“A Salvatore, perche in silenzio te lo avevo promesso”
1
INDICE
Introduzione generale
Estrogeni: generalità
STRUTTURA VIE BIOSINTETICHE DEGLI ORMONI GONADICI METABOLISMO
Recettori estrogeni
RECETTORI DI MEMBRANA o STRUTTURA o MECCANISMO D’AZIONE RECETTORI NUCLEARI o STRUTTURA o MECCANISMO D’AZIONE o ERα E ERβo DIFFERENZE CAVITA’ BINDING
Agonisti ed antagonisti a confronto
INTERAZIONI SITO BINDING SERMs: modulatori selettivi dei recettori degli estrogeni
SERBAs: agonisti selettivi per ERβ
DERIVATI NATURALI
DERIVATI BENZO-OSSAZOLICI
DERIVATI BENZO-PIRANICI Introduzione parte sperimentale
Sintesi dei composti 5, 6 e 12
Sintesi dei composti 7, 8 e 9
Sintesi dei composti 10, 11 e 13
Tentativo sintesi del composto 14
Tentativo sintesi del composto 15
Saggi di binding recettoriale
2
Parte Sperimentale
Materiali e metodi
Sintesi composti realizzati
Saggi di affinità per i recettori degli estrogeni
Metodi docking
Bibliografia
3
introduzione
generale
Introduzione generale
4
ESTROGENI
Generalità e struttura
Sino a pochi anni fa gli estrogeni erano considerati ormoni sessuali (con struttura steroidea a 18 atomi di carbonio) in grado di stimolare la proliferazione cellulare e indurre l’espressione dei recettori per il progesterone a livello uterino. Oggi, invece, è noto che gli estrogeni, oltre a promuovere lo sviluppo e il mantenimento dei caratteri sessuali secondari femminili, sono coinvolti in numerosi processi fisiologici. La funzione1 di entrambi gli ormoni è quella di promuovere:
nella donna la gametogenesi e lo sviluppo dei follicoli; nell’uomo la spermatogenesi.
Gli ormoni estrogenici presentano azioni diverse nei diversi distretti corporei:
Nel sistema nervoso centrale, ad esempio, agiscono sulle aree che preparano l’organismo per la riproduzione, aiutano a mantenere costante la temperatura corporea e proteggono le aree della memoria. Recenti studi clinici hanno permesso di ipotizzare un loro coinvolgimento anche nel morbo di Alzheimer2.
A livello del tessuto osseo svolgono un azione protettiva regolandone sintesi e degradazione, infatti mantengono la densità ossea riducendo il rischio di fratture.
Nel sistema cardiovascolare limitano gli accumuli di placche aterosclerotiche.
A livello del tratto gastro-intestinale, svolgono azione protettiva contro lo sviluppo del cancro al colon.
Infine, si ritiene possibile una loro azione anche sul sistema immunitario.Figura 1: Effetti degli estrogeni
Gli estrogeni sono coinvolti nel controllo del rilascio delle gonadotropine nell’asse ipotalamo-ipofisario. Il fattore ipotalamico3 di liberazione delle gonadotropine (GnRH) è un deca-peptide che stimola l’adenoipofisi (o ipofisi anteriore) a rilasciare l’ormone follicolo stimolante (FSH) e l’ormone luteinizzante (LH), che nella donna agiscono sull’ovaio, mentre nell’uomo sul testicolo.
Introduzione generale
5 L’FSH è l’ormone principale che si occupa del controllo della secrezione degli estrogeni, nella donna si occupa della conversione degli androgeni della granulosa in estrogeni; mentre nell’uomo agisce sulle cellule del Sertoli e stimola la produzione di una proteina legante gli androgeni.
L’LH invece, nella donna si occupa dell’ovulazione ed è il principale ormone che stimola la successiva secrezione di progesterone da parte del corpo luteo; nell’uomo invece stimola la secrezione di testosterone, agendo sulle cellule del Leyding
.
Figura 2: Regolazione ormonale
Tali ormoni risentono di un sistema di controllo a feed-back sia diretto che indiretto. I prodotti della secrezione delle gonadi esercitano un effetto a feedback negativo diretto, inibendo il rilascio di GnRH, FSH e LH. Quando invece le concentrazioni ematiche di estrogeni e androgeni sono basse, il controllo dell’asse ipotalamo-ipofisario è a feedback positivo, ossia si ha il rilascio di GnRH, e di conseguenza anche quello di LH e FSH.
Altri prodotti delle gonadi sono numerosi peptidi e proteine come, le inibine e le attivine che fanno parte della stessa famiglia dei fattori di regolazione della crescita. L’inibina1 è una glicoproteina che circola nel plasma e inibisce la secrezione ipofisaria di FSH indotta da GnRH. L’attivina esercita un’azione opposta e stimola pertanto la secrezione di FSH.
Non sempre, però, gli effetti degli estrogeni risultano vantaggiosi, in quanto possono favorire la proliferazione incontrollata di una popolazione cellulare già mutata ( e quindi
Introduzione generale
6 l’insorgenza di neoplasie), soprattutto in quei distretti, che rappresentano la principale sede della loro azione fisiologica, quali utero e seno.
Le donne in menopausa vanno in contro poi ad una diminuzione degli estrogeni che innesca una serie di disagi, come ad esempio piccoli shock termici (vampate di calore), incremento del colesterolo (LDL) ematico con conseguente aumento della probabilità di insorgenza di patologie cardiache e diminuzione della densità minerale ossea che può determinare l’insorgenza di patologie come l’osteoporosi.
Struttura
Gli estrogeni appartengono alla famiglia degli ormoni steroidei, alla quale appartengono anche androgeni e progestinici. Dal punto di vista chimico sono costituiti da un nucleo base comune, costituito da un sistema a quattro anelli condensati, che prende il nome di
ciclopentanoperidrofenantrene.
Figura 3: Struttura del Ciclopentanoperidrofenantrene
I principali estrogeni femminili sono l’estradiolo (17β-estradiolo), l’estrone e l’estriolo. L’estradiolo è il maggiore prodotto della secrezione ovarica, ma in parte viene anche prodotto dalla corteccia del surrene. Sebbene una certa quantità di estrone sia prodotta nell’ovaio, la maggior parte di questo ormone (e di estriolo) è sintetizzata nel fegato a partire dall’estradiolo, o in tessuti periferici a partire dall’androstenedione e da altri androgeni.
Estrone Estriolo Estradiolo
Figura 4: Ormoni estrogenici
Nell’estradiolo l’anello A è idrossilato in posizione C-3 e l’idrossile in posizione C-17 dell’anello a cinque termini conferisce l’attività estrogenica di tale ormone. L’assenza di quest’ultimo idrossile nell’estrone e la presenza invece anche di un altro ossidrile a livello del
Introduzione generale
7 C-16 nell’estriolo, determina una bassa attività ormonale di questi estrogeni e quindi una minore affinità per il recettore estrogenico.
Vie biosintetiche degli ormoni steroidei gonadici
Nei mammiferi la biosintesi degli estrogeni avviene principalmente a livello ovarico, in seguito a stimoli ipofisari ( FSH e LH), sottoposti, a loro volta, a regolazione ipotalamica. Entrambi i sessi utilizzano una via comune per la sintesi degli ormoni steroidei gonadici3. Questa via inizia dal colesterolo ed i geni che codificano la sintesi degli enzimi necessari e le loro caratteristiche e i cofattori indispensabili sono gli stessi presenti a livello delle ghiandole surrenali.
Nelle gonadi, il substrato iniziale della biosintesi1 è il colesterolo, che può essere generato
in situ dall’Acetil-CoA o captato dalle lipoproteine plasmatiche a bassa densità (LDL).
L’enzima 20,22 desmolasi (A) catalizza il distacco della catena laterale del colesterolo trasformandolo in ∆-5- pregnenolone. Nonostante il testosterone possa essere sintetizzato utilizzando due vie sintetiche parallele, la via predominante è la cosiddetta via Δ5
,che inizia con il pregnenolone. Il pregnenolone è trasformato in 17-OH-pregnenolone dalla
17-α-idrossilasi (B) ed in progesterone dagli enzimi 5-ese-3β-idrossisteroide deidrogenasi e 3-ossosteroide-4,5-isomerasi (C). Il 17-OH-pregnenolone subisce la conversione ad
deidroepiandrosterone ad opera della 17,20-liasi (D). L’ossidazione dell’anello A da parte del complesso enzimatico 3β-ol-deidrogenasi-isomerasi può avere luogo a ogni livello della sintesi, dal pregnenolone all’androstendiolo. L’androstendiolo viene convertito in testosterone ad opera della 17-β-idrossisteroide deidrogenasi (E). Solo una piccola quota di testosterone subisce una riduzione in posizione 5α ad opera della 5α-reduttasi (H), generando il diidrotestosterone. Nei testicoli si verifica un’ulteriore α- riduzione nella posizione 3-chetone, che genera il 5α-androstenediolo.
Gli androgeni rappresentano i precursori obbligati degli estrogeni. La reazione fondamentale nella conversione degli androgeni in estrogeni è l’aromatizzazione dell’anello A; azione fortemente favorita nell’ovaio e nella placenta. Pertanto, sia il testosterone che la sua forma ridotta il diidrotestosterone, insieme all’androstendione costituiscono i substrati sui quali agisce l’aromatasi (F).
Il complesso enzimatico aromatasi, che catalizza questa reazione, è una citocromo P-450 che determina:
idrossilazione del gruppo metilico in posizione 19 e sua ossidazione ad aldeide; idrossilazione della posizione 2 e, infine, formazione di un doppio legame tra le
posizioni 1 e 2 per riduzione.
Al termine di questa sequenza, l’atomo di carbonio in posizione 19 viene rimosso per decarbossilazione e si forma il caratteristico anello benzenico.
I due ormoni possono anche subire un interconversione per azione dell’enzima
Introduzione generale
8 SCHEMA 1: Sintesi degli ormoni sessuali a partire dal colesterolo
Colesterolo
∆-5-pregnenolone 17-OH-pregnenolone Deidroepiandrosterone
Progesterone 17-OH-progesterone Androstenedione
Diidrotestosterone Testosterone
Estrone Estradiolo A B B C C D D F E G H F
Introduzione generale
9
Metabolismo
L’estradiolo e l’estrone una volta in circolo si legano ad una α2
-globulina, detta SHBG (Sex-Hormone Binding Globulin), ma con affinità molto inferiore rispetto a quella del testosterone, pertanto, circolano per la maggior parte legati debolmente all’albumina e la loro clearance metabolica è relativamente molto alta.
Nelle donne in età feconda gran parte dell’estradiolo presente in circolo è prodotto dalla secrezione ovarica, solo una frazione minore deriva dal testosterone, per trasformazione nel tessuto adiposo, nel fegato e in altre sedi. L’estrogeno legato è relativamente indisponibile per la diffusione all’interno delle cellule. Pertanto, soltanto una piccola quota è libera ed è in grado di diffondere all’interno delle cellule, grazie alle proprietà idrofobiche del nucleo steroideo, quindi riesce ad essere fisiologicamente attiva.
Estradiolo Estrone
Estriolo 16-idrossiestrone
Figura 5 : Metabolismo dell’estradiolo
Buona parte dell’estrone circolante è derivato dall’estradiolo, per effetto delle deidrogenasi periferiche per i 17-idrossisteroidi4. L’estrone può essere idrossilato in posizione 16 e quindi essere ridotto a estriolo. I derivati solfatati e glucuronati di questi tre ormoni estrogenici vengono escreti nelle urine o mediante secrezione biliare.
Un’altra via metabolica degli estrogeni comprende la 2-idrossilazione e produce i così detti catecolestrogeni1. Questi composti assomigliano ai neurotrasmettitori noradrenalina e dopamina (catecolamine) per la presenza di anelli benzenici idrossilati. Poiché l’attività 2-idrossilasica è presente nell’ipotalamo, è stato ipotizzato che i catecolestrogeni prodotti nel
Introduzione generale
10 cervello possano modulare gli effetti dell’estradiolo sulla liberazione di GnRH. I catecolestrogeni si legano ai recettori per l’estradiolo, ma agiscono come sostanze antiestrogeniche naturali che possono incrementare la liberazione di GnRH mediante un meccanismo a feedback negativo.
Estradiolo Estrone 2-idrossiestradiolo 2-idrossiestrone
2-metossiestradiolo 2-metossiestrone
Introduzione generale
11
RECETTORI ESTROGENICI
Come detto precedentemente, l’estradiolo, entrato in circolo si lega alla α2-globulina, raggiunto il recettore target, il complesso estradiolo-SHGB4 si dissocia e l’estrogeno va ad interagire con il recettore di membrana. Gli estrogeni interagiscono con due tipi di recettori: recettori di membrana e recettori nucleari e si trovano principalmente nella mammella, nell’utero, nel cervello, nel fegato e nelle ossa. L’ estradiolo non complessato con SHGB diffonde liberamente attraverso la membrana e va ad interagire a livello del recettore nucleare.
Recettori di membrana
Struttura
I recettori trans-membrana sono proteine integrali di membrana, che interagendo con uno specifico ligando, mediano una risposta biochimica intracellulare, presentando un ruolo fondamentale nel processo di trasduzione del segnale. I recettori di membrana accoppiati a proteine G presentano sette domini trans membrana, nello specifico le GPCR sono costituite da una singola catena polipeptidica formata anche da 1100 residui. La caratteristica strutturale è rappresentata da 7 α-eliche trans- membrana, con un dominio extracellulare N-terminale di lunghezza variabile e un dominio intracellulare C-terminale. Il terzo lungo loop citoplasmatico del recettore rappresenta la regione della molecola che si accoppia alla proteina G.
Figura 7: Rappresentazione recettore di membrana accoppiato a proteine G e cascata di secondi messaggeri a seguito dell’attivazione.
Introduzione generale
12
Meccanismo
Le proteine G consistono di tre subunità α, β, γ e, quando l’agonista si lega al recettore, si attiva un cambiamento conformazionale, che coinvolge il dominio citoplasmatico. La subunità α si dissocia e va ad attivare un effettore, che in questo caso è la fosfolipasi C (PLC). Quest’ultimo porta alla formazione di IP3
(inositolo 1,4,5 trifosfato) che incrementa la concentrazione di Ca++ intracellulare, stimolando il suo rilascio dal reticolo sarcoplasmatico, e di DAG (diacilglicerolo) che a sua volta va ad attivare una proteina chinasi di membrana, la PKC (proteina chinasi C); tale proteina favorisce l’entrata di ioni calcio nella cellula. L’aumento della concentrazione di Ca++
intracellulare promuove l’attivazione delle NOS (NO-sintetasi) che rilasciano NO (ossido d’azoto) utilizzato nel SNC come neurotrasmettitore.
Recettori
nucleari
Struttura
I recettori nucleari sono strutture di natura proteica, si trovano nel citoplasma sottoforma di complessi oligomerici con particolari proteine stabilizzanti, dette Hsp90 (heat shock protein 90), che interagiscono con ligandi di natura ormonale o molecole specifiche.
Ogni recettore presenta 6 domini funzionali:
Una porzione –NH2 terminale A/B : responsabile del processo di dimerizzazione che segue all’interazione ligando-recettore, contenente la regione AF-1 coinvolti nell’attivazione della trascrizione.
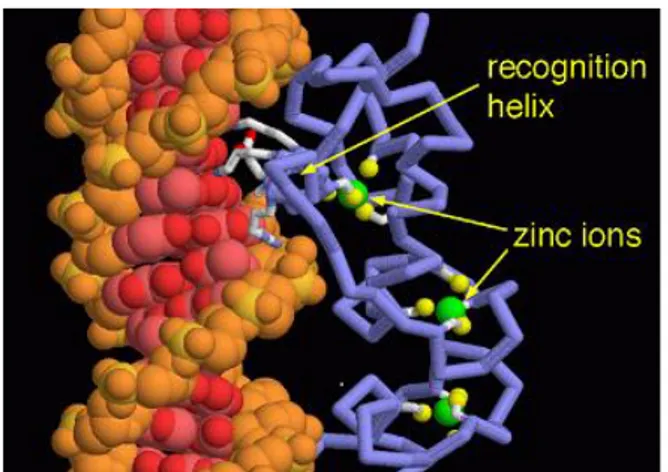
Una porzione centrale C che presenta affinità per specifiche sequenze di DNA, detta DBD (DNA binding domain). Questa sequenza recettoriale è costituita da aminoacidi basici in prevalenza che favoriscono il legame al DNA. La diversità di questa sequenza è la ragione della specificità di legame con le sequenze HRE (Hormone Responsive Elements). Sono anche presenti dei residui di cisteina ed istidina, che interagendo tramite legami di coordinazione con un atomo di zinco, costituiscono un dominio, definito “zinc finger”, che ha la funzione di mantenere il recettore nella conformazione necessaria per l’interazione con il DNA.
Introduzione generale
13
Una porzione E, che lega l’ormone (LBD),contenente una zona che attiva la trascrizione ormone-dipendente, detta AF-2. È formata da 12 α-eliche disposte a “sandwich”, che costituiscono la tasca idrofobica responsabile del legame con il ligando. LBD è formato da tre strati di α-eliche antiparallele: uno centrale a tre eliche (H5/6,H9 e H10), posto tra due strati addizionali di eliche (H1-4 e H7,H8, H11) ed infine possiede due tratti con struttura β-sheet (S1,S2) e un tratto α-elicoidale (H12).
Un dominio “ cerniera D” situato tra il DBD e l’LBD ( ligand-binding domain) che si occupa della variazione conformazionale a seguito del legame ligando-recettore.
Una porzione COOH-terminale F,che promuove la trascrizione da parte della RNA polimerasi.
Figura 9: Organizzazione strutturale del recettore nucleare
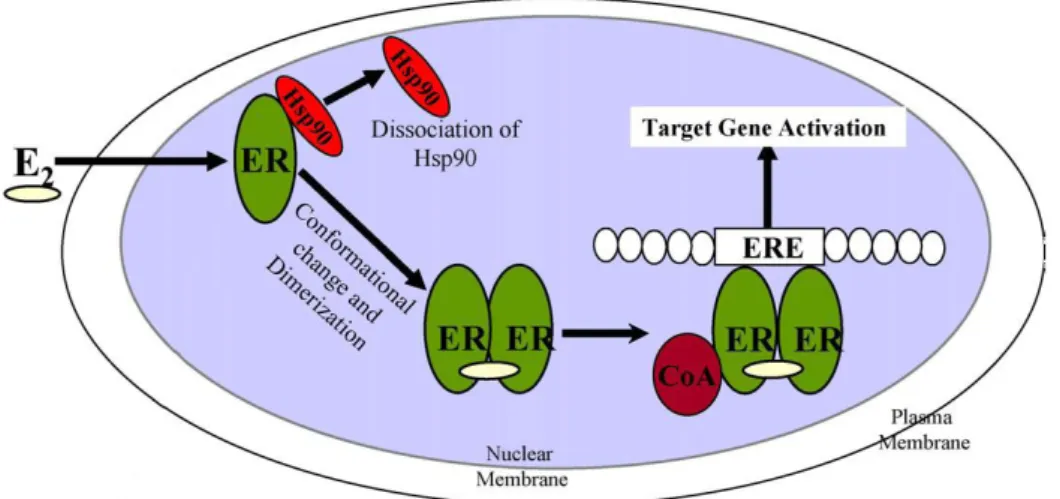
Meccanismo d’azione
L’ormone in forma libera proveniente dal plasma o dai tessuti interstiziali, passa nella cellula e si lega al recettore (dominio LBD) formando con esso un omo/etero-dimero. Il complesso ligando-recettore funge da fattore di trascrizione, il quale viene trasportato attivamente nel nucleo cellulare, dove il dominio C del recettore interagisce con specifiche sequenze palindrome di DNA, chiamate EREs (Estrogen Response Elements) e regola l’espressione genica. Le EREs sono localizzate a livello dell’estremità 5’ della regione
Introduzione generale
14 promotrice del gene. L’attivazione trascrizionale avviene mediante interazione diretta del complesso ligando-recettore con ERE e con i co-attivatori legati alla AF-2.
Figura 10: Rappresentazione del meccanismo d’azione dei recettori nucleari
Si ritiene che altre risposte cellulari all’azione degli estrogeni, relativamente rapide, siano mediate dall’attivazione di protooncogeni1
, come c-jun e c-fos, la cui espressione è indotta dal complesso ER-ligando. Questi fattori da soli o combinati possono facilitare le azioni successive all’attivazione della trascrizione.
Figura 11: Attivazione di proto-oncogeni
ERα e ERβ
Esistono due sottotipi recettoriali di recettori nucleari degli estrogeni ERα e ERβ. I recettori nucleari degli estrogeni sono i primi ad essere stati caratterizzati, ERβ5
addirittura solo nel 1996; sono costituiti da una sequenza1 che varia da 500 a 600 aminoacidi, che vengono codificati da geni diversi localizzati su cromosomi separati con una omologia6 variabile dal 22 al 96% . I domini leganti il DNA sono molto simili, hanno un omologia del 96%, mentre quelli leganti il ligando mostrano solo il 53% di omologia nella sequenza aminoacidica. Invece, per i domini A/B e D l’omologia è del 27% e del 26% rispettivamente.
Introduzione generale
15 Infine, la regione F è quella che presenta la minore omologia 22%. Il gene per il recettore α codifica per 595 amminoacidi, mentre quello per il recettore β codifica per 530 amminoacidi.
Figura 12: Differenze strutturali tra ERα ed ERβ
Le due forme del recettore hanno la stessa affinità per l’estradiolo, mentre quella per l’estrone e per numerosi agonisti e antagonisti di sintesi è molto diversa. La loro distribuzione tissutale mostra ampie regioni di sovrapposizione, ma anche localizzazioni specifiche.
Il recettore α è maggiormente espresso a livello dell’endometrio uterino, della mammella, delle cellule della granulosa del follicolo7, delle ossa, del cervello, del sistema cardiovascolare e del fegato.
Il recettore β si trova principalmente nel SNC, nell’apparato digerente, nei reni, nella prostata, nel tessuto polmonare, nell’endotelio vascolare, nel sistema immunitario, nelle ossa, nelle cellule della teca interna del follicolo, nell’utero, nelle cellule luteiniche del corpo luteo7 ed in piccola parte anche nella mammella.
La differente localizzazione tissutale6 ha fornito inseguito anche una spiegazione della diversa attivazione di questi recettori. È stato descritto che i complessi estradiolo-ERα attivano la trascrizione, mentre i complessi estradiolo-ERβ la reprimono6.
Differenze nella cavità di binding
Il dominio legante il ligando, LBDs, di ERα ed ERβ presenta solo il 59% di omologia nella sequenza aminoacidica ; la cavità di binding dei due sottotipi mostra solo piccole differenze nella struttura e nella composizione.
L’analisi strutturale dei due recettori complessati con l’estradiolo (E2) mostra che entrambi mantengono una forte interazione attrattiva che coinvolge il gruppo OH dell’anello fenolico di E2, il quale instaura un legame ad idrogeno con una molecola di acqua e si lega ulteriormente con due residui aminoacidici del dominio LBD di ER (Glu353 e Arg 394 in ERα, Glu305 e Arg346 in ERβ).
Introduzione generale
16 Figura 13: Interazioni dell’estradiolo a livello del sito di binding
In entrambi i recettori, inoltre, l’OH alcolico in posizione 17β del gruppo E2 presenta un legane ad idrogeno addizionale con His524 ( ERα) o His475 (ERβ). La cavità di binding ERα/ ERβ risulta essere composta da 23 residui aminoacidici che si trovano ad una distanza di 4Å dal ligando, e di questi 23, solo in due posizioni sono presenti differenti aminoacidi, le sostituzioni sono:
Leu384 e Met421in ERα ;
Met336 e Ile373 in ERβ.
La cavità di binding ERβ presenta un volume minore rispetto a quella di ERα8
.
AGONISTI ED ANTAGONISTI DEGLI ESTROGENI
La cristallizzazione del dominio responsabile dell’interazione dei ligandi dell’ER, ottenuta in presenza sia di un agonista (estradiolo) che di un antagonista (Tamoxifene) , ha fatto comprendere molti aspetti delle modificazioni conformazionali che si verificano a seguito della formazione del complesso ligando-recettore. La principale differenza sta nella posizione dell’elica 12 che è essenziale per il corretto legame dei co-attivatori in modo che si formi un complesso di trascrizione a livello del gene responsivo all’estrogeno.
Un agonista fa si che l’α-elica H126 si chiuda sull’α-elica H4, in modo che il ligando rimanga intrappolato all’interno di una tasca idrofobica del dominio di legame; questa conformazione permette il legame dei co-attivatori che determina l’attivazione della trascrizione.
Un antagonista impedisce invece la chiusura della tasca idrofobica che quindi rimane nella conformazione “aperta”, ne segue il mancato legame dei co-attivatori, di conseguenza non viene attivata la trascrizione. In questo caso la conformazione aperta è dovuta all’ingombro sterico determinato dalla catena laterale dell’antagonista, mancante nell’agonista, che nel caso del Tamoxifene è di tipo N,N-dimetilamminoetilica.
Introduzione generale
17 Figura 14: Differente disposizione di un agonista e di un antagonista a livello della cavità di binding.
Interazioni a livello del sito di binding
A livello del sito di binding di ER diverse sono le interazioni9 determinate da un antagonista o un agonista. Un agonista, considerando il caso dell’estradiolo, a livello del sito di binding determina la formazione di legami ad idrogeno che si instaurano tra l’ossidrile dell’anello fenolico A dell’estradiolo ed il Glu353, l’Arg394 e con una molecola di acqua. La presenza poi di un ulteriore gruppo OH sull’anello D determina la formazione di un nuovo legame ad idrogeno con l’His524; inoltre gli anelli condensati della molecola contribuisco alla formazione di interazioni di tipo idrofobico che stabilizzano la stessa a livello del sito di legame.
Figura 15: Presenza di un agonista a livello del sito di binding di ER.
Un antagonista, come il Raloxifene, presenta una struttura con gruppi funzionali importanti per l’aggancio al recettore: la prima è rappresentata dai due gruppi ossidrilici che, analogamente all’estradiolo, instaurano legami ad idrogeno con Glu353 e Arg394. I due ossidrili sono separati da un corpo centrale rigido, che mima la struttura steroidea degli
Introduzione generale
18 agonisti endogeni, contraendo interazioni idrofobiche con il sito. L’ossidrile dell’anello fenolico invece forma un legame ad idrogeno con His524. Un ulteriore sito di ancoraggio nel Raloxifene è rappresentato dalla catena laterale che termina con un anello piperidinico, il quale, a pH fisiologico, si presenta per lo più nella forma protonata, instaurando un nuovo legame ad idrogeno con la funzione carbossilica dell’Asp351, aumentando l’affinità per il recettore.
Figura 16: Presenza di un antagonista a livello del sito di binding di ER.
SERMs: MODULATORI SELETTIVI DEI RECETTORI ER
I SERMs10 sono una classe di farmaci che agiscono sui recettori degli estrogeni; la caratteristica che li distingue dagli agonisti ed antagonisti puri dei recettori, è il fatto che, a seconda dei vari tessuti dove agiscono, esplicano una differente attività, garantendo la possibilità di fungere da agonista su alcuni tessuti, e da antagonisti su altri.
Non è ancora del tutto chiaro come i SERMs possano agire da agonisti degli estrogeni in alcune cellule ed da antagonisti in altre, ma sono state proposte ipotesi diverse.
Una prima ipotesi riguarda la natura dei co-regolatori10
: alcuni tessuti potrebbero non essere in grado di produrre co-attivatori capaci di legarsi al complesso SERM-recettore, mentre altre cellule potrebbero presentare un set di coattivatori differenti o in parte modificati capaci di agganciare i recettori in conformazione anomala e dare il via ad un complesso di trascrizione funzionante11.
Un’altra ipotesi si basa sul fatto che i complessi formati dai SERMs siano incapaci di legare gli elementi di risposta agli estrogeni nei geni.
Infine non si può sottovalutare l’ipotesi che le isoforme α e β del recettore rispondano in maniera differente al legame dei SERMs, e che a seconda della distribuzione tissutale, vari la risposta generata.
Risulta da alcuni studi condotti sui topi che l’azione dei SERMs su ERα induca la trascrizione e che si abbia un’azione opposta su ERβ. Sembra addirittura che la stimolazione di ERβ porti all’inibizione di ERα6
Introduzione generale
19 Figura 17: Meccanismo d’azione dei SERMs.
Tamoxifene
Il Tamoxifene è un inibitore competitivo dell’estradiolo, con attività agonista parziale a livello del recettore per gli estrogeni. E’ un derivato trifeniletilenico e può essere considerato il capostipite della categoria dei SERMs. Nelle donne in post-menopausa mostra un attività di tipo estrogenico, creando un modesto rischio di cancro all’endometrio estrogeno-dipendente. Importante è il fatto che riduce il rischio di fratture all’anca e al polso, in quanto incrementa la densità ossea; causa anche una caduta del colesterolo legato alle LDL, ma non di quello legato alle HDL; ciò può portare ad una minore incidenza di coronaropatia12 negli individui ad alto rischio. Al contrario, il Tamoxifene causa un effetto antiestrogenico nel seno, prevenendo il tumore primario4.
Figura 18: Struttura chimica di: (A) Tamoxifene; (B) Raloxifene
Raloxifene
Il Raloxifene è un agonista-antagonista degli estrogeni. E’ un derivato benzotiofenico che ha dimostrato di abbassare il colesterolo LDL, ma di non aumentare quello HDL; mantiene la
Introduzione generale
20 densità ossea nelle donne in post-menopausa . E’ stato approvato negli USA per la prevenzione dell’osteoporosi e delle fratture spinali; la terapia fino ad oggi utilizzata è l’HRT (Hormon Replacement Therapy) . In questa terapia, l’effetto preventivo sul cancro al seno diventa, in qualche modo, un effetto collaterale positivo, anche perché non è accompagnato da stimolazione dell’endometrio. Non è efficace contro quei sintomi della menopausa, come sudorazione eccessiva e vampate di calore.
Clorotamoxifene
E’ derivato trifeniletilenico, analogo del Tamoxifene, utilizzato per il trattamento del cancro del seno in stadio avanzato nelle donne in post-menopausa. Il farmaco possiede anche proprietà estrogeniche in quanto abbassa i livelli di colesterolo e mantiene la densità ossea
.
Figura 19: Struttura chimica di: (C) Clotamoxifene;(D) Droloxifene; (E) Idoxifene.
Idoxifene e Droloxifene
Analoghi del Tamoxifene, capaci di mantenere la massa ossea; attualmente in fase di studio per la prevenzione dell’osteoporosi e del cancro al seno10
.
SERBAs: AGONISTI SELETTIVI PER ERβ
I SERBAs (Selective Estrogen Receptor Beta Agonists) sono molecole di natura non steroidea selettive per il recettore beta degli estogeni. Si ritiene che tali farmaci potrebbero presentare le caratteristiche ideali dei SERMs, come la capacità di promuovere effetti benefici a livello delle ossa, degli adipociti (regolare il metabolismo degli acidi grassi, favorendo la diminuzione del colesterolo nel sangue), del fegato, del SNC (come migliorare le capacità mnemoniche13), del sistema cardiovascolare ( prevenire l’ipertrofia del miocardio e i danni post-ischemici14), ma tuttavia essere privi dell’azione proliferativa mediata da ERα15. Infatti, è stata dimostrata l’utilità terapeutica di agonisti ERβ – selettivi nell’iperplasia/ cancro della prostata, nella demineralizzazione ossea ed anche alcune patologie infiammatorie16,17 che coinvolgono linfociti T- helper, alcune forme di asma, nell’artrite reumatoide e nell’infiammazione intestinale.
Introduzione generale
21 Sulla base di questi presupposti, la ricerca si è indirizzata verso lo sviluppo di nuovi e selettivi ligandi per ERβ, ma finora solo un numero limitato di composti hanno mostrato un buon livello di selettività, anche se diverse sono le classi di composti indagate.
Derivati naturali
Lo sviluppo dei nuovi composti ERβ, si è basato inizialmente su osservazioni effettuate su alcuni prodotti di origine naturale, come la genisteina, un isoflavone estratto dalla soia, che presentava un azione antiproliferativa a livello del tumore della ghiandola mammaria, ma era priva dei caratteristici effetti collaterali degli antiestrogeni. Tale azione era attribuita ad una elevata affinità e selettività per il sottotipo recettoriale β, confermata inseguito anche dai saggi di binding18 (Figura 20).
Figura 20: Genisteina
Questi dati sono stati la fonte di un aumentato interesse per il sottotipo recettoriale β e hanno ispirato la sintesi di alcuni derivati, tra cui il diaril-propion-nitrile (DPN) Figura 21, che si è dimostrato essere un potente e selettivo agonista ERβ19
. Tale composto, utilizzato inizialmente come miscela racemica, è risultato essere maggiormente affine per il sottotipo recettoriale β se utilizzato nella configurazione S20
, in quanto il gruppo CN aveva la possibilità di interagire positivamente con la Met336 di ERβ.
Figura 21: Diaril-propion-nitrile
Derivati benzo-ossazolici
Un’altra classe, che ha fornito composti di sintesi selettivi per ERβ, è stata quella dei benzo-ossazoli. Il capostipite di questa classe è ERB-041(Figura 22) per il quale sono state studiate le interazioni sia a livello di ERα che di ERβ, con lo scopo di valutare i sostituenti e le porzioni della molecola indispensabili.
RBA ERα = 0.017% RBA ERβ = 7.4% β/α = 440 RBA ERα = 0.25% RBA ERβ = 18% β/α = 78
Introduzione generale
22
Figura 22: Derivato benzo-ossazolico
Dall’analisi del docking21 è risultato che in entrambi i sottotipi recettoriali il sostituente 3-fluoro-4-idrossifenile si trova in prossimità di una coppia di amminoacidi Glu/Arg e il gruppo OH fenolico interagisce con quest’ultimi formando dei legami ad H, mentre l’altro gruppo OH presente sullo scaffold forma un altro legame ad H con un residuo di His presente alla fine della cavità di binding. La differente affinità di binding dei due sottotipi è fornita dall’interazione tra il sostituente vinilico con il residuo non conservato Met421 (ERα)/Ile373 (ERβ); infatti è stato dimostrato che il sostituente vinilico nel sottotipo ERα è bloccato a causa dall’ingombro sterico fornito dalla catena laterale del residuo di Met421; invece nel sottotipo β genera un’interazione idrofobica, che stabilizza il composto nel sito di binding (Figura 23).
Figura 23: Interazioni del composto ERB-041
Uno studio approfondito delle relazioni struttura-attività ha permesso di realizzare un notevole numero di derivati analoghi, comunque il composto ERB-041( IC50 = 1200 nM su ERα e di 5.4 nM su ERβ; β/α = 200) è risultato essere il derivato con il massimo livello di selettività ERβ22
di questa classe di composti.
Derivati benzopiranici
Questa classe di derivati sono stati ispirati ad alcune strutture molecolari di ligandi ERβ selettivi di origine naturale come la genisteina. Tra i primi composti23 sintetizzati vi è il derivato SERBAs-1(Figura 24), che si ottiene inizialmente come miscela racemica e presenta un anello addizionale, come il 3,4-ciclopentano, che ha rivelato di essere fondamentale per determinare il legame a ERβ.
Analoghi che presentavano un ingrandimento di tale anello condensato subivano una diminuzione sia dell’affinità che della selettività per ERβ.
Introduzione generale
23
Figura 24: Struttura SERBAs-1
Il composto SERBAs-1 è l’enantiomero (2R,3S,4R) e rispetto a SERBAs-2 l’enantionero (2S,3R,4S) ha dimostrato di possedere i valori migliori di selettività ed affinità per ERβ ( Ki = 0.19 nM per ERβ e 2.68 nM per ERα, β/α = 14). Tali dati sono stati convalidati dall’analisi strutturale24 ai raggi X del composto SERBAs-1 complessato sia con ERα che con ERβ, da cui si evince che il composto assume orientazioni differenti in base al sottotipo recettoriale, subendo addirittura una rotazione dell’asse di 180º. Le interazioni fondamentali di SERBAs-1 a livello di ERβ sono: un legame ad H tra il gruppo p-idrossi-fenilico e la coppia Arg346/Glu305; un altro legame ad H tra His475 e l’OH del fenolo condensato, mentre l’anello ciclopentanico occupa una piccola tasca idrofobica, chiusa esclusivamente in ERβ da un residuo di Ile373. Al contrario, in ERα la sostituzione dell’Ile373 con una Met421 riduce lo spazio della tasca idrofobica per l’inserimento dell’anello ciclopentanico, causando la rotazione di 180º dell’asse del bifenolo (Figura25)
i 24
Introduzione
parte
sperimentale
Introduzione parte sperimentale
25 Il mio lavoro di Tesi di Laurea è stato dedicato ad un progetto di ricerca, avviato da alcuni anni in collaborazione con il Prof. Katzenellenbogen, dell’Università dell’Illinois, il cui scopo è quello di identificare nuovi ligandi, in particolare piccole molecole organiche di natura non steroidea, selettivi per il recettore β degli estrogeni.
Figura 1: Anello A dell’estradiolo e pseudo ciclo A’ dei derivati salicilaldossimici
In particolare nel laboratorio presso il quale ho svolto la mia attività di Tesi si erano ottenuti buoni risultati con i derivati salicilaldossimici monoaril-sostituiti; dove il raggruppamento salicilaldossimico, riveste il ruolo di bioisostero del gruppo fenolico, presente nei derivati estrogenici, in quanto forma uno pseudo - anello (A’) a sei termini (Figura 1), grazie ad uno stabile legame ad idrogeno intramolecolare fra l’ossidrile fenolico e l’azoto ossimico.25
Questa classe di composti ha delle caratteristiche strutturali che sono in accordo con il modello farmacoforico per ligandi ERβ-selettivi11
descritto in Figura 2, delineato sulla base di analisi strutturali di alcuni composti selettivi per il recettore β di origine naturale come la genisteina,26 o di origine sintetica come i derivati indazolici.27
Genisteina Derivato indazolico
Figura 2: Composti utilizzati per definire il nuovo modello farmacoforico dei ligandi β-selettivi.
Introduzione parte sperimentale
26 Il composto 1 (Figura 3), risulta essere il primo derivato della classe dei derivati salicilaldossimici monoaril-sostituiti che nei saggi di binding si è dimostrato particolarmente selettivo per il sottotipo recettoriale β, nonostante la sua affinità per quest’ultimo non risulti particolarmente elevata.8
Figura 3: Composto 1 realizzato sul modello farmacoforico dei ligandi β-selettivi.
L’affinità di binding recettoriale di questi composti è stata valutata attraverso saggi radiometrici di spiazzamento competitivo del [3H]estradiolo sui sottotipi recettoriali ERα ed ERβ umani ricombinanti purificati. I dati delle affinità relative di binding (RBA) sono rapportati all’estradiolo (Kd = 0.2 nM sull’ERα e 0.5 nM sull’ERβ) la cui affinità è normalizzata al 100% su entrambi i sottotipi.30
Sullo scaffold del composto 1 sono state apportate delle modifiche strutturali al fine di migliorare l’affinità per ERβ, pur mantenendo la medesima selettività per lo questo sottotipo recettoriale. Una delle modifiche provate è stata l’introduzione di piccoli sostituenti (Cl, CN, CH3) in posizione 3 dell’anello aromatico centrale, ed in particolare il composto 2 (Figura 4), che presenta l’atomo di cloro in questa posizione, è stato quello che ha mostrato un’apprezzabile miglioramento dell’affinità per il recettore β, mantenendo simili valori di selettività. È stato inoltre introdotto un atomo di fluoro in posizione meta del sostituente arilico periferico (composto 3) che ha mostrato un’ulteriore aumento dell’affinità per il recettore β, mantenendo gli stessi valori di selettività (Figura 4).
Un ulteriore modifica apportata al composto 1 è stata quella di provare ad invertire la posizione della porzione ossimica con quella ossidrilica (composto 4).28 I risultati ottenuti sono stati molto gratificanti, poiché l’affinità per ERβ (ERβ-RBA = 2.64%) è stata incrementata rispetto al composto 1, sebbene non risultasse la migliore tra quelle rilevate fino a quello momento.
Per quanto riguarda i valori di selettività è da notare come questi, per il composto 4, risultino inferiori rispetto a quelli rilevati per il composto 1, mentre i derivati 2 e 3 hanno livelli di selettività paragonabili.
1 ERα = 0.007% ERβ = 0.553% β/α = 79
Introduzione parte sperimentale
27
Figura 4: Struttura chimicadei composti 2-4
Sulla base dei dati ottenuti, è stato pensato di combinare le modifiche strutturali sopra citate, che avevano portato a dei miglioramenti in termini di affinità e selettività per il recettore ERβ, ovvero l’inversione dei gruppi OH ed ossimico rispetto al composto 1, insieme all’inserimento di un atomo di cloro in posizione 3 dello scaffold centrale, ed uno di fluoro in posizione meta del sostituente periferico, che hanno portato alla sintesi del composto 5, e del composto 6 rispettivamente (Figura 5).
Figura 5: Struttura chimica dei composti 5 e 6
Questi due composti hanno fornito dei risultati molto interessanti. Addirittura il composto 5 ha rivelato un’affinità per il recettore β molto più elevata dello stesso estradiolo, nonostante i valori di selettività non siano entusiasmanti; invece il composto 6 ha mantenuto un’ottima affinità per il sottotipo β ed una discreta selettività recettoriale.29
RBA ERα =0.065% RBA ERβ = 4.21% β/α = 65 2 RBA ERα=0.114% RBA ERβ =7.01% β/α = 62 3 RBA ERα = 0.064% RBA ERβ = 2.64% β/α = 41 4 RBA ERα = 4.46% RBA ERβ = 130.3% β/α = 30 5 RBA ERα = 1.88% RBA ERβ =87.11% β/α = 46 6
Introduzione parte sperimentale
28 In particolare durante il mio lavoro di Tesi mi sono occupata dell’ottimizzazione della sintesi su larga scala dei composti 5 e 6 per poterli avere in quantità sufficienti all’esecuzione di un’ampia serie di saggi bio-farmacologici.
Per la sintesi dei nuovi composti si è pensato di verificare l’influenza di sostituenti di piccole dimensioni come il gruppo metilico in posizione opposta rispetto all’atomo di cloro dell’anello centrale del composto 5, che ha portato all’ottenimento del composto 7; si è poi pensato di invertire queste relative posizioni (Cl, CH3) ottenendo il composto 8 ed infine di introdurre un atomo di fluoro in posizione 3’ dell’anello arilico periferico ottenendo il composto 9. (Figura 6).
Figura 6: Struttura chimica dei composti 7-9
Si è poi pensato di valutare l’importanza della presenza dell’atomo di fluoro in posizione 3’ del sostituente arilico periferico; a tale scopo abbiamo pensato di sostituire l’atomo di fluoro con un atomo di cloro o con un gruppo metilico sullo scaffold dei composti 4 e 5. In particolare siamo riusciti a sintetizzare i composti 10, 11 e 12.
Il composto 10 presenta, sullo scaffold del composto 4 (Figura 7), un gruppo metilico in posizione 3’ del sostituente arilico periferico; il composto 11 presenta nella medesima posizione un atomo di cloro; infine il composto 12 è caratterizzato dalla presenza dell’atomo di cloro sia in posizione 3’ del sostituente arilico periferico, sia sullo scaffold centrale del composto (Figura 7).
Introduzione parte sperimentale
29 Figura 7: Derivazione strutturale dei composti 10-12 dai predecessori 4-5.
Un'ulteriore modifica apportata al composto 4 è rappresentata dallo spostamento del gruppo OH dalla posizione 4’ alla 3’ dell’anello arilico periferico, che ha fornito il composto 13 (Figura 8).
4
10 11
5
Introduzione parte sperimentale
30
Figura 8: Struttura chimica composto 13
Data la presenza di una porzione resorcinolica su alcuni derivati naturali che si sono dimostrati ottimi ligandi β–selettivi, come per esempio la genisteina (Figura 2), abbiamo pensato di introdurre sul nostro scaffold delle salicilaldossime monoaril-sostituite, la medesima porzione a livello del sostituente arilico periferico. A tale scopo abbiamo avanzato l’ipotesi di sintetizzare i composti 14 e 15 (Figura 9).
Figura 9: Struttura chimica composto 14 e 15
13 4
15 14
Introduzione parte sperimentale
31
Sintesi dei composti 5, 6 e 12
La sintesi dei derivati salicilaldossimici 5, 6 e 12 è mostrata nello schema 1 SCHEMA 1 2-metil-3-cloro anisolo 16 17 20 19 18 Reagenti e condizioni :
a) NBS, Bz2O2, CCl4, riflusso 3h; b) AcONa, AcOH, riflusso 3h; c) THF, MeOH, NaOH al 20% aq., riflusso 3h; d) PCC, CH2Cl2 1h; e) Br2 , AcOH, t.a., 2gg; f) acido 4-metossifenilboronico, aq. Na2CO3 2M, Pd(OAc)2, PPh3,
toluene/etanolo 1:1, 100 ºC, 24h; g) acido 3-fluoro-4-metossifenilboronico, aq. Na2CO3 2M, Pd(OAc)2, PPh3,
toluene/etanolo 1:1, 100 ºC, 24h; h) acido 3-cloro-4-metossifenilboronico, Na2CO3, Pd(OAc)2, PPh3, THF, 100
ºC, 24h; i) BBr3, CH2Cl2 anidro, -78 ºC t.a., 1h; l) NH2OH·HCl, etanolo, acqua, 50 ºC, 1h.
COMPOSTO R 5 H 6 F 12 Cl a b c d e f, g ,h i l 21, 23, 47 22, 24, 48
Introduzione parte sperimentale
32 Nello specifico, il 2- metil-3-cloroanisolo commerciale è stato sottoposto ad una reazione di bromurazione radicalica con N-bromosuccinimmide e perossido di benzoile in CCl4, ottenendo così il bromuro benzilico 16. Il composto 16 è stato trattato con acetato di sodio in AcOH formando, mediante una sostituzione nucleofila bimolecolare, il composto 17. Il derivato alcolico 18 è stato ottenuto per rimozione del gruppo acetilico mediante un idrolisi alcalina usando NaOH acquoso al 20% in THF e MeOH. La reazione di ossidazione controllata eseguita sul gruppo alcolico primario è stata ottenuta mediante l’utilizzo del piridinio cloro-cromato (PCC) in CH2Cl2, ottenendo così la benzaldeide 19. Quest’ultima è stata fatta reagire con bromo in AcOH dando origine al composto 20.
Il composto 20 è stato quindi suddiviso in tre aliquote e sottoposto a delle reazioni di cross-coupling nelle condizioni di Suzuki, utilizzando tre diversi acidi boronici: l’acido 4-metossifenilboronico ha fornito il prodotto monoaril-sostituito 21, l’acido 3-fluoro-4-metossifenilboronico ha fornito il composto 23, mentre l’acido 3-cloro-4-metossifenilboronico ha fornito il composto 47. Sui prodotti 21, 23 e 47 è stata eseguita la demetilazione con BBr3 in CH2Cl2 anidro per formare rispettivamente i composti 22, 24 e 48, che a loro volta sono stati trasformati nelle rispettive salicilaldossime 5, 6 e 12 per reazione con idrossilammina cloridrato in EtOH.
Introduzione parte sperimentale
33
Sintesi dei composti 7, 8 e 9
La sintesi dei derivati salicilaldossimici 7, 8 e 9 è mostrata nello schema 2. SCHEMA 2 Reagenti e condizioni:
a) allil-bromuro, K2CO3, acetonitrile, 80 ºC; b) 180 ºC; c) t-BuOK, DMSO, 55 ºC; d) OsO4,NaIO4, diossano,
H2O; e) Br2, AcOH, t.a., 2gg; f) MeI, K2CO3,acetone, RT; g) acido 4-metossifenilboronico o l’acido
3-fluoro-4-metossifenilboronico, Pd(OAc)2, PPh3, soluzione aq. Na2CO3 2M, toluene/etanolo 1:1, 100 ºC, 24h; h) BBr3,
CH2Cl2 anidro, -78 ºC t.a., 1h; i) NH2OH·HCl, etanolo, acqua, 50 ºC, 1h.
Il composto commerciale 5-cloro-2-metil-fenolo è stato sottoposto ad una reazione di alchilazione a livello del gruppo OH, utilizzando allil-bromuro in presenza di CH3CN e K2CO3, ottenendo così il composto 25. Tale composto viene sottoposto ad una reazione di
Composto R1 R2 R3 7 Cl CH3 H 8 CH3 Cl H 9 CH3 Cl F d c b a e f g h i 25, 33 26, 34 27, 35 28, 36 29, 37 30, 38 31, 39, 41 32, 40, 42
Introduzione parte sperimentale
34 trasposizione, scaldando ad alta temperatura, per fornire il composto 26, il quale a sua volta subisce una reazione di isomerizzazione utilizzando t-BuOK in DMSO, per avere il composto 27. Il composto 27 subisce una reazione di scissione ossidativa, per avere un gruppo aldeidico libero, utilizzando una soluzione di NaIO4 in H2O e diossano, con aggiunta di OsO4 al 25% in
t-BuOH ottenendo così il composto 28 che viene sottoposto ad una reazione di bromurazione
in posizione 4, utilizzando Br2 in AcOH, per fornire il composto 29. Il composto 29 viene metilato in posizione 1, utilizzando MeI, K2CO3 in acetone, ottenendo il composto 30. Tale composto è sottoposto ad una reazione di cross-coupling nelle condizioni di Suzuki utilizzando l’acido 4-metossifenilboronico, che ha fornito il prodotto monoaril-sostituito 31. Sul composto 31 viene eseguita la demetilazione con BBr3 in CH2Cl2 anidro per formare il composto 32, da cui si ottiene la saliciladossima 7, per reazione con l’idrossilammina cloridrato in EtOH.
La stessa via sintetica può essere seguita, utilizzando come prodotto di partenza commerciale il 2-cloro-5-metil-fenolo, al fine di ottenere l’ossima 8; sostituendo poi l’acido 4-metossifenilboronico della reazione di cross-coupling con il 3-fluoro-4-metossifenilboronico, si ottiene invece il composto 9.
Introduzione parte sperimentale
35
Sintesi dei composti 10, 11 e 13
La sintesi dei derivati salicilaldossimici 10, 11 e 13 è mostrata nello schema 3. SCHEMA 3 4-bromo-salicilaldeide Composto R 10 CH3 11 Cl Reagenti e condizioni:
a) Acido 3-metossifenilboronico, Pd(OAc)2, PPh3, soluzione aq. Na2CO3 2M, toluene/etanolo 1:1, 100 ºC, 24h; b) l’acido 3-metil-4-metossifenilboronico o l’acido 3-cloro-4-metossifenilboronico, Pd(OAc)2, PPh3, soluzione
aq. Na2CO3 2M, toluene/etanolo 1:1, 100 ºC, 24h; c) BBr3, CH2Cl2 anidro, -78 ºC t.a., 1h; d) NH2OH·HCl,
etanolo, acqua, 50 ºC, 1h.
La 5-bromo-salicilaldeide commerciale è stata sottoposta ad una reazione di cross-coupling nelle condizioni di Suzuki, utilizzando tre diversi acidi boronici su un ugual numero
a b c c d d 13 50 49 43, 45 44, 46
Introduzione parte sperimentale
36 di aliquote. L’acido 3-metossifenilboronico ha fornito il prodotto monoaril-sostituito 49, l’acido 3-metil-4-metossifenilboronico ha fornito il composto 43, e l’acido 3-cloro-4-metossifenilboronico invece ha fornito il composto 45.
Sui prodotti 43, 45, e 49 è stata eseguita la demetilazione con BBr3 in CH2Cl2 anidro per formare rispettivamente i composti 44, 46 e 50, che a loro volta sono stati trasformati nelle rispettive saliciladossime 10, 11 e 13 per reazione con idrossilammina cloridrato in EtOH.
Introduzione parte sperimentale
37
Tentativo di Sintesi del composto 14
Il tentativo di sintesi del composto 14 è mostrato nello SCHEMA 4
SCHEMA 4
5-bromo-salicilaldeide 51 52
14 Reagenti e condizioni:
a) Acido 2,4-dimetossifenilboronico, Pd(OAc)2, PPh3, soluzione aq. Na2CO3 2M, toluene/etanolo 1:1, 100 ºC,
24h; b) BBr3, CH2Cl2 anidro, -78 º C t.a., 1h; c) NH2OH·HCl, etanolo, acqua, 50 ºC, 1h.
La 5-bromo-salicilaldeide commerciale è stata sottoposta ad una reazione di cross-coupling nelle condizioni di Suzuki, utilizzando l’acido 2,4-dimetossifenilboronico al fine di ottenere il composto monoaril-sostituito 51. Tale composto è stato sottoposto a numerosi tentativi di demetilazione utilizzando BBr3 in CH2Cl2 anidro per ottenere il composto 52, che a seguito ripetuti tentativi di purificazione tramite colonna cromatografica e cristallizzazione frazionata non siamo riusciti ad isolare dal grezzo di reazione.
a
b
Introduzione parte sperimentale
38
Tentativo di sintesi del composto 15
Il tentativo di sintesi del composto 15 è mostrato nello SCHEMA 5
SCHEMA 5
20 53
15 Reagenti e condizioni:
a) Acido 2,4-dimetossifenilboronico, Pd(OAc)2, PPh3, soluzione aq. Na2CO3 2M, toluene/etanolo 1:1, 100 ºC,
24h; b) BBr3, CH2Cl2 anidro, -78 ºC t.a., 1h; c) NH2OH·HCl, etanolo, acqua, 50 ºC, 1h.
Un aliquota del composto 20 è stata sottoposta ad una reazione di cross-coupling nelle condizioni di Suzuki, utilizzando l’acido boronico 2,4-dimetossifenilboronico al fine di ottenere il composto monoaril-sostituito 53. Tale composto, come nel caso precedente, è stato sottoposto a numerosi tentativi di demetilazione utilizzando BBr3 in CH2Cl2 anidro per ottenere il composto 54, che a seguito ripetuti tentativi di purificazione tramite colonna cromatografica e cristallizzazione frazionata non siamo riusciti ad isolare dal grezzo di reazione.
a b
c
Introduzione parte sperimentale
39
Saggi di binding recettoriale
I composti sintetizzati sono stati sottoposti a prove di binding recettoriale mediante saggi radiometrici di spiazzamento competitivo del [3H]-estradiolo sui sottotipi recettoriali ERα e ERβ umani ricombinanti purificati. I dati delle affinità sono riportati come affinità relative (RBA, %) a quelle dell’estradiolo (Kd = 0.3 nM per ERα e 0.5 nM per ERβ) la cui affinità è normalizzata al 100% . La Tabella 1 riporta i dati di affinità recettoriale dei nuovi composti sintetizzati nella presente Tesi (7-13) insieme a quelli precedentemente riportati per i composti di riferimento (5 -6). Tabella 1 Struttura ERα umano-RBA (%) ERβ umano-RBA (%)
β/α
Estradiolo (100) (100) 1 5 4.46 130.3 30 6 1.88 87.11 46 7 0.07 0.576 9Introduzione parte sperimentale 40 8 1.50 18.27 10 9 0.392 7.90 20 10 0.035 0.763 25
Introduzione parte sperimentale 41 11 0.012 0.906 67 12 1.43 7.19 6.9 13 0.003 0.101 29
Dai risultati riportati in Tabella 4, si può notare come i composti 7, 8 e 9, che presentano un ulteriore sostituzione sull’anello centrale, non hanno mostrato soddisfacenti valori di selettività recettoriale nei saggi di binding. Per quanto riguarda le affinità per il recettore ERβ i composti 8 e 9 hanno mostrato valori apprezzabili; al contrario, il composto 7 si è dimostrato scarsamente affine.
Valutando inoltre i composti che presentano un sostituente diverso dall’atomo di fluoro, sull’anello arilico del sostituente periferico (composti 10, 11 e 12); si nota come il composto 12 è l’unico a mostrare buoni livelli di affinità recettoriale per ERβ, mentre i composti 10 e 11 risultano essere, nonostante meno affini, più selettivi per tale sottotipo recettoriale, in particolar modo il composto 11 che presenta un valore di selettività β/α di 67.
Introduzione parte sperimentale
42 Lo spostamento del gruppo OH dalla posizione 4’ alla 3’ dell’anello arilico del sostituente periferico (composto 13), non ha mostrato rilevanti valori né di affinità, né di selettività per il ERβ.
A questo punto, possiamo quindi affermare che su questo tipo di salicilaldossime, derivanti dal composto 4, l’introduzione dell’atomo di cloro sull’anello centrale e la presenza dell’atomo di fluoro in posizione 3’ del sostituente arilico periferico si siano dimostrate decisive in termini di affinità e selettività nei confronti di ERβ; infatti il composto 5 mostra un’eccezionale affinità recettoriale per ERβ, la più alta rispetto a tutti gli altri composti appartenenti a questa classe fino ad ora realizzati e allo stesso estradiolo. Tuttavia, rispetto alla notevole affinità per ERβ il composto 5 mostra una ridotta selettività; pertanto il composto più promettente resta 6, che mantiene un’ottima affinità per ERβ ed una buona selettività β/α pari a 46.
Introduzione parte sperimentale
43
Molecular Modeling
Per valutare le interazioni dei nuovi composti sintetizzati a livello dei sito recettoriale ER ci siamo avvalsi della collaborazione con il Dr. Tiziano Tuccinardi e il gruppo di ricerca del Prof. Adriano Martinelli che hanno realizzato gli studi di docking.
L’immagine riportata nella Figura 10 sottostante mostra le principali interazioni delle salicilaldossime 1, 4 e 5 nella cavità di binding ERβ.
Figura 10: Docking in ERβ dei composti 1(A), 4 (B), 5 (C).
Come si può notare, in tutte e tre le salicilaldossime, il sistema pseudo-ciclo/ossima forma un legame ad H con G305/A346/e una molecola d’acqua; per quanto riguarda l’OH dell’anello fenolico del sostituente periferico il discorso è più complesso, infatti, per la salicilaldossima 1 (appartenente alla prima serie di ossime non invertite) questo forma legame ad idrogeno con H475; invece per le salicilaldossime 4 e 5 (appartenenti alla serie delle ossime invertite) questo forma un legame ad idrogeno con T299. Dall’immagine si può notare ancora che l’atomo di cloro presente sulla salicilaldossima 5 sembra inserirsi molto bene in una tasca idrofobica, presente nella cavità di binding ERβ, delimitata da A302, T335, M336 e L339 e questo potrebbe spiegare i suoi eccezionali valori di affinità per tale recettore.
Nell’immagine riportata nella Figura 11 sono evidenziate le interazioni della salicilaldossima 6 nelle cavità di binding ERα/β, e quelle del composto 8 in ERβ.
Introduzione parte sperimentale
44 Figura 11: Docking del composto 6 in ERα (D) ed in ERβ (E) e del composto 8 in ERβ (F).
Come si può notare dall’immagine (Figura 11), la salicilaldossima 6, nella cavità di binding ERβ, presenta sostanzialmente le stesse interazioni della salicilaldossima 5 (Figura 10C). In ERα la sostituzione del residuo M336 con L384 causa una completa inversione della disposizione del composto 6 rispetto a quella mostrata nella cavità di ERβ. Infatti qui è l’OH fenolico del sostituente arilico periferico che interagisce con E353, R394 e una molecola di acqua, mentre il sistema pseudo-ciclo/ossima interagisce formando un legame ad H con H524. Probabilmente, nella cavità di binding ERα, dove non è presente la tasca idrofobica delimitata dalla M336, l’atomo di fluoro unito alla presenza dell’atomo di cloro fa sì che le interazioni della salicilaldossima in questione risultino meno stabili perché richiedono una configurazione a maggiore energia e, quindi, quest’ossima risulta essere meno affine per il sottotipo ERα, pertanto raggiungendo così dei livelli più elevati di selettività per il sottotipo recettoriale β.
Come si evince dalla Figura 11 la salicilaldossima 8, presenta nella cavità di binding ERβ, le medesime interazioni del composto 6, per quanto riguarda il gruppo OH fenolico del sostituente arilico periferico e il sistema pseudo-ciclo/ossima. Differente è invece l’interazione che forma l’atomo di cloro dello scaffold centrale, difatti esso non interagisce più con la tasca lipofila delimitata da A302, T335, M 336 e L339, dove adesso si inserisce il gruppo metilico, ma va ad inserirsi in una tasca idrofobica più piccola e delimitata dalla M340, in cui possono istaurarsi dei leggeri fenomeni di repulsione sterica. Questo spiega perché quest’ossima risulta mantenga una buona affinità per il recettore β, che comunque risulta inferiore a quella delle salicilaldossime 5 e 6.
i
45
Parte
Parte sperimentale
46
Materiali e metodi
La struttura di tutti i composti è stata verificata per mezzo della spettrometria di massa e ¹H-NMR.
Gli spettri di massa sono stati registrati con uno spettrometro GC/MS Trace GCQ Plus per introduzione diretta di un’energia nominale di 70 eV con programma del Dep da 50 a 350 ˚C. Gli spettri di risonanza magnetica nucleare protonica sono stati eseguiti con uno spettrometro Varian Gemini 200 operante a 200 MHz e riferiti al segnale residuo del solvente. I chimica shift δ sono espressi in ppm e le costanti di accoppiamento J sono espresse in Hz.
Le evaporazioni sono state eseguite sotto vuoto in evaporatore rotante.
Le TLC analitiche sono state eseguite usando lastrine di gel di silice 60 F254 (MERCK) contenenti un indicatore fluorescente; le varie macchie sono state evidenziate per mezzo di una lampada UV (254 nm).
Per le cromatografie su colonna è stato usato gel di silice 60 (0.040-0.063 mm) MERCK. Le procedure assistite dal micro-onde sono state effettuate con CEM Discove® LabMate™ Microwave.
Parte sperimentale
47
Sintesi del composto 16
2-Bromometil-3-cloroanisolo
Ad una miscela di 2-metil-3-cloroanisolo commerciale (2.0 g, 13 mmol), NBS (3.41 g, 19.2 mmol) e il perossido di benzoile (465 mg, 1.92 mmol) è addizionato il CHCl3 (200 ml) e la reazione è lasciata a riflusso per 5 h a 90-95 ˚C, sotto agitazione magnetica.
La miscela di reazione addizionata di una soluzione satura di tiosolfato di sodio (per neutralizzare l’eventuale Br2 formatosi) e lavata con una soluzione satura di bicarbonato di sodio ( per neutralizzare l’eventuale HBr liberato), viene filtrata ed infine evaporata.
Il grezzo ottenuto è purificato mediante cromatografia flash (Esano/AcOEt 9:1). Le opportune frazioni, riunite ed evaporate, hanno fornito 2.41 g (10.2 mmol) del composto 16 puro ( resa 80%).
¹H NMR (CDCl3) δ (ppm): 3.90 (s, 3H); 4.72 (s, 2H); 6.80 (d, 1H, J = 8.2 Hz); 7.00 (d, 2H, J = 8.0 Hz); 7.21 (t, 1H, J = 8.2 Hz).
Parte sperimentale
48
Sintesi composto 17
2-Cloro-6-metossibenzil-acetato
Ad una soluzione del composto 16 ( 2.41 g, 10.2 mmol) in AcOH (200 ml), è aggiunto AcONa (10.68 g, 130.3 mmol) e la miscela risultante è lasciata a riflusso per circa 3h sotto agitazione magnetica.
La miscela di reazione viene estratta con AcOEt, filtrata ed evaporata. Il grezzo ottenuto (2.54 g), dall’analisi ¹H NMR, è risultato essere costituito esclusivamente dal prodotto 17 puro (11.8 mmol). Di conseguenza, si è passati al passaggio successivo della sintesi senza ulteriore purificazione. (Resa > 99%).
¹H NMR (CDCl3) δ (ppm): 2.08 (s, 3H); 3.85 (s, 3H); 5.30 (s, 2H); 6.82 (d, 1H, J = 8.2 Hz); 7.02 (d,1H, J = 8.1 Hz); 7.26 (t, 1H, J = 8.1 Hz).
Parte sperimentale
49
Sintesi composto 18
(2-cloro-6-metossifenil)-metanolo
Ad una soluzione del composto 17 (2.54 g, 11.8 mmol) in THF (109 ml) e MeOH (73 ml) è stata aggiunta una soluzione di NaOH al 20% (18 ml). La reazione è portata a riflusso a 100 ºC per 3 h.
La miscela di reazione aggiunta di HCl 1N ( per neutralizzare l’NaOH in eccesso) e acqua, viene estratta con AcOEt, filtrata ed evaporata. Il grezzo ottenuto è purificato mediante cromatografia falsh (Esano/ AcOEt 8:2).
Le opportune frazioni, riunite ed evaporate, hanno fornito 1.81 g (10.5 mmol) del composto 18 puro (resa 82%).
¹H NMR (CDCl3) δ (ppm): 3.87 (s, 3H); 4.87 (s, 2H); 6.81 (d,1H, J = 8.2 Hz); 7.00 (d, 1H, J = 8.1, 0.9 Hz); 7.20 (t, 2H, J = 8.2 Hz).
Parte sperimentale
50
Sintesi composto 19
2-Cloro-6-metossibenzaldeide
Ad una sospensione di piridinio-cloro-cromato (PCC), (4.5 gr, 21 mmol) in CH2Cl2 (35 ml) viene addizionato il composto 18 (1.81 gr, 10.5 mmol) sciolto in CH2Cl2 e il tutto è lasciato per 50 minuti sotto agitazione magnetica.
La reazione è diluita con Et2O e filtrata su celite ed evaporata.
L’evaporazione ha fornito un grezzo (1.59 g) che, dall’analisi ¹H NMR, è risultato essere costituito esclusivamente dal prodotto 19 ( 9.32 mmol), (resa 80%). Di conseguenza, si è passati al passaggio successivo della sintesi senza ulteriore purificazione.
¹H NMR (CDCl3) δ (ppm): 3.92 (s, 3H); 6.91 (d,1H, J = 8.4Hz); 7.04(d, 1H, J = 8.1 Hz); 7.41 (t, 1H, J = .2 Hz); 10.50 (s, 1H).
Parte sperimentale
51
Sintesi composto 20
3-Bromo-2-cloro-6-metossibenzaldeide
Il composto 19 (1.59 g, 9.32 mmol) disciolto nella minima quantità di AcOH glaciale, è aggiunto di Br2 ( 0.52 ml) e la miscela è stata lasciata reagire a t. a., per 48 h , sotto agitazione magnetica.
La miscela di reazione è stata aggiunta di una soluzione di tiosolfato di sodio (per neutralizzare il Br2), estratta con AcOEt, filtrata ed evaporata. Il grezzo è stato purificato mediante cromatografia flash (Esano/AcOEt 8:2).
Le opportune frazioni, riunite ed evaporate, hanno fornito 1.87 g (7.50 mmol) del composto 20 puro (resa 80%).
¹H NMR (CDCl3) δ (ppm): 3.92 (s, 3H); 6.84 (d,1H, J = 9.0 Hz); 7.73(d, 1H, J = 9.0 Hz); 10.40 (s, 1H).
Parte sperimentale
52
Sintesi composto 21
2-Cloro-4,4’- dimetossibifenil-3-carbaldeide
In una fiala, sotto flusso di N2, sono stati aggiunti toluene ed etanolo assoluto (4.6 ml ciascuno), Pd(AcO)2 (13.5 mg, 0.06 mmol) e PPh3 (78.7 mg, 0.30 mmol), sotto agitazione magnetica. Dopo 10 minuti è stato aggiunto il composto 20, (500 mg, 1.99 mmol), una soluzione di Na2CO3 2M ( 4.55 ml) e infine l’acido 4-metossifenilboronico (363 mg, 2.39 mmol). La miscela di reazione è stata fatta reagire a 100 ˚C, per 24h , sotto agitazione magnetica.
La miscela di reazione viene estratta con AcOEt, filtrata ed evaporata. Il grezzo che è stato purificato mediante cromatografia flash (Esano/AcOEt 8:2) hanno fornito 238 mg ( 0.87 mmol) del composto 21 puro (resa 44%).
¹H NMR (CDCl3) δ (ppm): 3.86 ( s, 3H, OCH3); 3.95 (s, 3H, OCH3); 6.95-6.99 (m, 3H, H5, H3’, H5’); 7.31 (AA’XX’, 2H, JAX = 8.8 Hz, JAA’XX’ = 2.2 Hz, H2’, H6’); 7.45 ( d, 1H, J = 8.6 Hz, H6); 10.50 (s, 1H, CHO ).