un ruolo importante nella modulazione della sensibilità delle cellule tumorali alle radiazioni ionizzanti, agli agenti alchilanti e ai composti del platino (Hanigane and Pitot, 1985), ma i tentativi di dimostrare il ruolo della GGT nel controllo del GSH intracellulare e della resistenza GSH mediata ai farmaci antiblastici hanno dato risultati contrastanti.
E’ stato osservato che l’esposizione di cellule tumorali dell’ovaio al cisplatino (cis-diammino-dicloro-platino, CDDP) determina la comparsa di cellule CDDP-resistenti e la farmaco-resistenza è accompagnata da aumentati livelli di GGT e di GSH
(Godwin et al., 1992); analoghi risultati sono stati osservati in cellule di carcinoma dell’ovaio (Lewis et al., 1988).
Per contro, non è stata osservata alcuna correlazione tra CDDP-resistenza ed espressione della GGT in soggetti con tumori delle cellule germinali (Hanigan et al., 1999a) e nessuna induzione della GGT è stata osservata in pazienti affetti da tumori dell’ovaio in seguito ad una terapia basata su platino (Paolicchi et al., 1997). Inoltre nessuna apparente correlazione è stata osservata tra livelli di GSH e risposta al cisplatino in una serie di xenotrapianti di tumori umani CDDP-resistenti (Pratesi et al., 1995), e tra GGT e GSH intracellulare in cellule di carcinoma ovarico (Perego et al., 1998).
Anche l’indagine sulla relazione tra attività di GGT e maggiore livello intracellulare di GSH ha dato esiti negativi. E’ stato osservato che i cloni ottenuti da una linea cellulare di cheratociti di topo che era stata transfettata con il gene della GGT umana, hanno vantaggi in termini di abilità nel dar luogo a tumori e di crescita del tumore stesso quando iniettate in topi nudi¸ tuttavia i livelli di GSH dei tumori così ottenuti sono più bassi di quelli dei controlli (Warren et al., 1993). Analoghi risultati sono stati ottenuti mediante la transfezione del gene della GGT in una linea di cellule tumorali della prostata, che acquisiscono così vantaggi in termini di crescita e di resistenza al cisplatino, ma non mostrano aumenti significativi del GSH intracellulare (Hanigan et al., 1999b). Al contrario, studi condotti su due linee cellulari di ratto hanno mostrato che l’inibizione dell’attività di GGT con acivicina determina un aumento dei livelli intracellulari di GSH (Meredith and Williams, 1986).
Recenti ricerche hanno evidenziato che le basi dell’associazione tra elevata espressione di GGT e maggiore chemioresistenza e invasività potrebbero essere legate alla presenza di metaboliti rilasciati dal catabolismo extracellulare del GSH, ed in particolare della cisteinil-glicina che presenta una attività riducente molto più elevata del GSH. I risultati ottenuti in una linea cellulare di melanoma indicano che la GGT partecipa alla protezione della cellula contro il danno prodotto dal cisplatino attraverso l’inattivazione del farmaco, operata dalla cisteinil-glicina, già in ambiente extracellulare (Franzini et al., 2006).
1.4 Le Glutatione S-transferasi omega
1.4.1 La superfamiglia delle Glutatione S-transferasi
Le glutatione transferasi (GST) costituiscono una superfamiglia di enzimi detossificanti di fase II, che catalizzano la coniugazione del glutatione (GSH) con substrati elettrofili di natura esogena ed endogena. Tramite tale attività, le GST contribuiscono allo smaltimento di un gran numero di xenobiotici elettrofilici, quali tossici, cancerogeni chimici, inquinanti ambientali e farmaci. Molte GST possiedono inoltre altre attività, quali la riduzione GSH-dipendente dei perossidi, reazioni di isomerizzazione e sintesi di eicosanoidi (Townsend and Tew, 2003).
Esistono tre principali famiglie di proteine, ampiamente diffuse in natura, dotate di attività GSH transferasica. Due di queste famiglie, le GST citosoliche e le GST mitocondriali, comprendono enzimi solubili e sono solo lontanamente correlate tra di loro, mostrando qualche somiglianza nella loro conformazione tridimensionale (Robinson et al., 2004; Ladner et al., 2004). Le GST citosoliche derivano da un comune gene ancestrale, che nel tempo ha subito numerose modificazioni come duplicazioni, ricombinazioni genetiche e mutazioni, che hanno portato alla differenziazione tra le diverse GST (Townsend and Tew, 2003). La terza famiglia, strutturalmente diversa dalle due precedenti, è costituita dalle GST microsomiali ed è ora nota con la sigla MAPEG (Membrane-Associated Proteins in Eicosanoid and Glutathione metabolism) (Holm et al., 2002). Esiste anche un’ulteriore famiglia di transferasi, rappresentata dalle proteine batteriche FosA e FosB responsabili della resistenza alla fosfomicina (Armstrong et al., 2000).
Variazioni, determinate geneticamente, nei livelli dell’attività e/o dell’espressione di alcune GST sono identificate come fattori di rischio nello sviluppo di alcune forme tumorali (Strange and Fryer, 1999), nell’acquisizione della resistenza ai farmaci (Hayes and Pulford, 1995; O’Brien and Tew, 1996; Tew, 1994; Hall et al., 1994), e nell’insorgenza della malattia di Parkinson (Menegon et al., 1998).
Le GST sono di particolare interesse per farmacologi e tossicologi perché forniscono bersagli utili per le terapie antiasmatiche e antitumorali (Evans et al., 1991; Matsushita et al., 1998; Jakobsson et al., 1999a) e perché metabolizzano agenti chemioterapici, insetticidi, erbicidi, cancerogeni e prodotti dello stress ossidativo. Le GST citosoliche rappresentano la famiglia più numerosa di transferasi e funzionano in forma dimerica come omo o eterodimeri (questi ultimi si formano solo tra subunità della stessa classe)(Ladner et al., 2004).
Nell’uomo sono state identificate sette classi di GSTs citosoliche : α, μ, π, θ ,ζ , σ e Omega. Quest’ultima classe, indicata con la sigla GSTO, è di recente identificazione (Board et al., 2000) ed è stata oggetto di studio della presente tesi. Descriverò pertanto in maggior dettaglio le principali caratteristiche di questa nuova classe di glutatione transferasi.
1.4.2 La classe delle Glutatione transferasi Omega (GSTO)
La prima GSTO, identificata nel ratto come deidroascorbato reduttasi (DHAR), è stata purificata e caratterizzata nel laboratorio dove ho svolto la presente tesi (Maellaro et al., 1994; Ishikawa et al., 1998). La DHAR è stata purificata dal fegato del ratto in base alla sua capacità di catalizzare la riduzione, GSH dipendente,
dell’acido deidroascorbico ad acido ascorbico. La proteina è largamente espressa in tutti i tessuti del ratto, raggiungendo i livelli più alti nel fegato, nel rene e nell’apparato gastroenterico (Paolicchi et al., 1996). Successivamente, nel 1998, è stato clonato il cDNA di tale enzima e ne é stata approfondita la caratterizzazione molecolare (Ishikawa et al., 1998).
Nel 1999 un gruppo americano ha clonato il cDNA di una proteina murina di 28 KDa (p28) che risultava sovraespressa in una linea di linfoma dopo l’acquisizione della radioresistenza (Kodym et al., 1999). La sequenza aminoacidica della p28, formata da 240 aminoacidi, mostra un’identità del 81,2% con la sequenza della DHAR di ratto. Il relativo mRNA è espresso in molti tessuti e il fegato e il cuore ne contengono le più alte concentrazioni (Kodym et al., 1999). Gli Autori non sono stati in grado di identificare caratteristiche funzionali di tipo enzimatico della suddetta proteina, ma hanno ipotizzato, sulla base di analogie di sequenza, che sia una heath shock protein di basso peso molecolare. Infatti la p28 in seguito all’innalzamento della temperatura trasloca dal citoplasma, dove normalmente si trova, al nucleo. Gli stessi Autori hanno inoltre inserito in banca dati la sequenza di un’omologa proteina umana (GenBank accession number U90313) che ha un’identità del 76.5% con la DHAR del ratto.
Nel 2000 il gruppo di P.G. Board ha effettuato uno studio volto all’identificazione di nuovi membri della famiglia delle glutatione transferasi sulla base delle similitudini di sequenza. A tal fine gli Autori hanno svolto la loro ricerca su data base EST (Expressed Sequence Tag) contenente sequenze uniche espresse all’interno del genoma umano, usando come riferimento sequenze amminoacidiche delle GST ϑ e ζ
(Board et al., 2000). Questa ricerca ha portato all’identificazione del cDNA di una nuova proteina con caratteristiche tipiche della famiglia delle GST. Gli stessi Autori hanno notato però che una sequenza identica era già stata immessa in GenBanK (U90313) e che in altre banche dati si trovavano sequenze altamente simili relative a proteine di altre specie animali e, per l’esattezza, la sequenza della p28 del topo, quella della DHAR del ratto e quella di una proteina di un nematode (Caenorhabditis elegans) (Board et al., 2000; Kodym et al., 1999; Ishikawa et al., 1998). Queste sequenze avevano rispettivamente un’identità del 72%, 76% e 34% con quella della proteina umana.
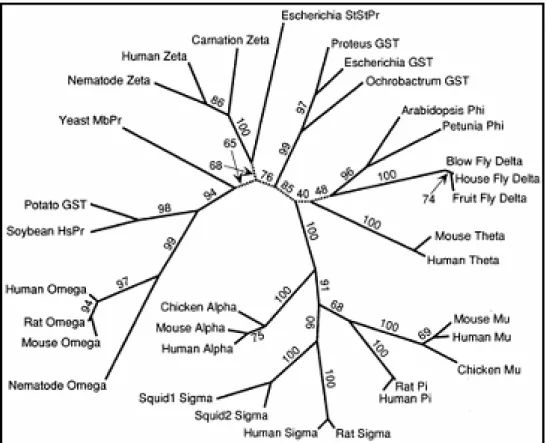
Eseguendo un’analisi filogenetica, basata sull’allineamento con sequenze rappresentative delle varie classi di GST, gli Autori (Board et al., 2000) hanno ricostruito un albero filogenetico in base al quale le suddette proteine vengono a costituire una nuova classe di GST, chiamata dagli Autori GST omega ed indicata con la sigla GSTO.
Figura 1.6 Correlazioni filogenetiche tra le varie classi di GST. (Board et al., 2000.)
Gli stessi autori hanno ulteriormente caratterizzato la GSTO 1 umana; in particolare hanno studiato la proteina ricombinante e la sua struttura cristallina (Board et al., 2000) .
La proteina ricombinante umana (27,5 kDa) mostra, alla cromatografia per esclusione molecolare, una massa di 56 kDa, indicando quindi che la proteina ricombinante in condizioni native forma un omodimero (GSTO 1-1).
La GSTO 1-1 mostra scarsa attività con i principali substrati delle GST, quale ad esempio il 1-cloro-2,4-dinitrobenzene. Al pari della omologa proteina del ratto mostra invece attività DHA reduttasica. L’attività tiol-transferasica (misurata con il
classico substrato, idrossietil disulfuro) risulta però di gran lunga la più alta. In conclusione la GSTO 1-1 umana mostra attività molto simili a quelle della glutaredossina, differenziandosi pertanto dalle altre GST (Board et al., 2000; Whitbread et al., 2003).
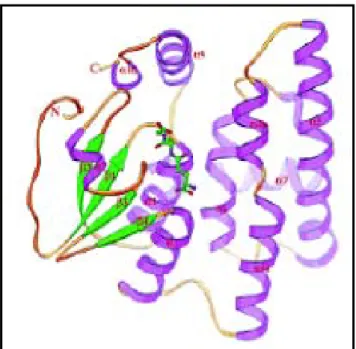
La struttura cristallina é stata determinata sul complesso covalente tra GSTO 1-1 e GSH (Board et al., 2000). Sebbene la GSTO 1-1 presenti una modesta identità di sequenza con le GST di cui é nota la struttura cristallina, essa adotta chiaramente la tipica conformazione delle GST. Vi sono due domini: un dominio N-terminale tioredossina-simile e un dominio C-terminale interamente strutturato in α-eliche. Il dominio N-terminale contiene 4 foglietti β, intercalati con 3 α-eliche (α1, α2, α3). Delle sette α -eliche del dominio C-terminale, cinque (α 4 – α 8) sono comuni alla maggior parte delle GST, ma le ultime due (α9 e α10) si piegano indietro sul dominio N-terminale, costituendo una struttura tipica di questa classe. Questa estensione C-terminale forma molti legami ad idrogeno con il dominio N-C-terminale, formando con esso una superficie continua.
Figura 1.7 Struttura della GSTO1-1 umana. Rappresentazione del monomero. (Board et
al., 2000.)
1.4.3 La regione G (G site) e il riconoscimento del GSH
Con il termine “G-site” si indica quella parte della proteina deputata al legame con il GSH. Due caratteristiche distinguono la GSTO 1-1 dalle tipiche GST per quanto concerne questa regione.
La prima consiste nella presenza di una cisteina in posizione 32 in grado di formare un disolfuro misto con il GSH. Non é però chiaro quale residuo del sito catalitico possa stabilizzare la forma tiolata del GSH, cosa necessaria per la classica funzione delle GST di trasferimento del GSH stesso su di un gruppo elettrofilo (Armstrong , 1997). Per la GSTM 2-2 é stato suggerito che lo spostamento di un residuo di arginina carico positivamente nella tasca catalitica promuova la ionizzazione del
sulfidrile del GSH (Patskovsky et al., 2000). Nella GSTO 1-1 non ci sono gruppi chimici in prossimità della cisteina 32 in grado di svolgere tale funzione.
La Cys-32 é localizzata nella parte N-terminale di α1, con il tiolo collocato precisamente sopra l’asse dell’elica. Questa posizione é esattamente quella della cisteina N-terminale del motivo Cys-Xaa-Xaa-Cys della tioredossina e della glutaredossina. E’ molto verisimile che questa sia la struttura determinante per il funzionamento della GSTO 1-1.
La seconda caratteristica originale della GSTO 1-1 é data dall’assenza di interazioni tra il GSH legato ad una catena polipeptidica e gruppi dell’altro polipeptide. Nelle altre strutture note di GST vi é un legame salino tra l’azoto N-terminale del GSH e un residuo acido sull’elica α4 della seconda subunità. Questo non avviene nella GSTO 1-1 perchè il residuo corrispondente é Lys-122.
Il dimero GSTO 1-1 presenta un’insolita configurazione aperta a forma di V. I contatti tra le due subunità sono limitati alle catene laterali di β4, α3 e α4. Gran parte delle interazioni alla interfaccia sono di tipo non polare; infatti sono presenti solamente due legami salini e mancano totalmente legami a idrogeno. L’orientamento dei monomeri all’interfaccia corrisponde a quello osservato in altre strutture cristalline di GST, ma l’interfaccia é più aperta cha negli altri dimeri di GST. L’area nascosta all’interfaccia misura 1960 Å2, mentre nella maggior parte dei dimeri delle altre GST misura 2700-3400 Å2.
Da tutti gli altri punti di vista, il legame del GSH é del tutto analogo a quanto si osserva nelle altre GST: tutte le interazioni tra GSH e proteina avvengono a livello
del dominio N-terminale e i residui che contribuiscono al legame con il GSH sono conservati o conservativamente sostituiti.
1.4.4 La regione H (H site)
La maggior parte delle GST catalizzano la coniugazione del gruppo tiolico del GSH con un gruppo elettrofilo, spesso componente di uno xenobiotico tossico con caratteri in parte idrofobici. La struttura tipica delle GST prevede una regione, chiamata regione H, che contiene un dominio idrofobo ed é situata in vicinanza del sito di legame al GSH, regione G. La regione H é formata da elementi sia del dominio N- che C-terminale e le variazioni a questo livello tra le varie classi di GST riflettono le diverse specificità di substrato.
La GSTO 1-1 possiede una ben definita cavità in corrispondenza della regione H presente nelle altre GST e si presume pertanto che rappresenti una zona di legame con altre molecole. Tuttavia, data la presenza dei residui Trp-222 e Arg-183, la tasca risulta molto meno idrofobica rispetto alle regione H delle altre GST.
In conclusione l’interfaccia particolarmente aperta tra le subunità e la natura relativamente polare della regione H, suggeriscono che il substrato della GSTO 1-1 potrebbe essere una molecola di grosse dimensioni e non altamente idrofobica. Il legame con un’altra proteina appare quindi possibile. Considerando che la principale attività osservata nella GSTO 1-1 é quella tiol-transferasica GSH-dipendente, si può ipotizzare che tra i substrati naturali vi siano peptidi S-tiolati. E’ noto, ad esempio, che in seguito ad uno stress ossidativo un gran numero di proteine forma addotti S-tiolati (disolfuri misti) con il GSH o la cisteina, con possibile inattivazione o
cambiamento di funzioni enzimatiche delle proteine interessate (Seres et al., 1996; Hanson et al., 1999; Ravichandran et al., 1994; Jahngen-Hodge et al., 1997). Una possibile funzione della GSTO 1-1 potrebbe pertanto essere quella di ridurre questi disolfuri, con ripristino della funzionalità delle proteine colpite (Board et al., 2000).
1.4.5 Organizzazione genomica delle Glutatione Transferasi Omega
Il gruppo di P.G. Board ha proceduto successivamente allo studio della organizzazione genomica della GSTO1 (Whitbread et al., 2003). Tramite la “fluorescence in situ hybridization” (FISH), il gene della GSTO1 é stato localizzato nel cromosoma 10 e, precisamente, nella regione 10q24.3. Nel corso di questi studi é stata evidenziata la presenza, sul cromosoma 10, di un secondo gene della classe omega, cui é stato dato il nome GSTO 2. Questo gene, al pari del GSTO 1, ha sei esoni e si trova 7,5 kb al di sotto del gene GSTO 1. E’ verisimile pertanto che sia il risultato di una duplicazione genica. Il relativo cDNA codifica per una proteina di 243 residui aminoacidici con il 64% di identità con la proteina GSTO 1 composta da 241 residui. E’ noto che i geni codificanti gli enzimi di ciascuna classe di GST tendono a trovarsi in “clusters” su cromosomi distinti. Ad esempio i geni che codificano per le GST di classe α sono localizzati nel cromosoma umano 6p12, quelli della classe μ in 1p13.3, quelli della classe ϑ in 22q11.2, ecc. Nessun gene di GST era stato fino ad ora localizzato sul cromosoma umano 10; pertanto la localizzazione di GSTO 1 e 2 in questo locus rappresenta un ulteriore dato a supporto della classificazione filogenetica delle GST omega come una classe distinta di GST.
Nel corso di questa ricerca é stato individuato un terzo gene di classe omega (GSTO 3p) sul cromosoma 3. Questo gene mostra un’alta omologia con la regione codificante di GSTO 1; tuttavia, data la sua mancata rappresentazione nella banca dati EST, l’assenza di sequenze introniche e la sua collocazione cromosomica diversa dalla GSTO 1 e dalla GSTO 2, sembra verosimile che si tratti di uno pseudogene.
1.4.6 Attivita’ enzimatiche delle GSTO umane Attività tioltransferasica
La tioltransferasi, conosciuta anche come glutaredossina, é un membro delle tiolo-disolfuro ossidoreduttasi. E’ una piccola proteina citosolica con un peso molecolare di 11,8 kDa. Catalizza in maniera specifica la riduzione di proteine tiolate dal GSH (PSSG) (Chrestensen , 1995) La reazione procede nel seguente modo:
RSSR’ + GSH --> RSH + GSSR GSSR’ + GSH --> GSSG + R’SH
Dove R e R’ rappresentano rispettivamente i tioli proteici e quelli non proteici. Pertanto la riduzione di PSSG avviene tramite formazione di GSSG che viene poi riciclato a GSH grazie all’azione della glutatione reduttasi.
Una significativa attività osservata nella GSTO1-1 è la capacità di agire, al pari della glutaredossina, come tioltransferasi GSH dipendente (Board et al., 2000). Tale capacità é mostrata, in misura pressochè simile, dalla GSTO2-2 (Schmuck et al., 2005). Sembra che questo tipo di attività dipenda dal fatto che la Cys-32 potrebbe
funzionare come centro nucleofilo, ipotesi basata sulla omologia strutturale con la tioredossina e la glutaredossina (Board et al., 2000).
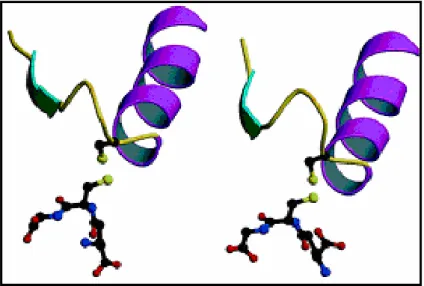
Figura 1.8 Comparazioni tra il sito di legame per il GSH nella glutaredossina (sinistra) e nella GSTO (destra).(Board et al., 2000).
Molte molecole contenenti legami disolfuri possono rappresentare potenziali substrati per questo tipo di attività, inclusi i polipeptidi S-tiolati. Per esempio, durante lo stress ossidativo, numerose proteine formano addotti S-tiolo con glutatione e cisteina (Seres et al., 1996; Hanson et al., 1999) con conseguente inattivazione o cambiamenti funzionali delle proteine (Ravichandran et al., 1994; Jahngen-Hodge et al., 1997). Quindi una probabile funzione della GSTO1-1 è di ridurre questi addotti S-tiolo ripristinando la funzionalità enzimatica. Come precedentemente descritto, la struttura cristallina della GSTO1-1 indica che il suo sito H è particolarmente aperto alla superficie e largo abbastanza per alloggiare una catena polipeptidica (Board et al., 2000).
Attività deidroascorbato reduttasica
Una caratteristica peculiare delle GSTO è quella di possedere attività DHA reduttasica (Maellaro et al., 1994; Board et al., 2000), cioè la capacità di ridurre il DHA, forma ossidata della vitamina C, ricostituendo l’acido ascorbico (AA).
L’AA è un chetolattone a sei atomi di carbonio, presente in tutti gli organismi viventi, sia vegetali che animali. Nel regno animale viene sintetizzato nel fegato degli anfibi, dei rettili, degli uccelli e dei mammiferi a partire dall’acido glucuronico derivante da glucosio (Chatterjee et al., 1975). Tra i mammiferi l’uomo, alcuni primati, la cavia e alcuni chirotteri hanno perso la capacità sintetica.
L’AA gioca un ruolo essenziale in tutti gli organismi viventi grazie alla sua capacità di andare facilmente incontro, nei sistemi biologici, ad ossidazione. Tale caratteristica gli consente di agire sia da scavenger diretto dei radicali liberi e delle specie attive dell’ossigeno, che da agente riducente in numerose tappe del normale metabolismo cellulare (biosintesi del collagene, della carnitina, della noradrenalina e attivazione di ormoni) (Padh, 1991). Il processo di ossidazione dell’acido ascorbico può avvenire attraverso il trasferimento di un elettrone, portando alla formazione del radicale semideidroascorbato oppure attraverso il trasferimento di due elettroni producendo il DHA; questo ultimo può formarsi inoltre anche per dismutazione di due AA⋅ secondo la seguente reazione:
AA. + AA. --> AA + DHA
E’ noto da tempo che gli organismi viventi sono dotati di attività DHA reduttasiche molto efficienti in grado di ripristinare l’AA utilizzando il GSH come donatore di equivalenti riducenti:
DHA + 2GSH --> AA + GSSG
Queste attività sono pertanto di grande importanza per mantenere costante la concentrazione di acido ascorbico a dispetto della continua ossidazione da parte dei radicali liberi.
I sistemi di riduzione del DHA però sono importanti non solo per riciclare la vitamina C: è ben documentato, infatti, che il DHA può produrre effetti tossici, a causa della sua capacità di legarsi ai gruppi amminici e sulfidrilici delle proteine. Ad esempio, alti livelli di DHA sono tossici per gli eritrociti (Bianchi and Rose, 1986), le isole pancreatiche (Pillsbury et al., 1973) e l’endotelio corneale (Wolff et al., 1987). La capacità di ridurre efficientemente il DHA ad AA può pertanto proteggere la cellula dagli effetti tossici del DHA.
Un’altra importante funzione dei sistemi riducenti il DHA è quella di contribuire all’uptake della vitamina C. L’AA e il DHA sono entrambi trasportati entro le cellule, ma con meccanismi diversi. L’AA è accumulato come tale attraverso un trasporto attivo carrier-mediato e saturabile, dipendente dal sodio e dalla sua stessa concentrazione (Welch et al., 1995). Il DHA penetra dentro le cellule, dove si accumula però come AA, attraverso un meccanismo di diffusione facilitata (Hughes and Maton, 1968; Bigley et al., 1981; Vera et al ., 1993 ; Welch et al., 1995), che sfrutta gli stessi trasportatori del glucosio (Vera et al., 1993). L’efficiente riduzione intracellulare del DHA, evitando il raggiungimento di un equilibrio tra
concentrazione extra- ed intra- cellulare del DHA, rende questa forma di uptake molto più efficiente dell’uptake diretto del AA.
In conclusione dai dati della letteratura possiamo dedurre che i sistemi di riduzione del DHA svolgono essenzialmente tre funzioni:
i) ripristinano la forma attiva della vitamina C ( AA); ii) impediscono gli effetti tossici del DHA;
iii) permettono l’uptake della vitamina C sotto forma di DHA.
I primi enzimi ad attività DHA reduttasica scoperti sono stati la glutaredossina e la disolfuro isomerasi (Wells et al., 1990). Successivamente, nel laboratorio dove ho preparato la tesi, è stata scoperta e caratterizzata la DHA reduttasi di ratto, poi inclusa nel gruppo delle GSTO (Maellaro et al., 1994). In base ad esperimenti di immunotitolazione, la capacità DHA reduttasica globale del citosol di fegato di ratto sembra dipendere in massima parte (almeno il 70%) dalla presenza di questo enzima (Paolicchi et al., 1996). Un’analoga attività deidroascorbato reduttasica è stata osservata successivamente nella forma umana (GSTO1-1) (Board et al., 2000). Infine un’attività particolarmente elevata è stata trovata nella forma umana GSTO 2-2. L’attività specifica di questa proteina (circa 100 volte superiore a quella della GSTO 1-1) è attualmente la più alta di qualsiasi altro enzima fin ad ora noto (Schmuck et al., 2005).
Attività monometilarseniato (MMAV) e dimetilarseniato (DMAV) reduttasica L’arsenico inorganico è un comune contaminante dell’acqua naturale in molti regioni del mondo ed è conosciuto come cancerogeno. La World Health
Organization ha stimato che circa 70 milioni di persone in Bangladesh sono a rischio di cancro perché bevono acqua contaminata dall’arsenico inorganico. Anche altre aree del mondo come la Romania, il Taiwan, la Cina, il Messico, la Mongolia e l’Ungheria hanno simili problemi (National Research Council Report, 1999; Arsenic Exposure and Health Effects, 1998 ).
La biotrasformazione dell’arsenico inorganico é un processo a più stadi catalizzato in successione dalla arseniato reduttasi (Radabaugh, and Aposhian, et al., 2000;), dalla arsenito metiltransferasi (Zakharyan et al., 1995; Wildfang et al., 1998), dalla monometilarseniato (MMAV) reduttasi (Zakharyan and Aposhian, 1999a), dalla monometilarsenito metiltransferasi e dalla dimetilarseniato (DMAV) reduttasi (Zakharyan et al., 1999b).
Figura 1.9 Tappe metaboliche della biotrasformazione dell’arsenico inorganico.
(Schmuck et al., 2005).
E’ discutibile se tale via metabolica debba considerarsi una via di detossificazione: alcuni dei metaboliti sono infatti cancerogeni e fortemente tossici (Cullen et al., 1989; Styblo et al., 1997). L’arsenito é metabolizzato dall’arsenito metiltransferasi a MMAV, che é un metabolita relativamente innocuo. Questo, però, deve essere ridotto a MMAIII, prodotto altamente tossico, prima che una seconda metilazione possa aver luogo per produrre il DMAV, che rappresenta, al contrario, il composto meno tossico tra tutti i metaboliti noti (Petrick et al., 2000; Mass et al., 2001). Una volta che sufficienti livelli di MMAV sono stati prodotti, la MMAV reduttasi può produrre il MMAIII, che può poi essere metilato dalla MMAIII metiltransferasi a DMAV . La DMAV, inoltre, può essere ridotta da una DMAV reduttasi a DMAIII, altro metabolita probabilmente molto tossico (Schmuck et al., 2005).
La reazione catalizzata dalla MMAV reduttasi è la tappa limitante nel metabolismo dell’arsenico inorganico, poiché la sua Km è nel range millimolare, mentre quelle delle metiltransferasi sono nel range micromolare (Zakharyan and Aposhian, 1999a). Esistono vari dati comprovanti la maggior tossicità del MMAIII rispetto agli altri metaboliti. Ad esempio Petrick et al. (2000) dimostrarono le seguenti tossicità relative nei confronti delle cellule Chang provenienti da fegato umano: MMAIII (LC50 = 6µM) > arsenito (LC50 = 68 µM) > arsenato (LC50= 1628 µM) > MMAV (LC50 = 8235 µM) > DMAV (LC50 = 9125 µM).
Dal momento che il DMAV é meno tossico e il MMAIII é più tossico rispetto all’arsenito inorganico, i possibili benefici della metilazione dell’arsenico come via di detossificazione devono essere valutati sulla base dell’accumulo dei vari metaboliti tossici.
Come abbiamo già detto, la MMAV reduttasi rappresenta la tappa limitante nel metabolismo dell’arsenico inorganico ed é quella che produce il composto più tossico (Zakharyan and Aposhian, 1999a). In seguito ad una parziale purificazione della MMAV reduttasi umana, é stato possibile determinare la sequenza aminoacidica del 92% della proteina (Zakharyan et al., 2001). Tale sequenza si é rivelata perfettamente identica a quella della GSTO1-1. In accordo con ciò, la GSTO 1-1 ricombinante umana mostra attività MMAV reduttasica con valori di Km e Vmax comparabili con quelli della MMAV reduttasi umana e, di converso, la MMAV reduttasi umana parzialmente purificata mostra capacità catalitiche analoghe alla GSTO 1-1 (attività tiol-transferasica e DHA reduttasica). Entrambi gli enzimi hanno un’assoluta specificità per il GSH. Infine la banda elettroforetica corrispondente alla
MMAV reduttasi umana viene riconosciuta, mediante Immunoblot, dall’antisiero verso la GSTO 1-1 umana. Gli Autori (Zakharyan et al., 2001), sulla base dei suddetti dati, giungono pertanto alla conclusione che la MMAV reduttasi umana e la GSTO 1-1 umana debbano essere considerate proteine identiche.
Recentemente, come abbiamo già descritto, uno studio sull’organizzazione genomica della GSTO 1 umana (Whitbread et al., 2003) ha portato alla identificazione di un secondo gene attivamente trascritto, cui é stata data la sigla GSTO 2. La proteina GSTO 2 ha una identità del 64% con la GSTO 1 per quanto riguarda la sequenza aminoacidica e conserva il residuo di cisteina in posizione 32, che si ritiene importante per il sito attivo della GSTO 1 (Whitbread et al., 2003). La successiva caratterizzazione della GSTO 2-2 umana (Schmuck et al., 2005) ha evidenziato che anche questo enzima ha un’attività MMAV reduttasica comparabile a quella della GSTO 1-1. Nel corso dello stesso studio é stato pure evidenziato che la GSTO 1-1 ha anche una buona attività DMAV reduttasica e può pertanto catalizzare anche l’ultima tappa nota nel metabolismo dell’arsenico. Pure la GSTO 2-2 mostra tale capacità, ma con un’attività specifica molto più bassa rispetto alla GSTO1-1.
Modulazione dell’attività dei canali intracellulari del calcio
Studi recenti hanno evidenziato un possibile ruolo della GSTO1-1 nella regolazione del Ca2+ intracellulare (Mariot et al., 2000; Pan et al., 2000). Studi condotti utilizzando la sequenza caratteristica delle GST di classe θ nel tentativo di trovare altri membri della famiglia GST, hanno infatti rivelato una significativa somiglianza di questa sequenza con i membri appartenenti al gruppo di canali proteici
intracellulari CLIC (Chloride Intracellular Channel), in particolar modo con la proteina NCC27 (CLIC 1) (Dulhunty et al., 2001). Ricerche dettagliate sull’allineamento tra la sequenza della GSTO1 e la sequenza di CLIC1 hanno rivelato la conservazione di alcuni residui fondamentali caratteristici della famiglia strutturale GSTO, in particolare nella regione N-terminale deputata al legame con il GSH e nel sito attivo dove é conservato il tipico residuo di cisteina delle GSTO (Dulhunty et al., 2001). Studi predittivi sulla conformazione della proteina indicano inoltre che la proteina CLIC 1 adotta il tipico modello conformazionale delle GST e che può quindi essere considerata un membro della famiglia strutturale delle GST (Dulhunty et al., 2001).
L’osservazione che le proteine CLIC probabilmente adottano la struttura terziaria tipica delle GSTO e che condividono con queste una significativa omologia di sequenza nella regione N-terminale e nel sito attivo, hanno indotto gli Autori a esaminare la possibilità che le GSTO abbiano attività analoghe alle proteine CLIC, che possano cioè agire come canali ionici e/o modulatori di canali ionici (Dulhunty et al., 2001).
Lo studio in vitro condotto su bilayer fosfolipidici contenenti la GSTO 1-1 ricombinante non ha evidenziato la capacità di tale proteina di formare canali ionici. Tuttavia, considerando che la GSTO 1-1 é fortemente espressa nel muscolo cardiaco e scheletrico, gli Autori hanno successivamente preso in esame l’eventuale capacità della proteina di modulare l’attività dei “Ryanodine Receptor” (RyRs), canali di rilascio del Ca2+ nei muscoli scheletrici e nel reticolo sarcoplasmatico cardiaco (Dulhunty et al., 2001).
I risultati indicano che la GSTO1-1 può sia inibire che potenziare i canali del calcio RyR . L’inibizione si osserva solo nei confronti di RyR2 e richiede il mantenimento della attività tiol-transferasica dell’enzima. L’attivazione, invece, si può osservare sia nei confronti di RyR2 che di RyR1 ed é indipendente dal mantenimento dell’attività enzimatica. Gli Autori suggeriscono che RyR2 abbia due siti di legame per la GSTO1-1, uno per l’attivazione del canale e l’altro per la sua inibizione. In effetti RyR ha un dominio citoplasmatico molto grande con diversi siti di legame per ligando, e sono noti ligandi con duplice effetto sull’attività del canale in seguito a legame su siti diversi. Esempi in tal senso sono il Ca2+ (Laver et al., 1995) e l’ATP (Kermode et al., 1998).
I canali RyR sono regolati da diversi fattori, incluse reazioni di ossidazione e riduzione (Dulhunty et al., 2000). La GSTO1-1 rappresenta un’ulteriore possibilità di regolazione (Dulhunty et al., 2001). Considerando che le proteine CLIC formano o modulano i canali ionici (Valenzuela et al., 1997; Qian et al., 1999) e hanno una struttura simile alle GST, si può ipotizzare che la modulazione dei canali ionici sia una proprietà generale di questa superfamiglia (Dulhunty et al., 2001).
Ruolo nella processazione post-traduzionale della interleuchina-1
L’interleuchina-1 (IL-1) é un mediatore proinfiammatorio prodotto in gran quantità da monociti e macrofagi dopo attivazione da parte del lipopolisaccaride (LPS) batterico (Dinarello, 1998). Diversamente da molte altre citochine prodotte da queste cellule, IL-1 non viene rilasciata costitutivamente. Per un efficiente rilascio di IL-1
occorre, infatti, che le cellule produttrici siano sottoposte all’azione di un effettore in grado di dare inizio ad un meccanismo di modifiche post traduzionali piuttosto insolito che porta in ultimo al rilascio della citochina (Hogquist et al., 1991; Perregaux et al., 1992; Rubartelli et al., 1990). E’ però necessario uno stimolo ulteriore e diverso per la secrezione, poiché l’iniziale prodotto traduzionale della IL-1 é privo di una sequenza segnale che diriga il polipeptide verso il reticolo endoplasmico (March et al., 1985; Auron et al., 1984; Walter and Johnson, 1994). Da qui tipicamente i polipeptidi destinati alla secrezione procedono verso la superficie cellulare attraverso un apparato secretorio che coinvolge il complesso di Golgi e le piccole vescicole secretorie. In mancanza di questo segnale di riconoscimento per entrare nel reticolo endoplasmico, la IL-1 di nuova sintesi si accumula nel compartimento citoplasmatico perdendo la sua funzione di mediatore extra-cellulare (Traub and Kornfeld, 1997; Singer et al., 1988; Stevenson et al., 1992).
Tra gli stimoli che, in vitro, promuovono un’efficiente modifica post-traduzionale delle IL-1, abbiamo l’ATP extra-cellulare (Hogquist et al., 1991; Perregaux and Gabel, 1994; Ferrari et al., 1997), la nigericina (ionoforo per il potassio)( Perregaux et al., 1992; Cheneval et al., 1998), tossine batteriche, quali la emolisina dell’Escherichia coli (Bhakdi et al., 1990) e le cellule T citotossiche (Hogquist et al., 1991; Perregaux et al.,1996).
L’IL-1 é un mediatore chiave di molti processi infiammatori. Studi preclinici su modelli animali e studi clinici sull’uomo hanno dimostrato l’efficacia terapeutica di vari inibitori dell’IL-1 . Recentemente é stata identificata una serie di inibitori
(CRID, Cytokine Release Inhibitory Drug), derivati dalla diarilsulfonilurea, che in maniera selettiva e con notevole efficacia inibiscono le modifiche post-traduzionali della IL-1 β (Laliberte et al., 2003).
Uno studio, intrapreso dagli Autori al fine di chiarire i meccanismi di tale inibizione, ha portato ad identificare la GSTO 1-1 come una proteina in grado di legare la diarilsulfonilurea e l’interazione tra i CRID e la GSTO 1-1 come la responsabile dell’inibizione del processo post-traduzionale della IL-1 β, indotto da ATP.
Le principali evidenze a supporto di tali conclusioni sono: i) CRID con diarilsulfonilurea, contenenti un gruppo epossidico marcato con 14C, si legano irreversibilmente alla GSTO 1-1 e tale legame correla con l’entità della inibizione nella produzione di citochine; ii) la GSTO 1-1 si lega reversibilmente ad una colonna di affinità contenente diarilsulfonilurea; iii) un addotto CRID-glutatione, somministrato a monociti intatti, inibisce le modifiche post-traduzionali della IL-1 β indotte da ATP e interagisce con la GSTO 1-1. Altri dati suggeriscono che l’inibizione si attua tramite il legame dei CRID con la cisteina 32, responsabile dell’attività catalitica della GSTO 1-1. Ad esempio la sostituzione della cisteina 32 con una alanina impedisce completamente il legame dell’inibitore con la GSTO 1-1. Il fatto che la GSTO 1-1 sia il target specifico attraverso cui si esplica l’azione inibitoria dei CRID suggerisce di conseguenza che tale proteina svolga un ruolo determinante nel contesto delle modifiche post-traduzionali della IL-1β indotte da stimoli vari Laliberte et al., 2003. Il meccanismo non é attualmente noto, ma gli Autori avanzano due ipotesi La prima prende spunto dalla somiglianza strutturale tra GSTO 1-1 e CLIC 1 di cui abbiamo parlato nel precedente capitolo. Gli Autori