1
DIPARTIMENTO DI SCIENZE PURE E APPLICATE_____________________________
CORSO DI DOTTORATO DI RICERCA IN SCIENZE DI BASE E APPLICAZIONI
Currriculum: SCIENZE CHIMICHE E FARMACEUTICHE
CICLO XXXI
TITOLO DELLA TESI
SVILUPPO DI MICRO-TECNICHE “GREEN” PER L’ANALISI CHIMICA
Settore Scientifico Disciplinare: CHIM 01
RELATORE DOTTORANDO
Chiar.mo Prof. Achille Cappiello Dott. Maurizio Piergiovanni
3
Sommario
INTRODUZIONE ... 7
TREND NELL’ANALITICA CONTEMPORANEA ... 9
GREEN ANALYTICAL CHEMISTRY ... 12
IL PROCESSO ANALITICO ... 17
PREPARAZIONE DEL CAMPIONE ED ESTRAZIONE ... 18
TECNICHE DI ESTRAZIONE CONVENZIONALI ... 20
TECNICHE DI ESTRAZIONE GREEN IN SCALA MICRO... 24
MEMBRANE SEMIPERMEABILI E ANALISI DIRETTE ... 33
METODI DI SEPARAZIONE CROMATOGRAFICA ... 38
SISTEMI DI RIVELAZIONE: LA SPETTROMETRIA DI MASSA ... 46
INTERFACCE LC-EI-MS E STATO DELL’ARTE ... 59
PARTE SPERIMENTALE ... 71
CAPITOLO 1: L’INTERFACCIA LIQUID-EI ... 72
ABSTRACT ... 72
INVENZIONE DELL’INTERFACCIA LIQUID ELECTRON IONIZATION ... 74
PRIMO PROTOTIPO ... 79
SECONDO PROTOTIPO ... 94
TERZO PROTOTIPO ... 113
USO DI UN’INTERFACCIA LIQUID-EI IN APPLICAZIONI CON APPROCCIO “DILUTE AND SHOOT” ... 121
ABSTRACT ... 121
ESPERIMENTI QUANTITATIVI IN FLOW INJECTION ANALYSIS ... 123
DETERMINAZIONE DI FLUNITRAZEPAM IN RESIDUI DI ALCOLICI ... 125
DETERMINAZIONE DI DIETILFTALATO IN PRODOTTI PER LA CURA DELLA PERSONA ... 129
DETERMINAZIONE DI IDROSSITIRSOLO E TIROSOLO IN CAMPIONI DI OLIO EXTRAVERGINE D’OLIVA . 132 MONITORAGGIO ONLINE DI REAZIONI CHIMICHE CON LA COMBINAZIONE DI MEMBRANE ED INTERFACCIA LIQUID-EI (CP-MIMS-LEI-MS/MS) ... 137
ABSTRACT ... 137
MONITORAGGIO ONLINE DI REAZIONI DI CHIMICA ORGANICA IN SOLVENTI NON ACQUOSI ... 139
CONCLUSIONI ... 149
CAPITOLO 2: SVILUPPO DI UN PROTOCOLLO GREEN PER L’ESTRAZIONE IN SCALA MICRO DI BENZODIAZEPINE DA RESIDUI DI BEVANDE ... 151
ABSTRACT ... 151
LA TEMATICA: I DRUG FACILITATED CRIMES ... 152
BENZODIAZEPINE COME ARMA DEL CRIMINE ... 153
CONCLUSIONI ... 166
COPYRIGHTS ... 167
BIBLIOGRAFIA ... 169
7
INTRODUZIONE
Dare delle definizioni è un’operazione ardua in quanto non è sempre possibile riuscire a sintetizzare in poche parole i contenuti ed i principi identificatori di qualcosa di
estremamente vasto ed articolato. Wikipedia, non è una delle fonti più autorevoli nel mondo scientifico ma certamente una delle più consultate dal pubblico generalista, definisce la chimica analitica come “la branca della chimica che copre le attività volte all'identificazione,
alla caratterizzazione chimico-fisica e alla determinazione qualitativa e quantitativa dei componenti di un determinato campione” (1). Questa definizione, per quanto efficace e pur
essendo tra le più flessibili, non riesce a trasmettere la complessità, la diffusione e la vastità dei campi di impiego della chimica analitica nella scienza. Nel 1994 Bruce Kowalski definì la chimica analitica come “the science of chemical information” (2) riassumendo la vera essenza di questa scienza in due parole, chimica ed informazione, le quali delimitano rispettivamente la sua caratteristica di base ed il suo obbiettivo. A seguito di un’ampia riflessione, Miguel Valcárcel ne riassunse tutta l’essenza affermando:
“Analytical chemistry is a metrological science that develops, optimizes and applies material, methodological and strategic tools of widely variable nature (chemical, physical,
mathematical, biochemical, biological, etc.) which materialize in measuring processes intended to derive quality (bio)chemical information of both a partial
[presence-concentration-structure of bio(chemical) analyte-species] and global nature on materials or systems of widely variable nature (chemical, biochemical and biological) in space and time in order to solve measuring problems posed by scientific, technical and social problems” (3).
Quest’ultima definizione, pur avendo oltrepassato i 20 anni, è tuttora quella maggiormente condivisa nel mondo scientifico in quanto estende l’essenza della chimica analitica anche al di fuori del suo campo di applicazione canonico. Oggigiorno varie tecniche appartenenti a questo ambito vengono impiegate con successo anche in settori quali le scienze naturali, la biologia molecolare, la diagnostica clinica ed anche in ambito forense; il prefisso stesso “chimica”, è forse superato. L’analitica, praticata soprattutto come chimica analitica ed a lungo identificata col lavoro stesso del chimico, si sta sviluppando come una disciplina a sé stante il cui ruolo copre ormai tutti i rami della scienza e della tecnologia. Questa natura
interdisciplinare è enfatizzata dalla diversa natura dei fenomeni utilizzati durante la fase di misurazione, quali ad esempio (4):
1) Chimica 2) Fisica 3) Informatica 4) Elettronica
5) Scienze dei materiali 6) Biologia
7) Chemiometria
I dati quali-quantitativi ricavati con le varie tecniche permettono una comprensione sempre più approfondita della problematica analizzata ed un più rapido avanzamento della
conoscenza e della tecnologia. Questo sottolinea due aspetti di estrema importanza:
1) La chimica analitica è sempre più centrale e riveste ruoli di elevata importanza in tutti gli ambiti di interesse, ben al di fuori della chimica. Questo fa sì che l’interesse nei suoi confronti stia diventando sempre più elevato e con ciò gli investimenti in ricerca e sviluppo; basti pensare che durante il secolo scorso, in quegli stati dove la sanità non godeva dei progressi tecnologici, la glicemia per la diagnosi del diabete veniva stimata “assaggiando” l’urina del paziente alla ricerca del sapore dolce indice della presenza di zuccheri (5). Oggi non solo questa è un’analisi di laboratorio routinaria, ma addirittura sono presenti in commercio strumenti portatili che permettono una misurazione in situ effettuata dal diretto interessato.
2) La distanza che separa i vari campi di applicazione richiede un’elevata versatilità per poter “rispondere” ad analiti, matrici e parametri molto diversi. In tutto ciò, la ricerca in ambito strumentale ha portato ad una iper-specializzazione con tantissime tecniche e strumentazioni dedicate in grado di dare elevatissime prestazioni per ogni
applicazione. Di conseguenza anche i parametri e le molecole analizzabili sono sempre maggiori, con quantità minime rilevabili ancora inferiori. Mentre in passato tutta l’analitica passava da beuta e buretta, oggi ogni campo di applicazione ha un vasto
pattern di tecniche da cui attingere informazioni.
Sulla base di quanto descritto in precedenza risulta chiaro come l’analitica sia una disciplina trasversale la quale deve coniugare prestazioni, rapidità ed affidabilità.
9
TREND NELL’ANALITICA CONTEMPORANEA
Nel XXI secolo l’uomo contemporaneo si trova davanti una serie di sfide antitetiche tra loro. Da una parte il momento storico che sta decorrendo identifica il proprio nemico nel tempo; ogni avanzamento ottenuto dalla tecnologia in qualsiasi campo ha permesso una forte velocizzazione del processo stesso. Questi miglioramenti sono sempre abbinati anche ad un incisivo miglioramento delle prestazioni o, quanto meno, ad un mantenimento delle stesse. Dall’altra parte la tematica ambientale ha preso piede andando a rivestire ruolo di forte centralità nello sviluppo dello stile di vita e dei beni materiali. Nessuna nuova tecnologia presentata può definirsi valida se prevede un impatto ambientale maggiore rispetto a quella che va a sostituire. La chimica, essendo tra i settori a maggior impatto, ha svolto un ruolo di protagonista; già nei primi anni 30 negli Stati Uniti un primo concetto di rivisitazione dei flussi operativi nell’ottica di moderare le ricadute sull’ambiente era stato proposto. Oggi che la chimica è stata completamente ripensata in modo da limitarne gli effetti sull’ambiente, anche l’analitica è stata rivisitata in chiave green con una particolare attenzione alla
sostituzione di solventi pericolosi ed inquinanti con altri meno dannosi e, soprattutto, sulla riduzione delle quantità impiegate nei vari step di lavoro. Questo secondo aspetto, ben più incisivo del primo, passa attraverso la minimizzazione della quantità di campione trattata, fattore che determina, di conseguenza, il consumo di tutto il resto.
Se da una parte i progressi sono volti al miglioramento di strumenti e metodi, sia dal punto di vista ambientale che dal punto di vista della produttività, la maggior parte degli sforzi sono dedicati al miglioramento delle prestazioni.
Quando si parla di prestazioni analitiche non si pensa alla massima grandezza analizzabile ma piuttosto al limite di rivelabilità (Limit Of Detection, LOD). Il limite di rivelabilità è la minima quantità di analita a cui può essere associato con certezza un segnale rivelato con un determinato metodo (6). Questo significa che al di sotto di questo valore non sarà più possibile distinguere dal rumore il segnale dovuto al composto investigato; di fatto,
solitamente nella maggior parte delle strumentazioni anche il “bianco” genera un segnale. Il suo concetto è strettamente correlato al limite di quantificazione (Limit Of Quantification, LOQ), definito come la minima quantità di analita associabile ad un dato quantitativo
affidabile. Il LOD strumentale è anche fortemente influenzato dalla procedura con cui viene calcolato. Due scuole di pensiero vengono ampiamente accettate dal mondo scientifico:
1) La IUPAC definisce il LOD come:
𝐿𝑂𝐷 = 3𝛼 𝑆
⁄
(LOD è il limite di rivelabilità, 𝛼 è la deviazione standard del segnale ed S è la pendenza della curva di calibrazione.
2) Il LOD è spesso definito come la quantità corrispondente ad un rapporto segnale/rumore (signal-to-noise ratio, S/N) maggiore o uguale a 3.
Grazie ai progressi permessi dallo sviluppo strumentale, spinto principalmente dai
miglioramenti in campo elettronico ed informatico, i limiti LOD e LOQ diventano sempre più bassi con ovvi vantaggi in termini di informazioni acquisibili da ogni campione. Di
conseguenza, anche gli aggettivi associati ai valori di concentrazione vanno rivisitati. La IUPAC propone la seguente classificazione (Tabella 1).
General name of analyte Analyte concentration Examples Sub-microtrace component
< 1 ppt Dioxins in various matrices
Ultra-microtrace component
< 1 ppb Trihalomethanes in drinking water or human urine.
Microtracce component
< 1 ppm Carbon monoxide in ambient air
Trace component < 100 ppm Methane in ambient air
Secondary component < 1% Carbon dioxide in ambient air
Primary component 1-100% Oxygen in waste gases or flue gases
11
Per le molte strumentazioni odierne determinare composti alla concentrazione dell’ordine dei ppb o ppt è parte della normalità; i LOD ed i LOQ di queste macchine sono varie migliaia di volte più piccoli rispetto ai sistemi precedenti. Analisi di componenti in ultra-micro tracce come, ad esempio, i trialometani nell’acqua potabile sono divenute di routine e non
richiedono particolari accorgimenti o preparazione da parte dell’operatore; così la definizione stessa di composto in tracce presentata dalla IUPAC appare obsoleta.
Grazie ai progressi nelle prestazioni è possibile una miglior comprensione del mondo microscopico con evidenti risvolti nel progresso della ricerca a tutti i livelli ed anche in ambito di sicurezza alimentare, ambientale o forense. L’identificazione di molecole come, ad esempio, i biomarkers in particolari patologie o di alcuni composti anomali nel plagio
alimentare è stata possibile solo grazie ai progressi della tecnologia descritti in precedenza.
Lo sviluppo dell’analitica moderna quindi persegue l’obbiettivo di mettere a disposizione strumenti e metodiche affidabili, riproducibili e performanti, nel massimo rispetto dell’ambiente e che garantiscano la massima produttività. Tra le tecniche che meglio riassumono l’insieme di queste caratteristiche emergono le tecniche ifenate (hyphenated, dal termine inglese hyphen corrispondente al trattino di unione usato per congiungere due parti di una parola composta) e, in particolare, la combinazione di cromatografia e
GREEN ANALYTICAL CHEMISTRY
L’attenzione alla sostenibilità delle attività umane iniziò ad emergere già nel periodo
compreso tra i due conflitti mondiali. La cosiddetta chemurgy prevedeva l’impiego di materie prime provenienti dalla produzione agricola rinnovabile in sostituzione dei derivati del
petrolio o dei prodotti dell’industria mineraria (7). Per comprendere l’incisività del concetto basti pensare che nel 1935 venne fondato il Farm Chemurgic Council tra i cui promotori figurava la Ford Motor Company di Henry Ford. Con l’avvento del secondo conflitto mondiale e dei successivi decenni di crescita e benessere l’idea “chemurgica” venne
accantonata fino ai primi anni 90. Il primo “mattone” venne deposto nel 1999, quando Paul Anastas pubblicò una serie di linee guida raccolte sotto il nome di “green chemistry” con l’obbiettivo di ripensare la chimica del futuro (8). Questi principi, divenuti oggi pietra miliare per la storia della chimica, sono riassunti nei 12 punti presentati di seguito:
1) Prevenzione
2) Rispetto dell’economia atomica 3) Riduzione delle sintesi pericolose 4) Progettazione di composti più sicuri
5) Uso di solventi e altri componenti ausiliari più sicuri 6) Efficienza energetica
7) Uso di materie prime rinnovabili
8) Riduzione degli step che generano ulteriori scarti 9) Uso di catalizzatori
10) Biodegradabilità
11) Analisi in tempo reale per prevenire l’inquinamento 12) Impiego di composti e reazioni nelle condizioni più sicure
La messa in pratica di queste regole ha avuto e sta a avendo risvolti decisivi nel presente e nel futuro, basti pensare all’importanza assunta dalle nanotecnologie e dalla “green chemical
engeneering”. Oggi la tematica green chemistry è così centrale che proprio quest’anno il
premio Nobel per la chimica è stato attribuito parimerito a Frances H. Arnold per i suoi studi su applicazioni di enzimi nella produzione industriale green (9).
13
Anche l’analitica, nonostante il suo limitato impatto ambientale, è stata fortemente influenzata dalla green-chemistry: cromatografia e preparativa sono stati gli ambiti in cui sono stati fatti i maggiori progressi a riguardo. Vari approcci metodologici per il
miglioramento del processo analitico sono mostrati in Tabella 2.
Trend Specific tendency Description Methodological Solventless sample
preparation techniques
It is an example of application of “green chemistry” principles into analytical practice
Development of speciation analytics
Speciation analytics in the process loading and determination of the different compounds and its different physical dorms and element
Application of sum parameters
Total parameters describe the total content and gives element in all the pollutants or in a particular
subgroup of pollutants in a sample under investigation Introduction of
biomonitoring and bioanalytics
In practice the following problem are discussed: - The use of results of chemical analytics of
biota samples to evaluate pollution of the abiotic part of the environment
- Faune and flore observation - Immunoanalysis and bioassays Simultaneous determination
of many analytes using one sample in one analytical cycle
High efficiency capillary columns used in chromatography are an excellent example of this approach
Tabella 2. Descrizione delle varie tendenze nel miglioramento di vari aspetti nel processo analitico.
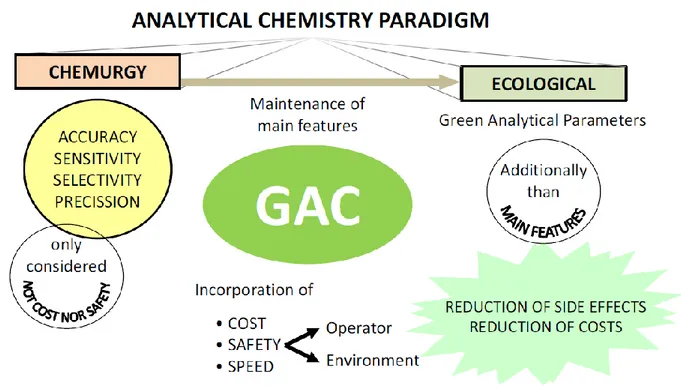
L’idea di affiancare un prototipo di green analytical chemistry (GAC) al concetto di green
chemistry risale al 2000 e agli studi di Namieśnik (10). Nonostante la sua ridotta incidenza in
termini assoluti, ci sono vari aspetti che evidenziano come l’applicazione di tecniche sicure per l’ambiente ha dato un importante contributo nel rendere green anche altri ambiti (11).
Figura 1. Evoluzione della green analytical chemistry, dal paradigma chemiurgico a quello ecologico (12).
La GAC è stata la chiave di volta nel passaggio dal paradigma chemiurgico al paradigma ecologico in analitica, creando uno strumento per la combinazione di analisi eco-friendly ed economiche (Figura 1). La sfida da affrontare era quella di raggiungere un compromesso tra il continuo miglioramento delle prestazioni e l’aumento della compatibilità ambientale. Partendo dai principi 1, 5, 6 ed 8 della green chemistry, una nuova serie di regole è stata sviluppata modellandola alle esigenze dell’analitica. Questi nuovi principi, 12 esattamente come per la green chemistry, sono i seguenti:
1) Analisi dirette devono essere impiegate per evitare pretrattamenti del campione. 2) Minimizzare dimensioni e numero di campioni deve essere un obbiettivo da perseguire. 3) Analisi in situ devono essere scelta preferenziale.
4) L’integrazione di processi analitici ed operazioni varie riduce gli sprechi di energia e l’uso di reagenti.
5) Metodi miniaturizzati ed automatizzati devono essere scelta preferenziale. 6) La derivatizzazione deve essere evitata quando possibile.
7) La produzione di grandi volumi di rifiuti deve essere evitata e la sua gestione deve essere effettuata compatibilmente con la natura stessa degli scarti.
15
8) Metodi multi-parametrici o multi-analita devono esser preferiti alle tecniche che monitorano un solo composto alla volta.
9) L’uso di energia deve essere evitato.
10) Reagenti prodotti da fonti rinnovabili devono esser preferiti a reagenti di sintesi. 11) Reagenti tossici devono essere eliminati o sostituiti.
12) La sicurezza dell’operatore deve essere continuamente incrementata.
Le analisi da remoto con misurazione diretta di campioni privi di trattamenti preliminari sono il sogno green dell’analista moderno; nonostante molto sia stato fatto in questa direzione, per molte applicazioni la fase preparativa risulta imprescindibile. Per questi motivi,
nonostante i concetti della GAC siano stati definiti da tempo, il passaggio da approccio qualitativo (definizione dei principi) a quello quantitativo (miglioramento misurabile dei vari parametri interessati) è stato ben più lento rispetto all’evoluzione della scienza in altri settori (12). La chiave di volta nell’implementazione dei principi della GAC nell’analitica
contemporanea è stata la miniaturizzazione: riduzione del volume di campione estratto o iniettato, riduzione del volume di solventi o fasi adsorbenti, riduzione del volume di scarti prodotto e del volume di fase mobile (come funzione del tempo necessario per la
separazione cromatografica) impiegato (13).
Questo processo, abbinato alla sostituzione di solventi pericolosi con altri sicuri, la riduzione di step energivori con la massima preferenza a trasformazioni a temperatura ambiente e tanti altri miglioramenti marginali, ha determinato le caratteristiche dell’analitica moderna che conosciamo (Figura 2).
17
IL PROCESSO ANALITICO
Come già ampiamente discusso nella parte precedente, l’analitica è una disciplina
ampiamente trasversale che copre applicazioni e condizioni di lavoro molto diverse tra loro. Ciononostante, come emerge andando a comparare metodiche analitiche di settori diversi reperibili in letteratura, l’iter seguito dalle procedure è praticamente il medesimo. La qualità del risultato di un’analisi dipende dalla accuratezza di tutte le procedure sperimentali che, a partire dalla matrice del campionare, portano al risultato finale. Gli errori compiuti nei vari step concorrono a determinare l’errore complessivo del risultato; risulta quindi necessario dedicare la massima attenzione a tutti gli aspetti e non solo all’analisi in quanto tale.
In maniera molto generale i principali passaggi in cui si articola il processo analitico sono i seguenti:
1. Campionamento
2. Preparazione del campione
3. Analisi
Il campionamento è il primo step di ogni procedura analitica e costituisce di fatto il prelievo dell’aliquota su cui verrà applicato il metodo. Ogni matrice presenta diverse problematiche ed accortezze da adottare ma, in generale, si possono ricordare alcuni principi generali. Il campione prelevato deve essere quanto più “fedele” possibile (significativo) della presenza e concentrazione degli analiti di interesse nella matrice esaminata e, per garantire questo requisito, va stabilito un punto di campionamento rappresentativo, vanno ridotte al minimo le disomogeneità al momento del prelievo e minimizzate le contaminazioni successive o i fenomeni di degradazione chimico-biologiche. Diverse strategie sono state studiate per ognuno dei punti elencati sopra ma la loro disamina esula dagli obbiettivi di questa tesi di dottorato e la si rimanda ad altre sedi.
PREPARAZIONE DEL CAMPIONE ED ESTRAZIONE
I campioni da analizzare, salvo casi estremamente particolari, sono composti da atomi di diversi elementi, presenti con diversi stati di ossidazione, e legati ad altri atomi a loro volta diversi e presenti in molecole diverse; l’insieme di tutte le molecole del campione a parte l’analita è definito matrice. Anche il più semplice dei campioni è composto da migliaia di molecole, tra le quali però, solo una o poche sono di interesse per l’analista. Queste molecole possono a loro volta contribuire a sopprimere o promuovere determinate
caratteristiche del composto di interesse che vengono impiegate nel processo di rivelazione. Questo fenomeno, largamente conosciuto come effetto matrice (matrix effect, ME) può essere così incisivo da emulare il segnale di un composto non presente (falso positivo) o sopprimere completamente la presenza di un composto presente (falso negativo). Contestualizzando questo fenomeno nella delicatezza dell’analitica nei suoi vari ambiti di applicazione, il risultato di questi errori può essere estremamente dannoso (14). L’effetto matrice è stato largamente studiato nel mondo scientifico e sono state messe a punto ed accettate diverse strategie per mitigarne le conseguenze, tra cui il metodo delle aggiunte standard e la calibrazione a standard interno. Matuzevsky (15) divulgò una semplice strategia per la quantificazione dell’effetto matrice che viene largamente accettata ed impiegata dalla comunità scientifica. Questa procedura prevede la quantificazione di due campioni a concentrazione nota recanti la stessa quantità di analita ma preparati, uno in matrice “post-extraction” e l’altro in solvente (il quale ci si aspetta che sia equiparabile a un bianco). Il rapporto tra in segnale registrato in presenza della matrice (S sample) e quello con il
solo solvente (S spiked blank) potrà differire solo per la compresenza di altre molecole. La
formula impiegata è la seguente:
𝑀𝑎𝑡𝑟𝑖𝑥 𝑒𝑓𝑓𝑒𝑐𝑡 (%) =
𝑆
sample
𝑆
spiked blank
∗ 100
L’effetto matrice percentuale potrà quindi essere sia maggiore che minore di 100 a seconda che ci si trovi, rispettivamente, in condizioni di signal enhancement o signal suppression (16).
Fatta questa doverosa premessa, utile per sottolineare la potenziale interferenza della matrice nel risultato analitico, appare evidente che la fase di preparazione del campione sarà volta a ridurne le conseguenze. Nella preparativa, spesso identificabile con l’estrazione, il
19
campione viene processato in modo da isolare il più possibile composto di interesse
all’interno del più piccolo volume possibile: questo sia per rimuovere eventuali interferenti che per aumentare la concentrazione analizzata. Ulteriori passaggi chimici o biochimici possono essere coinvolti ma si tratta di approfondimenti di minor interesse in questa panoramica. Tra le operazioni preliminari all’analisi vanno citate la centrifugazione, la filtrazione, la sedimentazione, la digestione, la distillazione, l’idrolisi, la diluizione e tante altre ancora. Ogni tecnica strumentale prevede delle procedure preparative dedicate in modo da rendere l’analisi efficace, prestazionale e riproducibile. Nella chimica delle piccole molecole organiche oggetto di questo lavoro di tesi, la preparativa più comune è
l’estrazione.
Estrarre un composto significa fondamentalmente rimuovere la matrice (17); questa definizione data da Janusz Pawliszyn appare particolarmente adeguata in quanto sposta il focus dall’analita a tutte le molecole che lo circondano.
Nell’analitica moderna la parte di preparazione del campione è stata oggetto di pesanti rivisitazioni; sempre più metodiche sono state sviluppate con approcci diretti o semi-diretti come il “dilute-and-shoot” o l’uso di tecniche cosiddette “ambient” dove nessuna
preparazione o solo qualche semplice passaggio come la diluizione sono previsti. Il trend generale prevede che ogni passaggio non strettamente fondamentale per il risultato finale venga rimosso o quantomeno semplificato. L’estrazione è infatti uno step nel quale non viene ottenuto nessun risultato analitico e che risulta spesso poco riproducibile essendo generalmente il primo imputato in caso di risultati insoddisfacenti.
TECNICHE DI ESTRAZIONE CONVENZIONALI
Ricollegandosi ai paragrafi precedenti, emerge come la fase estrattiva sia particolarmente limitante in termini di produttività in quanto esosa a livello di tempo, sia di lavoro che di tempi morti (come ad esempio digestioni o idrolisi enzimatiche). Inoltre, l’uso di fasi assorbenti di difficile smaltimento o di solventi inquinanti per le estrazioni va in forte contrapposizione con i principi della green analytical chemistry; anche eventuali processi termici o l’uso di microonde sarebbero da evitare o da sostituire con processi non energivori (18).
Attualmente, per la maggior parte delle metodiche analitiche, l’estrazione rimane una parte imprescindibile. Le principali tecniche per campioni liquidi sono suddivisibili, in due grandi famiglie:
1) Liquido – liquido 2) Solido – liquido
L’estrazione liquido – liquido (Liquid – Liquid Extraction, LLE) è una delle tecniche più antiche e conosciute; prevede l’impiego di un solvente da aggiungere al campione che quindi andrà ad arricchirsi delle molecole a lui affini. Il solvente estraente deve essere immiscibile con il solvente della matrice e soprattutto deve presentare una forte affinità per il composto di interesse. Il trasferimento dei soluti tra le due fasi è regolato dalle leggi di trasferimento di materia, dove uno dei parametri chiave è la superficie esposta; il sistema bifasico va infatti agitato per un tempo prestabilito in modo da formare una fine dispersione di solvente estraente e campione che permette di massimizzare il rapporto superficie/volume. Terminato questo step meccanico, le due fasi si separano in quanto immiscibili andando a posizionarsi l’una sopra all’altra sulla base del valore di densità. La fase estraente viene quindi ulteriormente trattata o anche iniettata direttamente se compatibile col sistema analitico utilizzato.
L’estrazione liquido-liquido è una tecnica veloce e che non richiede l’acquisto di apparecchiature dedicate (salvo l’imbuto separatore). I composti si trasferiscono dal campione alla fase estraente in base alla diversa affinità per i due solventi. Questa caratteristica è rappresentata dalla costante Pow ovvero coefficiente ottanolo – acqua o coefficiente di partizione; si tratta di un valore numerico calcolato per ogni composto a
21
condizioni di pressione e temperatura normali (1 atm e 25°C) che indica come questo si ripartisce in condizioni di equilibrio tra l’ottanolo (fase idrofoba) e l’acqua (fase idrofila). Per comodità la Pow è spesso utilizzata in forma logaritmica come segue:
log Pow = log
[𝑠𝑜𝑙𝑢𝑡𝑒]
octanol[𝑠𝑜𝑙𝑢𝑡𝑒]
waterIndicando [solute]octanol e [solute]water come le concentrazioni del composto rispettivamente
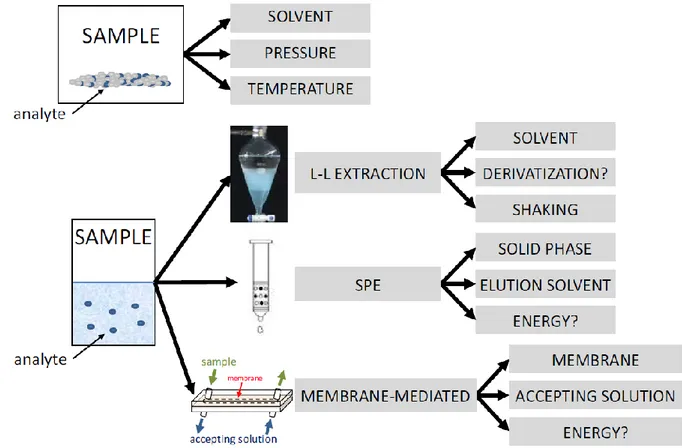
in ottanolo ed acqua, si ottiene un valore che indica idrofilicità o idrofobicità rispettivamente in base a quanto è minore o maggiore di zero. L’estrazione liquido – liquido è una tecnica ancora radicata nei laboratori di analisi in quanto economica e relativamente veloce nonostante siano stati sviluppate valide alternative in grado di ovviare ai suoi punti deboli. Estraendo con solvente matrici complesse come ad esempio molti campioni ambientali o biologici molte altre molecole verranno co-estratte assieme all’analita di interesse andando di fatto a limitare l’utilità di questo passaggio. I solventi impiegati per estrarre soluzioni acquose sono spesso nocivi e tossici, quindi in forte contrasto coi principi della GAC; inoltre, considerato che la quantità di estraente usata può essere anche varie volte il volume di campione, la problematica assume maggior rilevanza. Per i suddetti motivi la maggior parte dei migliori solventi sono stati infatti sostituiti con alternative più sicure ma anche meno efficaci. La LLE è dunque una tecnica estrattiva efficiente ed efficace solo per analiti molto affini al solvente estraente e che presentano polarità fortemente diversa dal resto dei componenti della matrice. In accordo coi principi della GAC varie tecniche alternative prive di solventi sono state studiate e messe a punto (Figura 3).
Tra le alternative presentate è presente anche l’estrazione solido – liquido (Solid – Phase
Extraction, SPE) citata in precedenza. I suoi principi di funzionamento sono analoghi alla
liquido – liquido ma con alcune differenze; in questo caso il composto estratto deve instaurare interazioni con una fase estraente solida che saranno di tipologia ed intensità diverse rispetto ad un solvente liquido (19). La procedura prevede che il campione,
preventivamente omogeneizzato e separato da residui solidi, venga fatto passare attraverso una fase adsorbente solida dedicata. Questo materiale verrà scelto in base alle interazioni instaurabili con gli analiti in modo da riuscire a trattenerli selettivamente a discapito della matrice. Di fatto questo passaggio appare come una via di mezzo tra una separazione
cromatografica (ma gli analiti non vengono eluiti in questa fase) ed una filtrazione dove la separazione è sulla base delle caratteristiche chimiche e non in base alle dimensioni.
Figura 3. Schema di tecniche alternative alla LLE per varie applicazioni che non necessitano di solventi (2).
Un primo condizionamento con lo stesso solvente di cui è composto il campione è
fondamentale come preliminare all’estrazione per pulire ed andar a ripristinare le condizioni nelle quali la fase adsorbente dovrà interagire con gli analiti. Una volta caricato il campione e fatto passare attraverso la fase solida, questa si sarà arricchita dei composti in grado di instaurare interazioni più forti con esso di quelle che ha con la matrice. La cartuccia viene quindi lavata con lo stesso solvente usato nel condizionamento (o una sua miscela piuttosto simile), asciugata con un flusso di gas ultrapuro ed inerte ed infine eluita. In quest’ultimo step, il solvente (o la miscela di solventi) usato per rimuovere i composti dalla fase solida deve instaurare con essi interazioni di maggior intensità di quelle presenti con la fase solida adsorbente, in modo da trasportarli col flusso di liquido nel volume di estratto. Quest’ultimo passaggio può esser ripetuto più volte per aver la certezza di pulire al meglio la cartuccia e recuperare tutto l’analita estratto. Tutta la suddetta procedura è schematizzata in Figura 4.
23
Figura 4. Schematizzazione delle operazioni fondamentali in un’estrazione SPE (20).
Comparata all’estrazione liquido – liquido, la solido – liquido riesce a rispettare meglio i principi della GAC: solo piccole quantità di solvente vengono utilizzate durante lavaggi ed eluzione e la fase adsorbente è riutilizzabile per varie decine di volte (21). La SPE risulta
particolarmente vantaggiosa per analiti in tracce in quanto, andando ad estrarre grandi volumi di campione, permette un’elevatissima pre-concentrazione tale da rendere la
concentrazione finale a valori rilevabili anche per tecniche strumentali a basse prestazioni; in questo caso, a differenza della LLE, non è necessaria l’evaporazione al rotavapor di grandi quantità di solvente dall’estratto, con ovvi vantaggi in termini di efficienza, costi e tempo. La SPE attualmente è ampiamente automatizzabile nella versione on – line: la cartuccia
estraente viene installata in serie in un sistema HPLC posizionata precedentemente alla colonna cromatografica. Con le opportune accortezze è possibile far fare tutta l’estrazione e la rivelazione comprensiva di separazione cromatografica direttamente nel sistema stesso senza tempi morti e senza rischio di perdere materiale durante le varie manipolazioni.
TECNICHE DI ESTRAZIONE GREEN IN SCALA MICRO
Lo sviluppo delle tecniche estrattive è stato portato avanti seguendo principalmente due delle dodici linee guida della “green analytical chemistry”: la sostituzione di solventi nocivi o comunque dannosi per l’ambiente con altri più sicuri e la riduzione delle quantità impiegate in termini di solventi, campione, scarti ed energia. Le variabili da tenere in considerazione per rendere un protocollo estrattivo green sono riassunte in Figura 5.
Figura 5. Aspetti da considerare nell’evoluzione di tecniche estrattive in ottica GAC (12).
Nell’ottica della GAC entrambe queste strategie sono ampiamente migliorabili, soprattutto in termini di prodotti di scarto (solventi estraenti e fasi esauste adsorbenti) e di quantità di campione impiegata. Proprio per rispondere ai suddetti principi, le tecniche liquido – liquido e solido – liquido sono state oggetto di un intenso processo di miniaturizzazione dal quale sono nate nuove varianti in grado di garantire prestazioni equivalenti, se non migliori, pur con quantità di campione ridotte di qualche ordine di grandezza.
La microestrazione in fase solida (Solid-Phase MicroExtraction SPME) è una tecnica basata sull’utilizzo di una fibra di silice fusa, rivestita esternamente da una piccola quantità di fase
25
stazionaria adsorbente. Questa tecnica è stata sviluppata da Pawliszyn e collaboratori nel 1989 (Arthur, Pawliszyn, 1990) e migliorata negli anni a seguire fino a diventare una delle strategie estrattive migliori sotto tutti i punti di vista (22). Il protocollo prevede che la fibra venga esposta al campione in moda da arricchirsi degli analiti a lei affini. Una volta
completata l’estrazione, gli analiti vengono desorbiti direttamente nello strumento analitico senza ulteriori passaggi. La SPME è stata sviluppata per rispondere alla crescente esigenza di metodi rapidi e affidabili, in grado di rispondere ai principi della GAC e di riassumere in un unico step l’estrazione e la purificazione. La SPME trova una perfetta combinazione nelle tecniche gascromatografiche in quanto, gli analiti vengono termo-desorbiti direttamente nell’iniettore e di fatto, si può considerare incorporata anche la determinazione analitica finale. Nel caso di accoppiamento con la cromatografia liquida (LC) deve essere
implementata una eluizione con solvente in modo da recuperare gli analiti che saranno poi iniettati; questo passaggio può essere automatizzato per aumentarne l’efficienza. I principali vantaggi della SPME possono essere riassunti come segue:
1. il consumo di solvente è ridotto al minimo se non azzerato;
2. per l’esecuzione dell’analisi è sufficiente una piccola quantità di campione;
3. gli step preliminari sono semplici e veloci;
4. il processo di preparazione del campione può essere automatizzato;
5. gli analiti vengono sia estratti che pre-concentrati da qualsiasi tipo di matrice;
6. sono possibili anche campionamenti on-site e in vivo.
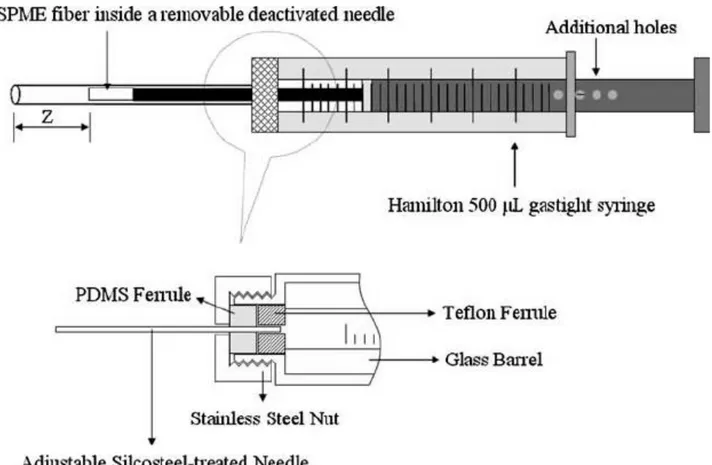
La fibra estraente è realizzata in silice fusa (lunga circa 1 cm e con diametro di 0,110 mm) ricoperta da una piccola quantità di fase estraente (nell’ordine di 1 µL con spessore
compreso tra 7 e 100 µm); questo è solitamente una fase polimerica ad alto peso molecolare (PM), simile a quelle impiegate per le fasi stazionarie nelle colonne GC, o un adsorbente solido altamente poroso, per aumentare l’area accessibile per l’adsorbimento. La fibra così rivestita è connessa ad un’astina che funge da pistone e viene fatta scorrere all’interno di un ago. Il sistema che ne risulta è commercializzato col nome di holder (Figura 6).
La procedura SPME si compone di una prima fase di estrazione in cui la fibra,
immersa nel campione liquido) da analizzare; in questo step, gli analiti vanno ad adsorbirsi sulla fibra fino al raggiungimento di un equilibrio con la concentrazione residua nel
campione. Il tempo di estrazione è un parametro fondamentale che deve essere
opportunamente ottimizzato. Terminato questo step, la fibra viene ritratta nell’ago e segue la fase di desorbimento, direttamente nell’iniettore per gascromatografia o in una camera di desorbimento se segue un’analisi con cromatografia liquida. Tutta la procedura estrattiva può essere automatizzata, ottenendo un miglior rispetto di tempi e volumi per una maggiore ripetibilità dell’analisi.
Figura 6. Sezione dell’holder SPME con dettaglio della fibra estraente e del suo posizionamento nell’ago (23).
Successivamente alla SPME un’altra tecnica microestrattiva solido – liquido è stata sviluppata, messa a punto e commercializzata. La Micro Extraction by Packed Sorbents (MEPS) è un’alternativa alla SPME basata su un dispositivo dotato di una cartuccia interposta tra l’ago ed il pistoncino di una siringa (manuale o meccanizzata); in questa maniera, durante il passaggio del campione aspirato si vengono a instaurare delle interazioni tra gli analiti e la fase adsorbente contenuta all’interno della cartuccia che ne permettono la separazione dal resto della matrice (Figura 7). Diversamente dalla SPME, nella MEPS la fase adsorbente è
27
posizionata dopo l’ago e richiede che il campione sia aspirato all’interno della siringa; nonostante la tipologia di interazioni col campione sia la stessa (di fatto le fasi adsorbenti sono le medesime), questo fattore determina un’efficienza e ripetibilità ben maggiore.
Figura 7. Cartucce MEPS e varie tipologie di siringhe, manuali, automatiche e semiautomatiche.
Infatti, nella MEPS il volume di campione trattato è dettato dal volume della siringa impiegata e l’interazione con gli analiti non è conseguenza dalla loro migrazione per
differenza di concentrazione dal bulk del liquido alla fase stazionaria, ma bensì è forzata dal suo passaggio attraverso la cartuccia (Figura 8). Sul campione può esser ripetuta l’estrazione più volte in modo da massimizzare l’efficienza estrattiva senza dover aspettare il
raggiungimento dell’equilibrio termodinamico come nelle tecniche solido – liquido.
Successivamente alla fase di adsorbimento, la cartuccia caricata di analiti viene prima lavata (come nella SPE/SPME) ed infine eluita con un’apposita miscela. Il lavaggio è importante per rimuovere quelle molecole della matrice che sono rimaste sulla superficie della fase
adsorbente e che potrebbero dare effetto matrice. L’eluizione può essere effettuata
direttamente all’interno dell’iniettore del GC/HPLC con ovvi vantaggi in termini di efficienza e produttività; ovviamente sia l’ago che il solvente utilizzato dovranno essere compatibili col sistema cromatografico impiegato.
Figura 8. Procedura generale di estrazione MEPS con siringa manuale (24).
Quando l’eluizione non coincide con l’iniezione lo stesso processo può esser ripetuto più volte in modo da aumentare l’efficienza ed evitare fenomeni di carry over (ossia presenza di molecole di analita non estratte dalla cartuccia che potrebbero essere eluite in analisi successive andando ad influenzarne i risultati).
La MEPS è una tecnica collaudata, automatizzabile e certamente selettiva vista la possibilità di scegliere tra più fasi adsorbenti; tra i suoi principali vantaggi vi è certamente la capacità di estrarre volumi estremamente ridotti di campione (attualmente 5 µL). A differenza della SPME però, il suo impiego è limitato ai soli campioni in fase liquida. Il fatto di dover far passare il campione attraverso la cartuccia impone però la necessità di doverlo preparare
29
con filtrazioni o centrifugazioni in modo da rimuovere il particolato solido che potrebbe ostruire la fase solida. Inoltre, campioni ad elevata viscosità sono inadatti a questa tecnica per l’impossibilità di far fluire il liquido a causa dell’eccessiva contropressione; questo limite può essere ovviato con un’opportuna diluizione ma questa scelta non sempre è compatibile con le prestazioni della strumentazione e la concentrazione del campione. Infine, la pre-concentrazione massima ottenibile utilizzando l’estrazione MEPS, estraendo con la siringa di massimo volume (1 mL) ed eluendo con la più piccola (5 µL), è di 200 volte, sensibilmente inferiore rispetto a quanto possibile con SPME e SPE (25).
Apparentemente in contrapposizione ai requisiti della GAC, che fortemente si oppongono all’uso di solventi e che quindi prediligono le tecniche di estrazione solido – liquido, esiste una variante green anche di tecniche liquido – liquido. La Dispersive Liquid – Liquid Micro
Extraction (DLLME) è una tecnica estrattiva liquido – liquido su scala micro presentata da
Assadi nel 2006 per la determinazione di idrocarburi (26) e pesticidi (27) nell’acqua. Oggi la DLLME è stata adattata ad applicazioni molto diverse tra loro andando ad adattarne il protocollo per massimizzarne l’efficienza ed enfatizzarne il caratterere green. Nonostante si tratti di una tecnica molto “giovane” è riuscita subito a catturare l’attenzione del mondo della ricerca grazie ai suoi indiscutibili vantaggi ed alla sua semplicità. Dal 2006 ad oggi la DLLME è stata oggetto di oltre 1300 pubblicazioni scientifiche e l’interesse nei suoi confronti risulta ancora in crescendo (28). Nel mondo scientifico viene ritenuta una tecnica green in quanto, diversamente dalla LLE convenzionale, nella DLLME le quantità in uso di solvente, campione ed altro vengono minimizzate a valori inferiori al mL. Ciò permette di ridurre ovviamente l’eventuale uso di energia e la produzione di rifiuti. La DLLME inoltre permette l’estrazione di più analiti con caratteristiche di polarità simili, unificando il recupero in un unico step. Il principio di estrazione DLLME, abbastanza simile a quello LLE, si basa sulla formazione di un sistema ternario composto dal campione acquoso in cui viene disciolto un sale ed a cui viene aggiunta una miscela di solventi organici definita fase estraente.
Figura 9. Punti chiave dell’evoluzione della DLLME a 10 anni dalla sua presentazione (28).
Quest’ultima è composta principalmente da (almeno) due componenti:
1) Extractor solvent: è il componente minoritario della miscela estraente, nonché quello che a fine processo verrà recuperato ed analizzato. Deve essere quanto più affine possibile agli analiti (idoneo indice Pow), non miscibile con l’acqua e possibilmente più denso della
matrice. I migliori solventi sotto il punto di vista delle prestazioni sono gli organoclorurati anche se la tendenza è l’impiego di liquidi provenienti da produzioni rinnovabili
compatibili con i principi della GAC. Visto comunque il loro impiego in quantità esigue, anche protocolli con solventi leggermente tossici sono considerabili come green (29). 2) Dispersive solvent: è il componente maggioritario della miscela estraente nonché quello
che serve a facilitare il trasferimento di materia tra solvente estraente e matrice. Il suo requisito principale è quello di essere miscibile sia con l’acqua che con l’extractor solvent. Vengono impiegati in questo ruolo solventi a media polarità, solitamente in grado di accettare un legame a idrogeno come CH3CN, acetone ed anche qualche alcol a media
catena. Il dispersive solvent viene impiegato in quantità maggiori e quindi la sua eco-compatibilità è requisito fondamentale.
31
Componente fondamentale è il sale che serve per garantire l’effetto di salting out effect necessario per aver maggior efficienza e migliori recuperi. Il salting out effect è un fenomeno di interazione elettrolita – non elettrolita in fase acquosa nel quale il non elettrolita diventa meno solubile ad elevate concentrazione di sale (30) (31). Va comunque sottolineato che esiste una condizione ottimale per favorire i recuperi e che soluzioni sature di sale non necessariamente danno risultati migliori. Stessa scelta di compromesso va attuata per la temperatura come dimostrato da Assadi (26) (32).
Figura 10. Esempio di procedura di estrazione DLLME generica (33).
Una volta sciolto il sale ed unita la miscela estraente al campione, il sistema viene agitato in modo tale da formare una fine dispersione di gocce di liquido organico all’interno della matrice acquosa così da massimizzare il rapporto area/volume e favorire il trasferimento di materia. Il tempo di agitazione viene definito in modo da permettere il raggiungimento dell’equilibrio termodinamico tra le due fasi; in questo senso la scelta e la quantità di dispersive solvent sono cruciali. Per ottimizzare proprio questa fase, che risulta essere il tallone d’Achille delle tecniche liquido – liquido, sono state impiegate con successo altre tecniche di agitazione tra cui l’utilizzo di ultrasuoni (Ultrasound Assisted Dispersive Liquid
Liquid Micro Extraction, UA-DLLME) (34).Gli ultrasuoni risultano particolarmente vantaggiosi
passaggio in fase organica (35). Segue una centrifugazione per ri-separare le due fasi e poter recuperare quella organica, ora arricchita degli analiti, nel fondo della provetta. L’estratto può essere quindi iniettato o sottoposto a derivatizzazione o tirato a secco per poter solubilizzare i composti in solvente idoneo.
I vantaggi della DLLME sono la sua versatilità, la sua semplicità (pochi passaggi, tutti a temperatura ambiente o simile), la sua economicità (non sono richieste attrezzature
dedicate) e la non-necessità di trattamenti preliminari particolari. Inoltre, il fatto che i volumi di sovente estrattore siano inferiori al volume di campione garantisce un fattore di
arricchimento fondamentale per il miglioramento dei limiti di rivelabilità (LOD) e di quantificazione (LOQ) (36). D’altro canto il vincolo di non miscibilità in acqua del solvente estrattore ne limita la gamma di analiti estraibili e questo, unito alla difficile
automatizzabilità del metodo ne ostacola la diffusione nella pratica routinaria.
Tra le tecniche microestrattive, oltre alle tre citate sopra, molte altre sono state sviluppate e messe a punto anche se il loro campo di applicazione è storicamente rimasto limitato a pochi ambiti di nicchia. In questo lavoro di dottorato l’attenzione è stata focalizzata in particolare su MEPS e DLLME in quanto, nonostante siano tecniche legate a principi chimico – fisici diversi, sono accumunate dalla potenzialità di estrarre efficientemente una quantità ridotta di campione coerentemente con i principi della green chemistry. Grazie a queste
caratteristiche sono tecniche di estremo interesse per quelle applicazioni, solitamente forensi, in cui il campione è in quantità particolarmente limitata.
33
MEMBRANE SEMIPERMEABILI E ANALISI DIRETTE
Esistono tecniche strumentali in grado di acquisire il dato analitico in maniera diretta, spesso senza ricorrere alle estrazioni: la più diffusa di esse è il Purge-and-Trap. Questo sistema è particolarmente utile per composti volatili solubilizzati in matrici liquide come ad esempio i trialometani nelle acque o gli aromi negli alimenti; per poter trattare sia i composti volatili che i non voltatili, invece, si stanno recentemente affermando le membrane semipermeabili. Le membrane in realtà sono ampiamente studiate da vari decenni e da oltre 35 anni
vengono usate per il monitoraggio in tempo reale di composti volatili.
Il punto di forza delle membrane è la permeoselettività ossia la loro capacità di permettere il passaggio di determinati analiti e, selettivamente, di impedirlo ad altri (37). Posizionando un materiale con queste caratteristiche come parte di un capillare esposta al campione da un lato, e ad un flusso di fluido affine agli analiti, chiamato fase accettrice (acceptor phase, AP), dall’altro, è possibile estrarre solo le molecole con le determinate caratteristiche con anche un’elevata pre-concentrazione. Le membrane cave di questo tipo sono definite come Hollow
Fiber Membrane (HFM) (38). La fase accettrice può essere sia un solvente liquido che un gas,
in base alle caratteristiche dei composti investigati e del detector utilizzato. Il flusso
arricchito degli analiti viene quindi direzionato al detector per una misurazione del segnale in tempo reale. Ottimi detector da accoppiare a strumenti basati sulle membrane
semipermeabili sono quelli basati sulla spettrometria di massa; la configurazione che ne consegue, conosciuta col nome di MIMS (Membrane Introduction Mass Spectrometry), riesce a dare la necessaria selettività analitica ad analisi in continuo dove gli analiti non sono
separati tramite cromatografia e quindi privi di un’informazione caratterizzante come il tempo di ritenzione. Alcuni dei possibili accoppiamenti tra membrane e spettrometri di massa sono riportate in Figura 11. Le membrane attuali possono esser scelte tra diverse varianti di materiali e selettività sterica e permettono l’analisi su matrici gassose, liquide e solide (39).
Figura 11. Alcune configurazioni nell’interfacciamento MIMS per campionamenti su gas e su liquidi (40).
Le membrane maggiormente utilizzate in configurazione MIMS sono realizzate in materiali polimerici di sintesi come il polidimetilsilossano (PDMS). Questo materiale è stato
ampiamente utilizzato sin dai primi prototipi di membrane, in quanto già ampiamente studiato e conosciuto per la sua stabilità, robustezza e capacità di trasferire selettivamente solo composti idrofobici (41). Il PDMS da i migliori risultati con composti volatili
(storicamente i primi studiati in configurazione MIMS) mentre spesso necessita di
modificazioni con i meno volatili. Considerando che uno dei punti di forza delle membrane è la misurazione in tempo reale, risulta evidente che il trasferimento di materia deve avvenire fino al raggiungimento dell’equilibrio termodinamico durante il tempo di contatto; per questo motivo i composti altobollenti, ad elevato peso molecolare nonché
abbondantemente funzionalizzati da gruppi polari, sono più ostili ad allontanarsi dalla matrice acquosa e richiedono accorgimenti particolari (42). In questi casi una strategia utilizzata con composti volatili e AP gassosa è l’incremento della temperatura; con composti altobollenti e AP temperature troppo elevate possono dare problemi quali la formazione di bolle, il trasferimento di vapore acqueo e la degradazione del materiale. Altri materiali usati per la realizzazione delle membrane sono il Naflon® (38), il Polivinilidenfloruro (44) e tutte le
35
varianti ottenute andando a modificare il rapporto tra i monomeri costituenti il polimero ed altri parametri strutturali. Un’altra strategia per la misurazione di composti di natura
biologica è la possibilità di immobilizzare degli enzimi sulle superficie esterna della membrana in modo da trasformare analiti non permeabili in prodotti permeabili e quindi rilevabili (45).
Figura 12. Comparazione tra una tecnica di campionamento/preparativa discontinua come il Purge and Trap ed una in tempo reale come l’accoppiata membrane-spettrometria di massa nel monitoraggio di toluene in tracce nell’atmosfera. Emerge chiaramente come con le membrane sia possibile registrare tutte le informazioni e dare
una caratterizzazione molto più accurata (40).
Oggi le membrane sono una delle migliori tecniche per l’analisi di composti target in grado di soddisfare i requisiti della GAC in quanto permettono di evitare i due step tipici di molti protocolli a più elevato impatto ambientale, preparativa e cromatografia (40). La capacità di misurare il segnale analitico in maniera diretta e continua garantisce un’incrementata risoluzione temporale rispetto alle tecniche a monitoraggio discreto e quindi riesce a dare maggiori informazioni (Figura 12). Strumenti basati sulle MIMS sono ottimi per monitoraggi
ambientali in situ, riducendo drasticamente il tempo di maneggiamento del campione e la possibilità di errori; la loro diffusione in questo senso è stata ostacolata dalle dimensioni (e costo) dell’apparecchiatura completa. Oggi, la riduzione delle quantità impiegate nel processo analitico richieste dalla GAC ha determinato una miniaturizzazione anche delle strumentazioni, le quali hanno dimensioni compatibili a quelle di un device portatile.
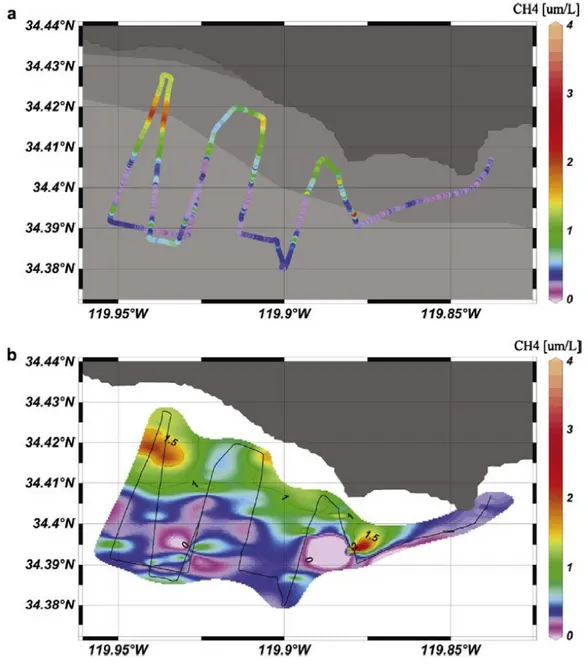
Figura 13. Caratterizzazione della quantità di metano superficiale in un fiume sotterraneo nel canale di Santa Barbara (California, USA) misurata con un sistema MIMS-EI (40).
Utilizzando sistemi MIMS è possibile caratterizzare nel dettaglio varie matrici ambientali di qualsiasi tipo come aria, mare, corsi d’acqua e falde sotterranee (Figura 13). Le loro
37
in itinere delle condizioni operative. Vari tentativi sono stati fatti su sistemi biologici e su reazioni organiche ma i risultati ottenuti sono limitati a composti volatili (per accoppiamenti con AP gassose) o prodotti di idrolisi enzimatica. Per il monitoraggio on-line di reazioni chimiche di composti non volatili sono necessarie tecniche di ionizzazione in fase liquida efficienti e prive di effetto matrice come la spettrometria di massa a ionizzazione elettronica (EI-MS). Questa sorgente, per quanto altamente prestazionale per un’applicazione di questo tipo, non viene impiegata in quanto operante in condizioni di alto vuoto e con molecole gassose. Per poterla impiegare con AP liquide è necessario l’impiego di apposite interfacce EI-MS, tematica di ricerca che verrà approfondita in seguito.
METODI DI SEPARAZIONE CROMATOGRAFICA
La cromatografia è una tecnica di separazione degli analiti presenti in una miscela omogenea fatti passare attraverso un supporto con il quale instaurano interazioni di differente entità. La diversa affinità dei vari composti per le due fasi, una fissa (stazionaria) ed una corrente (mobile) fa sì che questi oppongano una diversa “resistenza” all’avanzamento (46). Ne consegue che i vari composti usciranno dalla fase stazionaria con un ritardo direttamente proporzionale alle interazioni instaurate; questa differenza è il tempo di ritenzione.
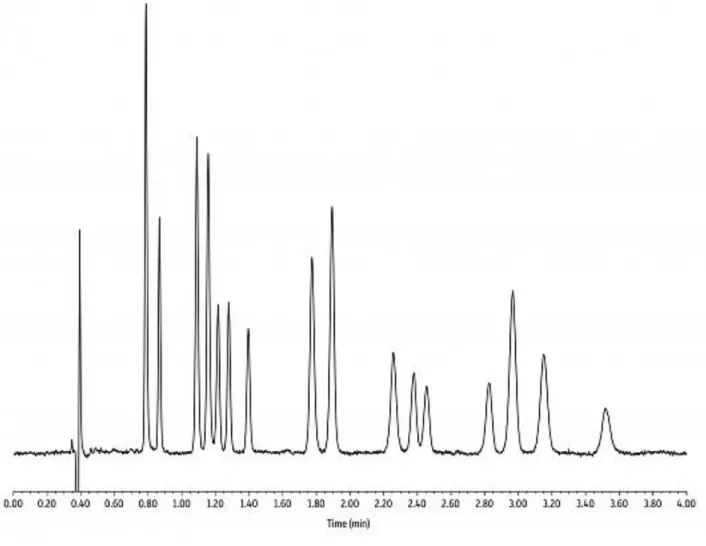
Figura 14. Cromatogramma UHPLC – UV di una miscela standard di 16 cannabinoidi con colonna Restek ARC-18® (47). I composti separati sono (in ordine di eluzione): Cannabidivarinic acid (CBDVA), Cannabidivarin (CBD),
Cannabidiolic acid (CBDA), Cannabigerolic acid (CBGA), Cannabigerol (CBG), Cannabidiol (CBD), Tetrahydrocannabivarin (THCV), Tetrahydrocannabivarinic acid (THCVA), Cannabinol (CBN), Cannabinolic acid
(CBNA), Δ9-Tetrahydrocannabinol (Δ9-THC), Δ8-Tetrahydrocannabinol (Δ8-THC), Cannabicyclol (CBL), Cannabichromene (CBC), Tetrahydrocannabinolic acid A (THCA-A), Cannabichromenic acid (CBCA).
39
Il risultato di una separazione cromatografica è riportato in un grafico definito
cromatogramma che associa al tempo (ascissa) il segnale degli analiti (ordinata). Un esempio di cromatogramma è riportato in Figura 14. Parlando genericamente di cromatografia, infatti, si intende sia la tecnica di purificazione di composti tipica della chimica organica, che il processo di separazione del campione usato in campo analitico. Siccome anche in questo passaggio nessun segnale correlabile agli analiti viene acquisito, è ragionevole pensare alla cromatografia come alla parte finale della preparativa. La fase stazionaria è raccolta all’interno di un cilindro di vario diametro e lunghezza definito colonna; qui avviene il contatto tra fasi e si ha la separazione. Le interazioni instaurate tra composti e fase
stazionaria variano in base alle loro differenti dimensioni ed all’affinità chimica degli uni per l’altra (principalmente forze di coesione dovute alla diversa polarità ma anche interazioni di carattere elettrostatico come le π tipiche degli anelli aromatici). Il primo esperimento di cromatografia mai realizzato viene attribuito al botanico italo-russo Michail Semënovič Cvet (erroneamente trascritto come Tswett) il quale separò una miscela di pigmenti naturali usando una fase stazionaria composta da carbonato di calcio ed una fase mobile basata su una miscela di etanolo ed etere di petrolio; è per questo che il nome della tecnica è una traslitterazione della parola greca “khrôma” (colore) (48).
La cromatografia come tecnica analitica strumentale, invece, ha avuto origine negli anni 40 in concomitanza con l’emergere della cromatografia di ripartizione e della teoria dei piatti da parte di Martin e Synge nel 1941 (49). Secondo la suddetta teoria, la colonna cromatografica può essere approssimata ad una colonna per la distillazione di liquidi ovvero suddivisa in stadi di equilibrio tra le fasi chiamati piatti; mentre nella distillazione la fase vapore è in equilibrio col proprio liquido, nella colonna la fase mobile è in equilibrio con quella
stazionaria. Quanti più stadi di equilibrio sono presenti quanto più efficiente è la separazione quindi, per avere ottime prestazioni con colonne di lunghezza ragionevole, sono necessari piatti “bassi”; da qui la definizione del parametro Altezza Equivalente del Piatto Teorico (Height Equivalent to a Theorethical Plate, HETP) con cui si misura la prestazione di una fase stazionaria. Lo sviluppo della cromatografia di ripartizione è valsa per Martin e Synge la vincita del Premio Nobel per la chimica nel 1952 (50). Circa 15 anni dopo, la teoria dei piatti è stata completata da van Deemter con la correlazione tra efficienza cromatografica e velocità lineare della fase mobile (51). La conseguente rate theory riassume in un’unica
equazione iperbolica chiamata equazione di van Deemter, gli effetti di tutti i fenomeni fisici, cinetici e termodinamici che interessano la cromatografia.
𝐻𝐸𝑇𝑃 = 𝐴 +
𝐵
µ
+ 𝐶µ
Nell’equazione di van Deemter sono presenti vari termini, ognuno rappresentativo di un fenomeno diverso della cromatografia:
- µ è la velocità longitudinale della fase mobile ed è espressa in m.
- A è il parametro che rappresenta l’effetto della presenza di cammini preferenziali dovuti a difetti nell’impaccamento (conosciuto anche come coefficiente di Eddy). La presenza di queste imperfezioni non è dovuta alla velocità della fase mobile, quindi il suo valore è espresso in m.
- B è il coefficiente di diffusione longitudinale ovvero nel senso della corrente di fase mobile. Logicamente la sua incidenza è maggiore a basse velocità, quindi lo si trova inversamente proporzionale alla velocità della fase mobile ed espresso come m2s-1.
- C è la “resistenza” al trasferimento di materia, ossia l’effetto del mancato raggiungimento dell’equilibrio termodinamico tra le due fasi. La sua incidenza è ovviamente maggiore con flussi veloci quindi viene correlato linearmente alla velocità ed espresso come s.
41
Questa equazione, caratteristica di ogni colonna con determinata geometria e fase stazionaria, tende asintoticamente a HETP infinito per v che tende a zero ed ha infinito, mentre ha un valore di v per il quale HETP raggiunge il minimo e rappresenta il flusso di massima efficienza (Figura 15). L’equazione di van Deemter è stata raffinata nel corso degli anni andando ad aggiungere termini correttivi in grado di massimizzarne l’accuratezza ma la struttura teorica è rimasta la stessa ed è, ad oggi, il modello di riferimento.
Dal punto di vista pratico è possibile calcolare l’altezza del piatto reale H come rapporto tra la lunghezza della colonna L ed il numero di piatti N.
𝐻 =
𝐿
𝑁
A sua volta vi sono vari modi di calcolare il numero di piatti andando a sfruttare le caratteristiche del picco gaussiano di un composto noto; una delle più usate si basa sul rapporto tra tempo di ritenzione tR ed ampiezza del picco alla base Wbase.
𝑁 = 16 (
𝑡
𝑅𝑊
𝑏𝑎𝑠𝑒)
2
Altri parametri fondamentali in cromatografia sono il fattore di capacità (k’) ed il coefficiente di selettività (α).
𝑘
′=
𝑡𝑅− 𝑡𝑀𝑡𝑀
; 𝛼 =
𝑘′𝑏 𝑘′𝑎Il fattore di capacità k’ è un parametro che relaziona il tempo di ritenzioni di un analita col tempo morto tM, dove quest’ultimo è il tempo di ritenzione per un composto non ritenuto. Il
fattore di selettività è il rapporto tra i k’ di due composti ed indica, a parità di condizioni cromatografiche, se è possibile separare efficacemente due composti.
In funzione della condizione di fase mobile e della natura degli analiti sono state sviluppate molte varianti di cromatografia in grado di coprire un’ampia gamma di applicazioni. Tra le tipologie più importanti vanno citate:
- Cromatografia a Esclusione Sterica (SEC): Usata per polimeri e macromolecole, gli analiti vengono fatti passare attraverso una fase stazionaria inerte con pori di dimensione compresa in un intervallo ben preciso. I composti escono in ordine di dimensioni
decrescenti (53). Se effettuata su gel è conosciuta come Gel Permeation Chromatography (GPC) per sostanza solubili in acqua, o Gel Filtration Chromatography (GFC) per sostanze non solubili in acqua.
- Cromatografia ionica (IC): Usata per separare ioni dello stesso segno. Il campione interagisce con una fase stazionaria ionizzata che ritiene gli analiti in base alla densità di carica. L’eluizione viene effettuata con una fase mobile a gradiente di forza ionica (54). Richiede la rigenerazione della fase stazionaria tra una corsa e l’altra, oltre all’uso di rivelatori specifici.
- Gascromatografia (GC): Usata per separare molecole volatili. Il campione viene
vaporizzato all’interno dell’iniettore posto ad elevata temperatura, e spinto in colonna da una corrente di gas inerte (career gas). La colonna, composta da un capillare lungo qualche decina di metri la cui superficie interna è ricoperta da un film di fase stazionaria, trattiene le molecole facendole uscire all’aumentare della temperatura in base alla loro volatilità (55). Usata per la chimica organica e ambientale, impiega come rivelatori tipici il FID (Flame Ionization Detector), l’ECD (Electron Capture Detector) e lo spettrometro di massa a ionizzazione elettronica (EI-MS).
- Cromatografia liquida (LC): Usata per separare campioni in fase liquida a prescindere dal solvente e con ampia gamma di caratteristiche chimico – fisiche degli analiti. La fase stazionaria è composta da particelle con diametro nell’ordine dell’unità di µm e porosità variabile; qui, gli analiti vengono ritenuti in base all’affinità per la fase stazionaria
attraverso un gradiente di composizione della fase mobile. La cromatografia liquida si divide in fase normale (fase stazionaria polare e fase mobile apolare) e fase inversa (fase stazionaria apolare e fase mobile polare). Data la robustezza strutturale delle colonne, in LC è possibile lavorare a flussi e pressioni elevate che permettono separazioni veloci e performanti; da qui la sigla HPLC (High Performance Liquid Chromatography) usata frequentemente per intendere la cromatografia liquida. I detector per eccellenza sono il rivelatore UV e la spettrometria di massa con sorgenti API (56).
In particolare per la cromatografia liquida, la variante di maggior interesse in questa tesi, importanti passi avanti sono stati fatti verso il rispetto dei principi della GAC. Un sistema
43
HPLC convenzionale opera a flussi dell’ordine di varie centinaia di µL/min fino a vari mL/min; contestualizzando questo consumo su base giornaliera, il volume di scarico supera il litro di liquidi.
Grazie al progredire della tecnologia nuovi sistemi HPLC sono stati sviluppati in grado di dare elevatissime prestazioni anche con flussi dell’ordine delle decine di µL/min (micro - HPLC), unità di µL/min (capillary - HPLC) e centinaia di nL/min (nano – HPLC). Una suddivisione di sistemi HPLC in base al flusso operativo è riassunta in Tabella 3.
Categoria Diametro interno colonna (mm) Condizioni operative Preparativa > 10 > 20 mL/min Convenzionale 5 – 4 1 – 10 mL/min Micro 1 – 2 50 – 100 µL/min Capillare 0.1 – 1 4 – 20 µL/min Nano 0.025 – 0.1 1 – 0.1 µL/min
Tabella 3. Suddivisione dei sistemi HPLC in base alla scala dimensionale di diametro colonna e flusso operativo.
I sistemi nano - HPLC hanno assunto sempre più rilevanza nel corso degli ultimi anni; questa espansione è stata fortemente sostenuta dallo sviluppo di detector dedicati ad alte
prestazioni e dal carattere green dello strumento. Le colonne impiegate, chiamate nano colonne, sono basate su capillari in silice fusa riempiti con la stessa fase stazionare delle colonne per sistemi convenzionali (57); oltre ai vantaggi dal punto di vista della GAC come riduzione di flusso, scarti prodotti e volume di iniezione, il diametro interno di 50 – 100 volte inferiore permette un abbassamento del HETP, una riduzione della banda cromatografica ed un miglioramento del rapporto S/N (58). La relazione tra caratteristiche geometriche della colonna e prestazioni è riassunta dalla seguente formula (59):
𝐷 =
𝐶
𝑖𝐶
𝑚𝑎𝑥=
[𝜀𝜋𝑟
2(1 + 𝑘′)√2𝜋𝐿𝐻]
𝑉
𝑖𝑛𝑗Le sigle riportate in formula indicano:
- ε: porosità della fase stazionaria; - r: raggio interno della colonna; - k’: fattore di capacità;
- L: lunghezza della colonna;
- H: altezza equivalente del piatto teorico; - Vinj: volume iniettato.
Al netto dei vantaggi sopra citati, la nano – HPLC ha tutta una serie di limiti che le hanno impedito di sostituire i cromatografi a flussi convenzionali. Nelle connessioni tra componenti e tenute possono crearsi piccoli spazi vuoti nei quali la corrente va a depositarsi e
rimescolarsi (Figura 16); questi cosiddetti volumi morti, se presenti post colonna, determinano un allargamento della banda cromatografica con conseguente perdita di prestazioni.
Figura 16. Sezione dell’accoppiamento colonna – capillare dove si vedono i siti in cui potrebbero formarsi volumi morti (60).
45
L’entità dei volumi morti risulta essere molto più incisiva in sistemi a ridotta portata. La nano – HPLC è globalmente una tecnica meno affidabile dal punto di vista meccanico in quanto la miniaturizzazione dei vari componenti provoca più problemi nel mantenimento delle tenute ed anche le colonne possono facilmente bloccarsi con campioni sporchi (61). Infine, al diminuire della “scala” del cromatografo, il tempo necessario per ottenere la separazione aumenta; nonostante a parità di fase stazionaria l’altezza dei piatti teorici non varia considerevolmente, a causa del diametro interno inferiore operando con la velocità della fase mobile si ottengono contropressioni spesso superiori al massimo consentito. Oggi questo limite è stato in parte arginato dai nano – UHPLC (Ultra High Performance Liquid
Chromatography) i quali operano a pressioni superiori a 1000 bar in modo da garantire una
velocità del flusso adeguata. Considerate le sue caratteristiche ed il continuo affermarsi delle tecniche ifenate LC-MS, la cromatografia nano – HPLC è risultata essere la perfetta
combinazione per le sorgenti ESI nano-electrospray (Figura 17).
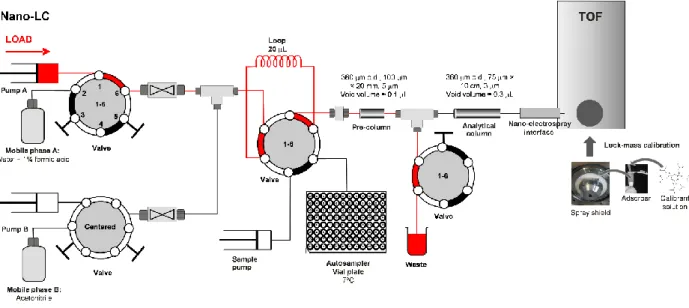
Figura 17. Schema di un sistema nano LC-MS con sorgente nano-electrospray (62).
Allo stato attuale però questa tecnologia resta relegata alle attività di ricerca in quanto inadatta ad analisi di routine.
SISTEMI DI RIVELAZIONE: LA SPETTROMETRIA DI MASSA
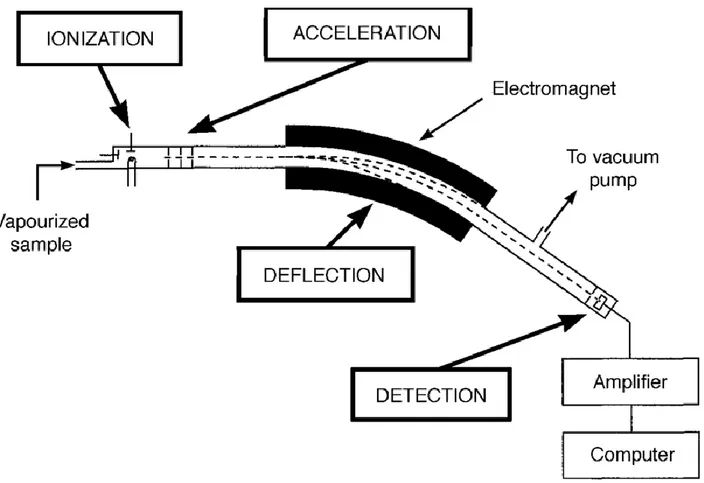
La spettrometria di massa è da quasi 50 anni la tecnica strumentale di riferimento per l’analisi di molecole organiche e macromolecole biologiche; oggi, la sua evoluzione ne ha esteso l’utilizzo anche per la mappatura della superficie di tessuti (imaging) e l’analisi di metalli (ICP-MS). Nella spettrometria di massa gli analiti vengono resi carichi (ioni) ed è quindi misurato il loro rapporto massa/carica (m/z); in caso di energia residua, questa può andare a rompere i legami più deboli in modo da frammentare la struttura dello ione.L’insieme dei segnali derivanti dalla molecola ionizzata e dai suoi frammenti è riassunta in un grafico chiamato spettro di massa, dove nell’asse delle ordinate c’è l’intensità relativa e sull’asse delle ascisse il m/z. Nello spettro di massa l’intensità è espressa come normalizzata sul rapporto m/z più intenso chiamato picco base. Lo ione corrispondente alla molecola tal quale è chiamato ione molecolare mentre i segnali successivi a rapporti m/z inferiori sono imputabili ai suoi frammenti. Dalla lettura dello spettro è possibile ricavare alcune
informazioni strutturali della molecola ionizzata (63). A causa della ionizzazione, gli analiti non possono essere recuperati e quindi la spettrometria di massa è una tecnica distruttiva, diversamente da molte tecniche moderne quali NMR, Raman o IR. Il funzionamento dello spettrometro di massa è basato su una serie di processi comuni riportati in Figura 18.
Figura 18. Schema a blocchi riportante i tre passaggi chiave del funzionamento degli spettrometri di massa.
Nella spettrometria di massa le molecole alimentate allo spettrometro vengono trasformate in ioni, ossia vengono dotate di una carica; questo primo passaggio, fa sì che il composto carico risponda alla presenza di campi elettrici e/o magnetici. Questi ioni possono essere basati su atomi, cluster, molecole o frammenti.