Dipartimento di Medicina Sperimentale Sezione Fisiopatologia Medica ed Endocrinologia XXXII ciclo di Dottorato in “Scienze Endocrinologiche”
Ruolo degli assi ormonali-metabolici
nell’ipertensione arteriosa polmonare idiopatica:
stratificazione del rischio ed impatto prognostico.
Relatore Dottorando
Ch.mo Prof. Stefania Basili Dr. Alberto Maria Marra
Matricola N. 1744351
Ad Alessandra Alla mia Famiglia
“Nella vita non bisogna mai rassegnarsi, arrendersi alla mediocrità, bensì uscire da quella zona grigia in cui tutto è abitudine e rassegnazione passiva. Bisogna coltivare il coraggio di ribellarsi.”
Sommario
1. INTRODUZIONE ... 4
1.1 IPERTENSIONE POLMONARE ... 4
1.2 CLASSIFICAZIONE EMODINAMICA E FISIOPATOLOGICA DELL’ IPERTENSIONE POLMONARE ... 5
TABELLA 1 CLASSIFICAZIONE CLINICA IPERTENSIONE POLMONARE ... 7
1.3 EPIDEMIOLOGIA DELL’ IPERTENSIONE ARTERIOSA POLMONARE ... 8
FIGURA 1. SOPRAVVIVENZA DIVERSE FORME DI IPERTENSIONE POLMONARE ... 12
1.4 FISIOPATOLOGIA DELL’IPERTENSIONE ARTERIOSA POLMONARE: LA VASCULOPATIA POLMONARE ... 13
FIGURA 2. FISIOPATOLOGIA DELLA VASCULOPATIA POLMONARE ... 15
1.5 FISIOPATOLOGIA DELL’IPERTENSIONE ARTERIOSA POLMONARE: IL VENTRICOLO DESTRO . 16 FIGURA 3. DALLA VASCULOPATIA POLMONARE ALLO SCOMPENSO CARDIACO DESTRO ... 19
1.6 L’IPOTESI ORMONALE NELL’IPERTENSIONE ARTERIOSA POLMONARE ... 20
FIGURA 4. L’IPOTESI ORMONALE NELLE MALATTIE CRONICHE ... 21
2. OBIETTIVI DEL PROGETTO DI DOTTORATO ... 22
3. MATERIALI E METEODI ... 23
3.1 DISEGNO DELLO STUDIO ... 23
3.2 PROCEDURE ... 24
3.2 ANALISI STATISTICA ... 27
4. RISULTATI ... 28
TABELLA 2 CARATTERISTICHE DELLA POPOLAZIONE ... 28
4.1 PREVALENZA DEFICIT ORMONALI NELL’IPERTENSIONE POLMONARE IDIOPATICA ... 29
FIGURA 5. DISTRIBUZIONE DEL NUMERO DI DEFICIT NELLA POPOLAZIONE ... 29
FIGURA 6. PREVALENZA DEI SINGOLI DEFICIT ORMONALI ... 30
4.2 DEFICIT ORMONALI MULTIPLI E PARAMETRIC CLINICI NELL’ÌPERTENSIONE ARTERIOSA POLMONARE ... 31
TABELLA 3 CONFRONTO TRA PAZIENTI CON DEFICIT ORMONALI MULTIPLI E PAZIENTI SENZA DEFICIT O CON DEFICIT ISOLATO. ... 31
FIGURA 7 DIFFERENZA IN TERMINI DI DISTANZA PERCORSA AL TEST DEL CAMMINO IN 6 MINUTI IN BASE ALLA GRAVITÀ DEI DEFICIT ORMONALI MULTIPLI ... 33
4.3 IMPATO SUGLI OUTCOMES DEI DEFICIT ORMONALI MULTIPLI NELLA PAH ... 34
FIGURA 8 CURVA DI KAPLAN-MAYER PER IL COMPOSITO DI MORTALITÀ E OSPEDALIZZAZIONE 35 FIGURA 9 CURVA DI KAPLAN-MAYER PER MORTALITÀ DA TUTTE LE CAUSE ... 36
5. DISCUSSIONE ... 37
5.1 I DEFICIT ORMONALI MULTIPLI NELL’IPERTENSIONE ARTERIOSA POLMONARE: SIGNIFICATO CLINICO E MECCANISMI SOTTOSTANTI. ... 38
5.2 I DEFICIT ANABOLICI NELLE MALATTIE CARDIOVASCOLARI ... 42
5.3 IMPLICAZIONI CLINICHE ... 44
5.4 LIMITAZIONI ... 45
6. CONCLUSIONI ... 46
7. REFERENZE ... 47
1. Introduzione
1.1 Ipertensione Polmonare
L’Ipertensione Polmonare (IP) è una condizione clinica che si definisce con la
presenza di un aumento della pressione arteriosa polmonare media (PAPm) a riposo maggiore o uguale a 25 mmHg, misurata invasivamente tramite esecuzione di
cateterismo cardiaco destro1.
L’ipertensione polmonare può essere il risultato di molteplici condizioni cliniche e non
definisce de facto nessuna singola patologi esclusiva2. Come risultato di tale
condizione, si verifica, a prescindere dal fattore scatenante, un aumento del post-carico del ventricolo destro che porta irrimediabilmente ad una condizione di
scompenso cardiaco franco e morte del paziente3.
Le manifestazioni cliniche e la sopravvivenza a tale condizione clinica sono in tal guisa strettamente associate alla capacità del cuore destro di adattarsi all’aumento
delle pressioni nel circolo polmonare4. Caratteristica dell’IP in tutte le sue fasi è,
infatti, un progressivo deterioramento strutturale e funzionale delle sezioni cardiache destre con evoluzione verso l’insufficienza cardiaca manifesta che porta il paziente
affetto da tale condizione inesorabilmente a morte3. Parallelamente, il cronico
elevarsi della pressione arteriosa polmonare a causa di un progressivo
rimodellamento a carico delle strutture vascolari del circolo polmonare, il quale non è fisiologicamente predisposto a tollerare regimi pressori elevati, trattandosi di un
sistema ad elevata capacità ma bassa resistenza al flusso ematico5. Dunque tutte le
condizioni patologiche che possono alterare la meccanica di questo sistema sono possibili fonti eziopatogenetiche d’ Ipertensione polmonare: da qui l’esigenza di
definire e classificare l’ipertensione polmonare non solo da un punto di vista meramente emodinamico ma, soprattutto, in base al profilo etiopatogenetico e fisiopatologico, ciascuno dei quali caratterizzato da implicazioni prognostiche e possibilità terapeutiche diverse.
1.2 Classificazione emodinamica e fisiopatologica dell’ ipertensione polmonare
Secondo le ultime ed attuali linee guida dell’European Society of Cardiology (ESC) l’IP si definisce come una condizione emodinamica e fisiopatologica caratterizzata da una PAPm ≥ 25 mmHg a riposo documentata mediante cateterismo cardiaco
destro (RHC)1. Secondo dati storici presenti in letteratura si può definire come
normale una PAPm di 14 ± 3 mmHg6. Motivo per il quale risultano attualmente da
considerarsi anomali i soggetti che presentano valori compresi fra 21 e 24 mmHg, i quali rappresentano (in particolar modo quelli con associata sclerosi sistemica) un
subset di pazienti che presentano un iniziale esordio di patologia2. Infatti, è stato
dimostrato ampiamente come questi pazienti siano caratterizzati da maggiore
progressione verso le forme franche di IP7, peggiore adattamento del ventricolo
destro allo stress fisico8 e aumentata mortalità9. Per quanto riguarda la misurazione
delle pressioni polmonari durante sforzo fisico, la definizione di IP da sforzo basata sul riscontro di valori di PAPm > 30 mmHg non è confermata dai dati pubblicati in letteratura, ma risulta essere comunque una condizione caratterizzata da peggiori
outcomes10. Sul piano emodinamico si fa poi distinzione tra IP pre-capillare,
caratterizzata da una pressione polmonare di incuneamento (PCWP) ≤ 15 mmHg con una gittata cardiaca normale o ridotta, e IP post-capillare, con PCWP > 15
due categorie, ipertensione polmonare primitiva e secondaria, in base alla presenza di fattori di rischio e/o cause specifiche. Nel 1998, in occasione del Secondo
Simposio Internazionale sull’IP tenutosi ad Evian, venne introdotta una
classificazione clinica dell’ipertensione polmonare al fine di individuare differenti categorie di IP accomunate da simili rilievi patologici, caratteristiche emodinamiche paragonabili e, infine, simile gestione terapeutica. Questo sistema è stato
progressivamente rielaborato nel corso dei meeting successivi fino ad arrivare al Sesto Simposio Mondiale tenutosi a Nizza nel 2018 dove è stata stabilita la versione
tutt’ora utilizzata della suddetta classificazione2. Questa comprende 5 categorie
cliniche: ipertensione arteriosa polmonare (PAH, Gruppo 1), ipertensione polmonare da malattie del cuore sinistro (Gruppo 2), ipertensione polmonare da malattie
polmonari e/o ipossia croniche (Gruppo 3), ipertensione polmonare cronica post-tromboembolica (Gruppo 4) ed ipertensione polmonare dalla patogenesi
multifattoriale e/o con meccanismi patogenetici non ancora del tutto noti (Gruppo 5). Il Gruppo 1 contiene a sua volta due sottogruppi, 1’ e 1”, riconducibili rispettivamente alla malattia veno-occlusiva polmonare o emangiomatosi capillare polmonare e alla ipertensione polmonare persistente del neonato (PPHN), quest’ultima introdotta in tale categoria a seguito del Simposio di Nizza del 2013 in modo tale da ottenere una classificazione omnicomprensiva comune per l’adulto ed il bambino. Di
conseguenza, l’ipertensione polmonare da cardiomiopatie congenite o da lesioni ostruttive congenite o acquisite del cuore sinistro sono state aggiunte al gruppo 2.
Tabella 1 Classificazione Clinica Ipertensione Polmonare
1 Ipertensione Arteriosa Polmonare (PAH)
1.1 PAH idiopatica 1.2 PAH familiare
1.3 PAH associata a farmaci o tossine 1.4 PAH associata a:
1.4.1 malattie del tessuto connettivo 1.4.2 HIV
1.4.3 Ipertensione Portale
1.4.4 Malattie congenite cardiache 1.4.5 Schistosomiasi
1.5 PAH responsiva ai calcio antagonisti (table 4)
1.6 PAH associata a malattia polmonare veno-occlusiva/emangiomatosi polmonare
1.7 Ipertensione polmonare persistente del neonato
2 . Ipertensione Polmonare (IP) da Insufficienza Cardiaca Sinistra
2.1 IP da insufficienza cardiaca a ridotta frazione di eiezione 2.2 IP da insufficienza cardiaca a conservata frazione di eiezione 2.3 Malattie valvolari
2.4 Malattie congenite cardiache o cardiomiopatie associate a IP post-capillare
3 IP da malattie respiratorie o ipossia
3.1 BPCO
3.2 Malattie restrittive
3.3 Malattie con pattern misto restrittivo/Ostruttivo 3.4 Ipossia senza malattia polmonare
3.5 Malattie polmonari dello sviluppo
4 IP da ostruzione dell’albero vascolare
4.1 IP cronica post trombo-embolica (CTEPH) 4.2 Altre ostruzioni arteria polmonare
5 IP con meccanismo non chiaro
5.1 malattie ematologiche
5.2 Malattie Sistemiche e metaboliche 5.3 Altre
5.4 Malattie Cardiache Complesse
1.3 Epidemiologia dell’ Ipertensione arteriosa polmonare
L’ipertensione arteriosa polmonare (PAH) appartiene al Gruppo 1 della
classificazione clinica di Nizza. Nella PAH il primum movens è l’innescarsi di una
vasculopatia polmonare12, che accomuna tutte le forme di PAH: le forme idiopatiche,
forme ereditarie, forme legate all’assunzione di farmaci o all’esposizione a tossine, forme associate a malattie sistemiche quali le connettiviti, ad infezione da HIV, forme correlate all’ipertensione portale o, sia pur meno frequentemente, a schistosomiasi. Alcuni pazienti presentano una PAH con una rimarabile storia familiare, e sono legati
a specifiche mutazioni (BMPR2, ALK-1, ENG, SMAD9, CAV1, KCNK313. Tutti questi
quadri sono accomunati sul piano emodinamico da un profilo di tipo pre-capillare,
con PCWP ≤ 15 mmHg13. Sebbene la PAH sia definibile come una malattia rara, con
una prevalenza stimata di 15-50 casi per milione, la prevalenza di questa stessa
condizione all’interno di gruppi a rischio è considerevolmente più elevata14. Ad
esempio, nei pazienti infetti da HIV la prevalenza è dello 0,5%, mentre nei pazienti
con sclerosi sistemica questa si attesta tra il 7% e il 12%14. Nell’80% delle famiglie
con casi multipli di PAH, è possibile identificare mutazioni della proteina BMPR2,
appartenente alla superfamiglia del TGF-beta13, mentre nel 5% dei casi sono state
scoperte rare mutazioni in altri geni appartenenti alla superfamiglia del TGF-beta:
ALK1, ENG e SMAD913. In circa il 20% delle famiglie non è invece possibile
identificare mutazioni in geni attualmente associati alla patologia15.
Per quanto concerne le forme di PAH correlate all’esposizione a farmaci o tossine sono state identificate diverse categorie di rischio sulla base della forza delle evidenze, distinguendo associazioni definite, verosimili, possibili.
Il Benfluorex è una sostanza ad azione ipolipidemizzante ed ipoglicemizzante derivata dalla fenfluramina ed il suo principale metabolita è la norfenfluramina.
Questo farmaco è stato ritirato dal commercio nel 1998 in tutti i paesi europei eccetto la Francia dove il Benfluorex è stato commercializzato fino al 2009 ed è stato
frequentemente utilizzato tra il 1998 e il 2009 in alternativa all’Isomeride, altro
farmaco a scopo anoressizzante16. Il meccanismo con il quale questi farmaci
contribuiscano al determinismo della PAH è sconosciuto, ma è probabilmente legato alle interazioni con i recettori serotoninergici espressi sia a livello del SNC che a livello della parete vasale, dove si rendono responsabili dell’attivazione di processi
fibrogenici17. Studi analoghi sono stati condotti circa la verosimile associazione tra
dasatinib, un inibitore di tirosina chinasi impiegato nella terapia della leucemia mieloide cronica (CML), e casi di PAH. Tra il novembre del 2006 e il settembre del 2010, 9 casi trattati con dasatinib hanno sviluppato una PAH diagnosticata tramite cateterismo cardiaco destro e si sono osservati miglioramenti sul piano clinico,
funzionale ed emodinamico a seguito della sospensione del trattamento18. Alla luce
di tali osservazioni, il dasatinib è pertanto considerato un verosimile fattore di rischio per PAH. In sintesi, diversi nuovi farmaci sono stati recentemente identificati come definiti, verosimili o possibili fattori di rischio per PAH. Per poter migliorare
l’identificazione di farmaci potenzialmente in grado di favorire l’insorgenza di PAH è cruciale sottolineare l’importanza di ottenere una storia dettagliata della presente e della passata esposizione a farmaci o tossine in ogni paziente con PAH.
Malattie sistemiche quali le connettiviti e l’infezione da HIV sono altri due noti fattori
di rischio per lo sviluppo di PAH7,19. Come si è detto in precedenza, la prevalenza
prognosi in questo subset di pazienti resta infausta ed è peggiore se comparata agli altri sottogruppi di PAH. La mortalità ad 1 anno in pazienti con PAH idiopatica è approssimativamente il 15% contro il 30% dei pazienti con PAH associata a
sclerodermia20. Per quanto riguarda la PAH associata ad HIV, invece, prima dello
sviluppo della terapia HAART e di specifici farmaci per il trattamento della PAH, la prognosi della PAH HIV-relata era estremamente infausta, con un tasso di mortalità
del 50% ad 1 anno4. Trend invertitosi successivamente all’impiego dei suddetti
farmaci, con un tasso di sopravvivenza a 5 anni superiore al 70%21.
Importanti sono anche le forme di PAH associata a cardiopatie congenite nell’adulto. Queste comprendono difetti del cuore associati ad ampi shunt sinistro-destro (difetti del setto interatriale) e anomale comunicazioni tra i grandi vasi (pervietà del dotto
arterioso)4. Il numero di bambini con malattie cardiache congenite che sopravvive
fino all’età adulta è oggigiorno notevolmente aumentato, in virtù dei progressi compiuti nella diagnosi e nel management delle cardiopatie congenite nelle ultime
decadi22. Si stima che circa il 10% degli adulti con cardiopatia congenita possa
sviluppare PAH23. In particolare, pazienti con un fenotipo specifico caratterizzato da
overload di pressione e di volume risultano essere molto più a rischio di sviluppare
PAH rispetto ai pazienti con sovraccarico volumico isolato4. Vista la prevalenza della
PAH tra gli adulti con cardiopatia congenita, è lecito ipotizzare che ogni paziente con cardiopatia congenita meriti un assessment per stabilire se la PAH sia effettivamente presente1.
L’ipertensione polmonare associata all’ipertensione portale, talvolta definita come ipertensione porto-polmonare o sindrome porto-sistemica, è verosimilmente dovuta alla presenza di shunt tra circolazione portale e sistemica che comportano la
epatico, a livello del circolo polmonare, con conseguente produzione di citochine ed
endotossine, più che a fattori puramente emodinamici24. L’ipertensione
porto-polmonare (POPH) si riscontra in una percentuale che va dal 2% al 6% dei pazienti con ipertensione portale e la prognosi a lungo termine è naturalmente correlata alla
severità della cirrosi e alla funzione cardiaca di questi pazienti24.
Un profilo emodinamico simile a quello caratterizzante la POPH si riscontra nei pazienti con PAH associata a schistosomiasi. Questa tipologia di PAH è stata inclusa nel gruppo I nel 2008. Precedentemente era allocata nel gruppo 4. Oggi, la PAH associata a schistosomiasi è potenzialmente la causa più prevalente di PAH nel
mondo19. La schistosomiasi colpisce infatti oltre 200 milioni di persone, dei quali il
10% sviluppa una schistosomiasi epatosplenica e il 5% dei suddetti può sviluppare
infine PAH19. Il tasso di mortalità della PAH schistosomiasi-relata può raggiungere il
15% a 3 anni. Sembra che questi pazienti possano peraltro beneficiare, oltre che del
trattamento specifico della patologia di base, delle terapie indicate per la PAH25.
La PAH idiopatica ha un’incidenza annuale di circa 1-2 casi per milione negli Stati
Uniti e in Europa ed è dalle 2 alle 4 volte più frequente nelle donne26. L’età media
alla diagnosi è di 45 anni, sebbene l’esordio dei sintomi possa avvenire a qualsiasi età. I pazienti possono presentare inizialmente sintomi aspecifici quali astenia, palpitazioni, scarsa resistenza allo sforzo fisico; con la progressione della malattia
compariranno dispnea e angina da sforzo14. A causa della natura poco specifica dei
primi sintomi, la PAH è sfortunatamente diagnosticata quando i pazienti hanno
raggiunto uno stadio avanzato di malattia, in classe funzionale WHO III e IV27. La
PAH è molto più frequente nel sesso femminile, sebbene quando colpisca il sesso maschile ha una prognosi più infausta. I pazienti affetti da forme idiopatiche sono quelli meno caratterizzati sia fenotipicamente che fisiopatologicamente.
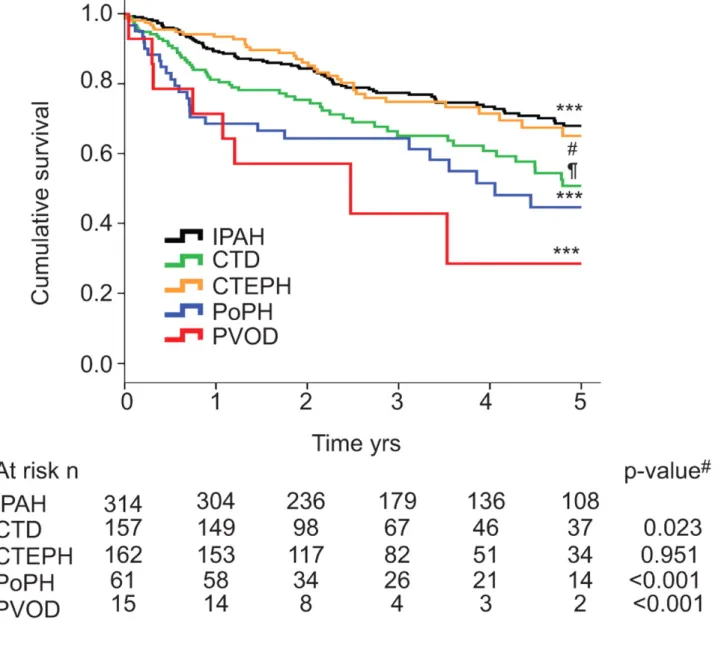
Figura 1. Sopravvivenza diverse forme di ipertensione polmonare
Differente aspettativa di vita nelle diverse forme di ipertensione arteriosa polmonare. Fonte28
1.4 Fisiopatologia dell’Ipertensione Arteriosa Polmonare: la vasculopatia polmonare La fisiopatologia dell’ Ipertensione Arteriosa polmonare non è ancora del tutto compresa. Diverse teorie sono state proposte negli anni e molte vedono come primum movens, la presenza di una lesione vascolare scatenata da trigger sconosciuti corroborata dalla comparsa di un infiltrato cellulare perivascolare costituito da macrofagi, linfociti T CD4+ e cellule B che a loro volta producono
anticorpi29. Oltre all'infiltrazione perivascolare, l'apoptosi delle cellule endoteliali (EC)
viene attivata su base autoimmune30,31. Leucociti infiltranti, EC disfunzionali e
matrice extracellulare alterata (ECM) possono indurre cellule vascolari muscolari lisce (VSMC) e EC stesse a trasformare il loro fenotipo in quello di cellule
mesenchimali, che successivamente migrano nell’intima ed iniziano a produrre in maniera incontrollata matrice extracellulare. Il risultato finale di questo processo risulta nella formazione del "neointima", tipico segno distintivo della vasculopatia
polmonare che va quindi a realizzarsi32–34. Questa transizione da endoteliale a
mesenchimale è suggerita da un insieme coerente di evidenze. Secondo Arciniegas et al., le cellule endoteliali sono in grado di perdere marker vascolari come
l'E-caderina e di esprimere marker tipici dei miofibroblasti come l'α-SMA e il collagene di tipo I (Arcinegas E et al. 2005). Il fattore di crescita β (TGF-β) potrebbe essere
responsabile di questa induzione di questa trasformazione35. L'occlusione del piccolo
vaso non è bilanciata da un aumento compensativo dell'angiogenesi e della
vasculogenesi. Un aumento dell'espressione del recettore solubile Flt-1, che a sua volta lega e riduce le proprietà angiogeniche del fattore di crescita endoteliale
vascolare (VEGF) è stato riportato in SSc patiens36,37. La vasculopatia precede
l'attivazione dei fibroblasti tissutali con una produzione di ECM costitutiva e
collegare il danno vascolare all'attivazione dei fibroblasti. La sovraespressione del TGF-β e del fattore di crescita del tessuto connettivo (CTGF) indotta dai macrofagi, inghiottendo EC apoptotiche, porta alla differenziazione miofibroblasti e alla
produzione di ECM38. Oltre a questi fenomeni, la sovrapproduzione del fattore di
crescita piastrino-derivato da piastrine attivate, innescate a loro volta da lesioni endoteliali, potrebbe contribuire alla fibrosi. La riduzione della perfusione tissutale provoca ipossia e ischemia cronica dovuta all'obliterazione microvascolare e porta
ad un circolo vizioso di ischemia-reperfusione che potrebbe aggravare la fibrosi39–41.
Il pool di cellule che producono la CEM è costituito da cellule residenti che assumono un fenotipo fibroblastico e da una quantità di cellule progenitrici circolanti (come le cellule derivate dal midollo osseo) o fibrociti che vengono reclutati dal flusso
sanguigno39. Una secrezione incontrollata di fattori di crescita, chemochine e
citochine sia nella moda autocrina che paracrina, aumenta e sostiene la produzione
1.5 Fisiopatologia dell’Ipertensione Arteriosa Polmonare: il ventricolo destro Una volta innescatosi il processo fisiopatologico che porta alla vasculopatia polmonare si verifica l’aumento delle resistenze vascolari polmonari con
conseguente aumento clinicamente manifesto della pressione in arteria polmonare42.
Questo processo procede lentamente fino ad arrivare ad un inevitabile coinvolgimento delle sezioni cardiache destre.
La previsione dell’outcome in pazienti con ipertensione polmonare è stata ampiamente studiata sia in trials con ampie coorti sia in studi di più piccole
dimensioni aggiungendo parametri di imaging43. Un ritrovamento ricorrente tra questi
studi è che la sopravvivenza nei pazienti con PAH è strettamente collegata
all’adattamento del ventricolo destro all’aumentato carico pressorio cui questi risulta
essere sottoposto44. Studi emodinamici hanno dimostrato il valore predittivo della
pressione atriale destra e dell’indice cardiaco45. Studi ecocardiografici hanno poi
evidenziato il valore predittivo del TAPSE, delle dimensioni atriali e ventricolari destre
e della presenza di versamento pericardico46. In particolare, come dimostrato da
Ghio S. et al., il TAPSE, il grado di insufficienza tricuspidalica, il grado di
collabimento della vena cava inferiore, l’indice di eccentricità del ventricolo sinistro sono tutti parametri associati in maniera indipendente alla sopravvivenza di pazienti
con ipertensione polmonare idiopatica46. Pazienti con TAPSE ≤ 15 mm ed indice di
eccentricità ≥ 1.7 hanno mostrato il più elevato tasso di eventi (51.7 % anni/persona), mentre pazienti con TAPSE > 15 mm ed insufficienza tricuspidalica lieve o assente
hanno manifestato un outcome sensibilmente migliore46. In un altro studio, Ghio S. et
al.,hanno analizzato la rilevanza clinica e prognostica della geometria ventricolare
cateterismo cardiaco destro in 72 pazienti con diagnosi di PAH idiopatica; la durata media del follow-up è stata di 38 mesi, con 22 pazienti deceduti nel corso dello
stesso48. E’ stato così possibile osservare come il tasso di mortalità fosse
significativamente superiore nei pazienti con diametro ventricolare destro > 36.5 mm che non in pazienti con diametro inferiore o uguale a 36.5 mm: 15.9% vs 6.6 % di eventi anno/persona. Al contrario, il tasso di mortalità è risultato essere simile nei pazienti con spessore della parete del ventricolo destro superiore o inferiore al valore medio. Tuttavia, tra i pazienti con spessore di parete > 6.6 mm, un diametro
ventricolare destro > 36 mm non si è dimostrato esser associato ad una prognosi peggiore. Questo studio ha dunque permesso di osservare come, nei pazienti con PAH idiopatica, un più elevato diametro ventricolare destro sia indicatore di cattiva prognosi ma un maggior spessore di parete permette di ridurre il rischio di morte tra i pazienti che presentano un ventricolo destro dilatato. La valutazione della funzione ventricolare destra (RV) raccoglie informazioni prognostiche cruciali in diverse
condizioni cardio-respiratorie e ovviamente nella PAH49,50.
La risposta della RV e della circolazione polmonare a fattori di stress come
l'esercizio fisico o l'uso di farmaci inotropi raccoglie informazioni cliniche rilevanti, in
misura maggiore rispetto alla valutazione a riposo della funzione di RV10,51. La
riserva contrattile RV in PAH è un parametro emergente che è stato studiato da diversi gruppi negli ultimi anni: Grünig et al. e Blumberg et al.hanno dimostrato che la riserva contrattile ventricolare destra era un predittore indipendente della mortalità
nei PAH52,53 e della capacità di esercizio53. Chia et al. hanno dimostrato che i
pazienti SSc con PAP a riposo normale avevano una riserva contrattile RV smussata
rispetto ai controlli54 e Hsu et al. hanno dimostrato che la riserva contrattile RV è
La compliance polmonare arteriosa (PAC) è un parametro fisiopatologico molto importante per descrivere il post-carico polmonare e che rappresenta circa il 25%
del carico totale di ritorno della RV56. Esso diminuisce precocemente nel corso della
PAH57, correla in modo stretto con la gravità della PAH58 , inducendo una
vasculopatia proliferativa distale25,59 e aumenta il carico di lavoro pulsatile del RV44.
Questo porta alla disfunzione del RV e al fallimento del disaccoppiamento della RV
con l’arteria polmonare60,61. Inoltre, il PAC ha dimostrato di essere un miglior
1.6 L’ipotesi ormonale nell’Ipertensione Arteriosa Polmonare
Alla luce dei dati epidemiologici indicati risulta chiaro come risulti necessario identificare dei sottogruppi di pazienti affetti da ipertensione polmonare che
necessitino di maggiore attenzione e investigazioni più profonde da destinare a più
stretto follow-up clinico e strategie terapeutiche più aggressive14. A questo proposito,
è importante menzionare il crescente interesse, registrato nelle ultime due decadi anno, per lo squilibrio anabolico/catabolico nelle malattie cardiovascolari, soprattutto
nell'insufficienza cardiaca cronica sinistra64–67 che rappresenta la prima causa di
ipertensione polmonare. Gli assi ormonali anabolici (testosterone,
deidroepiandrosterone, ormone della crescita/fattore di crescita insulino-simile 1, insulino-resistenza) sono comunemente depressi nell’Insufficienza Cardiaca sinistra,
e associati a condizioni cliniche compromesse e mortalità aumentata68–72. Da notare
che studi preliminari hanno dimostrato che la terapia ormonale sostitutiva potrebbe
migliorare i marcatori prognostici surrogati nei pazienti con tale condizione73–75.
Deficit ormonali anabolici (AD) sono probabilmente presenti anche negli PAH per diversi motivi. In primo luogo, la presenza di chiara miopatia nei pazienti affetti
PAH76, tenendo conto del fatto che il muscolo scheletrico è un tessuto bersaglio degli
ormoni anabolizzanti. In secondo luogo, il ruolo centrale svolto dall'attivazione
infiammatoria nella PAH che potrebbe infine risultare in un danno anabolizzante61. In
terzo luogo, la presenza del cosiddetto obesity paradox77, che è un'ipotesi
fisiopatologica, che sostiene che l'obesità, potrebbe essere protettiva e associata a una maggiore sopravvivenza e potrebbe rispecchiare la presenza di uno squilibrio anabolico.
Tuttavia, la prevalenza, la associazione con variabili cliniche e l'impatto sugli outcomes dei deficit ormonali-anabolici nella PAH non è stato finora oggetto di studio.
2. Obiettivi del progetto di dottorato
L’obiettivo di questo progetto di dottorato è stato quello di esplorare con un’analisi retrospettiva e hypothesis-generating, il ruolo dei deficit ormonali-metabolici multipli nell’ipertensione arteriosa polmonare idiopatica. Gli obiettivi specifici di questo studio sono stati quindi quelli di:
-stimare la prevalenza dei deficit ormonali metabolici singoli e multipli in una popolazione affetta da ipertensione arteriosa polmonare idiopatica
-esplorare l’associazione dei deficit ormonali metabolici singoli e multipli con
parametri di esercizio fisico, classe funzionale, parametri emodinamici rilevati tramite cateterismo cardiaco destro, indici di architettura e funzione ventricolare destra misurati tramite risonanza magnetica cardiaca.
-valutare l’impatto dei deficit anabolici singoli e multipli sull’outcome composito di mortalità ed ospedalizzazione.
3. Materiali e meteodi
3.1 Disegno dello studio
Questa analisi è stata condotta in maniera retrospettiva su pazienti affetti da ipertensione arteriosa polmonare idiopatica afferenti in maniera consecutiva dal 1 gennaio 2009 al 1 gennaio 2013, presso l’Unità operativa dipartimentale dell’Azienda ospedaliera-universitaria policlinico Umberto I di Roma, diretta dal prof. Carmine Dario Vizza (Dipartimento di Scienze Cardiovascolari, Respiratorie, Nefrologiche, Anesteriologiche e Geriatriche, Università “Sapienza” di Roma). Pazienti con
diagnosi di ipertensione arteriosa polmonare secondo le attuali linee guida europee, effettuata con cateterismo cardiaco destro e che erano stati sottoposti a test del cammino dei sei minuti (6MWT), risonanza magnetica cardiaca, valutazione della classe funzionale secondo i criteri della World Health Organization sono stati inclusi nell’analisi.
Sono stati esclusi dall’analisi pazienti con significativa malattia cardiaca sinistra, significativa malattia respiratoria, presenza di scintigrafia ventilatoria/perfusionale suggestiva di malattia trombo-embolica. Sono stati inoltre esclusi i pazienti che non possedevano valori dei 5 ormoni che sono stati scelti per lo studio, nello specifico: -Testosterone
-Dheidroandrostenedione solfato (DHEA-S) -Insulin-like growth factor 1 (IGF-1)
-Insulina e Glicemia
-Ormoni tiroidei (t3,t4 e TSH)
Le ospedalizzazioni considerate come evento sono state considerate nel computo dell’analisi solo al verificardi delle seguenti condizioni: 1) una riduzione della capacità
di esercizio (-15%) rispetto ai precedenti 6MWT, confermati da 2 prove effettuate ≤ 2 settimane) più il classe funzionale WHO, ricovero ospedaliero non selettivo per PAH (necessità di farmaci diuretici o inotropi per via endovenosa, necessità di nuovi farmaci specifici per la PAH, trapianto polmonare, septostomia), o mortalità da tutte le cause.
3.2 Procedure
Cateterismo Cardiaco Destro
L'esame a riposo è stato eseguito come precedentemente descritto7 in posizione
supina utilizzando l'accesso transgiugulare con un set di introduttore da 8F (MXI100, MEDEX, Smiths Group PLC, UK). La cateterizzazione è stata effettuata con cateteri per termodiluizione 7F-Swan-Ganz a triplo lume 7F-Swan-Ganz di Edwards
Lifesciences (REF:131F7, Edwards Lifesciences LLC, Irvine, CA, USA). Le pressioni sono state continuamente registrate e mediate su diversi cicli respiratori durante la respirazione spontane. La gittate cardiaca (CO) è stata misurata mediante
termodiluizione tramite la media di tre misurazioni che non presentassero una variazione inferiore al 10% tra i valori misurati. Il punto di riferimento zero per le registrazioni di pressione è stato fissato a 1/3 del diametro toracico al di sotto della
superficie toracica anteriore secondo le linee guida ESC/ERS 20151. Le resistenze
vascolare polmonari (PVR) è stato calcolato con la formula PVR=(mPAP-PWP)/CO. La conformità dell'arteria polmonare è stata calcolata come il rapporto tra il volume dell'ictus e la pressione dell'impulso (SV/PP): SV/PP (ml/mmHg) = (volume del colpo) / (pressione sistolica polmonare - pressione diastolica).
Risonanza Magnetica Cardiaca (CMR)
Tutti gli studi CMR sono stati eseguiti con un'unità 1.5-T (Avanto Siemens, Erlangen, Germania) utilizzando un software cardiaco dedicato, bobina del ricevitore di
superficie a schiera di fase e attivazione dell'ECG, entro 1 settimana dall’esecuzione del RHC.
Le masse e i volumi biventricolari sono stati determinati utilizzando una tecnica CMR in cine free-precession, a stato stazionario a tenuta di respiro costante, acquisita sia su piani orizzontali che sull’asse lungo che su piani ad asse corto che abbraccia completamente entrambi i ventricoli con una pila di fette contigue.
Tutti gli studi CMR sono stati analizzati off-line con il consenso di due osservatori esperti utilizzando una postazione di lavoro dedicata con software cardiaco. (CMR 42, Circle Canada).
Cine CMR è stato utilizzato per quantificare i volumi RV e del ventricolo sinistro (LV) (utilizzando pile a 4 camere), RV e la frazione di eiezione (EF) e le masse RV e LV. Contorni di immagini sono stati tracciati semiautomaticamente a fine diastole e fine sistole da piani orizzontali asse lungo, corrispondenti misure con immagini ad asse corto quando necessario.
Per la misurazione della massa della RV, abbiamo preso in considerazione lo spessore della parete libera della RV e 1/3 dello spessore della parete libera della RV setto interventricolare. Muscolo papillare e trabeculazioni sono stati inclusi nella cavità, mentre particolare cura è stata presa a escludere l'atrio dai contorni in fette di base.
Analisi Ormonali
I campioni di sangue sono stati prelevati per puntura venosa dopo il digiuno notturno. Per ottenere siero e plasma, i campioni sono stati centrifugati entro 1 ora, congelati e conservati a -80° C fino all'analisi. Gli ormoni sierici sono stati analizzati in un
laboratorio core-lab dedicato (IRCSS-SDN, Napoli, Italia) in collaborazione con il Centro di Coordinamento Clinico. L'insulina e il fattore di crescita dell'insulina-1 (IGF-1) sono stati analizzati con un test immunometrico chemiluminescente marcato da un enzima (IMMULITE 2000; IGF-1, variazione inter-test= 5, 7% CV, Siemens Medical Solutions Diagnostics). Il testosterone totale è stato misurato con un kit DPC Coat-A-Count RIA. Il DHEA-S è stato misurato con un test immunoenzimatico
chemiluminescente competitivo in fase solida. I deficit ormonali sono stati definiti come segue: i) Deficit di IGF-1: livelli sierici di IGF-1 inferiori al 33° percentile di una popolazione di controllo sano: 122 ng/ml (età < 55 anni); 109 ng/ml (55 anni < età < 64,9 anni); 102 ng/dl (65 anni < età < 74 anni).9 anni); 99 ng/dl (età > 75 anni) [8]; ii) deficit di testosterone (TD): livelli sierici di testosterone inferiori a 300 ng/dl nel maschio e inferiori a 25 ng/dl nella femmina; deficit di S: livelli sierici di DHEA-S inferiori a 80 µg/dL; basso T3: T3 libero nel siero inferiore a 2 pg/mL (3.).1
mmol/L); insulino-resistenza (IR): presenza di diabete mellito di tipo 2 o HomeOstasis Model Assessment (HOMA-Index) superiore a 2,5 (secondo la formula: IR= insulina (mcU/mL) x glucosio (mmol/L)/22.5.I pazienti sono stati ulteriormente divisi in due gruppi che riflettono il successivo numero di carenze anabolizzanti: (1) pazienti con una carenza anabolizzante o senza defict (MHDS-) e (2) pazienti con due o più carenze anabolizzanti (cioè: MHDS).
3.2 Analisi statistica
Le variabili continue normalmente distribuite sono state espresse come media ± Deviazione Standard (SD), mentre i dati continui con distribuzioni non normali sono stati espressi come mediana e intervallo interquartile [IQR]. Le variabili categoriche sono state espresse come conteggi e percentuali. La distribuzione delle variabili è stata testata con il test Kolmogorov-Smirnov. Il test t di Student per dati non appaiati è stato utilizzato per valutare la differenza nei parametric clinici tra pazienti MHDS- e MHDS+.
Le associazioni tra le variabili analizzate e la sopravvivenza sono state stabilite utilizzando le analisi di regressione del rischio proporzionale Cox. Per valutare i potenziali predittori di sopravvivenza sono stati utilizzati sia modelli lineari univariati che multivariabili.
Nelle analisi univariate, sono stati inclusi parametri demografici, biochimici, correlati alla malattia e clinici e strumentali. Per quanto riguarda lo stato anabolizzante, abbiamo considerato il singolo deficit, la somma di deficit (da 0= nessuna carenza a 5= carenza in tutti e 5 gli assi) o la presenza di un MHDS (0= no MDHS, 1= presenza di MHDS). Inoltre, la gravità di MHDS è stata valutata e classificata come segue: (1 deficit= MHDS lieve, 2 deficit= MDHS media, 3 o più deficit= MHDS grave). Le analisi statistiche sono state effettuate utilizzando la versione R 3.0 (http: //www.r-project.org) e la versione 25.0 (SPSS Inc, Chicago, Illinois, USA). Un valore di p<0,05 è stato considerato statisticamente significativo.
4. Risultati
Un totale di 47 pazienti sono stati inclusi nello studio. Dopo averne esclusi 16 in quanto non presente la misurazione di tutti gli assi ormonali, la popolazione finale è stata composta da 30 pazienti. La tabella 2 descrive le caratteristiche generali di questa popolazione di pazienti.
Tabella 2 Caratteristiche della popolazione
Parametro
BMI (Kg/mq) 26.53 ± 5.36
Sesso femminile (n) 20
PAS (mmHg) 114.66 ± 11,51
PAD (mmHg) 74.66 ± 9.73
classe WHO (II/III/IV) 12/15/3 Frequenza cardiaca (bpm) 75.83 ± 18.33
6MWD (m) 436.43 ± 76.36
Borg dopo 6MWT (unità arbitrarie) 6.46 ± 1.83 sPAP (mmHg) 69.13 ± 22.24 mPAP (mmHg) 45.20 ± 13.98 dPAP (mmHg) 29.23 ± 10.31 wedge (mmHg) 9.7 ± 3.48 PAC (ml/mmHg) 1.68 ± 0.76 RV-EDV (ml) 185.61 ± 52.76 RV-ESV (ml) 112.75 ± 56.28 RV-EF (%) 33.92 ± 12.96 IGF-1 (ng/ml) 152.86 ± 69.45 DHEA-S (µg/dl) 83.32 ± 126.59 Testosterone Totale (ng/dl) 2.24 ± 3.51 HOMA (µU*mmol/dl) 3.34 ± 2.36 Low t3 (n) 7
BMI: indice massa corporea; PAS: pressione arteriosa sistolica, PAD: pressione arteriosa diastolica, 6MWD: distanza percorsa al test del cammino di sei minuti, PAP: pressione polmonare, s: sistolica, d: diastolica, m: media, PAC: compliance arteria polmonare, RV: ventricolo destro, EDV: volume telediastolico, EDS: volume telesistolico, EF:
4.1 Prevalenza deficit ormonali nell’Ipertensione Polmonare Idiopatica
Secondo I cut-offs precedentemente definiti 1 paziente non ha presentato nessun deficit (3.3%), 16 pazienti un singolo deficit (53.3%), 5 pazienti 2 deficit (16.7%), 6 pazienti presentavano 3 deficit (20.0%), 1 paziente presentava 4 deficit (3,3%) ed infine 1 paziente era affetto da 1 deficit (3.3%).
Per quanto riguarda i singoli deficit, 5 (16.7%) pazienti presentavano deficit di IGF-1, 23 (76.7%) pazienti erano affetti da deficit di DHEA-S, 12 pazienti presentavano deficit di testosterone (40%), 6 (20%) pazienti risultavano insulino resistenti mentre la lowt3 syndrome era presente in 7 (23.3%) pazienti.
4.2 Deficit ormonali multipli e parametric clinici nell’ìpertensione arteriosa polmonare
Nella tabella 3 sono riportate le differenze nei principali parametri clinici esaminati tra i pazienti con nessun deficit ormonale o con un singolo deficit (MHDS-) più deficit ormonali concomitanti (≥2, deficits MHDS+).
Tabella 3 Confronto tra pazienti con deficit ormonali multipli e pazienti senza deficit o con deficit isolato.
Parametro MHDS- MHDS+ p value
BMI (Kg/mq) 26.62 ± 5.02 26.26 ± 6.58 ns
classe WHO (II,III,IV) 10,6,1 2,9,2 0.03
6MWD (m) 456.91 ± 70.35 380.12 ± 66.11 0.012 sPAP (mmHg) 64.95 ± 22.73 80.62 ± 17.11 ns mPAP (mmHg) 40.77 ± 12.81 57.37 ± 9.33 0.001 dPAP (mmHg) 25.09 ± 8.47 40.62 ± 4.77 0.0001 Cardiac Index (l/mq) 2.59 ± 0.465 2.14 ± 0.64 0.04 wedge (mmHg) 9.00 ± 3.51 11.62 ± 2.72 ns PVR (Woods Units) 7.07 ± 2.99 12.62 ± 3.26 0.001 PAC (ml/mmHg) 1.81 ± 0.72 1.37 ± 0.84 ns RV-EDV (ml) 172.01 ± 48.77 214.53 ± 51.95 ns RV-ESV (ml) 93.7 ± 48.91 153.25 ± 51.34 0.01 RV-EF (%) 37.30 ± 10.07 26.76 ± 16.05 0.05 RV-massa (g) 80.28 ± 20.75 100.31 ± 18.23 0.03
MHDS= sindrome ormonale metabolica multipla, BMI: indice massa corporea; 6MWD: distanza percorsa al test del cammino di sei minuti, PAP: pressione
polmonare, s: sistolica, d: diastolica, m: media, PAC: compliance arteria polmonare, RV: ventricolo destro, EDV: volume telediastolico, EDS: volume
telesistolico, EF: Frazione di eiezione, ns: non significante
I pazienti con MHDS+ presentavano più frequentemente una classe funzionale WHO più svantaggiosa. Parallelamente una differenza media di circa 70 m al test del cammino di 6 minuti veniva riscontrata tra pazienti con deficit multipli e pazienti con deficit isolato/assenza di deficit (p= 0.01). Per quanto riguarda l’emodinamica del circolo polmonare i pazienti MHDS+ presentavano peggiori pressioni medie e diastoliche, un ridotto indice cardiaco. Inoltre la differenza in termini di resistenze vascolari polmonari risultava essere altamente significativa (p <0.001) con una differenza di circa 5 WU fra i due gruppi.
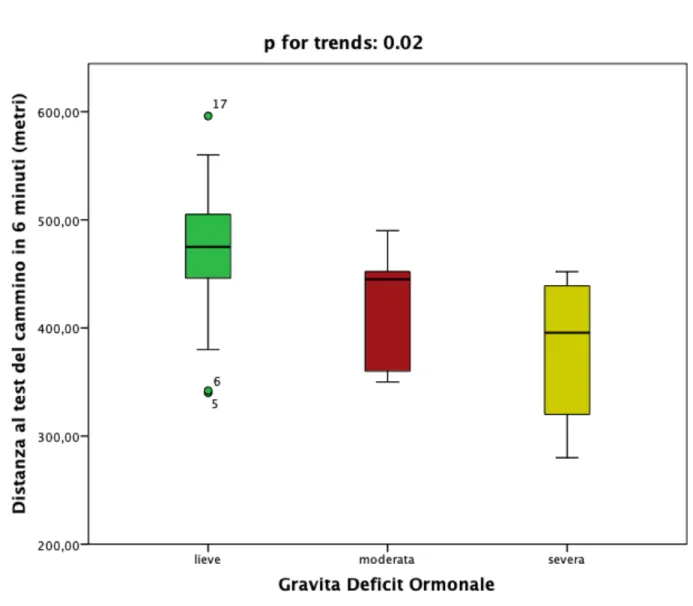
In merito ai dati emersi dall’esame Risonanza Magnetica cardiaca, i pazienti con deficit multipli risultavano avere volumi ventricolari destri più aumentati, con lieve riduzione della funzione di pompa e maggior grado di ipertrofia ventricolare destra. Dividendo in base alla gravità dei deficit ormonali (1 deficit= MHDS lieve, 2= MDHS media, 3 o più deficit= MHDS grave) una differenza statisticamente significativa risultava per quanto riguarda la distanza percorsa al test del cammino (467.94 ± 70.62 vs 419.40± 61.33 vs 380.12±66.12 in lieve, moderata e lieve, rispettivamente, p:0.02) (Figura 7)
Figura 7 differenza in termini di distanza percorsa al test del cammino in 6 minuti in base alla gravità dei deficit ormonali multipli
Una simile differenza veniva riscontrata sia in riguardo alle resistenze vascolari polmonari (figura 8) (7,28±2,98 vs 8,59±4,38 vs 14,2 ±0,49 in lieve moderata e severa rispettivamente, p: 0.003) sia in merito ai volumi ventricolari destri
telediastolico (170.44±53.85 vs 178.15±30.26 vs 249.85±43.46, p: 0.02), telesistolico (91,14±48,05 vs 111,0±53,6 vs 186,5 ±10,91, in lieve moderata e severa
rispettivamente, p: 0.007) e la frazione di eiezione del ventricolo destro (37,75±8,97 vs 34,28±14,85 vs 20,7±14,59, in lieve moderata e severa rispettivamente, p:0.05) misurati tramite risonanza magnetica.
4.3 Impato sugli outcomes dei deficit ormonali multipli nella PAH
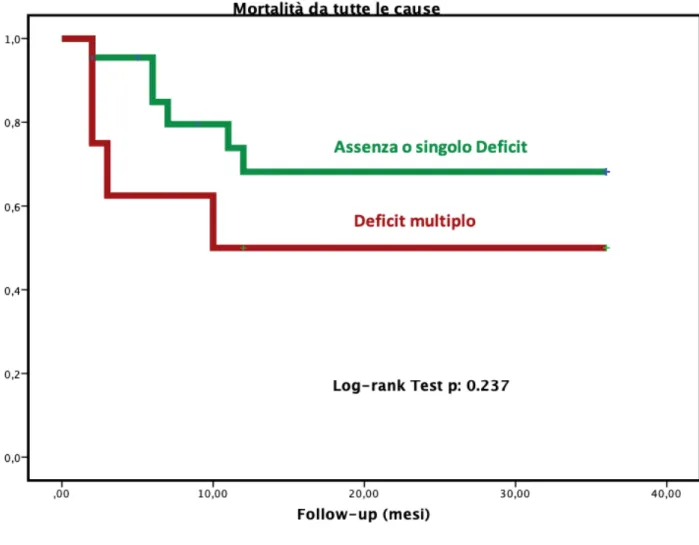
Nel corso del follow-up un totale di 14 eventi sono stati registrati, di cui dieci morti e quattro ospedalizzazioni. Nello specifico, fra i pazienti del gruppo senza o con singolo deficit (n:22), 6 (27%) sono deceduti e 3 sono stati ospedalizzati (per un totale di 9 eventi). Nel gruppo con deficit multiplo (n:8) invece 4 pazienti sono
deceduti (50%) mentre 1 paziente è stato ospedalizzato. Nonostante una differenza nell’end-point combinato di circa il 20% fra i due gruppi ed una differenza del 23% nell’occorrenza dell’evento morte, il basso sample size non ha permesso il
raggiungimento della significatività statistica (p: 0.278 per il composito di mortalità ed ospedalizzazione, p:0.237 per la mortalità).
5. Discussione
Volendo riassumere I principali risultati del presente studio :
1) I deficit ormonali sono molto comuni nell’ipertensione arteriosa polmonare idiopatica. Nel nostro campione di 30 pazienti, il 97% presentava almeno deficit di almeno uno degli assi ormonali studiati, il 43% era affetto da deficit ormonali
concomitanti. Tra I singoli deficit quello di DHEA-S è stato di più frequente riscontro nella popolazione.
2) I deficit ormonali multipli identificano un sottogruppo di pazienti caratterizzati da peggiore status clinico, capacità di esercizio fisico, emodinamica polmonare. Inoltre I pazienti con più di due deficit concomitanti mostravano volumi ventricolari aumentati e peggiore funzione sistolica del ventricolo destro.
3) Nonostante siano stati registrati un maggior numero di eventi (sia composito di mortalità ed ospedalizzazione, sia mortalità da tutte le casi) nel gruppo di pazienti con deficit ormonale multiplo, tale differenza non ha raggiunto una significatività statistica. Tale dato potrebbe essere stato probabilmente inficiato dalla bassa numerosità campionaria di tale studio.
5.1 I deficit ormonali multipli nell’ipertensione arteriosa polmonare: significato clinico e meccanismi sottostanti.
A nostra conoscenza, il presente rappresenta il primo studio che ha
sistematicamente analizzato un pannello esteso di assi ormonali nella PAH. Secondo I nostri dati la presenza di un deficit ormonale multiplo si associa a una peggiore classe funzionale WHO e a una peggiore capacità di esercizio fisico nel paziente con PAH. Inoltre i pazienti con deficit ormonali multipli hanno un peggior profilo emodinamico caratterizzato da maggiori pressioni polmonari, e ridotta gittata cardiaca; il tutto che quindi si traduce in resistenze polmonari più elevate. Un altro dato rilevante è la presenza di maggiori volumi e ridotta funzione sistolica
ventricolare destra nei pazienti con deficit ormonali multipli, che indicano maggiore overload delle camere cardiache destre e con iniziale compromissione della funzione sistolica. Un dato oltremodo interessante è che con l’aumentare dei deficit ormonali si assiste a un progressivo peggioramento di questi importanti parametri clinici. In definitiva la presenza di deficit ormonali concomitanti denota la presenza di un sottogruppo di pazienti (secondo la nostra casistica circa il 43%) con peggiori profili emodinamici, ventricolare destro e ridotta capacità di esercizio fisico. La presenza di aumentate resistenze vascolari polmonari è senz’altro di notevole importanza, considerando che si tratta del più forte predittore indipendente di mortalità secondo i
dati del registro americano REVEAL, il più grande del mondo78. Tale parametro
costituisce una valutazione diretta del grado di vasculopatia polmonare, che è la
noxa patogena che innesca successivamente l’avanzamento della malattia12.
Successivamente è facile ipotizzare come l’aumento del post-carico polmonare si
destro-circolo polmonare è un regime a basso regime pressorio ma elevato flusso80. All’instaurarsi delle aumentate pressioni il ventricolo destro non ha la capacità di adattarsi così come la controparte sinistra, motivo per il quale è molto più facile che
vada in contro a dilatazione e successivamente scompenso clinico franco44. Infatti
l’insorgenza dello scompenso cardiaco destro è spesso l’evento che peggiora la
prognosi dei pazienti con PAH81. La presenza di deficit anabolici multipli nella PAH
può sottendere la pronunciata attivazione infiammatoria presente in questi pazienti. Elevati livelli di mediatori pro-infiammatori circolanti, come l'interleuchina (IL)-1b, IL-2, IL-4, IL-5, IL-6, IL-6, IL-8,
IL-10, e IL-12p70 e fattore di necrosi tumorale (TNF)-a, sono stati riscontrati sia in
modelli animali che in pazienti con PAH82,83 e sono stati associati a peggiore
prognosi82.
La compromissione dell'esercizio fisico è una caratteristica ben nota della PAH. Tradizionalmente questa caratteristica era attribuita allo status di bassa portata cardiaca o alla disfunzione dello scambio respiratorio. Tuttavia, diversi studi hanno
evidenziato una vasta gamma di anomalie di entrambi i muscoli scheletrici76,84 e
respiratori84 nei pazienti con PAH che possono contribuire già precedente
menzionata rilevante limitazione dell'esercizio. La disfunzione muscolare nella PAH ha le seguenti caratteristiche: un cambio delle Fibre da un fenotipo "resistente" (tipo I) a fibre "veloce" (tipo II), ridotta densità capillare muscolare, minore attività
enzimatica aerobica, alterata biogenesi mitocondriale / funzione, e aumento della degradazione delle proteine muscolari mediata dal sistema di protezione
dell'ubiquitina/proteasome (UPS), anomalie mitocondriali, e modificato
Un possibile meccanismo alla base della compromissione muscolare PAH può essere ricondotto all'inibizione degli assi ormonali/metabolici, che a loro volta sono essenziali per promuovere la sintesi proteica e protezione delle proteine muscolari
dalla degradazione85.
Molti assi anabolici infatti agiscono soprattutto attraverso l'attivazione del recettore dell'insulina substrato (IRS)/fosfatidilinositolo 3 chinasi (PI3K)/Akt con un ampio
spettro di risultati a valle86. Il signaling insulinico è compromessa da un'attivazione
infiammatoria cronica86. Infatti, TNF-a e IL-6 possono ridurreil segnale insulinico
attraverso l'inibizione del pathway IRS/PI3K/Akt 87anche nelle cellule muscolari76.
Una ridotta attivazione di Akt e un aumento dell'atrogin-1 e l'attività MURF-1 sono
stati riportati in campioni muscolari di pazienti con IPAH84. Si può ipotizzare che il
deterioramento del pathway IRS/PI3K/ Akt e l’attivazione infiammatoria sono entrambi
legato all'attivazione del sistema dell’Ubiquitina/Proteasoma in PAH che contribuisce al consumo muscolare nella PAH. Dato interessante è che il Diabete Mellito in
generale sembrerebbe essere un fattore prognostico negativo nel decorso della
PAH88. L’IGF-1 ha un pathway di azione molto simile all’insulina ed è considerato il
più forte ormone anabolizzante nel corpo umano89. L’associazione tra il deficit di
IGF-1 è ridotta funzione cardiaca destra è stata già dimostrata in passato70. Un dato
interessante proviene inoltre da studi nell’animale che dimostrano come IGF-1 attraverso l’induzione di iNOS rivesta un ruolo protettivo sulla vascolarizzazione
polmonare in modelli animali di PAH90. Uno dei mediatori più studiati nella PAH è il
DHEA. Recentemente uno studio ha mostrato in maniera inequivoca come i bassi livelli di DHEA-S siano associati a peggiore prognosi nei pazienti con PAH
Similarmente un ruolo protettivo sul letto vascolare polmonare è stato mostrato in
modelli animali92. Recentemente anche il ruolo delle malattie tiroidee nella PAH è
5.2 I deficit anabolici nelle malattie cardiovascolari
Negli ultimi due decenni la comunità scientifica ha prestato molta attenzione ai rapporti fra ormoni e sistema cardiovascolare. Le prime evidenze risalgono infatti ai primi anni 90 quando è stato dimostrato come i pazienti ipopituitarici morissero più
frequentemente di patologie cardiovascolari94. Le maggiori attenzioni sono state,
infatti, rivolte nei confronti dell’Insufficienza Cardiaca Cronica (CHF). Una delle tesi suggerite da diversi autori è che la storia naturale dell'insufficienza cardiaca non solo è secondaria alla sovraespressione di molecole biologicamente attive appartenenti al sistema adrenergico, renina-angiotensina e citochine, ma anche all'abbassamento
della regolazione degli assi ormonali cardines67. Diversi studi indipendenti
documentano, infatti, che la mancanza di diversi ormoni siano associate non solo ad una compromissione delle prestazioni fisiche, ma anche ad una ridotta
sopravvivenza. In particolare, è stata condotta una notevole serie di studi preliminari focalizzati sul ruolo delle singole anomalie ormonali o deficit in CHF. In breve, il ruolo giocato da ogni elemento del cosiddetto asse somatotropo (IGF-1 e GH) è stato ampiamente studiato negli ultimi 20 anni, con diversi gruppi indipendenti hanno riferito che livelli sierici IGF-1 più bassi sono associati ad una maggiore attivazione
infiammatoria95, aumento della sarcopenia96, riduzione della classe funzionale e della
capacità di esercizio69,97, e tassi di sopravvivenza ridotti69,98,99. Inoltre, la mancanza
di GH è anche associata a uno stato clinico alterato, al rimodellamento del VL, alla
disfunzione del VR e ad un aumento della mortalità globale70. Allo stesso modo, la
ridotta carenza di testosterone è anche associata ad una significativa
fisico e di scarsi risultati nei pazienti con CHF100. Allo stesso modo, anche la
sindrome T3 bassa101 e la resistenza all'insulina102 sono state associate ad un
aumento della mortalità in CHF con ridotta frazione di eiezione.
Questo corpus coerente di prove ha rappresentato la base scientifica per uno studio che ha generato ipotesi condotto da Jankowska e colleghi nel 2006. In una coorte di 208 pazienti maschi con CHF, questo studio è stato il primo a dimostrare che la concomitante carenza di tre assi anabolizzanti (Testosterone, DHEA-S e IGF-1) è
associata indipendentemente con una ridotta sopravvivenza72.
Il nostro studio potrebbe essere considerato il primo step per l’implementazione di studi simili anche nel contesto della PAH.
5.3 Implicazioni cliniche
A differenza di tanti altri biomarcatori utilizzati e proposti abitualmente nella gestione
quotidiana della PAH103, le carenze ormonali hanno l'ulteriore vantaggio di essere
potenzialmente anche un valido obiettivo terapeutico, attraverso l'applicazione clinica della terapia ormonale sostitutiva. Diversi gruppi indipendenti hanno dimostrato che
la terapia sostitutiva con GH73,74 o Testosterone75,104 è associata a un notevole
miglioramento di alcuni solidi endpoint surrogati come il picco VO2. Inoltre, l’insulino
resistenza sta emergendo come promettente obiettivo terapeutico in CHF105,106. In
sintesi, il nostro studio ha due principali implicazioni cliniche. Da un lato, i nostri dati evidenziano l'importanza di valutare lo stato ormonale in pazienti con PAH al fine di stratificare il rischio per implementare strategie terapeutiche più aggresssive. D'altra parte, i dati dello studio attuale forniscono una solida base per l'implementazione di studi con terapia sostitutiva che potrebbe migliorare lo stato clinico di questi pazienti.
5.4 Limitazioni
La principale limitazione di questo studio è sicuramente la natura retrospettiva dell’osservazione. Inoltre lo studio è fortemente limitato dalla bassa numerosità campionaria. Tuttavia la PAH è una malattia rara ed il campione presentato è quello solitamente presentato in numerosi studi. La limitazione del sample size inoltre non ha reso possibile cercare di valutare differenze negli outcomes clinici valutati. Inoltre un’altra limitazione è che non sono stati compiuti test dinamici per valutare lo status ormonale dei pazienti. Inoltre non sono presenti valutazioni omronali ripetute nel tempo.
Infine non sono state effettuate misurazioni come citochine, mediatori
dell’infiammazione o funzione muscolare che avrebbero potuto chiarire meglio il ruolo fisiopatologico dei deficit ormonali multipli nella PAH.
Tuttavia lo scopo di questo studio era quello di generare un’ipotesi in merito al ruolo dei deficit ormonali nella PAH, senza lo scopo di trovare alcun link meccanicistico per spiegare questo fenomeno clinico.
Studi futuri condotti in maniera prospettiva e coinvolgenti un numero maggiore di pazienti potranno essere condotti sulla base di queste evidenze.
6. Conclusioni
In base alle nostre osservazioni, i deficit ormonali sono molto comuni nell’Ipertensione Arteriosa Polmonare idiopatica. I deficit ormonali multipli
caratterizzano un sottogruppo di pazienti che presenta peggiore capacità di esercizio fisico, emodinamica polmonare, dimensione e funzione ventricolare destra. Tali parametri risultavano essere ancor di più compromessi quando il deficit ormonale era di gravità maggiore. Studi futuri dovranno confermare questi dati su campioni di pazienti più grandi cercando possibilmente di valutare l’eventuale associazione tra deficit ormonali multipli e mortalità.
7. Referenze
1. Galiè N, Humbert M, Vachiery J-L, et al. 2015 ESC/ERS Guidelines for the
diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2015;46(4):ehv317. doi:10.1093/eurheartj/ehv317
2. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions
and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. doi:10.1183/13993003.01913-2018
3. Rudski LG, Gargani L, Armstrong WF, et al. Stressing the Cardiopulmonary
Vascular System: The Role of Echocardiography. J Am Soc Echocardiogr. 2018;31(5):527-550.e11. doi:10.1016/j.echo.2018.01.002
4. Peacock AJ, Naeije R, Rubin LJ. Pulmonary Circulation : Diseases and Their
Treatment. https://www.crcpress.com/Pulmonary-Circulation-Diseases-and-
Their-Treatment-Fourth-Edition/Peacock-Naeije-Rubin/p/book/9781498719919. Accessed October 3, 2017.
5. Bossone E, Dellegrottaglie S, Patel S, et al. Multimodality Imaging in
Pulmonary Hypertension. Can J Cardiol. 2015;31(4):440-459. doi:10.1016/j.cjca.2015.02.012
6. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure
during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888-894. doi:10.1183/09031936.00145608
7. Condliffe R, Kiely DG, Peacock AJ, et al. Connective Tissue Disease–
associated Pulmonary Arterial Hypertension in the Modern Treatment Era. Am J Respir Crit Care Med. 2012;179(2). doi:10.1164/rccm.200806-953OC
8. Nagel C, Marra AM, Benjamin N, et al. Reduced right ventricular output
reserve in patients with systemic sclerosis and mildly elevated pulmonary arterial pressures. Arthritis Rheumatol. January 2019. doi:10.1002/art.40814
9. Douschan P, Kovacs G, Avian A, et al. Mild Elevation of Pulmonary Arterial
Pressure as a Predictor of Mortality. Am J Respir Crit Care Med. 2018;197(4):509-516. doi:10.1164/rccm.201706-1215OC
10. Lewis GD, Bossone E, Naeije R, et al. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation.
2013;128(13):1470-1479. doi:10.1161/CIRCULATIONAHA.112.000667 11. Rosenkranz S, Lang IM, Blindt R, et al. Pulmonary hypertension associated
with left heart disease: Updated Recommendations of the Cologne Consensus Conference 2018. Int J Cardiol. 2018;272:53-62.
doi:10.1016/j.ijcard.2018.08.080
12. Tuder RM, Archer SL, Dorfmüller P, et al. Relevant Issues in the Pathology and Pathobiology of Pulmonary Hypertension. J Am Coll Cardiol.
2013;62(25):D4-D12. doi:10.1016/j.jacc.2013.10.025
13. Song J, Eichstaedt CA, Viales RR, et al. Identification of genetic defects in pulmonary arterial hypertension by a new gene panel diagnostic tool. Clin Sci (Lond). 2016;130(22):2043-2052. doi:10.1042/CS20160531
14. Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186(8):790-796. doi:10.1164/rccm.201203-0383OC
15. Olschewski A, Berghausen EM, Eichstaedt CA, et al. [Pathobiology, pathology and genetics of pulmonary hypertension: Recommendations of the Cologne
Consensus Conference 2016]. Dtsch Med Wochenschr. 2016;141(S 01):S4-S9. doi:10.1055/s-0042-114520
16. Gorter TM, van Veldhuisen DJ, Bauersachs J, et al. Right heart dysfunction and failure in heart failure with preserved ejection fraction: mechanisms and management. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2018;20(1):16-37. doi:10.1002/ejhf.1029
17. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443-455. doi:10.1038/nrcardio.2011.87
18. Galié N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61(2):227-237.
http://www.ncbi.nlm.nih.gov/pubmed/14736539. Accessed July 7, 2016. 19. Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of
pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-41. doi:10.1016/j.jacc.2013.10.029
20. Humbert M, Yaici A, de Groote P, et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: Clinical characteristics at diagnosis and long-term survival. Arthritis Rheum. 2011;63(11):3522-3530. doi:10.1002/art.30541
21. Parikh R V., Scherzer R, Nitta EM, et al. No Title. 2014;28(4). doi:10.1097/QAD.0000000000000124
22. Voges I, Al-Mallah MH, Scognamiglio G, Di Salvo G. Right Heart-Pulmonary Circulation Unit in Congenital Heart Diseases. Heart Fail Clin. 2018;14(3):283-295. doi:10.1016/j.hfc.2018.02.005
23. Hirschfeld S, Meyer R, Schwartz DC, Kofhagen J, Kaplan S. The
echocardiographic assessment of pulmonary artery pressure and pulmonary vascular resistance. Circulation. 1975;52(4):642-650.
http://www.ncbi.nlm.nih.gov/pubmed/1157277. Accessed August 13, 2015. 24. Salzano A, Sirico D, Golia L, et al. [The portopulmonary hypertension: an
overview from diagnosis to treatment]. Monaldi Arch chest Dis = Arch Monaldi per le Mal del torace. 2013;80(2):66-68. doi:10.4081/monaldi.2013.81
25. Dodson MW, Brown LM, Elliott CG. Pulmonary Arterial Hypertension. Heart Fail Clin. 2018;14(3):255-269. doi:10.1016/j.hfc.2018.02.003
26. Humbert M, Sitbon O, Yaïci A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36(3):549-555. doi:10.1183/09031936.00057010
27. Swift AJ, Rajaram S, Hurdman J, et al. Noninvasive Estimation of PA Pressure, Flow, and Resistance With CMR Imaging. JACC Cardiovasc Imaging. 2013;6(10):1036-1047. doi:10.1016/j.jcmg.2013.01.013
28. Escribano-Subias P, Blanco I, López-Meseguer M, et al. Survival in pulmonary hypertension in Spain: insights from the Spanish registry. Eur Respir J.
2012;40(3):596-603. doi:10.1183/09031936.00101211
29. Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad Dermatol. 1980;2(2):161-170. doi:10.1016/s0190-9622(80)80396-3
30. Albert ML, Pearce SFA, Francisco LM, et al. Immature dendritic cells
phagocytose apoptotic cells via αvβ 5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188(7):1359-1368.
doi:10.1084/jem.188.7.1359
surface tissue factor procoagulant activity. Lab Invest. 1996;75(2):281-289. http://www.ncbi.nlm.nih.gov/pubmed/8765328. Accessed September 6, 2019. 32. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition
(EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3). doi:10.1186/ar1963
33. Meyer G, Vicaut E, Danays T, et al. Fibrinolysis for patients with intermediate-risk pulmonary embolism. N Engl J Med. 2014;370(15):1402-1411.
doi:10.1056/NEJMoa1302097
34. Matucci-Cerinic M, Krieg T, Guillevin L, et al. Elucidating the burden of
recurrent and chronic digital ulcers in systemic sclerosis: long-term results from the DUO Registry. Ann Rheum Dis. 2016;75(10):1770-1776.
doi:10.1136/annrheumdis-2015-208121
35. Akashi YJ, Musha H, Kida K, et al. Reversible ventricular dysfunction takotsubo cardiomyopathy. Eur J Heart Fail. 2005;7(7):1171-1176. doi:10.1016/j.ejheart.2005.03.011
36. Allanore Y, Borderie D, Périanin A, Lemaréchal H, Ekindjian OG, Kahan A. Nifedipine protects against overproduction of superoxide anion by monocytes from patients with systemic sclerosis. Arthritis Res Ther. 2005;7(1):R93-100. doi:10.1186/ar1457
37. Avouac J, Wipff J, Goldman O, et al. Angiogenesis in systemic sclerosis: impaired expression of vascular endothelial growth factor receptor 1 in
endothelial progenitor-derived cells under hypoxic conditions. Arthritis Rheum. 2008;58(11):3550-3561. doi:10.1002/art.23968
38. Lauber K, Bohn E, Kröber SM, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113(6):717-730. doi:10.1016/s0092-8674(03)00422-7
39. Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther. 2013;15(3):215.
doi:10.1186/ar4230
40. Manetti M, Milia AF, Guiducci S, Romano E, Matucci-Cerinic M, Ibba-Manneschi L. Progressive loss of lymphatic vessels in skin of patients with systemic sclerosis. J Rheumatol. 2011;38(2):297-301.
doi:10.3899/jrheum.100767
41. Carrier M, Righini M, Djurabi RK, et al. VIDAS D-dimer in combination with clinical pre-test probability to rule out pulmonary embolism. A systematic review of management outcome studies. Thromb Haemost. 2009;101(5):886-892. http://www.ncbi.nlm.nih.gov/pubmed/19404542. Accessed January 9, 2018.
42. Jacobs W, van de Veerdonk MC, Trip P, de Man F, Heymans MW, Marcus JT, Kawut SM, Bogaard HJ, Boonstra A VNA. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest. 2014;145(6):1230-1236. doi:10.1378/chest.13-1291
43. Marra AMAM, Egenlauf B, Ehlken N, et al. Change of right heart size and function by long-term therapy with riociguat in patients with pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Int J Cardiol. 2015;195:19-26. doi:10.1016/j.ijcard.2015.05.105
44. Vonk Noordegraaf A, Westerhof BE, Westerhof N. The Relationship Between the Right Ventricle and its Load in Pulmonary Hypertension. J Am Coll Cardiol. 2017;69(2):236-243. doi:10.1016/j.jacc.2016.10.047
Heart Failure With Preserved and Reduced Ejection Fraction. JACC Cardiovasc Imaging. October 2018. doi:10.1016/j.jcmg.2018.08.020
46. Ghio S, Temporelli PL, Klersy C, et al. Prognostic relevance of a non-invasive evaluation of right ventricular function and pulmonary artery pressure in patients with chronic heart failure. Eur J Heart Fail. 2013;15(4):408-414. doi:10.1093/eurjhf/hfs208
47. Ghio S, Guazzi M, Scardovi AB, et al. Different correlates but similar
prognostic implications for right ventricular dysfunction in heart failure patients with reduced or preserved ejection fraction. Eur J Heart Fail. 2017;19(7):873-879. doi:10.1002/ejhf.664
48. Ghio S, Recusani F, Klersy C, et al. Prognostic usefulness of the tricuspid annular plane systolic excursion in patients with congestive heart failure secondary to idiopathic or ischemic dilated cardiomyopathy. Am J Cardiol. 2000;85(7):837-842. http://www.ncbi.nlm.nih.gov/pubmed/10758923. Accessed March 19, 2019.
49. Ferrara F, Gargani L, Armstrong WF, et al. The Right Heart International Network (RIGHT-NET): Rationale, Objectives, Methodology, and Clinical Implications. Heart Fail Clin. 2018;14(3). doi:10.1016/j.hfc.2018.03.010 50. Olsson KM, Halank M, Egenlauf B, et al. Decompensated right heart failure,
intensive care and perioperative management in patients with pulmonary hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol. 2018;272:46-52.
doi:10.1016/j.ijcard.2018.08.081
51. Grünig E, Tiede H, Enyimayew EO, et al. Assessment and prognostic relevance of right ventricular contractile reserve in patients with severe pulmonary hypertension. Circulation. 2013;128(18):2005-2015.
doi:10.1161/CIRCULATIONAHA.113.001573
52. Marra AM, Grünig E. Assessment of pulmonary vascular response to exercise with Doppler-echocardiography: State of the art? J Thorac Dis. 2017;9(10). doi:10.21037/jtd.2017.09.45
53. Blumberg FC, Arzt M, Lange T, Schroll S, Pfeifer M, Wensel R. Impact of right ventricular reserve on exercise capacity and survival in patients with
pulmonary hypertension. Eur J Heart Fail. 2013;15(7):771-775. doi:10.1093/eurjhf/hft044
54. Chia E-M, Lau EMT, Xuan W, Celermajer DS, Thomas L. Exercise testing can unmask right ventricular dysfunction in systemic sclerosis patients with normal resting pulmonary artery pressure. Int J Cardiol. 2016;204:179-186.
doi:10.1016/j.ijcard.2015.11.186
55. Hsu S, Houston BA, Tampakakis E, et al. Right Ventricular Functional Reserve in Pulmonary Arterial Hypertension. Circulation. 2016;133(24):2413-2422. doi:10.1161/CIRCULATIONAHA.116.022082
56. Mancini D, Katz S, Donchez L, Aaronson K. Coupling of hemodynamic
measurements with oxygen consumption during exercise does not improve risk stratification in patients with heart failure. Circulation. 1996;94(10):2492-2496. http://www.ncbi.nlm.nih.gov/pubmed/8921793. Accessed June 21, 2016. 57. Bossone E, Gargani L, Kasprzak JD, et al. No Title. Heart Fail Clin.
2018;14(3). https://linkinghub.elsevier.com/retrieve/pii/S1551713618300151. Accessed October 17, 2018.
58. Sanz J, Dellegrottaglie S, Kariisa M, et al. Prevalence and correlates of septal delayed contrast enhancement in patients with pulmonary hypertension. Am J