_______________________________________________________________________________________________________________________________ 486 http://www.mjms.mk/ http://www.id-press.eu/mjms/
ID Design 2012/DOOEL Skopje, Republic of Macedonia
Open Access Macedonian Journal of Medical Sciences. 2017 Jul 25; 5(4):486-489.
Special Issue: Global Dermatology
https://doi.org/10.3889/oamjms.2017.138 eISSN: 1857-9655
Case Report
Trichorhinophalangeal Syndrome
Mario Vaccaro1,Georgi Tchernev2, Uwe Wollina3, Torello Lotti4, Claudio Guarneri5*
1
Universita degli Studi di Messina, Policlinico Universitario, Via Consolare Valeria, Messina, Sicilia 98125, Italy; 2Medical Institute of the Ministry of Interior, Dermatology, Venereology and Dermatologic Surgery; Onkoderma, Private Clinic for Dermatologic Surgery, Dermatology and Surgery, Sofia 1407, Bulgaria; 3Krankenhaus Dresden-Friedrichstadt, Department of Dermatology and Venereology, Dresden, Sachsen, Germany; 4Universitario di Ruolo, Dipartimento di Scienze Dermatologiche, Università degli Studi di Firenze, Facoltà di Medicina e Chirurgia, Dermatology, Via Vittoria Colonna 11, Rome 00186, Italy; 5Universita degli Studi di Messina, Clinical and Experimental Medicine, Section of Dermatology, Messina 98122, Italy
Citation: Vaccaro M, Tchernev G, Wollina U, Lotti T, Guarneri C. Trichorhinophalangeal Syndrome. Open Access Maced J Med Sci. 2017 Jul 25; 5(4):486-489. https://doi.org/10.3889/oamjms.2017.138
Keywords: Trichorhinophalangeal syndrome; congenital; skeletal abnormalities; radiological imaging; differential diagnosis.
*Correspondence: Claudio Guarneri. Universita degli Studi di Messina, Clinical and Experimental Medicine, Section of Dermatology, Messina 98122, Italy. E-mail: [email protected]
Received: 22-Apr-2017; Revised: 05-May-2017; Accepted: 08-May-2017; Online first: 22-Jul-2017 Copyright: © 2017 Mario Vaccaro, Georgi Tchernev, Uwe Wollina, Torello Lotti, Claudio Guarneri. This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (CC BY-NC 4.0).
Funding: This research did not receive any financial support.
Competing Interests: The authors have declared that no competing interests exist.
Abstract
Trichorhinophalangeal syndrome (TRPS) is the collective name of three rare congenital conditions characterised by craniofacial and skeletal abnormalities. The three known types of TRPS have different modalities of genetic transmission: namely, TRPS I and III are inherited as an autosomal dominant disease, while the cases of TRPS II are essentially sporadic.The diagnosis of the different types of TRPS is based on clinical and radiological findings, eventually integrated by genetic analysis, particularly useful in some cases with the non-classical clinical presentation. Alopecia and structural abnormalities of the nose and the hands should be considered as clinical hallmarks, whereas endocrine disorders, renal alterations, ureteral reflux, heart pathology and bone dysplasia have been documented, in the setting of a multisystem involvement.
Introduction
Trichorhinophalangeal syndrome (TRPS) is the collective name of three rare congenital conditions characterised by craniofacial and skeletal abnormalities. The three known types of TRPS have different modalities of genetic transmission: namely, TRPS I and III are inherited as an autosomal dominant disease, while the cases of TRPS II are essentially sporadic.The diagnosis of the different types of TRPS is based on clinical and radiological findings, eventually integrated by genetic analysis, particularly useful in some cases with the non-classical clinical presentation. Alopecia and structural abnormalities of the nose and the hands should be considered as clinical hallmarks, whereas endocrine disorders, renal alterations, ureteral reflux, heart pathology and bone dysplasia have been documented, in the setting of a multisystem involvement. Pigmentation disorders have never been reported in TRPS patients.
Herein is described a case of skin discoloration occurring in a boy affected by TRPS: it was only a fortuitous association?
Case report
A 16-year-old boy presented to our department with a complaint of an asymptomatic dirt-like hyperpigmentation of the upper chest and abdomen, which had been present for about six months (Fig. 1). The condition became progressively more evident after holidays at the seaside, despite the routinary hygiene and the application of various emollients and oils.
The patient was apparently health, although his hairs were referred as slowly growing and being, in fact, thin and sparse. General physical examination was notable for pectus excavatum (Fig. 1), short and
Vaccaro et al. Trichorhinophalangeal syndrome ______________________________________________________________________________________________________________________________
_______________________________________________________________________________________________________________________________ Open Access Maced J Med Sci. 2017 Jul 25; 5(4):486-489. 487 stubby hands with lateral deviations and deformations
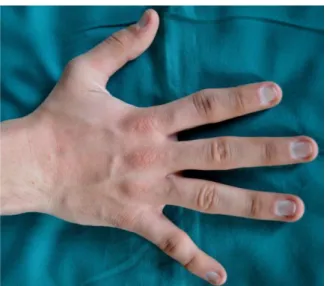
of the interphalangeal joints (Fig. 2). About these, he presented the report of a previous x-ray examination, showing cone-shaped epiphyses of the second to the fifth middle phalanges of both hands and bilateral brachymetacarpalia of the metacarpal IV. The short stature had also been strictly checked by his paediatrician, as well as the flat feet, the arched palate and the hypoplastic mandible.
Biochemical and endocrinological
investigations failed to demonstrate abnormal pituitary gland, related to the GH-IGF-1, hypothalamic-pituitary-thyroid, -adrenal and -gonadal axis, and prolactin secretion.
Figure 1: Physical examination showing skeletal abnormalities (pectus excavatum and left hand) and darkening of the skin of the lateral thorax
On mother repeated interviewing, it emerges that other two members of the family (the father and the sister of the patient) presented similar skull features, having both triangular face, bulbous pear-shaped nose, elongated philtrum, thin upper lip, horizontal groove on the chin and protruding ears. In particular, the sister, who was 20, was previous diagnosed with TRPS type I in 2008 through cytogenetic analysis. About the cutaneous concern, anamnesis was not significant for possible underlying
diseases (diabetes, obesity or
supplementations/medications assumption) and only the sister manifested similar hyperpigmentation of the umbilicus and axillae. He also denied local trauma, eczema or excessive use of cosmetics before.
Figure 2: Particular of the hand: note the lateral deviations and deformations of the interphalangeal joints
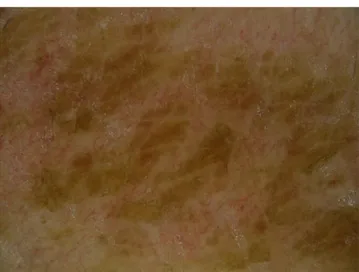
On physical examination, abnormal brown to dark discoloration of the lateral part of the thorax was apparent (Fig. 3). Dermoscopy of the lesions showed large polygonal plate-like brown scales arranged together giving an irregular mosaic pattern (Fig. 4).
Figure 3: Brown to dark discoloration of the lateral part of the thorax.
Tentative rubbing with 70% isopropyl alcohol lead to the partial removal of the hyperpigmented patch (Fig. 5), and the patient refused any further diagnostic procedure (e.g. punch biopsy).
Discussion
Trichorhinophalangeal syndrome type I is a malformation syndrome characterised by craniofacial and skeletal abnormalities [1]. Inherited in an autosomal dominant manner with high penetrance and variable expressivity, TRPS have sparse and slow-growing hair, pear-shaped nose, elongated philtrum, thin upper lip, protruding ears and bone deformities, including cone-shaped epiphyses of the phalanges, hip malformations and short stature as typical clinical features [2].
Case Report
_______________________________________________________________________________________________________________________________
_______________________________________________________________________________________________________________________________ 488 http://www.mjms.mk/ http://www.id-press.eu/mjms/
Figure 4: Dermoscopy of the affected area (original magnification x60)
Although there is a great variability in the clinical findings with considerable overlap, even among subjects of the same family, three subgroups of TRPS can be distinguished: TRPS I, associated with alterations of chromosome 8 band q24.1, TRPS II caused by a larger deletion, involving the TRPS 1 and EXT 1 (hereditary multiple exostoses) genes (8q24.11-q24.13), and TRPS III which results from missense mutations in exon 6 of TRPS1 [3].
Short stature is usual in TRPS type I [4]. Winged scapulae, multiple cartilaginous exostoses, redundant skin, and mental retardation are distinctive characteristics of TRPS type II, while severe brachydactyly, due to short metacarpals, and severe short stature are typical of TRPS type III [1, 5].
The diagnosis of the different types of TRPS is based on clinical and radiological findings, eventually integrated by genetic analysis, particularly useful in some cases with non-classical clinical presentation; other syndromes that include alopecia and structural abnormalities of the nose and the hands should be considered in differential diagnosis (Larsen’s syndrome, oral-facial-digital syndrome, Coffin-Siris syndrome, alopecia-onycho- dysplasia-hypohidrosis-deafness syndrome, trichoonychodental dysplasia, Clouston’s syndrome, Ellis-van Creveld syndrome) [1, 2, 6, 7].
TRPS may be associated with endocrine disorders, renal alterations, ureteral reflux, heart pathology and bone dysplasia: thus, a follow-up has to be set up in order to make an early diagnosis of a possible multisystemic involvement, a Perthes-like disease (dysplasia of the hip in >70% of the cases) [9], because of the frequent evolution in avascular necrosis of the femoral head (Legg-Perthes-Calvé disease) [9], or the appearance of exostoses because of a possible, although rare, sarcomatous degeneration [2]. Radvanyi et al. also showed that the TRPS1 gene is overexpressed in more than 90% of breast cancers [10].
Apart from the above mentioned cutaneous involvement, no other skin abnormalities have been reported in TRPS. In particular, no pigmentation disorders have been noted, according to with the updated literature.
With regard to our case, dermatological findings should not strictly considered in the field of pigmentation disorders, since Terra Firma Forme Dermatosis (TFFD), or Duncan ‘dirty’ disease, implies Incomplete maturation of keratin squames, combined with retention of melanin and build-up and compaction of scales, sebum and dirt through inadequate cleansing [11]. In facts, TFFD is an idiopathic benign condition that may represent an exasperating skin defect, as it involves exposed areas in psychologically more fragile patient categories (mainly children and adolescents) with the features of ‘dirt’ and giving the picture of poor body hygiene [12]. Atopy seems to be a predisposing condition [13].
Figure 5: Partial removal of the pigmentation
By excluding other dirty-appearing disorders, and using some ancillary tools (e.g. dermoscopy), the diagnosis is easy [13], thus avoiding inappropriate exams and unnecessary treatments [11, 12].
The occurrence of some cases presenting ‘topographic’ disease (Unal E et al., unpublished data) and the anecdotal association with genetic diseases (Guarneri C et al., unpublished data) may suggest the possibility of a ‘syndromic’ phenotype, as in this case, and it is worthy of attention in future cases of these conditions.
However, in clinical practice, TFFD is more frequent than the only 44 cases reported in literature lead to expect [14].
In conclusion, TRPS is a group of rare congenital conditions characterised by craniofacial and skeletal abnormalities, Type I and III being more frequent and inherited as an autosomal dominant
Vaccaro et al. Trichorhinophalangeal syndrome ______________________________________________________________________________________________________________________________
_______________________________________________________________________________________________________________________________ Open Access Maced J Med Sci. 2017 Jul 25; 5(4):486-489. 489 character. As the diagnosis is based on clinical and
radiological findings, eventually integrated with genetic analysis, the role of a dermatologist is pivotal, through recognising the typical hallmarks and managing the further in-depth analyses.
Specialist’s should also be aware of unexpected clinical signs, thus eventually expanding the phenotype of the disease and/or characterising new variants.
References
1. Vaccaro M, Guarneri C, Blandino A. Trichorhinophalangeal syndrome. J Am Acad Dermatol. 2005;53:858-60.
https://doi.org/10.1016/j.jaad.2005.06.003 PMid:16243138 2. Maas S, Shaw A, Bikker H, Hennekam RCM.
Trichorhinophalangeal Syndrome. 2017 Apr 20. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017.
3. Seitz CS, Lüdecke HJ, Wagner N, Bröcker EB, Hamm H. Trichorhinophalangeal syndrome type I: clinical and molecular characterization of 3 members of a family and 1 sporadic case. Arch Dermatol. 2001;137(11):1437-42.
https://doi.org/10.1001/archderm.137.11.1437 PMid:11708946 4. Hazan F, Korkmaz HA, Yararbaş K, Wuyts W, Tükün A. Trichorhinophalangeal syndrome type II presenting with short stature in a child. Arch Argent Pediatr. 2016;114(6):e403-e407. PMid:27869420
5. Unglaub F, Hahn P, Spies CK. Type I Trichorhinophalangeal Syndrome - Rare, but Catchy. Handchir Mikrochir Plast Chir. 2017;49(1):58-59. PMid:28423447
6. Carrington P R, Chen H, Altick J A. Trichorhinophalangeal syndrome, type I. J Am Acad Dermatol. 1994;31:331-336.
https://doi.org/10.1016/S0190-9622(94)70166-0
7. Vaccaro M, Guarneri F, Barbuzza O, Gaeta M, Guarneri C. A familial case of trichorhinophalangeal syndrome type I. Pediatr
Dermatol. 2009;26(2):171-5.
https://doi.org/10.1111/j.1525-1470.2009.00905.x PMid:19419465
8. McGuire KJ, Westacott S, MacEwen GD. Trichorhinophalangeal syndrome: evolution of Perthes-like changes in the hips.
Orthopedics. 2000;23(8):855-6. PMid:10952053
9. Minguella I, Ubierna M, Escola J, Roca A, Prats J, Pintos-Morel G. Trichorhinophalangeal syndrome, type I, with avascular necrosis of the femoral head. Acta Paediatr. 1993;82:329-30.
https://doi.org/10.1111/j.1651-2227.1993.tb12676.x PMid:8495099 10. Radvanyi L, Singh-Sandhu D, Gallichan S et al. The gene associated with trichorhinophalangeal syndrome in humans is overexpressed in breast cancer. Proc Natl Acad Sci USA.
2005;102:11005-10. https://doi.org/10.1073/pnas.0500904102
PMid:16043716 PMCid:PMC1182410
11. Duncan WC, Tschen JA, Knox JM. Terra firma-forme dermatosis. Arch Dermatol. 1987;123(5):567-9.
https://doi.org/10.1001/archderm.1987.01660290031009
PMid:3579334
12. Guarneri C, Guarneri F, Cannavò SP. Terra firma-forme dermatosis. Int J Dermatol. 2008; 47(5):482-4.
https://doi.org/10.1111/j.1365-4632.2008.03516.x PMid:18412867 13. Solak B, Erdem T. Terra firma-forme dermatosis: wipe first. Int
J Dermatol. 2015;54(12):e548-9. https://doi.org/10.1111/ijd.12938
PMid:26341247
14. Unal E, Guarneri C, Chokoeva AA, Wollina U, Tchernev G. Terra firma-forme dermatosis. Wien Med Wochenschr.
2017;167(3-4):66-69. https://doi.org/10.1007/s10354-016-0519-1