CAPITOLO 3: MATERIALI E METODI
3.1 BIOLOGIA MOLECOLARE
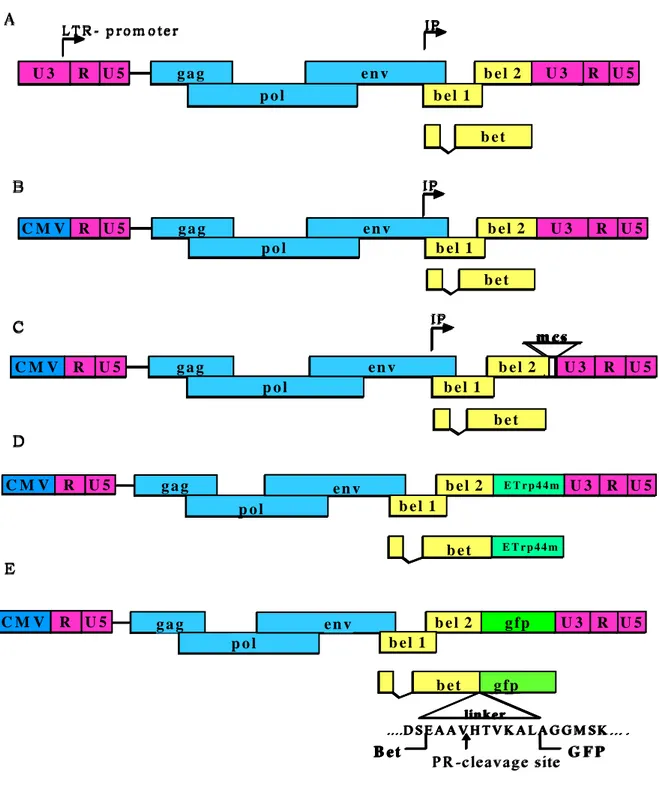
3.1.1 Vettori FFV
I vettori replicazione-competenti utilizzati per l’espressione del peptide ETrp44m e della GFP sono stati realizzati e forniti dal Professor Loechelt (German Cancer Research Center, University of Heidelberg, Heidelberg, Germany). I vettori sono stati costruiti a partire dal clone wild-type del FFV (pFeFv-7) in cui la regione U3 dell’LTR in 5’ è stata sostituita col promotore forte del citomegalovirus (CMV-IE).
Il clone risultante, pCF-7 è stato usato per sostituire parte della sequenza U3 del promotore nell’LTR in 3’ (da –308 a –725) con un corto multiple cloning site (mcs).
Il clone ottenuto è il pCF-7∆U3 che è stato utilizzato per clonare il peptide 44mer in frame con il gene bet originando così il costrutto pCF- Bet-FIV44mer o pCF-Bet-GFP contenente il gene reporter GFP al posto di ETrp44m. In entrambi i casi il codone di stop del gene bet è stato deleto e fra le sequenze codificanti il gene bet e il transgene è stata inserita una sequenza linker che contiene un sito di taglio per una proteasi cellulari per far sì che la forma wild-type venga rilasciata. L’organizzazione genomica del costrutto wild-type e gli intermedi sono mostrati in Fig. 3.1.
Oltre a questi vettori replicazione-competenti abbiamo utilizzato anche i rispettivi costrutti SIN nei quali la regione U3 dell’LTR in 3’ è stata deleta da -725 a -18 comprensiva quindi della TATA box e di tutte le regioni necessarie all’attacco dei vari fattori di trascrizione cellulari come mostrato in Fig. 3.2
In questi sono stati inseriti, come nei costrutti replicazione-competenti, il peptide ETrp44m (Fig. 3.2) e la GFP (Fig. 3.2).
U 3 R U 5 b e l 1 b e l 2 g a g p o l e n v L T R - p ro m o te r IP b e t R U 5 U 3 C M V R U 5 g a g U 3 R U 5 p o l e n v b e l 1 b e l 2 b e t IP b e t p o l e n v p o l e n v b e t b e t b e l 2 E T r p 4 4 m b e l 1 R U 5 U 3 C M V R U 5 E T r p 4 4 m g a g b e t g fp lin k e r ....D S E A A V H T V K A L A G G M S K … . B e t G F P g a g p o l e n v g a g p o l e n v b e t b e t g fp lin k e r ....D S E A A V H T V K A L A G G M S K … . B e t b e l 2 g fp b e l 1 R U 5 U 3 P R -cleav ag e site C M V R U 5 A B C M V R U 5 g a g U 3 R U 5 p o l e n v b e l 1 b e l 2 b e t m c s IP C D E U 3 R U 5 b e l 1 b e l 2 g a g p o l e n v L T R - p ro m o te r IP b e t R U 5 U 3 U 3 R U 5 U 3 R U 5 b e l 1 b e l 2 g a g p o l e n v L T R - p ro m o te r IPIP b e t R U 5 U 3 R U 5 U 3 C M V R U 5 g a g U 3 R U 5 p o l e n v b e l 1 b e l 2 b e t IP C M V R U 5 g a g U 3 R U 5 p o l e n v g a g p o l e n v b e l 1 b e l 2 b e t IP IP b e t p o l e n v p o l e n v b e t b e t b e l 2 E T r p 4 4 m b e l 1 R U 5 U 3U 3 R U 5 C M V R U 5 E T r p 4 4 m g a g b e t g fp lin k e r ....D S E A A V H T V K A L A G G M S K … . B e t G F P g a g p o l e n v g a g p o l e n v b e t b e t g fp lin k e r ....D S E A A V H T V K A L A G G M S K … . B e t b e l 2 g fp b e l 1 R U 5 U 3 R U 5 U 3 P R -cleav ag e site C M V R U 5 C M V R U 5 A B C M V R U 5 g a g U 3 R U 5 p o l e n v b e l 1 b e l 2 b e t m c s IP C D C M V R U 5 C M V R U 5 g a g U 3 RR U 5U 5 p o l e n v b e l 1 b e l 2 b e t m c s IP IP C D E
Figura 3.1: Organizzazione genomica di: A) FFV wild-type B) clone pCF-7 C) clone pCF-7∆U3 D) pCF-Bet-FIV44m E) pCF-Bet-GFP
U3 TATA R U5 SIN -18 -725 bel2/bet U3 TATA R U5 SIN -18 -725 bel2/bet A) CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag pol env IP SIN R U5 IP CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag
pol env 44mer R U5
IP CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag pol env gfp R U5 B) C) D) CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag pol env IP SIN R U5 CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag pol env IP SIN R U5 R U5 R U5 IP CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag
pol env 44mer R U5
IP CMV R U5R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag
pol env 44mer R U5R U5R U5
IP CMV R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag pol env gfp R U5 IP CMV R U5R U5 PR-RT RH IN Elp SU TM bel 1 bel 2 bet gag pol env gfp R U5R U5R U5 B) C) D)
Figura 3.2: (A) Regione della 3’ LTR in cui è stata effettuata la delezione
della TATA box e altri fattori di trascrizione dando origine al costrutto in (B); (C) costrutto SIN con il peptide ETrp44m; (D) costrutto SIN con la GFP.
3.1.2 Trasformazione
Per crescere i plasmidi contenenti i vettori è stato utilizzato il ceppo JM109 di E.coli reso competente nel nostro laboratorio e conservato in aliquote da 200 µl a –80°C. Tutte le trasformazioni sono state eseguite aggiungendo 10-50 ng di DNA plasmidico alla soluzione contenente i batteri e lasciando incubare la miscela in ghiaccio per 30 minuti.
Dopo l’incubazione le cellule sono sottoposte a shock termico tenendole per 45 secondi a 42°C, in modo da alterare la permeabilità della membrana plasmatica e favorire l’entrata del DNA plasmidico. Le cellule sono state poi trasferite rapidamente in ghiaccio per 2 minuti.
A questo punto ciascuna miscela viene posta in agitazione per un’ora a 37°C previa aggiunta di 1 ml di terreno SOC (2% Triptone, 0,5% di
estratto di lievito, NaCl 10 mM, KCl 2,5 mM, MgCl2 10 mM, MgSO4
10mM e glucosio 20 mM).
Le cellule vengono infine piastrate sul terreno LB–agar (10 g/l Triptone, 5 g/l di estratto di lievito, 10 g/l NaCl a pH 7,0, 1,5% di agar) a cui è stata aggiunta ampicillina 50 µg/ml. Poiché i plasmidi pCF7 contengono il gene che conferisce resistenza a questo antibiotico, è possibile la selezione delle cellule che hanno acquisito il plasmide.
3.1.3 Estrazione dei plasmidi
Per effettuare l’estrazione del DNA plasmidico, le colonie selezionate sono state fatte crescere overnight a 37°C in 2 ml di terreno liquido LB addizionato con ampicillina 50 µg/ml e il DNA è stato estratto attraverso una lisi alcalina. A questo scopo 1.5 ml di coltura sono stati sedimentati centrifugando per 5 minuti a 12000 rpm a 4°C. Una volta eliminato il surnatante, il pellet è stato risospeso in 100 µl di soluzione I (glucosio 50 mM, 25mM Tris HCl a pH 8,0, 10 mM EDTA pH 8,0). Per la lisi della parete sono stati aggiunti 200 µl di soluzione II alcalina (0,2N
NaOH, 1%SDS) e 150 µl di soluzione III neutralizzante (3M potassio acetato, 5M acido acetico). Dopo un’incubazione di 3-5 minuti in ghiaccio, il lisato batterico è stato centrifugato 5 minuti a 12000 rpm a 4°C e il surnatante, contenente il DNA plasmidico, è stato trasferito in un tubino nuovo. Per eliminare le proteine contenute nel surnatante è stato aggiunto un volume di fenolo/cloroformio seguito da centrifugazione a 12000 rpm a 4°C per 5 minuti.
La centrifugazione porta alla formazione di due fasi, quella inferiore contenente fenolo/cloroformio, quella superiore contenente il DNA che è stato prelevato e precipitato con l’aggiunta di 0.7 volumi di isopropanolo seguita da una centrifugazione di 15 minuti a 12000 rpm.
Dopo un lavaggio con 100-200 µl di etanolo al 70% e un’ultima centrifugazione a 12000 rpm per 5 minuti è stato aspirato il surnatante. Il pellet, una volta asciugato, è stato risospeso in 20 µl di H2O e RNasi A
(un decimo del volume). La RNasi A è stata lasciata agire per 30 minuti a 37°C.
Per ottenere una quantità di materiale maggiore, le colonie sono state cresciute in 50 ml di LB addizionato con ampicillina 50 µg/ml e il DNA è stato estratto secondo il protocollo Midi plasmid Kit (Qiagen Hilden, Germany). Dopo estrazione, il DNA è stato controllato su gel d’agarosio e quantificato allo spettrofotometro misurando la densità ottica alla lunghezza d’onda 260 nm, dalla quale è ricavabile la concentrazione dell’acido nucleico in ng/µl.
3.1.4 Elettroforesi su gel d’agarosio
L’agarosio è stato sciolto in tampone TAE 0,5X (242 g/l di Tris base, 57,1 ml di EDTA 0.5 M pH 8), scegliendo la concentrazione in base alla risoluzione desiderata, dopodichè è stato aggiunto bromuro di etidio (50 ng/ml) e la miscela è stata fatta solidificare in una vasca elettroforetica. I campioni vengono mescolati con loading buffer 10X (0,25% di blu di
bromofenolo, 0,25% xilene cianolo, 30% glicerolo in H2O) in rapporto
10:1, e quindi caricati su gel a cui è stata applicata una differenza di potenziale di 60-90 V. A corsa terminata i gel sono stati fotografati mediante transilluminatore ad UV (Ultra Violet Products).
3.2 BIOLOGIA CELLULARE
3.2.1 Cellule
La linea cellulare utilizzata per la trasfezione e l’infezione dei costrutti FFV derivati è rappresentata da CrFK, cellule aderenti mantenute in coltura a 37°C in atmosfera umidificata, con il 5% di CO2 in terreno
Dulbecco Modified Eagle Medium (DMEM) addizionato con 10% di siero fetale bovino (FCS), 1% di glutammina, 1% di streptomicina e 1% di penicillina.
Al raggiungimento della confluenza viene rimosso il terreno e dopo un lavaggio con PBS (PBS 10X: 80 g/l di NaCl, 2 g/l di KCl, 14,4 g/l di Na2HPO4, 2,4 g/l di KH2PO4, HCl a pH 7,4) le cellule vengono staccate
con tripsina (Eurobio, Francia). Le piastre sono lasciate per qualche minuto a 37° per permettere alla tripsina di agire, viene poi aggiunto il terreno in rapporto 1:5 (tripsina:terreno) perché il siero contenuto all’interno è in grado di disattivare l’enzima.
Le cellule vengono poi sedimentate tramite centrifugazione a 1200 rpm per 7 minuti e, dopo aver eliminato il surnatante e risospeso il pellet, sono nuovamente seminate in opportune diluizioni in nuove piastre.
3.2.2 Preparazione DNA per trasfezione
Il DNA da utilizzare nella trasfezione viene precipitato in 1/10 di volume di sodio acetato che viene aggiunto al volume di templato, contenente la quantità di DNA desiderata, insieme a due volumi di etanolo assoluto freddo. I campioni così preparati vengono lasciati a –20°C overnight ed il giorno dopo, a precipitazione avvenuta, vengono centrifugati a 12000
rpm per 30 minuti a 4°C. Una volta eliminato il surnatante sono stati aggiunti 200 µl di etanolo al 70% ed i campioni sono stati nuovamente centrifugati a 12000 rpm per 10 minuti a 4°C. Il pellet ottenuto è stato separato dal surnatante e asciugato.
3.2.3 Trasfezione con il metodo del calcio fosfato
Ventiquattro ore prima di effettuare la trasfezione 2,5X106 CrFK sono
state trasferite in piastre da 100mm per colture cellulari contenenti in totale 10 ml di terreno DMEM + 10% FBS.
La quantità di cellule è stata scelta in modo tale che al momento della trasfezione si abbia una confluenza del 60-80%. Un’ora circa prima della trasfezione il terreno è stato sostituito con terreno fresco. La quantità di DNA utilizzata per la trasfezione è di 20 µg (per piastre con pozzetti da 100mm) o 10µg (per piastre con pozzetti da 60mm). Il DNA è stato precipitato e risospeso in 450 µl di buffer TE (Tris 1 mM, EDTA 0.1 mM a pH 8.0) fino a completa solubilizzazione. A questi sono stati poi aggiunti 50 µl di cloruro di calcio (250 mM) e aggiunti poi a 500µl di
HBS 2x (280 mM NaCl, 10mM KCl, 1,5 mM Na2HPO4, 12mM destrosio,
50 mM Tampone Hepes pH 7,2). Dopo aver miscelato fino a completa omogeneizzazione della soluzione, è stata fatta un’incubazione di 20-30 minuti ed in seguito la soluzione contenente DNA è stata distribuita uniformemente goccia a goccia sulla piastra contenente le cellule da trasfettare. La piastra è stata poi successivamente incubata a 37°C in presenza di CO2 al 5% per 6-8 ore al termine delle quali è stato rimosso
3.2.4 Infezione
Tre giorni dopo la trasfezione il surnatante delle cellule trasfettate è stato raccolto, chiarificato con centrifugazione a 1700 rpm per 10 minuti per eliminare eventuali cellule presenti in sospensione e poi usato per infettare circa 40000 CrFK seminate ventiquattro ore prima in piastre da 24 pozzetti contenenti ciascuno 1 ml di terreno DMEM completo.
3.2.5 Lettura al citofluorimetro
Questo saggio è stato utilizzato per valutare l’efficienza di trasfezione e la percentuale di cellule trasdotte basandoci sull’espressione del gene reporter GFP. A vari tempi le cellule da usare per la lettura sono state staccate con tripsina, è stato effettuato un lavaggio con FACS buffer (2g /l BSA 0,2%, 1g/l Sodio azide 0,1%, PBS), seguito da centrifugazione di 7 minuti a 1200 rpm, è stato eliminato il surnatante e le cellule risospese sono state fissate con FACS FIX (50 ml FACS buffer, 500mg paraformaldeide 1%). L’analisi al citofluorimetro (FACScan, Becton Dickinson) è stata eseguita sfruttando la metodica della citofluorimetria a flusso.
I dati sono stati interpretati andando ad osservare lo spostamento delle cellule esprimenti GFP verso valori più alti di fluorescenza rispetto alle cellule di controllo.
3.2.6 Western blot
Per valutare l’espressione della proteina Bet alla quale sono fusi Etrp44m e GFP nei due costrutti vettore è stato effettuato un western blot sulle cellule trasfettate e trasdotte. Circa 1X105 cellule sono state
0,15mM, Nonidet 0,5%), a cui sono stati poi aggiunti un ugual volume del colorante SDS Sample Buffer 2X (Tris-HCl 0,5 M pH 6.8, glicerolo 10%, SDS 10%, β-mercaptoetanolo, 0.05% blu di bromofenolo). I campioni così preparati sono stati bolliti per 7 minuti e caricati su gel di acrilammide/bisacrilammide al 12%. Lo stacking gel è stato preparato con il 30% di mix acrilammide/bisacrilammide (Acrilamide/Bis Solution, 29:1 (Bio-Rad Laboratories, Hercules, CA, USA), Tris-HCl 0.5mM pH 6.8, 10% SDS, 10% APS, TEMED (Sigma Aldrich). Il resolving gel è stato preparato con il 30% di acrilammide/bisacrilammide (BioRad Laboratories), Tris-HCl 1.5 mM pH 8.8, 10% SDS, 10% APS, TEMED. La corsa elettroforetica è stata effettuata in tampone running buffer 1X (Tris base 25 mM, 0,1% SDS, 1,44% glicina), ad una differenza di potenziale di 90 volt. Il trasferimento delle bande sulla membrana di nitrocellulosa (Pfizer) è stato effettuato in presenza del buffer di trasferimento (0.3% Tris-base, 1.44% glicina, 20% metanolo) ad una differenza di potenziale di 100 volt, dopodiché è stato effettuato il bloccaggio con PBS-skim milk al 3%. La membrana è stata ibridata con siero Pet (1:200) e α-Bet (1:2000) in PBS Tween-skim milk all’1% e lasciata overnight in agitazione. Il giorno seguente è stato aggiunto l’anticorpo secondario, rispettivamente α-cat perossidato (1:1000) e α-rabbit (1:5000) in PBS Tween-skin milk allo 0.5%. La rivelazione delle bande è stata effettuata con un substrato contenente diaminobenzidina (DAB 40 mg/ml), Tris HCl 100mM, perossido di idrogeno, NiCl2 80 mg/ml.
3.2.7 Saggio di competizione
Per valutare che la banda del peptide ETrp44m che si rivela col siero tramite western blot è effettivamente la banda di nostro interesse, è stato eseguito un saggio di competizione. In questo saggio si esegue in parallelo un western blot col siero e uno col siero preincubato col peptide libero (10µg per colonna) per un’ora a 37°C. Con questo metodo si osserva la sparizione della banda in corrispondenza a quella che
invece viene rivelata esclusivamente dal siero a dimostrazione che il peptide libero sequestra gli anticorpi presenti nel siero e specifici per il peptide.
3.2.8 Saggio di infettività
Per valutare l’infettività dei costrutti, Bet-GFP e Bet-FIV44mer è stato eseguito un saggio di infettività, un metodo semplice e rapido per la titolazione quantitativa del virus. Questo saggio prevede l’utilizzo di una particolare linea cellulare adatta a questo scopo, le FeFAB (Foamy virus-activated Gal expression cells) nelle quali l’espressione della β-galattosidasi può essere attivata dall’infezione con FFV. Le FeFAB-cells sono cellule CrFK-derivate che hanno integrato stabilmente il gene β-gal sotto il controllo del promotore LTR del FFV. Se le cellule vengono infettate il genoma si integra e comincia a esprimere Bel1 che è un forte transattivatore il quale si lega all’LTR e induce l’espressione della β-galattosidasi. Questo enzima scinde uno specifico substrato cromoforo, l’X-gal, dando una reazione colorimetrica.
Poiché un segnale di localizzazione nucleare è inserito a valle del gene β-gal un intenso color blu si ritrova nei nuclei cellulari dove il β-gal si è accumulato e attraverso il quale è possibile andare a contare i foci di infezione, risalendo alle FFU presenti nel surnatante sulla base della diluizione effettuata (Yu and Linial; 1993).
X-Gal gag
pol env bel1bel2 bet LTR LTR LTR-Promoter IP LTR FFV Β-galattosidasi Β-galattosidasi Reazione colorimetrica
FeFAB
Bel1 X-Gal gagpol env bel1bel2 bet
LTR gag LTR
pol env bel1bel2 bet LTR LTR LTR-Promoter IP LTR FFV Β-galattosidasi Β-galattosidasi Reazione colorimetrica
FeFAB
Bel1 Bel1Il giorno prima della titolazione si seminano in una piastra da 24 pozzetti circa 30000 FeFAB per pozzetto con 1 ml di terreno DMEM in modo che le cellule aderiscano bene al substrato. Il surnatante delle cellule infettate, raccolto 2-3 giorni dopo la prima infezione, si centrifuga per 5 minuti a 1500 rpm per pellettare i detriti cellulari. Si aggiunge 1 ml di surnatante dei rispettivi costrutti al primo pozzetto della prima fila (FeFAB 100). Si aggiungono 110 µl di surnatante al
secondo pozzetto e si mescola per ripetuto pipettamento (FeFAB 10-1);
da questo pozzetto si prendono 110µl e si aggiungono al successivo (10 -2) e così per i successivi pozzetti. In questo modo si ha una diluizione
con fattore 1:10. Normalmente sono sufficienti per la titolazione 6 diluizioni (7 pozzetti in tutto).
Le cellule vengono lasciate incubare 2-3 giorni e poi si effettua il saggio: si leva il terreno dai pozzetti, si aggiunge 1ml di PBS/1mM MgCl2 per
ciascun pozzetto per lavare le cellule, si leva il PBS, si aggiunge 1ml di fixation solution (25ml PBS/MgCl2 1mM, 200µl Glutaraldeide al 25%,
ambiente, poi si elimina la soluzione fissativa e si lavano le cellule 3 volte con PBS. Si toglie il PBS e si aggiungono 300µl di staining solution (10ml PBS/MgCl2 1mM, 80µl Kaliumhexacyanoferrat (II) 0,5M, 80µl Kaliumhexacyanoferrat (III) 0,5M, 40µl X-Gal 100mg/ml in DMF) per ciascun pozzetto e si incuba la piastra per 1 ora a 37°C. Dopo 1 ora si leva la staining solution e si lavano le cellule 2 volte con PBS o acqua. La titolazione si effettua al microscopio dove si contano le cellule blu per ciascuna diluizione. Il titolo è dato dagli spot blu contati per il reciproco della diluizione.