INDICE
1. Riassunto………..
22. Introduzione………..
4Criteri classificativi
Caratteristiche delle Sindromi Overlap
Connettivite Mista
- Cenni Storici
- Epidemiologia
- Eziopatogenesi
- Anatomia patologica
- Clinica
- Esami bioumorali
- Criteri classificativi di MCTD
- Prognosi
- Terapia
Sindrome da anticorpi anti-sintetasi
- Quadro clinico
- Diagnosi
- Prognosi
- Terapia
Scleromiosite
Sindrome Rhupus
Sclerolupus
Sclerosi Sistemica/ Artrite Reumatoide
LES/ Sindrome di Sj gren
Conclusioni
3. Scopo dello studio………
404. Pazienti e metodi………..
405. Risultati………..
426. Discussione………..
557. Appendice……….
588. Bibliografia………
669. Ringraziamenti……….
71Riassunto
Le diverse malattie autoimmuni del tessuto connettivo, denominate “connettiviti sistemiche” sono malattie caratterizzate da un processo infiammatorio cronico che coinvolge estesamente vari organi e tessuti dell’organismo. I soggetti affetti da tali connettiviti sistemiche presentano manifestazioni cliniche estremamente eterogenee, non “malattia-specifiche” che rendono spesso difficile la distinzione tra le differenti patologie. Per tale motivo sono stati istituiti degli strumenti per classificare i diversi quadri di connettivite, detti Criteri Classificativi, che hanno valore, principalmente, per studi epidemiologici e per la ricerca clinica.
Talvolta accade che alcuni pazienti soddisfino i criteri di più di una connettivite, dando luogo a forme caratterizzate dall’associazione di due o più malattie autoimmuni in uno stesso individuo. Prendono il nome di “Sindromi Overlap definite”. Può anche accadere che alcuni soggetti non soddisfino il numero minimo di criteri per una determinata forma, ma che comunque presentino manifestazioni cliniche specifiche di più malattie autoimmuni definite, condizioni denominate “Sindromi Overlap indefinite”.
Lo scopo di questa tesi è stato così quello di individuare, presso la UO di Reumatologia della Azienda Ospedaliera Universitaria
Pisana, i pazienti affetti da Sindrome Overlap e di descriverne le principali caratteristiche cliniche e sierologiche.
Sono stati così studiati un totale di 93 soggetti (10 maschi e 83 femmine) affetti da Sindrome Overlap che all’esordio, come manifestazioni cliniche principali, hanno presentato artralgie (52%), fenomeno di Raynaud (37%) e artrite (26%) e nel tempo ne hanno sviluppate ulteriori, quali xerostomia (63%) e xeroftalmia (60%).
Per quanto riguarda il profilo sierologico hanno mostrato positività per gli autoanticorpi ANA nel 91% dei casi e a seguire positività del Ra test nel 43% dei casi.
Gli obiettivi che ci proponiamo sono quelli di aumentare la casistica di pazienti affetti da Sindrome Overlap, per poter procedere a un confronto tra le varie tipologie di Sindromi Overlap definite, al confronto tra Sindromi Overlap definite e Connettiviti definite isolate e alla ricerca, all’ interno del gruppo delle Sindromi Overlap indefinite di qualcosa (sintomo/segno, anticorpo ecc. ), che le unifichi come entità nosologica a sé stante. Il fine ultimo è quello di giungere nel tempo ad un corretto raggruppamento delle malattie autoimmuni sistemiche.
Sindromi Overlap o Sindromi
da Sovrapposizione
La diagnosi di una malattia è strettamente legata alla conoscenza della sua eziologia e dei meccanismi patogenetici che ne sono alla base.
Le diverse malattie autoimmuni del tessuto connettivo, che prendono il nome di “connettiviti sistemiche” sono malattie caratterizzate da un processo infiammatorio cronico che coinvolge estesamente vari organi e tessuti dell’organismo. Tali affezioni hanno in comune aspetti di ordine diverso.
Dal punto di vista epidemiologico condividono ad esempio un elevato rapporto femmine/maschi e un’elevata prevalenza in giovane età.
Alla base di tali patologie si riscontrano inoltre disordini immunologici, quali la attivazione policlonale di linfociti B, la produzione di autoanticorpi e l’aumento dei livelli di immunocomplessi circolanti.
Per quanto riguarda l’immunogenetica è stata osservata l’associazione con specifici geni coinvolti nella regolazione della risposta immunitaria, come le molecole HLA di classe 1 e 2.
Infine tali affezioni condividono numerosi aspetti clinici, come sarà evidenziato nelle pagine seguenti.
Al di là di queste osservazioni non esiste, attualmente, una chiara e completa conoscenza dell’eziopatogenesi delle connettiviti sistemiche.
Si propende per un’ eziopatogenesi di tipo multifattoriale, nella quale sembrano giocare un ruolo fondamentale la predisposizione genetica, sulla quale vanno ad agire fattori di tipo ambientale(infettivi, farmacologici ecc), responsabili dell’innesco dell’espressione della malattia autoimmune.
Criteri classificativi
Le malattie autoimmuni sistemiche sono caratterizzate dalla presenza di manifestazioni cliniche estremamente eterogenee, spesso non “malattia-specifiche” e quindi presenti in diverse condizioni.
Inoltre il quadro clinico e l’impegno di organo possono variare, all’interno di una stessa malattia, sia tra pazienti sia nello stesso paziente nel corso del follow-up.
Tale estrema variabilità può rendere difficile la distinzione delle differenti patologie.
Di fronte a tale difficoltà è venuta a delinearsi l’esigenza di creare degli strumenti per classificare i diversi quadri di connettivite e sono stati istituiti dei Criteri Classificativi riconosciuti a livello internazionale (vedi appendice).
I criteri classificativi permettono alle casistiche di vari centri di ricerca di uniformarsi e di poter procedere al paragone tra gruppi di pazienti provenienti da differenti centri, che hanno intrapreso indagini diagnostiche e trials terapeutici.
Hanno quindi valore, principalmente, per studi epidemiologici e per la ricerca clinica e sono invece inadeguati per la stima di una prognosi. Non si propongono l’inclusione o l’esclusione di un singolo paziente da una determinata diagnosi, anche se attualmente, nella pratica clinica si utilizzano ampiamente anche per la diagnosi.
Bisogna tuttavia ricordare che anche i più raffinati criteri classificativi costituiscono comunque delle approssimazioni di meccanismi patogenetici che sono complesse interazioni tra fattori genetici, endocrini e ambientali.
La struttura di questi criteri si fonda infatti su caratteristiche cliniche e sull’individuazione degli aspetti sierologici e immunogenetici più comuni di una data malattia, con l’esclusione di altre manifestazioni meno frequenti. Sono stati
creati con tentativi di selezionare ragionevoli combinazioni di sensibilità e specificità.
Tra i più grossi limiti di questi criteri, che portano ad ampi raggruppamenti c’è quello di non consentire l’analisi di sottoinsiemi omogenei di pazienti nei quali, presi a sé, potrebbero operare meccanismi patogenetici distinti. Talvolta accade che alcuni pazienti, pur presentando quadri suggestivi di una connettivite, non possano essere inquadrati in maniera precisa in una di queste malattie. Questo può accadere per due motivi:
1) I pazienti non soddisfano il numero minimo di criteri per una determinata forma e siamo quindi di fronte ad entità cliniche sfumate che prendono il nome di “Connettiviti indifferenziate” (UCTD).
2) I pazienti soddisfano i criteri di più di una connettivite dando luogo a forme caratterizzate dall’ associazione di due o più malattie autoimmuni in uno stesso individuo. Prendono il nome di “Sindromi Overlap”.
Le combinazioni più frequenti sono le seguenti.
La “Connettivite mista o MCTD”, caratterizzata dall’associazione tra Lupus Eritematoso Sistemico (LES), Sclerosi Sistemica (SSc) e Polimiosite/Dermatomiosite (PM/DM).
La compresenza di LES e Artrite Reumatoide (AR), detta “Sindrome Rhupus”.
Lo "Sclerolupus", caratterizzato dall’associazione di Sclerosi Sistemica e Lupus Eritematoso Sistemico.
La combinazione tra Sclerosi Sistemica, Sindrome di Sj gren (SS) e Cirrosi Biliare Primitiva (CBP).
Infine l’associazione della Sindrome di Sj gren con l’Artrite Reumatoide oppure con la Sclerosi Sistemica.
La presenza di queste forme cliniche, siano esse UCTD o Sindromi Overlap, sembra confermare l’ipotesi di comuni meccanismi eziopatogenetici, tra le diverse tipologie di connettiviti sistemiche.
Caratteristiche delle sindromi overlap
Una Sindrome Overlap si identifica sulla base di due elementi che sono l’interessamento clinico e/o la presenza di particolari specificità autoanticorpali.
Riguardo all’interessamento clinico devono essere presenti manifestazioni abbastanza specifiche di malattia, quali ad esempio un’artrite erosiva, suggestiva di Artrite Reumatoide, evidenziabile all’esame radiografico o una sclerosi cutanea/sclerodattilia suggestiva di Sclerosi Sistemica.
La presenza di manifestazioni meno specifiche, quali il fenomeno di Raynaud o un’alveolite non possono invece essere usate, da sole, per definire una sindrome overlap, sono infatti presenti in un ampio numero di malattie reumatiche.
Alcuni studi hanno messo in evidenza come l’artrite erosiva possa occasionalmente presentarsi anche in corso di Lupus Eritematoso Sistemico e di Sclerosi Sistemica e si può pertanto ipotizzare che caratteristiche simil-Artrite Reumatoide siano, in qualche paziente, manifestazioni della sua malattia di base.
Un altro caso è quello della Sindrome di Sj gren: tutte le malattie reumatiche possono essere accompagnate dai sintomi e segni di secchezza oculare e secchezza delle fauci, tipici della suddetta malattia che viene in questi casi definita ‘Sindrome di Sj gren secondaria’ al disturbo reumatico in questione.
Caratteristica che si osserva nelle malattie reumatiche sistemiche è la presenza, nel sangue, di anticorpi antinucleo (ANA): immunoglobuline specifiche per antigeni self (acidi nucleici o proteine leganti gli acidi nucleici, BP). Sono autoanticorpi non organo specifici che raggiungono frequenza e livelli elevati proprio nelle connettiviti e mostrano ben definite specificità antigeniche.
I seguenti autoanticorpi sono osservabili in alcune Sindromi Overlap (vedi tabella 1):
- anti-U1 RNP: il marker sierologico della Connettivite Mista;
- anti-tRNA (il più frequente è l’anti Jo-1): associato ad un quadro caratterizzato da PM/DM, artrite e fibrosi polmonare;
- anti-PM/Scl: in pazienti con overlap PM/DM e SSc; - anti-La/SSB: in pazienti con overlap LES e SS.
L’importanza di questi anticorpi, oltre che fornire un punto di partenza per paragonare tra di loro studi, è quella di far presupporre l’esistenza di meccanismi eziopatogenetici comuni, seppur ancora sconosciuti, alla base dell’ insorgenza della patologia in individui diversi. E la conoscenza e la scoperta di tali meccanismi giustificherebbe l’utilizzo del termine ‘diagnosi’ nella descrizione di queste sindromi.
Tabella 1. Sindromi Overlap e relativi autoanticorpi. Connettivite mista (MCTD) LES + SSc +
PM/DM
Anticorpi anti-U1 RNP
Sindrome da anticorpi anti-sintetasi PM/DM + SSc + AR
Anticorpi anti-Jo1 Scleromiosite PM + SSc Anticorpi
anti-PM/Scl
Sindrome Rhupus AR + LES FR, ANA, anticorpi anti-SSA
Sclerolupus SSc + LES ANA Sclerosi sistemica/ Artrite
reumatoide
SSc + AR FR Lupus Eritematoso Sistemico/
Sindrome di Sjögren
CONNETTIVITE MISTA (MCTD)
Cenni storici
La Connettivite Mista (MCTD) è la più frequente e la più studiata tra le Sindromi Overlap.
Combina insieme manifestazioni del Lupus Eritematoso Sistemico, della Sclerosi Sistemica e della Polidermatomiosite associate a un marker sierologico, l’anticorpo anti-U1 RNP, per definizione presente nel 100% dei casi e generalmente a titolo elevato.
Tra il 1961 e il 1969, nella clinica reumatologica dell’Università di Stanford furono dosati gli anticorpi antinucleo (ANA) nel monitoraggio di alcuni pazienti, al fine di valutare la risposta di tali soggetti alla terapia e nacquero in questo modo le prime osservazioni dal confronto tra pazienti affetti da LES e pazienti affetti da MCTD.
Si osservò che nei pazienti portatori di LES il titolo degli ANA si riduceva oppure gli ANA diventavano negativi, durante i periodi di remissione della malattia; al contrario permanevano elevati titoli di tali anticorpi, sia nei periodi di attività della malattia che nei periodi di remissione, in quei pazienti che possedevano manifestazioni cliniche sia di LES, sia di PM/DM sia di SSc . Nel 1972 Sharp e i suoi colleghi coniarono il termine “Connettivite Mista” e sostennero la tesi che essa
rappresentasse una entità clinica distinta, sulla base della somiglianza delle caratteristiche cliniche e sierologiche in questo gruppo di pazienti e della buona prognosi che sembrava accompagnare questo tipo di sindrome.
Negli anni seguenti furono intrapresi e pubblicati molti studi, la maggior parte di tipo retrospettivo, allo scopo di rispondere alla domanda se la MCTD fosse davvero una entità clinica distinta, come sostenevano Sharp e i suoi colleghi. Tali studi portarono a risultati contrastanti e visioni diverse circa l’esistenza e la natura di MCTD: molti conclusero che, per la maggior parte dei pazienti, la MCTD fosse una fase intermedia nella progressione geneticamente determinata verso un connettivite distinta, che più di frequente è la SSc o il LES. Altre osservazioni in contrasto con la tesi di unità clinica distinta sono le seguenti :
1) non esistono, come vedremo in seguito, dei criteri classificativi universalmente accettati per la MCTD poiché molti pazienti soddisfano i criteri classificativi di altre connettiviti, quali il LES, la SSc e la AR.
2) la prognosi della MCTD non è così buona come sembrava in un primo momento, a causa dello sviluppo di complicanze quali l’ipertensione polmonare e non si discosta di tanto dalla prognosi dei pazienti affetti da LES.
3) Gli anticorpi anti-U1 RNP non sono in realtà diagnostici perché si riscontrano anche in altre connettiviti.
4) Gli anticorpi anti-U1 RNP non sempre sono presenti in pazienti che presentano un quadro clinico tipico di MCTD.
5) non è stato ancora dimostrato alcun ruolo patogenetico degli anticorpi anti-U1 RNP.
D’altro canto altri studiosi continuano a sostenere la tesi che la MCTD sia una entità clinica distinta e a supporto di tale convinzione è l’osservazione dell’esistenza di un gruppo di pazienti che sviluppa e mantiene un insieme di sintomi e segni più miti, appartenenti a diverse connettiviti e invece non possiede sintomi e segni più specifici di malattia (esempi sono le lesioni a carico del sistema nervoso centrale o le lesioni renali per il LES e la sclerosi prossimale per la SSc).
La contraddittorietà di tali risposte probabilmente permarrà fintanto che non saremo giunti ad una migliore comprensione dell’eziologia e della patogenesi a monte del quadro clinico e del quadro sierologico.
Epidemiologia
La prevalenza di MCTD è sconosciuta. Si riscontra una prevalenza per il genere femminile con un rapporto femmine/maschi di 8:1.
Non è stato dimostrato l’interessamento di particolari gruppi etnici, in linea con la SSc e al contrario di quanto accade nel LES e nella PM/DM.
Eziopatogenesi
L’eziologia della MCTD è ancora poco conosciuta, probabilmente è multifattoriale comprendente fattori individuali di ordine genetico e fattori ambientali quali agenti infettivi, fisici e chimico-farmacologici che svolgono un ruolo scatenante per un ipotetico processo di mimetismo molecolare tra autoantigeni e antigeni esogeni.
Il ruolo dell’ereditarietà è stato confermato attraverso ripetuti rilievi di una associazione significativa tra malattia e geni del sistema maggiore di istocompatibilità. E’ stata infatti riscontrata una associazione con gli aplotipi HLA-DR4, in particolare con il sottotipo Dw4.
Non risultano, invece significativamente presenti, gli aplotipi HLA-DR2 e HLA-DR3, che sono gli antigeni associati al LES né gli antigeni DR3 e DR5, associati alla SSc, né l’antigene DR3 associato alla PM/DM.
La patogenesi è di tipo autoimmunitario e la presenza dell’anticorpo anti-U1 RNP è, tra i disordini immunologici riscontrati, il più importante e costante.
L’U1 RNP è un complesso di ribonucleoproteine ricche in uridina, implicato nello splicing del RNA messaggero ; è costituito da 8 polipeptidi che legano il RNA nucleare , tre di questi (quelli da 68 KDa, 33 KDa e 22 KDa) portano gli epitopi riconosciuti dal siero di pazienti con MCTD. Gli altri polipeptidi, in particolar modo quelli da 28 KDa e da 16 KDa, reagiscono invece, con anticorpi anti-Sm.
Raramente possiamo riscontrare anticorpi anti-U1 RNP anche nel LES, dove sono generalmente a basso titolo ed è altrettanto raro trovare anticorpi anti-Sm e anti-dsDNA (ANA tipici del LES) in soggetti con MCTD e, qualora presenti, sono di solito transitori.
Un altro disordine immunologico che conferisce una caratteristica distintiva tra pazienti con MCTD e pazienti con LES o AR riguarda la risposta sierologica nei confronti di un’altra proteina che è associata allo spliceosoma, la hnRNP-A2 (heterogeneous nuclear RNP-A2). I soggetti con MCTD presentano anticorpi diretti verso i suoi domini RBD1 e RBD2 (RNA binding domains), mentre i pazienti con LES e AR presentano anticorpi soltanto verso il dominio RBD2.
Anatomia patologica
Nella MCTD è frequente una vascolopatia che interessa sia le piccole sia le grandi arterie.
Questa vascolopatia è caratterizzata da proliferazione dell’ intima e da ipertrofia della media, scarsa infiltrazione cellulare e assenza dei fenomeni di necrosi e fibrosi. Queste caratteristiche la rendono in parte diversa da altri quadri vasculopatici presenti in altre malattie. Nella SSc ritroviamo infatti, un’importante fibrosi, scarsa proliferazione dell’ intima e scarsa ipertrofia della media; nel LES riscontriamo invece, fenomeni necrotici e infiltrazione cellulare e, sia nella SSc sia nel LES sono interessate, principalmente, le piccole arterie.
Per quanto riguarda le grandi arterie, nella MCTD, l’interessamento dell’arteria polmonare è tra i più frequenti ed è responsabile dell’ipertensione polmonare, una delle più temibili complicanze di questa sindrome overlap.
Altra caratteristica istopatologica della MCTD, è la presenza di alterazioni a carico di polmoni, esofago e intestino, simili a quelle della Sclerosi Sistemica, seppur di minore entità. Queste sono rappresentate da una fibrosi interstiziale polmonare, preceduta da alveolite, suscettibile di una progressiva compromissione della funzione ventilatoria e la sostituzione fibrosa della muscolatura liscia parietale di esofago e intestino responsabile di una ipotonia ingravescente, con marcata riduzione e compromissione della peristalsi.
Clinica
L’età di esordio di malattia è simile a quella delle altre connettiviti, presentandosi di solito tra la seconda e la terza decade d’età. La sovrapposizione di sintomi e segni clinici di più di una connettivite può avvenire in uno stesso periodo o anche in periodi diversi (talora a distanza di anni) e, quest’ultima possibilità spiega come spesso, in questi soggetti, venga fatta dapprima diagnosi di LES o SSc o PM/DM e, solo in seguito di MCTD. Le manifestazioni d’esordio sono in genere aspecifiche e costituite da astenia, anoressia, calo ponderale, febbre, linfadenopatia, artromialgie; caratteristico è il riscontro, talvolta, di neuropatia trigeminale. Tali manifestazioni in alcuni pazienti persistono per anni, sostanzialmente invariate, in altri, nel corso del tempo progrediscono e vengono a sommarsi nuove componenti che portano così alla diagnosi di MCTD. Più raramente l’esordio è acuto con febbre alta, miosite, sierosite, artrite, meningite asettica.
Manifestazioni Vascolari
Il fenomeno di Raynaud è descritto quasi nel 100% dei casi, costituisce il reperto clinico più costante ed è spesso il primo sintomo che compare.
Di frequente riscontro sono edema a carico delle mani, soprattutto a livello delle dita con aspetto “a salsicciotto”(si riscontra in più del 2/3 dei pazienti), sclerodattilia, teleangectasie e calcinosi.
La più alta prevalenza di tale manifestazione, quando rappresenta il sintomo di una sottostante malattia (fenomeno di Raynaud “secondario”) è osservata, oltre che in pazienti affetti da MCTD, anche in pazienti affetti da SSc e sembra essere meno severo nel primo gruppo di soggetti rispetto al secondo. Tale forma secondaria sembra avere alla base della sua insorgenza una vascolopatia strutturale concomitante.
All’esame capillaroscopico si riscontrano alterazioni microvasali dello “scleroderma pattern disorder” di tipo lento, tipiche di PM/DM e SSc, caratterizzate da ingrandimenti irregolari a carico di alcuni capillari, in assenza o con una minima perdita di tessuto capillare. In pazienti affetti da MCTD si osservano i caratteristici megacapillari o capillari giganti, emorragie del fondo ungueale e capillari distrofici, ramificati, simili a cespugli. Per quanto riguarda la specificità anticorpale è plausibile che gli anticorpi anti-U1 RNP rappresentino un marker per il processo vascolare che causa fenomeno di Raynaud o che questi anticorpi giochino un ruolo nella vascolopatia strutturale ad esso associata. Tali anticorpi sono infatti fortemente correlati col fenomeno di Raynaud e lo sono anche con alterazioni
polmonari, in particolar modo con l’ipertensione polmonare. Infine la loro presenza correla anche con alterazioni capillaroscopiche appartenenti allo scleroderma pattern lento.
Manifestazioni Muco-Cutanee
Molto raramente possono presentarsi alterazioni muco-cutanee quali eritema a farfalla e lupus discoide e può essere presente fotosensibilità.
Il trattamento consiste in misure preventive : mancata esposizione ala luce solare e utilizzo di creme solari ad alta gradazione.
Sempre con bassissima frequenza possiamo trovarci di fronte anche a una sclerodermia diffusa o a papule di Gottron.
Manifestazioni Articolari E Tendinee
Il 100% di questi soggetti presenta poliartralgie che nel 75% dei casi evolvono in artrite franca, di solito una poliartrite non deformante e non erosiva. Talvolta l’artrite può assomigliare a quella che si osserva nell’artrite reumatoide, con erosioni visualizzabili alle immagini radiografiche (il più delle volte piccole e marginali) e può essere deformante, con deformità articolari come deviazione ulnare delle dita “a colpo di vento”, dita ‘en boutonnière’, dita a ‘collo di cigno’, pollice a ‘Z’.
L’interessamento tendineo è più frequente a carico degli estensori e dei flessori delle dita delle mani e può condurre anch’esso a vari gradi di deformità.
In circa il 40% dei pazienti si possono riscontrare piccoli noduli peritendinei, prevalentemente in prossimità dei gomiti e a livello di mani e piedi; istologicamente essi non mostrano segni di infiammazione specifica e non presentano la tipica morfologia dei noduli reumatoidi. I noduli reumatoidi presentano una zona centrale di necrosi fibrinoide, una zona intermedia formata da uno o più strati di cellule allungate la cui esatta natura è ancora da accertare e una zona esterna di tessuto di granulazione infiltrato da linfociti e plasmacellule.
Manifestazioni Muscolari
Mialgia e miosite si osservano in 35%-75% dei casi. Il sintomo più caratteristico della miosite è l’astenia a carico dei muscoli prossimali degli arti e dei cingoli con difficoltà nell’esecuzione di alcuni semplici movimenti come accavallare le gambe, alzarsi in piedi dalla posizione seduta, salire le scale, pettinarsi o appendere gli abiti.
Agli esami ematochimici c’è il riscontro di una elevazione degli enzimi muscolari CPK, LDH e ALDOLASI, all’elettromiografia sono presenti alterazioni miopatiche e la biopsia muscolare è
positiva per degenerazione delle fibre e presenza di infiltrati infiammatori.
Manifestazioni Gastroenteriche
In più del 50% dei pazienti vi è il riscontro di anomalie alla manometria esofagea, è infatti descritta una dismotilità esofagea, dovuta ad interessamento della muscolatura liscia. Spesso la disfunzione esofagea, in un primo momento è subclinica e in seguito i pazienti sviluppano pirosi, rigurgito e disfagia.
Soprattutto la pressione a carico dello sfintere esofageo inferiore (SEI), in questi pazienti è nettamente più bassa rispetto ai controlli, così come la forza della peristalsi nel tratto più distale dell’ esofago. Viene comunque descritta una riduzione della pressione anche a livello dello sfintere esofageo superiore (SES), seppur di più lieve entità.
Tali alterazioni pressorie, insieme alle alterazioni della motilità esofagea sono le dirette responsabili dei ricorrenti episodi di aspirazione polmonare di cui soffrono questi soggetti.
Segni meno frequenti sono l’ipomotilità, la dilatazione, il malassorbimento o la sclerosi intestinale, la diarrea secretiva, l’epatite cronica attiva , la pancreatite acuta e la formazione di pseudocisti pancreatiche.
Apparato Respiratorio
L’impegno polmonare è molto comune, il più delle volte è presente una fibrosi interstiziale (tra il 20% e il 60% dei pazienti) quasi sempre bilaterale e più marcata alle basi ; nelle prime fasi della malattia l’interessamento è asintomatico e poi caratterizzato da dispnea per la progressione del quadro, che comporta insufficienza ventilatoria di tipo restrittivo. Oltre alla dispnea si riscontrano rantoli bilaterali alle basi e può essere presente dolore toracico pleuritico.
L’interessamento fibrotico polmonare può essere evidenziato, oltre che alla radiografia del torace (alterazioni della trama interstiziale con infiltrati, ispessimento della pleura e versamento pleurico), anche con le prove di funzionalità respiratoria (PFR), nelle quali si assiste ad una riduzione della capacità di diffusione del monossido di carbonio (DLCO).
In un 15-20% dei casi è descritta la più temibile delle complicanze della MCTD, l’ipertensione polmonare, complicanza che si presenta soprattutto negli stadi più avanzati della malattia. E’ sempre necessario ricercarla con le PFR, l’ecocardiogramma e la TC del torace. Talvolta essa è secondaria ad una rapida progressione della fibrosi interstiziale polmonare, ma più spesso essa è un evento primitivo associato alla proliferazione dell’intima a livello delle arteriole polmonari.
Un progressivo declino della DLCO sembra poter essere un campanello d’allarme importante, visto che aumenta, in pazienti con Connettivite Mista, il rischio di sviluppare ipertensione polmonare.
La prognosi è severa per chi ne è affetto, l’ ipertensione polmonare può infatti rapidamente progredire in insufficienza ventricolare destra che è la prima causa di morte in soggetti con MCTD.
Manifestazioni Neurologiche
Sono aspecifiche e di scarsa entità, presenti in 10-55% dei casi. Le più comuni sono la neuropatia sensitiva del trigemino, la sindrome del tunnel carpale e la cefalea vasomotoria.
La neuropatia sensitiva del trigemino, la più frequente tra le manifestazioni neurologiche sopraelencate, quando presente, costituisce una delle manifestazioni di esordio della MCTD.
Manifestazioni Renali
Venivano descritte da Sharp come tipicamente assenti, ma possono invece presentarsi nel 25% dei casi, soprattutto in quei casi in cui l’esordio della malattia è in età pediatrica.
La più frequente manifestazione di impegno renale è la glomerulonefrite membranosa (di classe V nel LES), la quale si contrassegna per il diffuso e irregolare ispessimento della membrana basale glomerulare.
Il più delle volte, tale glomerulonefrite è asintomatica e raramente può evolvere in una sindrome nefrosica, comunque abbastanza responsiva a dosi elevate di corticosteroidi.
Talvolta possono presentarsi casi di crisi ipertensive reno-vascolari, il cui trattamento d’elezione è a base di ACE inibitori.
Manifestazioni Cardiache
Può esserci il riscontro di una pericardite, la manifestazione clinica più comune ma comunque rara, che di rado si complica in tamponamento cardiaco.
Il miocardio può essere interessato da una miocardite o, più di frequente può essere danneggiato secondariamente all’ipertensione polmonare.
Si riscontrano alterazioni elettrocardiografiche in un 20% di questi pazienti ed esse sono: ipertrofia ventricolare destra, dilatazione atriale destra e alterazioni della conduzione atrio-ventricolare.
Esami bioumorali
Alla biochimica è frequente il riscontro di una ipergammaglobulinemia policlonale.
I livelli del complemento nel siero sono generalmente nella norma, anche se è descritta una riduzione lieve-moderata in quei soggetti che sviluppano nefrite.
Durante le riacutizzazioni della MCTD abbiamo un’elevazione degli indici di flogosi, i quali si normalizzano durante le fasi di remissione.
All’emocromo in 1/3 dei casi si ritrovano anemia e leucopenia; più rara, invece, la trombocitopenia, evenienza più comune nei bambini.
Di fronte a lieve anemia e leucopenia non è richiesta alcuna terapia farmacologica. Nei casi più rari di anemia emolitica autoimmune e trombocitopenia sono necessarie alte dosi di corticosteroidi. In soggetti resistenti a tale terapia possiamo adottare un trattamento con immunoglobuline per via endovenosa oppure farmaci immunosoppressivi.
Talvolta possono presentarsi rarissimi casi con porpora trombotica trombocitopenica che vanno trattati con farmaci immunosoppressori e concomitante plasmaferesi.
Il profilo anticorpale è caratterizzato dall’alto titolo degli anticorpi anti-nRNP . Esso persiste invariato per diversi anni sia nei periodi di attività sia di inattività della malattia e risulta, invece, non dosabile, solo in seguito ad una prolungata remissione. Nel 70% dei pazienti è presente positività del fattore reumatoide (FR), spesso ad alto titolo e frequente è il riscontro di immunocomplessi circolanti.
Negli anni 70 Sharp, insieme al suo gruppo di lavoro definì che la positività per anticorpi anti-Sm e anti-dsDNA fosse criterio di esclusione per la diagnosi di MCTD; attualmente invece, l’orientamento è cambiato ed è quello di non escludere categoricamente tale diagnosi in loro presenza, in particolar modo se il titolo riscontrato è basso.
Criteri classificativi di MCTD
Nel 1986 si tenne a Tokyo un simposio internazionale, avente per tema la Connettivite Mista e gli Anticorpi anti-nucleo.
Allo scopo di facilitare studi prospettici, al termine dell’incontro furono selezionate tre serie di criteri classificativi per la MCTD, nelle quali requisito fondamentale ma non sufficiente per la classificazione della Connettivite Mista è un alto titolo di anticorpi anti-U1 RNP, sebbene il loro ruolo nella patogenesi del danno tissutale non sia ancora stato chiarito.
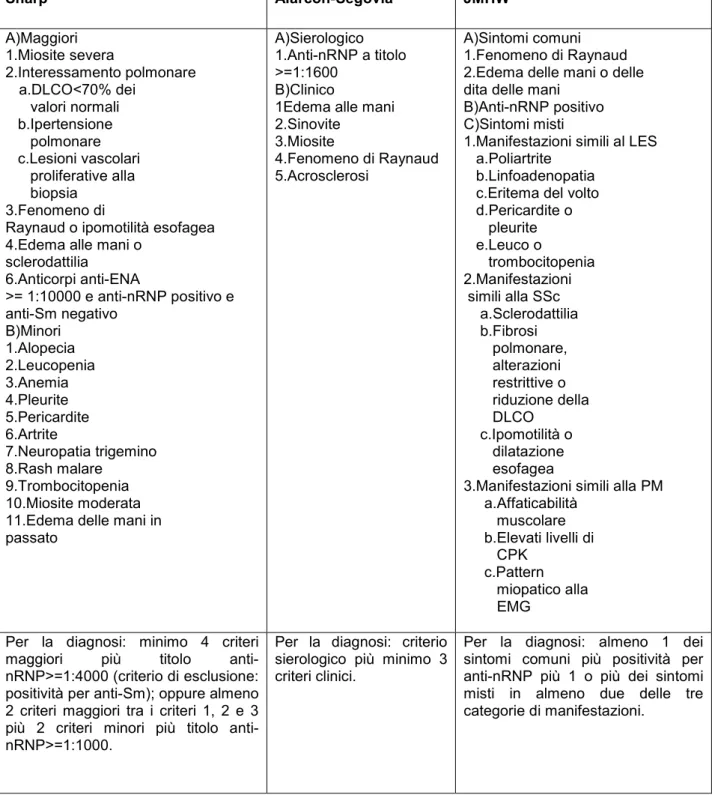
Queste tre serie di criteri classificativi sono i criteri di Sharp, già esistenti a partire dall’inizio degli anni 70, i criteri di Alarcòn-Segovia rivisti da quelli originali del 1976 e infine i criteri più recenti della JMHW ( The Research Committee of Japanese Ministry of Health and Welfare for MCTD), riportati nella tabella 2.
Negli anni seguenti, a partire dal 1987 fino al 1998 sono stati condotti numerosi studi volti ad esaminare la sensibilità e la specificità di queste tre serie di criteri (Tabella 3).
I risultati hanno attribuito ai criteri di Alarcòn-Segovia il maggior grado di sensibilità e specificità e ai criteri di Sharp i più bassi valori di specificità.
Attualmente quindi i criteri di Alarcòn-Segovia sembrano essere quelli di più semplice utilizzo per la classificazione generale della MCTD e per differenziarla da altre connettiviti ; i criteri di JMHW sembrano invece essere i più utili per analizzare ogni sintomo e segno della MCTD e i criteri di Sharp sono attualmente i meno utilizzati.
Tabella 2. Criteri classificativi di Sharp, di Alarcòn-Segovia e della JMHW. Sharp Alarcòn-Segovia JMHW A)Maggiori 1.Miosite severa 2.Interessamento polmonare a.DLCO<70% dei valori normali b.Ipertensione polmonare c.Lesioni vascolari proliferative alla biopsia 3.Fenomeno di
Raynaud o ipomotilità esofagea 4.Edema alle mani o
sclerodattilia 6.Anticorpi anti-ENA >= 1:10000 e anti-nRNP positivo e anti-Sm negativo B)Minori 1.Alopecia 2.Leucopenia 3.Anemia 4.Pleurite 5.Pericardite 6.Artrite 7.Neuropatia trigemino 8.Rash malare 9.Trombocitopenia 10.Miosite moderata 11.Edema delle mani in passato
A)Sierologico 1.Anti-nRNP a titolo >=1:1600
B)Clinico
1Edema alle mani 2.Sinovite 3.Miosite 4.Fenomeno di Raynaud 5.Acrosclerosi A)Sintomi comuni 1.Fenomeno di Raynaud
2.Edema delle mani o delle dita delle mani
B)Anti-nRNP positivo C)Sintomi misti
1.Manifestazioni simili al LES a.Poliartrite
b.Linfoadenopatia c.Eritema del volto d.Pericardite o pleurite e.Leuco o trombocitopenia 2.Manifestazioni simili alla SSc a.Sclerodattilia b.Fibrosi polmonare, alterazioni restrittive o riduzione della DLCO c.Ipomotilità o dilatazione esofagea
3.Manifestazioni simili alla PM a.Affaticabilità muscolare b.Elevati livelli di CPK c.Pattern miopatico alla EMG Per la diagnosi: minimo 4 criteri
maggiori più titolo anti-nRNP>=1:4000 (criterio di esclusione: positività per anti-Sm); oppure almeno 2 criteri maggiori tra i criteri 1, 2 e 3 più 2 criteri minori più titolo anti-nRNP>=1:1000.
Per la diagnosi: criterio sierologico più minimo 3 criteri clinici.
Per la diagnosi: almeno 1 dei sintomi comuni più positività per anti-nRNP più 1 o più dei sintomi misti in almeno due delle tre categorie di manifestazioni.
Tabella 3. Studi di sensibilità e specificità dei criteri classificativi di Sharp, di Alarcòn-Segovia e della JMHW.
Anno Indagine Numero di pz studiati Malattie Alarcòn Segovia Sharp JMHW 1987 JMHW Sensib:81 Specif:261 MCTD SLE/SSc/ PM/DM 88% 87% 1989 Alarcòn-Segovia Sensib:80 Specif:513 MCTD SLE/SSc/ AR/PM/ DM/SS 100% 99-100% 100% 55-100% 96% 99-100% 1991 Doria Sensib:32 Specif:75 MCTD SLE/SSc/ PM 87% 94% 1996 Amigues Sensib:45 Specif:25 pz con anti-RNP positivo AR/LES/ SSc/Ov 62.5% 86.2% 100% 38% 56.2% 65.5% 1998 Smolen Sensib:26 MCTD 100% 70% 92% Prognosi
La MCTD può andare incontro a due tipi di evoluzione: nella maggior parte dei pazienti la malattia va in remissione o viene controllata con basse dosi di corticosteroidi, ma in 10-20% dei casi evolve verso una delle tre connettiviti che si trovano sovrapposte nella sua storia. Nella stragrande maggioranza dei casi evolve in una severa Sclerosi Sistemica: questo si spiega col fatto che le manifestazioni di LES e PM/DM presentano generalmente una buona responsività alla terapia corticosteroidea, mentre quelle sclerodermiche possono persistere, nonostante i corticosteroidi, e progredire.
I soggetti affetti da MCTD richiedono, in ogni caso, un attento follow-up a lungo termine, che può essere di aiuto nel prevedere e quindi nel prevenire eventuali riprese di malattia; in alcuni pazienti infatti, una caduta della conta leucocitaria (soprattutto la conta dei linfociti), un aumento della VES e dei livelli totali di immunoglobuline circolanti, possono essere importanti segnali d’allarme.
Il follow-up consiste nella rilevazione della pressione arteriosa, nell’esecuzione dell’emocromo, nella rilevazione degli indici di flogosi, nel dosaggio degli enzimi muscolari, nella ricerca di proteinuria e in esami più specifici qualora sia sospettato o già conosciuto un impegno di altro tipo, come ad esempio quello polmonare.
Ad intervalli di tempo più ampi vengono anche ridosati gli autoanticorpi, inclusi gli anti-Sm e gli anti-dsDNA per scongiurare un’eventuale evoluzione della malattia verso il LES. Fattori prognostici negativi sono: l’impegno renale, l’ipertensione polmonare e infezioni conseguenti alla terapia immunosoppressiva.
Le più frequenti cause di morte associate alla MCTD sono l’ipertensione polmonare e lo scompenso cardiaco e più raramente sepsi con seguente CID, tamponamento cardiaco o
perforazione intestinale; ancora più rara è la morte per insufficienza renale.
Terapia
Il trattamento è interamente dipendente dall’entità e dalla tipologia di impegno clinico. Di fronte a quadri sindromici estremamente eterogenei è indispensabile adottare una terapia specifica alla persona, al suo interessamento d’organo e alla severità dell’attività di malattia.
In generale le manifestazioni infiammatorie di MCTD che si sovrappongono a LES e PM/DM mostrano una buona risposta alla terapia con Corticosteroidi, i cui dosaggi vengono adattati a seconda dell’impegno d’organo e saranno ridotti non appena le condizioni cliniche lo consentano. Occasionalmente si rende necessario l’utilizzo di agenti citotossici quali gli Antimalarici, la Ciclofosfamide, il Methotrexate ecc, da dare in associazione alla terapia corticosteroidea.
Al contrario le manifestazioni sclerodermiche spesso non sono responsive al trattamento con corticosteroidi; una eccezione importante è fornita dall’interessamento esofageo che mostra, invece, quasi sempre beneficio dalla terapia.
Anche manifestazioni quali le neuropatie periferiche, la sindrome nefrosica e l’artrite severa deformante sono spesso resistenti a tale trattamento.
In quei pazienti in cui sia prevalente la componente sclerodermica è consigliato l’impiego di tutti i farmaci utilizzati nella SSc ad eccezione della D-Penicillamina, perché sembra che possa scatenare o aggravare la componente lupica della malattia. Si tratta di farmaci vasoattivi quali i calcio-antagonisti, gli ACE inibitori, i sartanici e i prostanoidi (Iloprost) oltre ai farmaci immunosoppressori quali la ciclosporina, il methotrexate e la ciclofosfamide; la plasmaferesi è stata utilizzata in quei soggetti con forme cliniche severe a rapida evoluzione.
SINDROME DA ANTICORPI ANTI-SINTETASI
E’ una sindrome caratterizzata sul piano clinico da alcune manifestazioni della Polidermatomiosite, della Sclerosi Sistemica e dell’Artrite Reumatoide e sul piano sierologico dalla positività degli anticorpi anti-tRNA sintetasi.
Le tRNA sintetasi sono una serie di enzimi citoplasmatici implicati nella sintesi proteica, dove hanno il compito di addizionare amminoacidi all’RNA transfert durante l’assemblaggio dei polipeptidi. Il target più comune del sistema
autoimmunitario finora descritto è l’istidil tRNA sintetasi, antigene al quale è stato dato il nome Jo-1.
L’eziopatogenesi della malattia non è conosciuta, sebbene ci siano delle evidenze di interazione tra ospite e virus (enterovirus, coxsackie B virus) su un substrato di suscettibilità genetica, che potrebbero essere responsabili dell’attivazione autoanticorpale.
La positività degli anticorpi anti-Jo1 è associata agli antigeni HLA di classe2 DR3 e DRw52.
Quadro clinico
I pazienti affetti da questa sindrome presentano tre caratteristiche principali che sono: miosite, pneumopatia interstiziale e poliartralgie/poliartrite.
L’esordio si caratterizza più spesso con fenomeno di Raynaud (descritto nel 50-100% dei pazienti) o con poliartralgie/poliartrite (85-100% dei pazienti) generalmente di modesta entità e autolimitantesi, in 10-15% dei casi può essere presente sinovite erosiva; le articolazioni maggiormente interessate sono le interfalangee prossimali e le matacarpofalangee, i polsi, i gomiti, le caviglie e metatarsofalangee.
Miosite e interstiziopatia polmonare si sviluppano generalmente dopo un periodo variabile di 18-36 mesi.
La miosite, presente nell’ 86-100% dei pazienti può assumere il quadro della polimiosite in taluni e della dermatomiosite in altri.
L’interstiziopatia polmonare si riscontra in 75-100% dei pazienti. Altre manifestazioni cliniche di frequente riscontro in questa sindrome sono manifestazioni cutanee simil-sclerodermiche, quali sclerodattilia, teleangectasie e “mani da meccanico”, quadro piuttosto insolito caratterizzato da ipercheratosi sulla superficie palmare delle dita delle mani.
Infine, alcuni pazienti lamentano xeroftalmia e xerostomia e in 1/3 di questi si associa una positività degli anticorpi anti-Ro/SSA e anti-La/SSB (il reperto di anti-La isolati è “eccezionale).
Diagnosi
Si fa diagnosi di sindrome da anticorpi anti-sintetasi quando un paziente con un quadro clinico indicativo di questa sindrome è positivo per l’ anticorpo anti-Jo1.
La diagnosi differenziale dovrà essere posta con tutte le connettiviti da sovrapposizione, in particolar modo con la MCTD, la più frequente tra di esse. Alcuni studi hanno visto come la miosite e la pneumopatia interstiziali siano manifestazioni più frequenti nei pazienti positivi per anti-Jo1 rispetto a soggetti affetti da MCTD e anche come la prognosi dell’alveolite fibrosante sia migliore nei pazienti affetti da questa sindrome rispetto a quelli positivi per anti-nRNP.
Prognosi
L’elemento prognostico più sfavorevole è rappresentato dalla interstiziopatia polmonare: i pazienti presentano infatti dispnea, da sforzo o anche a riposo e può sopravvenire la morte per insufficienza respiratoria. Anche un coinvolgimento muscolare resistente alla terapia è elemento prognostico negativo, in quanto comporta grave inabilità con perdita dell’autosufficienza e della capacità di deambulare.
Terapia
Il trattamento si avvale dell’impiego di cortisonici e, solo in caso di manifestazioni polmonari e muscolari resistenti a tale terapia, viene effettuato un trattamento aggiuntivo con immunosoppressori.
SCLEROMIOSITE (PM/SSc)
E’ una sindrome nella quale riscontriamo la sovrapposizione delle manifestazioni della Sclerosi Sistemica e della Polimiosite/Dermatomiosite ed è caratterizzata da positività per anticorpi anti-PM/Scl e da miosite e pneumopatia interstiziale, entrambi tardivi.
Queste manifestazioni cliniche, presenti anche nella sindrome da anticorpi anti-sintetasi sono meno severe e più responsive al
trattamento con corticosteroidi e immunosoppressori nella scleromiosite. La prognosi è infatti favorevole.
SINDROME RHUPUS (AR/LES)
E’ una sindrome data dalla sovrapposizione di alcune manifestazioni dell’ Artrite Reumatoide e del Lupus Eritematoso Sistemico.
E’ caratterizzata, per quanto riguarda il quadro artritico da un’artrite simmetrica, erosiva e deformante e per quanto riguarda il quadro lupico da manifestazioni mucocutanee e alterazioni ematologiche, raro è l’impegno neurologico.
Il quadro immunologico mostra una frequente positività del fattore reumatoide e degli anticorpi anti-nucleo e talvolta anche il riscontro dell’anticorpo anti Ro/SSA.
SCLEROLUPUS (SSc/LES)
E’ una sindrome nella quale riscontriamo sovrapposizione tra Sclerosi Sistemica e Lupus Eritematoso Sistemico.
Generalmente le manifestazioni sclerodermiche precedono la comparsa di quelle lupiche.
Sono comuni il fenomeno di Raynaud, la sclerosi cutanea diffusa e l’ipomotilità esofagea, meno frequente è la fibrosi polmonare.
La componente lupica è caratterizzata, il più delle volte, da alterazioni mucocutanee, sierositi, disordini ematologici e positività degli anticorpi anti-nucleo.
SCLEROSI SISTEMICA/ ARTRITE REUMATOIDE
In questa sindrome sono le manifestazioni dell’AR che precedono quelle sclerodermiche.
Frequente è il fenomeno di Raynaud. Le manifestazioni artritiche sono caratterizzate da una poliartrite generalmente grave, simmetrica ed erosiva, più la positività per il fattore reumatoide. Per quanto riguarda le manifestazioni sclerodermiche, costanti sono la fibrosi polmonare e l’ipomotilità esofagea.
LES/SINDROME DI SJÖGREN
Spesso l’ampia varietà di manifestazioni cliniche e immunologiche di pazienti con Sindrome di Sjögren primitiva crea qualche problema nel differenziare una Sindrome di Sjögren primitiva, una Sindrome di Sjögren associata a un’altra
connettivite o una connettivite con presentazione simil- Sjögren. Il più alto grado di sovrapposizione della Sindrome di Sjögren si ha con il Lupus Eritematoso Sistemico : gli anticorpi indirizzati verso l’antigene La(SSB) sono infatti spesso presenti in entrambe le sindromi e ancora più frequenti in quei pazienti che presentano caratteristiche sia dell’una sia dell’altra patologia. Il quadro clinico è caratterizzato da manifestazioni oculari, salivari e extraghiandolari. Quest’ultime sono manifestazioni tipiche del LES e sono rash, artrite, fenomeno di Raynaud, leucopenia e trombocitopenia; caratteristica è la presenza in un 30% di pazienti di un rash purpureo ipergammaglobulinemico. Rara è la nefrite, mentre può essere presente un’acidosi tubulare renale asintomatica.
La terapia si effettua in gran parte con farmaci sintomatici: lacrime artificiali e sostituti/fluidificanti della saliva. Per le manifestazioni extraghiandolari possono essere usati corticosteroidi da soli o in associazione con antimalarici o azatioprina.
CONCLUSIONI
La diagnosi di una delle Sindromi Overlap invece di un’altra non crea grosse differenze per quanto riguarda il trattamento farmacologico, ma può essere di grande aiuto al medico per
prevenire determinati complicanze, di più frequente riscontro in alcune forme rispetto ad altre.
La descrizione delle Sindromi Overlap in termini di autoanticorpi è invece basata sul presupposto che un determinato pattern autoimmunitario rifletta la sottostante causa della malattia. Le eziologie attualmente sono sconosciute ma la loro conoscenza, un giorno potrà finalmente giustificare completamente l’utilizzo del termine ‘diagnosi’.
SCOPO DELLO STUDIO
Lo scopo del lavoro è stato quello di individuare, presso la UO di Reumatologia della Azienda Ospedaliera Universitaria Pisana, i pazienti affetti da Sindrome Overlap e di descriverne le principali caratteristiche cliniche e sierologiche.
L’ obiettivo auspicabile è quello di arrivare ad un corretto raggruppamento delle malattie autoimmuni sistemiche, obiettivo di importanza centrale per lo svolgimento di studi epidemiologici, clinici e di patogenesi.
PAZIENTI E METODI
Sono stati selezionati 93 soggetti ( 83 femmine e 10 maschi) afferenti all’ UO di Reumatologia della Azienda Ospedaliera Universitaria Pisana dal 1974 ad oggi, 77 pazienti ancora in follow-up, 15 persi dal follow-up, 1 deceduto per arresto cardiocircolatorio.
La ricerca dei pazienti affetti da Sindrome Overlap è stata effettuata utilizzando i seguenti ausili:
• il data base della attività ambulatoriale disponibile presso la clinica (totale pazienti 30.000, affetti da connettiviti 4200, affetti da reumatismo articolare infiammatorio 4100).
• i registri dei ricoveri
• i data base relativi alle singole malattie
• intervistando i medici del reparto
I dati epidemiologici, clinici, di laboratorio e relativi alla terapia dei pazienti identificati sono stati raccolti in una cartella, creata per lo studio.
La diagnosi di Sindrome Overlap è stata posta in base ai seguenti criteri
- la compresenza di due o più malattie autoimmuni sistemiche definite, denominate “Sindromi Overlap definite”
oppure
- la presenza di manifestazioni cliniche specifiche di più malattie autoimmuni definite, ma non sufficienti a soddisfare i criteri
classificativi per una connettivite definita (CTD). In particolare sono state ritenute specifiche di CTD le seguenti manifestazioni cliniche: artrite erosiva, miosite, alveolite, ipertensione polmonare, sclerosi cutanea, fenomeno di Raynaud con alterazioni capillaroscopiche tipiche per PM/DM-SSc pattern.Inoltre la presenza di anticorpi anti-fosfolipidi (anticorpi anticardiolipina
eanticoagulanti lupici) a titolo medio-elevato in associazione a livedoreticularis, corea, piastrinopenia, vegetazioni valvolari è stata considerata specifica per lo spettro della Sindrome da Anticorpi Anti-fosfolipidi, come suggerito da Asherson.Tali condizioni sono denominate “Sindromi Overlap indefinite”.
RISULTATI
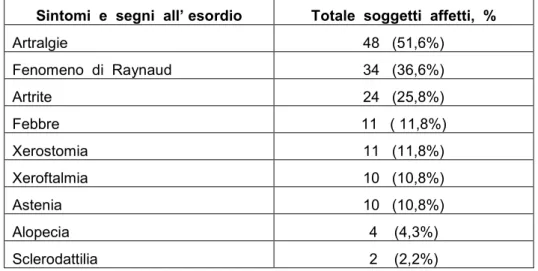
Sono stati valutati 93 pazienti, 10 maschi (M) e 83 femmine (F), età media 56 ± 16 anni (min-max : 26-88; mediana : 56), età media di esordio 36 ± 15 anni (min-max : 9-73; mediana=35), durata media di malattia nei soggetti in follow-up 17,5 ± 9,6 anni. Come sintomi e segni all’esordio hanno manifestato artralgie (51,6%), fenomeno di Raynaud (36,6%), artrite (25,8%), febbre (11,8%), xerostomia (11,8%), xeroftalmia (10,8%), astenia (10,8%), alopecia (4,3%), sclerodattilia (2,2%) come riportato in tabella 4.
Tabella 4. Sintomi e segni all’esordio del totale di 93 pazienti.
Sintomi e segni all’ esordio Totale soggetti affetti, %
Artralgie 48 (51,6%) Fenomeno di Raynaud 34 (36,6%) Artrite 24 (25,8%) Febbre 11 ( 11,8%) Xerostomia 11 (11,8%) Xeroftalmia 10 (10,8%) Astenia 10 (10,8%) Alopecia 4 (4,3%) Sclerodattilia 2 (2,2%)
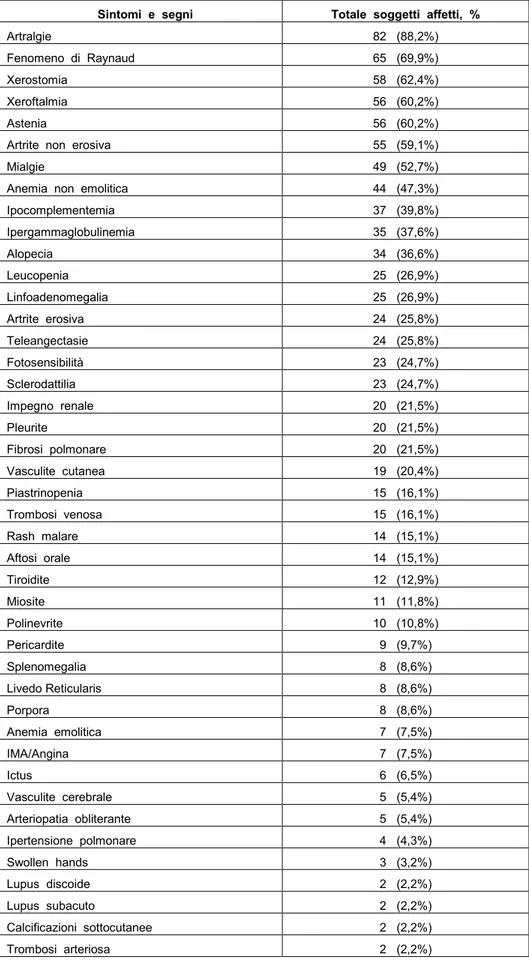
Le manifestazioni cliniche osservate nei 93 pazienti includono : artralgie (88,2%), fenomeno di Raynaud (69,9%), xerostomia (62,4%), xeroftalmia (60,2%), astenia (60,2%), artrite non erosiva (59,1%), mialgie (52,7%), anemia non emolitica (47,3%), febbre (44,1%).
Il totale delle manifestazioni cliniche presentate è riportato nella tabella 5.
Tabella 5. Sintomi e segni presentati dai 93 soggetti studiati.
Sintomi e segni Totale soggetti affetti, %
Artralgie 82 (88,2%)
Fenomeno di Raynaud 65 (69,9%)
Xerostomia 58 (62,4%)
Xeroftalmia 56 (60,2%)
Astenia 56 (60,2%)
Artrite non erosiva 55 (59,1%)
Mialgie 49 (52,7%)
Anemia non emolitica 44 (47,3%)
Ipocomplementemia 37 (39,8%) Ipergammaglobulinemia 35 (37,6%) Alopecia 34 (36,6%) Leucopenia 25 (26,9%) Linfoadenomegalia 25 (26,9%) Artrite erosiva 24 (25,8%) Teleangectasie 24 (25,8%) Fotosensibilità 23 (24,7%) Sclerodattilia 23 (24,7%) Impegno renale 20 (21,5%) Pleurite 20 (21,5%) Fibrosi polmonare 20 (21,5%) Vasculite cutanea 19 (20,4%) Piastrinopenia 15 (16,1%) Trombosi venosa 15 (16,1%) Rash malare 14 (15,1%) Aftosi orale 14 (15,1%) Tiroidite 12 (12,9%) Miosite 11 (11,8%) Polinevrite 10 (10,8%) Pericardite 9 (9,7%) Splenomegalia 8 (8,6%) Livedo Reticularis 8 (8,6%) Porpora 8 (8,6%) Anemia emolitica 7 (7,5%) IMA/Angina 7 (7,5%) Ictus 6 (6,5%) Vasculite cerebrale 5 (5,4%) Arteriopatia obliterante 5 (5,4%) Ipertensione polmonare 4 (4,3%) Swollen hands 3 (3,2%) Lupus discoide 2 (2,2%) Lupus subacuto 2 (2,2%) Calcificazioni sottocutanee 2 (2,2%) Trombosi arteriosa 2 (2,2%)
Secondo i criteri descritti in “Pazienti e Metodi”, i 93 soggetti selezionati sono stati suddivisi nei gruppi delle “Sindromi Overlap definite” e delle “Sindromi Overlap indefinite”.
I soggetti affetti da “Sindrome Overlap definita” sono in numero di 68, 7 maschi (M) e 61 femmine, età media 60 ± 15 anni (min-max : 26-88; mediana : 61,5), età media all’ esordio 39 ± 16 anni (min-max 9-73; mediana : 38).
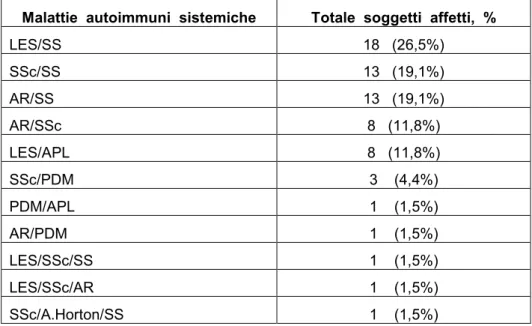
Le diagnosi formulate in tali pazienti, secondo i criteri esistenti in letteratura sono riportate in tabella 6.
Tabella 6. Diagnosi dei soggetti affetti da “Sindromi Overlap definite”. Malattie autoimmuni sistemiche Totale soggetti affetti, %
LES/SS 18 (26,5%) SSc/SS 13 (19,1%) AR/SS 13 (19,1%) AR/SSc 8 (11,8%) LES/APL 8 (11,8%) SSc/PDM 3 (4,4%) PDM/APL 1 (1,5%) AR/PDM 1 (1,5%) LES/SSc/SS 1 (1,5%) LES/SSc/AR 1 (1,5%) SSc/A.Horton/SS 1 (1,5%)
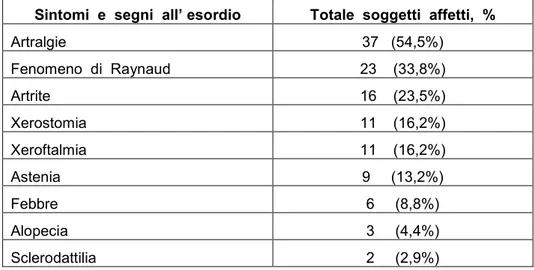
I sintomi e segni presentati da tali pazienti all’esordio di malattia erano artralgie (54,5%), fenomeno di Raynaud (33,8%), artrite (23,5%), xerostomia (16,2%), xeroftalmia (16,2%), astenia
(13,2%), febbre (8,8%), alopecia (4,4%), sclerodattilia (2,9%), come riportato in tabella 7.
Tabella 7. Sintomi e segni all’ esordio dei 68 pazienti affetti da “Sindrome Overlap definita”.
Sintomi e segni all’ esordio Totale soggetti affetti, %
Artralgie 37 (54,5%) Fenomeno di Raynaud 23 (33,8%) Artrite 16 (23,5%) Xerostomia 11 (16,2%) Xeroftalmia 11 (16,2%) Astenia 9 (13,2%) Febbre 6 (8,8%) Alopecia 3 (4,4%) Sclerodattilia 2 (2,9%)
I soggetti affetti da “Sindrome Overlap indefinita” sono in numero di 25, 3 maschi (M) e 22 femmine, età media 44 ± 13 anni (min-max : 26-74; mediana : 43,5), età media all’ esordio 29 ± 12 anni (min-max 15-61; mediana : 27,5).
Come sintomi e segni all’ esordio hanno manifestato fenomeno di Raynaud (48%), artralgie (44%), artrite (28,6%), febbre (20%), astenia (4%), alopecia (4%), come riportato in tabella 8.
Tabella 8. Sintomi e segni all’ esordio dei 25 pazienti affetti da “Sindrome Overlap indefinita”.
Sintomi e segni all’ esordio Totale soggetti affetti, % Fenomeno di Raynaud 12 (48%) Artralgie 11 (44%) Artrite 8 (28,6%) Febbre 5 (20%) Astenia 1 (4%) Alopecia 1 (4%)
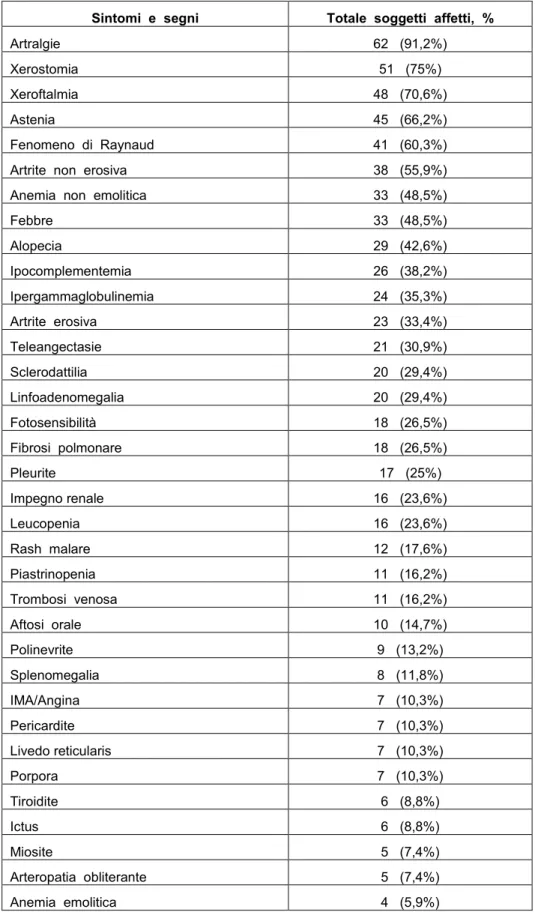
Le manifestazioni cliniche osservate nei 68 pazienti affetti da “Sindrome Overlap definita” includono: artralgie (91,2%), xerostomia (75%), xeroftalmia (70,6%), astenia (66,2%), fenomeno di Raynaud (60,3%). Il totale delle manifestazioni cliniche presentate è riportato in tabella 9.
Tabella 9. Sintomi e segni presentati dai 68 soggetti affetti da “Sindrome Overlap definita”.
Sintomi e segni Totale soggetti affetti, %
Artralgie 62 (91,2%)
Xerostomia 51 (75%)
Xeroftalmia 48 (70,6%)
Astenia 45 (66,2%)
Fenomeno di Raynaud 41 (60,3%)
Artrite non erosiva 38 (55,9%)
Anemia non emolitica 33 (48,5%)
Febbre 33 (48,5%) Alopecia 29 (42,6%) Ipocomplementemia 26 (38,2%) Ipergammaglobulinemia 24 (35,3%) Artrite erosiva 23 (33,4%) Teleangectasie 21 (30,9%) Sclerodattilia 20 (29,4%) Linfoadenomegalia 20 (29,4%) Fotosensibilità 18 (26,5%) Fibrosi polmonare 18 (26,5%) Pleurite 17 (25%) Impegno renale 16 (23,6%) Leucopenia 16 (23,6%) Rash malare 12 (17,6%) Piastrinopenia 11 (16,2%) Trombosi venosa 11 (16,2%) Aftosi orale 10 (14,7%) Polinevrite 9 (13,2%) Splenomegalia 8 (11,8%) IMA/Angina 7 (10,3%) Pericardite 7 (10,3%) Livedo reticularis 7 (10,3%) Porpora 7 (10,3%) Tiroidite 6 (8,8%) Ictus 6 (8,8%) Miosite 5 (7,4%) Arteropatia obliterante 5 (7,4%) Anemia emolitica 4 (5,9%)
Vasculite cerebrale 4 (5,9%) Ipertensione polmonare 3 (4,4%) Calcificazioni sottocutanee 2 (2,9%) Trombosi arteriosa 2 (2,9%) Swollen hands 1 (1,5%) Lupus discoide 1 (1,5%) Lupus subacuto 1 (1,5%)
Le manifestazioni cliniche osservate nei 25 pazienti affetti da “Sindrome Overlap indefinita” includono : artralgie (80%), fenomeno di Raynaud (80%), artrite non erosiva (68%), BPCO (56%), astenia (44%), anemia non emolitica (44%), ipergammaglobulinemia (44%), mialgie (40%), ipocomplementemia (40%). Il totale delle manifestazioni cliniche presentate è riportato in tabella 10.
Tabella 10. Sintomi e segni presentati dai 25 soggetti affetti da “Sindrome Overlap indefinita”.
Sintomi e segni Totale soggetti affetti, %
Artralgie 20 (80%)
Fenomeno di Raynaud 20 (80%)
Artrite non erosiva 17 (68%)
Astenia 11 (44%)
Anemia non emolitica 11 (44%)
Ipergammaglobulinemia 11 (44%) Ipocomplementemia 10 (40%) Leucopenia 9 (36%) Febbre 8 (32%) Xerostomia 7 (28%) Miosite 7 (28%) Xeroftalmia 7 (28%) Tiroidite 6 (24%) Alopecia 5 (20%) Linfoadenomegalia 5 (20%) Fotosensibilità 5 (20%) Vasculite cutanea 5 (20%) Impegno renale 4 (16%) Piastrinopenia 4 (16%) Trombosi venosa 4 (16%) Aftosi orale 4 (16%) Teleangectasie 3 (12%) Sclerodattilia 3 (12%) Pleurite 3 (12%) Pericardite 2 (8%) Anemia emolitica 2 (8%) Fibrosi polmonare 2 (8%) Swollen hands 2 (8%) Artrite erosiva 1 (4%) Polinevrite 1 (4%) Ulcere vasculitiche 1 (4%) Livedo reticularis 1 (4%) Porpora 1 (4%)
IMA/Angina 1 (4%) Rash malare 1 (4%) Vasculite cerebrale 1 (4%) Lupus discoide 1 (4%) Lupus subacuto 1 (4%) Ipertensione polmonare 1 (4%)
Il profilo anticorpale osservato nei 68 pazienti affetti da “Sindrome Overlap definita” è riportato nella tabella 11.
Tabella 11. Profilo anticorpale dei 68 soggetti affetti da “Sindrome Overlap definita”.
Positività anticorpale Totale soggetti, %
ANA 62 (91,2%) ACLA IgM 42 (61,8%) ACLA IgG 41 (60,3%) Ra test 36 (52,9%) SSA 26 (38,2%) ACA 12 (17,6%) Clif 11 (16,2%) SSB 9 (13,2%) Scl70 7 (10,3%) RNP 4 (5,9%) AMA 4 (5,9%) ASMA 3 (4,4%) Jo1 1 (1,5%) CCP 1 (1,5%) antiB2GPI 1 (1,5%) ANCA 0 (0%) Sm 0 (0%)
Il profilo anticorpale osservato nei 25 pazienti affetti da “Sindrome Overlap indefinita” è riportato nella tabella 12.
Tabella 12. Profilo anticorpale dei 25 soggetti affetti da “Sindrome Overlap indefinita”.
Positività anticorpale Totale soggetti, %
ANA 23 (92%) RNP 23 (92%) ACLA IgG 11 (44%) ACLA IgM 11 (44%) SSA 5 (20%) Ra test 4 (16%) Sm 4 (16%) Clif 3 (12%) SSB 3 (12%) CCP 1 (4%) ASMA 1 (4%) ACA 0 (0%) Scl70 0 (0%) AMA 0 (0%) Jo1 0 (0%) ANCA 0 (0%) antiB2GPI 0 (0%)
I soggetti affetti da “Sindrome Overlap definita” sono stati sottoposti a trattamento medico con steroidi < 10 mg (88,2%), calcio vitaminaD (66,2%), inibitori di pompa protonica (64,7%), plaquenil (61,8%), immunosoppressori (60,3%), bisfosfonati
(47,1%), Steroidi > 10mg (45,6%), Calcioantagonisti (41,2%). Le diverse terapie sono riportate in tabella 13.
Tabella 13. Terapia effettuata dai 68 pazienti affetti da “Sindrome Overlap definita”.
Trattamento medico Pazienti sottoposti, % Steroidi < 10 mg 60 (88,2%) Calcio VitaminaD 45 (66,2%) IPP 44 (64,7%) PQN 42 (61,8%) Bisfosfonati 32 (47,1%) Steroidi > 10 mg 31 (45,6%) Calcioantagonisti 28 (41,2%) Antiaggreganti 27 (39,7%) MTX 25 (36,8%) AZA 19 (27,9%) Pentossifillina 18 (26,5%) CyA 13 (19,1%) ACE inibitori 13 (19,1%) CS boli 11 (16,2%) Anticoagulanti 11 (16,2%) CFX 500 mg ev 10 (14,7%) Iloprost 8 (11,8%) Statine 6 (8,8%) CFX 1g ev 6 (8,8%) CFX os 5 (7,4%) IVIG 5 (7,4%) MMF 3 (4,4%) Nitroderivati 2 (2,9%)
I soggetti affetti da “Sindrome Overlap indefinita” sono stati sottoposti a trattamento medico con steroidi < 10 mg (84%),
plaquenil (72%), immunosoppressori (60%), steroidi > 10 mg (44%), calcio vitaminaD (36%), inibitori di pompa protonica (36%), antiaggreganti (32%). Le diverse terapie sono riportate in tabella 14.
Tabella 14. Terapia effettuata dai 25 pazienti affetti da “Sindrome Overlap indefinita”.
Trattamento medico Pazienti sottoposti, % Steroidi < 10 mg 21 (84%) PQN 18 (72%) Steroidi > 10 mg 11 (44%) Calcio VitaminaD 9 (36%) IPP 9 (36%) Antiaggreganti 8 (32%) MTX 7 (28%) Calcioantagonisti 7 (28%) CS boli 6 (24%) Bisfosfonati 5 (20%) AZA 4 (16%) CyA 4 (16%) Anticoagulanti 4 (16%) CFX 500 mg ev 4 (16%) CFX os 4 (16%) Iloprost 3 (12%) Statine 3 (12%) MMF 2 (8%) Nitoderivati 2 (8%) Pentossifillina 2 (8%) IGIV 1 (4%) ACE inibitori 0 (0%) CFX 1 g ev 0 (0%)
DISCUSSIONE
Il termine “Sindromi Overlap” individua condizioni largamente eterogenee, a partire dalla simultanea presenza di due o più malattie autoimmuni sistemiche ben definite (che soddisfano cioè i Criteri Classificativi), fino ad arrivare a condizioni caratterizzate dalla presenza di manifestazioni cliniche estremamente specifiche per connettiviti definite (CTD), che tuttavia non soddisfano alcuno dei Criteri Classificativi.
Nella popolazione di pazienti analizzata in questa tesi abbiamo identificato 68 pazienti con Sindrome Overlap da sovrapposizione di due o più malattie autoimmuni sistemiche (“Sindrome Overlap definita” ) e 25 pazienti affetti da Sindrome Overlap per la presenza di sintomi e segni specifici per connettiviti definite ( “Sindrome Overlap indefinita” ).
I pazienti che non soddisfano il numero minimo di criteri per una determinata forma di connettivite vengono spesso individuati in letteratura con il termine di “Connettiviti indifferenziate” (UCTD).
Tali Connettiviti indifferenziate sono caratterizzate dalla presenza di manifestazioni cliniche lievi e aspecifiche, da un andamento clinico benigno e da un profilo anticorpale semplificato e possono persistere invariate per un lungo periodo di tempo o anche per tutta la durata della vita oppure possono
rappresentare l’ esordio di una connettivite definita che subentra in genere entro 2-3 anni dopo l’ inizio della sintomatologia.
Ecco che quei soggetti che presentano manifestazioni cliniche specifiche per CTD ma che appunto non abbracciano il numero minimo di Criteri Classificativi per una malattia autoimmune sistemica, proprio per questa loro ultima caratteristica, vengono anch’ essi individuati in letteratura tra le UCTD.
La differenza sostanziale risiede, però, nel fatto che l’ analisi delle caratteristiche cliniche di questi soggetti evidenzia la presenza di segni e sintomi severi e specifici di connettiviti definite o la presenza di autoanticorpi specifici, tali da suggerire meccanismi patogenetici diversi oltre che richiedere un differente approccio diagnostico e terapeutico rispetto alle “vere” Connettiviti indifferenziate.
Queste condizioni piuttosto che “indifferenziate” sembrano quindi appartenere allo “spettro” di connettiviti definite.
Allo scopo di giungere ad un corretto raggruppamento delle malattie autoimmuni sistemiche, punto cruciale per lo svolgimento di studi epidemiologici, studi clinici ed associazioni in biomarkers e studi di tipo patogenetico, appare auspicabile quindi inserire tali condizioni nello “spettro” delle malattie autoimmuni definite e separarle in tal modo dalle malattie autoimmuni indifferenziate.
La definizione di “Sindrome Overlap indefinita” potrebbe risolvere tale confusione.
Ulteriori studi sono in corso con il fine di aumentare la casistica dei pazienti studiati presso la nostra Unità Operativa e di procedere al confronto tra le varie tipologie di Sindromi Overlap definite (Esempio: LES/AR con LES/SSc ); al confronto tra Sindromi Overlap definite e Connettiviti definite isolate (Esempio: LES/AR con LES e con AR ) e alla ricerca, all’ interno del gruppo delle Sindromi Overlap indefinite di manifestazioni cliniche e/o sierologiche che possano unificarle come entità nosologiche a sé stanti.
Appendice. Criteri per la classificazione e la diagnosi delle malattie reumatiche
Artrite Reumatoide
Devono essere soddisfatti almeno 4 dei seguenti 7 criteri:
1) Rigidità articolare mattutina 2) Artrite in 3 o più articolazioni
3) Artrite a livello delle articolazioni della mano 4) Artrite simmetrica
5) Noduli reumatoidi
6) Positività del fattore reumatoide 7) Alterazioni radiografiche
I primi quattro criteri devono essere presenti per almeno 6 settimane.
Spondiloartrite Dolore spinale infiammatorio
Oppure Sinovite
-asimmetrica o
-prevalente a carico degli arti inferiori e uno o più dei seguenti
1) anamnesi familiare positiva 2) psoriasi
3) malattia infiammatoria cronica intestinale
4) storia di uretrite, infiammazione della cervice uterina o diarrea acuta un mese prima della comparsa di artrite
5) dolore a carico della regione glutea con alternanza tra area destra e sinistra
6) entesopatia 7) sacro ileite
Febbre reumatica Manifestazioni
maggiori
Manifestazioni minori Evidenze di precedente infezione streptococcica - Cardite - Poliartrite - Corea - Eritema marginato - Noduli sottocutanei Cliniche: - Artralgia - Febbre Laboratoristiche: - Indici di flogosi elevati (VES e PCR) -Prolungato intervallo PR all’ ECG
- Positività della coltura faringea o del test rapido all’antigene streptococcico - Elevato titolo anticorpale anti- streptococcico
Lupus Eritematoso Sistemico
Per fare diagnosi di LES è necessaria la presenza di almeno 4 dei seguenti 11 criteri. 1- Rash malare 2- Lupus discoide 3- Fotosensibilità 4- Ulcere orali 5- Artrite 6- Sierosite
7- Alterazioni renali (proteinuria persistente > 0,5 grammi/die oppure riscontro di globuli rossi, emoglobina, cellule tubulari nell’urina)
8- Alterazioni neurologiche
9- Disordini ematologici: anemia emolitica con reticolocitosi oppure leucopenia (< 4000/mm^3 in due o più riscontri oppure linfopenia (< 1500/mm^3 in due o più riscontri oppure trombocitopenia (< 100000/mm^3 in assenza di terapia farmacologia favorente)
10- Disordini immunologici: Positività degli anticorpi anti-dsDNA oppure Positività degli anticorpi anti-Sm oppure Positività degli anticorpi antifosfolipide