Research Doctorate School in Biological and Molecular Science
Program in Molecular Biotechnology
XXVI Cycle (2011-2013)
The peculiar structure of the olive (Olea europaea
L.) genome as shown by massively parallel
sequencing data
Supervisor: Candidate:
The olive tree (Olea europaea L.) is poorly characterized at genetic and genomic level compared to other fruit tree crops. In the frame of the Italian project OLEA, aimed to obtain the complete sequence of the olive genome, we performed a deep analysis of the repetitive component of this genome, using NGS techniques (454-Roche and Illumina).
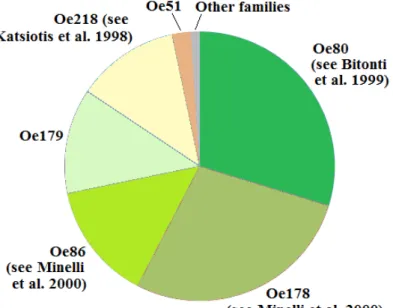
In a first work, we described different computational procedures for isolating and characterizing olive repeated sequences. These procedures were used to determine the structure of the genome and the composition of its repetitive fraction. Our analyses showed the peculiar landscape of the olive genome, related to the massive amplification of tandem repeats, which represent about 31% of the whole genome, more than that reported for any other sequenced plant genome. Tandem repeats are represented by six main families of different length, two of which were firstly discovered in these experiments. The other large redundant class in the olive genome is represented by transposable elements, especially LTR-retrotransposons (LTR-REs). Similar procedures were concurrently applied to the genome of another species, the sunflower, and an article on this species is reported as appendix # 2.
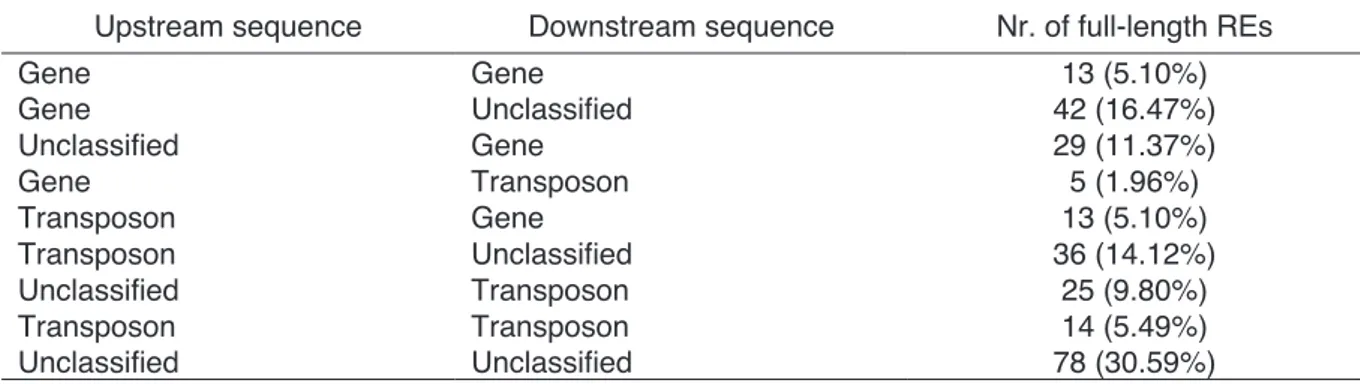
In a second work, we provided a characterization of 255 unique full-length LTR-REs, identified scanning a number of BAC clone sequences. Copia elements resulted more numerous than Gypsy ones (162 vs. 81), 12 elements were not assigned to any superfamily because lacking of distinctive domains. Mapping a large set of Illumina reads onto the LTR-REs revealed that Gypsy families are made of more members than
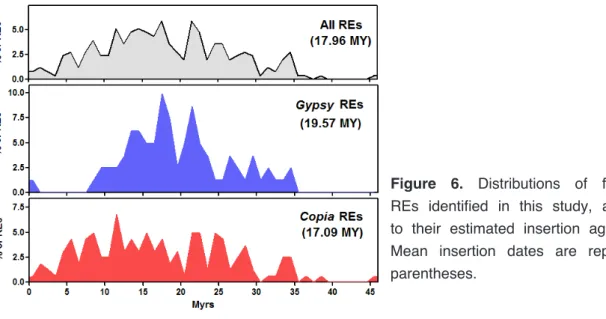
Copia ones. Four RE families resulted composed especially by solo-LTRs. The insertion time of intact retroelements, measured by sister LTRs divergence, showed that the mean insertion age of the isolated REs is around 18 million years (MY), although some isolated elements inserted relatively recently. Gypsy and Copia REs showed different waves of transposition, with Gypsy elements especially active between 10 and 25 MY and nearly inactive in the last 7 MY.
In the third work, using a specific bioinformatic pipeline on olive BAC clone sequences, we identified 418 olive Short Interspersed Nuclear Elements (SINEs), that constitute one of the first SINE collection in a dicotyledonous species. The identified SINEs represent 0.48% of the olive genome and their length ranges from 62 to 588 bp. The vast majority of identified SINEs resulted low or medium redundant, often in association with genic sequences. Analysis of sequence similarity allowed to identify ten major families. Our results demonstrate the suitability of the pipeline employed for SINE identification and will favour further analyses on these relatively unknown elements to be performed in other plant species.
Introduction 1
The olive tree 3
Development of molecular markers and genomic resources 4
Procedures for whole genome sequencing and assembly 5
The repetitive component of the genome 7
Methods for the detection of repetitive sequences in the genomes 12
The project OLEA 13
Aim of the thesis 13
References 14
The peculiar landscape of repetitive sequences in the olive (Olea europaea L.)
genome 21
Abstract 21
Introduction 21
Materials and Methods 23
Illumina and 454 sequencing 23
Graph-based clustering of sequences 24
Sequence assembly procedure 24
Estimation of sequence abundance 25
Annotation of redundant contigs 25
Preparation, sequencing and annotation of a small insert library 26
Sequence analyses of tandem repeats 26
Reconstruction and analysis of full-length LTR-retrotransposon sequences 27
Results 27
Graph-based sequence clustering of olive genome 27
De novo assembly of genomic DNA 29
Estimation of average coverage of assembled contigs 29
Analysis of the repetitive fraction of WGSAS 30
The composition of the olive genome 32
Analysis of a Sanger-sequenced short insert library 34
Analysis of tandem repeats 36
Analysis of LTR-retrotransposons 38
Discussion 38
Production of a set of olive repetitive sequences 38
The structure of olive genome 41
Acknowledgements 43
References 44
LTR-retrotransposon dynamics in the evolution of the olive (Olea europaea)
genome 51
Abstract 51
Introduction 51
Materials and methods 53
Sequencing of BAC clones 53
Sequence analysis 54
Estimation of sequence abundance 54
Phylogenetic analyses 55
Insertion age calculation of full-length LTR-REs 55
Results 56
Phylogenetic relationships among isolated LTR-REs 59
Putative insertion dates of LTR-REs 59
Discussion 63
Acknowledgements 66
References 67
OLEASINE: a Collection of Olive Tree (Olea europaea L.) Short Interspersed
Nuclear Elements 73
Abstract 73 Introduction 73
Materials and methods 76
Sequencing of BAC clones 76
SINEs characterization 76
Estimation of sequence abundance 77
Phylogenetic analyses 77
Identification of modules 77
Results and discussion 78
Conclusions 82 Acknowledgements 83 References 83 Conclusions 89 Appendix 1 Supplementary material 1 95 Supplementary material 2 100 Supplementary material 3 101 Supplementary material 4 102 Supplementary material 5 140 Appendix 2
The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads
The Olive tree
The cultivated olive (Olea europaea L.) is one of the oldest crops grown in Italy and in the Mediterranean area and plays a vital role in agricultural economy and food habits.
Olive is the most important species of the Oleaceae family which comprise about 600 species and 24 genera (Johnson, 1957; Rohwer, 1996; Wallander and Albert, 2000; Green, 2004).
Olive is a diploid species (2n=2x=46) and the genome size ranges between 2.90 pg/2C and 3.07 pg/2C, with 1C = 1,400-1,500 Mbp (Loureiro et al., 2007).
Olea europaea subsp. europaea is present in two forms, namely wild (O. europaea subsp. europaea var. sylvestris) and cultivated (O. europaea subsp. europaea var
europaea) and has conserved a very wide germplasm estimated in more than 1,200 cultivars (Bartolini et al., 2008).
The cultivation of olive trees dates back to ancient time in the Mediterranean region where it started in the third millennium B.C. (Loukas and Krimbas, 1983).
The cultivated olive is an evergreen, out-crossing, vegetatively propagated tree with a very wide genetic patrimony that is the result of both plant longevity and the scarcity of genotype turnover through centuries of cultivation.
Olive is the second most important oil fruit crop cultivated worldwide after oil palm and its cultivation covers over eight million hectares of land, predominantly concentrated in the Mediterranean basin, where 70% of the olive oil produced is consumed (Baldoni and Belaj, 2009).
Several studies have highlighted the beneficial effects of olive oil on human health (Keys, 1995; Pérez-Jimenéz et al., 2007). Some minor constituents, named secoiridoids and representing the most important class of the olive phenolics, seem to be responsible for the major effect on health. These components are exclusively present in the Oleaceae family, and seem to have a protective activity against chronic degenerative diseases and tumors and are also responsible for the agreeable sensory properties of virgin olive oil (Servili et al., 2004).
Despite its economic, cultural and ecological importance in the Mediterranean area, now extending to other regions, the olive is a poorly characterized species at genetic and genomic level compared to other fruit tree crops. Therefore, the inheritance of most genes controlling the agronomical performance and quality traits, still remains unknown though, in the last thirty years, a wide molecular survey has been performed on the olive germplasm (Rugini et al., 2011).
As many woody plants, olive requires several growth seasons from seed germination to first flowering (juvenile phase). This long juvenile phase had severely hampered the generation of new breeding cultivars that could replace ancient ones still under cultivation (Bellini, 1993). Moreover, new cultivars adapted to the new trends in olive growing, i.e. shorter juvenile periods (Moreno-Alias et al., 2010), tree architecture
suitable for hedgerow orchards and mechanical harvesting (Rallo et al., 2008), or increased oil content and organoleptic profiles tailored to different markets (Leòn et al., 2008), are needed. For this purpose new research programs on structural and functional genomics of olive tree are to be developed as already done for other important crops.
Development of molecular markers and genomic resources
Many efforts were made in the last years, as consequence of the increased interest for the olive tree, to produce molecular data especially related to the application of molecular markers to the analysis of genetic variability in the genus Olea and to develop efficient molecular tools for the oil origin traceability.
Due to their simplicity of application and low cost, RAPDs (Random Amplified Polymorphic DNA) were the first markers used to evaluate the genetic variability in olive (Bogani et al.,1994). Subsequently many DNA-based molecular markers have been applied in Olea europaea studies, such as AFLPs (Amplified Fragment Length Polymorphism), SCARs (Sequence Characterised Amplified Region), ISSRs (Inter Simple Sequence Repeats), SNPs (Single Nucleotide Polymorphism). These markers were applied for the DNA fingerprint of cultivars (Bogani et al., 1994; Angiolillo et al., 1999; Baldoni et al.,2009; Reale et al., 2006), for the detection of intra-cultivar variability (Gemas et al., 2000; Belaj et al., 2004), for the construction of linkage maps (De la Rosa et al., 2004; Wu et al., 2004), for cultivar traceability in olive oil (Pasqualone et al., 2001, Muzzalupo and Perri, 2002; De la Torre et al., 2004; Martins-Lopez et al., 2008) and for phylogenetic studies (Hess et al., 2000; Baldoni et al., 2006; Besnard et al., 2001, 2007).
Also the polymorphism of ribosomal and cytoplasmic DNA, such as internal transcribed spacer (ITS) and intergenic spacer (IGS), were widely used for phylogenetic studies (Hess et al., 2000; Besnard et al., 2009).
With regard to genomic research, in the last years efforts were made for the identification of expressed sequence tags, with particular interest in those sequences expressed in pollen allergens (Rodriuez et al., 2002; Hammam-Khalifa et al., 2008) and during fruit development, especially those involved in lipid and phenolic metabolism (Alagna et al., 2009; Galla et al., 2009).
Important results in olive genomic studies have been the sequencing of the entire chloroplast genome of the Italian cultivar “Frantoio” (Mariotti et al., 2010), wich provided new information on the olive nucleotide sequence, opening the olive genomic era.
Very recently the de novo assembly and functional annotation of the Olea europaea transcriptome (Muñoz-Mérida et al., 2013) made available important informations on olive functional genomics, nevertheless the structure of the olive genome remains largely uncharacterized. A first rough draft of the whole genome sequence has been recently produced by the structural genomics group of the Italian project OLEA, an
initiative, mainly supported by Italian Minister of Agricultural, Food and Forestry Policies. Concerning repeated sequences, which represent a substantial portion of the genome, only a few data are available for the olive. The most characterized repetitive sequences of olive are four tandem repeats, isolated from genomic libraries and, in some instances, localized by cytological hybridization on chromosomes (Katsiotis et al., 1998; Bitonti et al., 1999; Minelli et al., 2000; Lorite et al., 2001; Contento et al., 2002). Also putative retrotransposon fragments have been isolated and sequenced (Stergiou et al., 2002; Natali et al., 2007), but a comprehensive picture of repetitive elements landscape in the olive genome is still lacking.
Procedures for whole genome sequencing and assembly
The sequencing of the genome is a very important step towards understanding its functioning and evolution. Crop genome sequencing, even at the current levels of completeness, have had a major impact on crop research and improvement in a relatively short time ( Bolger et al., 2014).
One of the main advantages of whole genome sequencing is the availability of high-density molecular markers which can be used to quickly map agronomically desirable traits and to identify candidate genes within a region of interest. These traits, once characterized, can be bred into elite varieties. High throughput genotyping by sequencing are options for efficient marker assisted breeding, for genomic selection, and for efficient management of genetic resources at a higher resolution than is otherwise possible (Cabezas et al., 2011).
Reconstructing a complete genome from a set of sequences can be metaphorically described as the process of assembling a jigsaw puzzle. In the case of the largest, most repetitive plant genomes, it could be described as assembling a large jigsaw consisting of blue sky (represented by repeated sequences) separated by nearly indistinguishable wisps of white clouds of genes.
Some of the biggest technical challenges in sequencing eukaryotic genomes are caused by repetitive DNA, i.e., sequences that are similar or identical to sequences elsewhere in the genome (Alkan et al., 2011). Such challenges are even more difficult when using the next generation sequencing (NGS) procedures, that produce sequences of shorter length than the classical Sanger sequencing method. From the computational perspective, repeats contained in short sequences create ambiguities in alignment and in genome assembly (Treangen and Salzberg, 2012).
The first step in characterizing and sequencing large genomes should be a genome survey, from which important information about common repeat sequences can be obtained. NGS methods allows performing relatively inexpensive and detailed genome surveys without the necessity of cloning, bacteria or vector libraries (Swaminathan et al., 2007), through the production of large numbers of short sequences.
Actually, the major computational task in both de novo and reference guided assembling of millions of reads is to decide what to do with multi-reads, i.e. reads that map to multiple locations and/or contain highly repeated k-mers (Alkan et al., 2011; Treangen and Salzberg, 2012). In the reference guided assembly, an algorithm has three choices for dealing with multi-reads (Simola and Kim, 2011). The first is to ignore them, meaning that all multi-reads are discarded. The second option is the best match approach, in which the alignment with the fewest mismatches is reported. If there are multiple, equally good best match alignments, then an aligner will either choose one at random or report all of them. The third choice is to report all alignments up to a maximum number, regardless of the total number of alignments found. To simplify the analysis, some alignment protocols prefer the ‘ignore’ strategy for multi-reads. However, this strategy limits analysis to unique regions in the genome, discarding many multi-gene families as well as all repeats, which might result in biologically important variants being missed.
The other two strategies can fill in repetitive regions, although only the best match approach will provide a reasonable estimate of coverage (Treangen and Salzberg, 2012). Allowing multi-reads to map to all possible positions avoids making a possibly erroneous choice about read placement.
All de novo assemblers fall into one of two classes: overlap-based assemblers and de Bruijn graph assemblers, both of which create graphs (of different types) from the read data. The algorithms then traverse these graphs in order to reconstruct the sequence. From a technical perspective, repeats cause branches in these graphs, and assemblers must then make a guess as to which branch to follow. Incorrect guesses create false joins (chimaeric contigs) and erroneous copy numbers. If the assembler is more conservative, it will break the assembly at these branch points, leading to an accurate but fragmented assembly with fairly small contigs.
The most common error is that an assembler will create a chimaera by joining two repeats that are not close in the genome. To resolve chimaeras the only hint of a problem is found in the paired-end links. Paired-end reads are generated from a single DNA fragment of a fixed size, from which both ends are sequenced. An assembler uses both the expected distance and the orientation of the reads when reconstructing a sequence. If the sequence data do not contain paired ends that span a particular repeat, then it might be impossible to assemble the data unambiguously.
In either an overlap graph or a de Bruijn graph, all copies of a repeat will initially be represented by a single node. Repeat boundaries and sequencing errors show up as branch points in the graph, and complex repeats appear as densely connected ‘tangles’ (Pevzner et al., 2001). Assemblers use two main strategies to resolve these tangles. First and most importantly, they use mate-pair information from reads that were sequenced in pairs.
of coverage for each contig. These statistics do not tell assemblers exactly how to assemble each repeat, but they do identify the repeats themselves. In order to make use of this information, assembly programs must assume that the genome is uniformly covered; this means that if a genome is sequenced to 50-fold (50×) coverage, then the assembler assumes that most contigs should also be covered at 50×. A repetitive region, by contrast, will have substantially deeper coverage, which allows the algorithm to identify it as a repeat and to process it differently. In particular, repeats are usually assembled after unique regions, and assemblers may require multiple paired ends to link a repetitive contig to a unique one.
When sequencing a genome, a combination of strategies have to be used for resolving problems that are caused by repetitive DNA, including sequencing strategies that use fragment libraries of varying sizes (Wetzel et al., 2011), post-processing software that is designed for detecting misassemblies (Phillippy et al., 2008), analysing coverage statistics and detecting and resolving tangles in a de Bruijn graph. None of these strategies completely solves the problems and the ultimate solution may require much longer read lengths. Such problems are less stringent, however, when the objective of the research is to obtain not a complete genome but merely single sequence families. In the case of repetitive DNA families, a smaller coverage can allow to reduce redundancy of reads and hence assembly them into contigs (see for example Rasmussen and Noor, 2009; Straub et al., 2011).
The repetitive component of the genome
Early genome sequencing projects discovered that the functional genome, which predominantly consists of protein-coding genes, represents only a small percentage of the genome while the non-coding regions represent a higher and variable portion of the complete genome.
The non coding genome is made mainly of repeated sequences, which arise from a variety of biological mechanisms that result in extra copies of a sequence being produced and inserted elsewhere into the genome. They can be widely interspersed repeats, tandem repeats or nested repeats, and occur even in millions of copies, ranging in size from 1–2 bases to thousands of bases.
A number of different repetitive sequences have been reported in eukaryotic genomes and these can be classified into classes based on structure and sequence composition such as Transposable Elements, Tandem Repeats, Pseudogenes and ribosomal DNAs, which encode the structural RNA components of ribosomes.
Transposable elements (TEs) are dispersed repeat DNA sequences that have the ability to move from one position to another in the genome, reason why they were designed as mobile DNAs.
found in almost all eukaryotic organisms, including all plant species that have been investigated.
Depending on the organism, the proportion of TEs can be highly variable, for example 3% in yeast (Kim et al., 1998), 15% in Drosophila (Dowsett and Young, 1982); 45% in human (Lander et al., 2001) and more than 80% in maize (Schnable et al., 2009). TEs are very aboundant in organisms with a very large genome, indicating their role in the expansion of the host’s genome size.
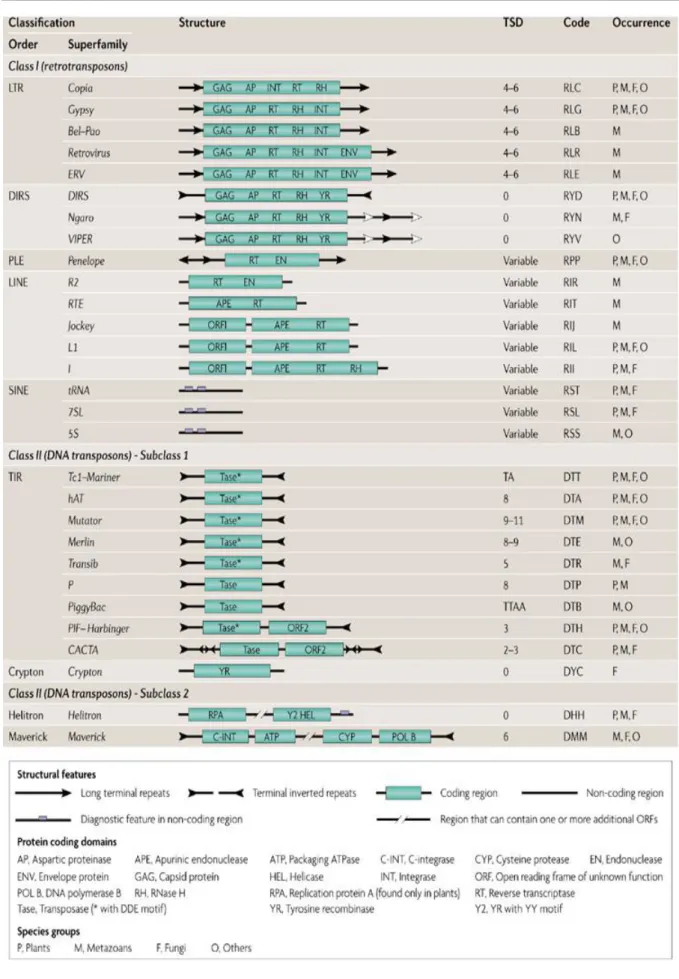
Different types of TEs have been identified and several attempts were made to classify them (Wicker et al., 2007; Kapitonov and Jurka, 2008). Globally, two main classes have been described according to their transposition intermediates: Class I elements use an RNA intermediate while Class II elements use a DNA intermediate.
Class I TEs (Retrotransposons) transpose through an RNA intermediate which is transcribed from a genomic copy and then reverse-transcribed into DNA by a TE-encoded reverse transcriptase (RT). Hence, these elements do not excise when they transpose. Instead, they make a copy that inserts elsewhere.
Retrotransposons can be divided into five orders on the basis of their mechanistic features, organization and reverse transcriptase phylogeny: LTR retrotransposons,
DIRS-like elements, Penelope-like elements (PLEs), LINEs and SINEs (Wicker et al.,
2007).
Among Class I elements, retrotransposons that contain direct long terminal repeats (LTR) are less abundant in animals, but are the predominant order in plants.
LTR-retrotransposons vary in size from several hundred bases to over 10 kb, with LTRs that are usually a few hundred bases to several thousand bases in length. LTR retrotransposons encode an integrase (IN) function that allows them to incorporate the circular product of reverse transcription into the chromosome. The two major subclasses of LTR-retrotransposons are called Gypsy and Copia elements, and they differ in the position of integrase within the encoded polyprotein(Figure 1).
The long interspersed nuclear elements (LINEs) lack LTRs, can reach several kilobases in length and are found in all eukaryotic kingdoms. Autonomous LINEs encode at least an RT and a nuclease in their pol ORF for transposition (Eickbush et al., 2002; Ostertag and Kazazian, 2005). A gag-like ORF is sometimes found 5’ to pol, but its role remains unclear. Only members of superfamily I contain RNaseH. At their 3’ end, they can display either a poly(A) tail, a tandem repeat or merely an A rich region.
Although LINEs generally form Target Site Duplications (TSD) upon insertion, truncated 5’ ends make them difficult to find (Eickbush and Malik, 2002). The truncations probably result from premature termination of reverse transcription (Eickbush and Malik, 2002; Petrov and Hartl, 1998). LINEs vary in prevalence and diversity in eukaryotes, but predominate over the LTR retrotransposons in many animals. The L1 family for instance made about 20% of the human genome.
from accidental retrotransposition of various polymerase III (Pol III) transcripts (Kramerov and Vassetzky, 2005). Unlike retroprocessed pseudogenes, they possess an internal
Pol III promoter, allowing them to be expressed. They rely on LINEs for trans-acting transposition functions such as RT (Kramerov and Vassetzky, 2005; Kajikawa and Okada, 2002). Some ‘stringent’ SINEs have a unique, obligatory partner (Dewannieux et al., 2003) whereas others are generalists (Kajikawa and Okada, 2002).
The ‘head’, which harbours the Pol III promoter, defines SINE superfamilies and reveals their origin: tRNA, 7SL RNA and 5S RNA. SINE internal regions (50–200 bp) are family-specific and of variable origin, sometimes deriving from SINE dimerization or trimerization. The origin of the 3’ region, although it can sometimes be a LINE, is generally obscure. It can be either A- or AT-rich, harbour 3–5-bp tandem repeats, or contain a poly(T) tail, the Pol III termination signal (Kramerov and Vassetzky, 2005). The best known SINE is the Alu element, which is present in at least 500,000 copies in the human genome (Rowold and Herrara, 2000).
In plants, SINEs are relatively rare in most genomes that have been investigated
(Bennetzen 1993; Grandbastien, 1992).Because these RNA polymerase III-transcribed
genes carry a promoter specified within the RNA itself, a newly inserted element can usually be transcribed in any active part of the genome, thus creating a high potential for amplification (Maraia, 1995).
In addition to the classical class I TEs, two new groups have recently been described: the DIRS-like and the Penelopelike (PLE) elements (Figure 1). Members of the DIRS (Cappello et al., 1985) order contain a tyrosine recombinase gene instead of an INT, and therefore do not form TSDs. Their termini are unusual, resembling either split
direct repeats (SDR) or inverted repeats. These features indicate a mechanism of
integration that is different from the LTR and LINE TEs. Nevertheless, their RT places them in class I. Members of this order have been detected in diverse species, ranging from green algae to animals and fungi (Goodwin and Poulter, 2004).
The PLE order was first found in Drosophila virilise (Evgen’ev et al.,1997). Its absence in other sequenced genomes reflects its patchy distribution, although it has been detected in more than 50 species, including unicellular animals, fungi and plants (Evgen’ev et al., 2005). PLE encodes an RT that is more closely related to telomerase than to the RT from LTR retrotransposons or LINEs, and an endonuclease that is related both to intron-encoded endonuclease and to the bacterial DNA repair protein UvrC. Some members contain a functional intron. Members also have LTR-like sequences that can be in a direct or an inverse orientation.
Class II TEs (DNA Transposons) transpose trough a “cut and paste” mechanism which provide a DNA intermediate. Like class I elements they are ancient and found in almost all eukaryotes. Class II elements are also found in prokaryotes in simple forms called insertion sequences (IS) or as part of more complex structures (Chandler et Mahillon, 2002).
Based on the number of DNA strands that are cut during transposition, Class II elements are grouped in two subclasses (Wicker et al., 2007).
Subclass 1 comprises classical ‘cut-and paste’ TEs of the order TIR, characterized by their terminal inverted repeats (TIRs) of variable length. The known superfamilies are distinguished by the TIR sequences and the TSD size. These TEs can increase their numbers by transposing during chromosome replication from a position that has already been replicated to another that the replication fork has not yet passed (Greenblatt et Brink, 1962). Alternatively, they can exploit gap repair following excision to create an extra copy at the donor site (Capy et al., 1998; Nassif et al., 1994). Transposition is mediated by a transposase enzyme that recognizes the TIRs and cuts both strands at each end. This subclass includes Tc1-Mariner, hAT, Mutator, Merlin, Transib, P,
PiggyBac ,CACTA, PIF–Harbinger and Crypton superfamilies.
Subclass 2 holds DNA TEs that undergo a transposition process that entails replication without double-stranded cleavage, sharply different from that of subclass 1. These copy-and-paste TEs transpose by replication involving the displacement of only one strand such as Helitrons and Maveriks (Wicker et al., 2007).
Helitron appear to replicate via a rolling-circle mechanism, with only one strand cut (Kapitonov et Jurka, 2001), and do not generate TSDs. Helitron ends are defined only by TC or CTRR motifs (where R is a purine) and a short hairpin structure lying a few nucleotides before the 3’ end, although this does not seem to be a true diagnostic feature. Autonomous Helitrons encode aY2-type tyrosine recombinase, with a helicase domain and replication initiator activity. They can also encode a single-strand binding protein or other proteins.
The miniature inverted-repeat transposable elements (MITEs) compose another abundant class of small transposable elements in plants. MITEs have a structure indicating that they are likely to be DNA transposable elements (Bureau et al., 1994; Wessler et al., 1995).
The MITEs are unusual in having no identified autonomous elements (Wessler et al., 1995). The existence of undiscovered autonomous elements that encode MITE-specific transposases is likely, although it is also possible that these tiny elements utilize a trans-acting transposition function that is not itself encoded on a mobile DNA. Such an activity might be specified by a standard ‘host’ gene involved in some other cellular process (e.g. DNA replication, recombination or repair), although there are no obvious candidates at this time.
During the years since their discovery the status of Transposable Elements has moved from simple junk DNA to major players in evolution (Biémont, 2010). TEs have a significant impact on genome evolution and along with polyploidy on the evolution of genome size (Kidwell and Lisch, 2000; Biémont and Vieira, 2006).
Plant transposable elements have a range of possible activities, all of them associated with possible alterations in genome and/or gene structure and function. The repetitive nature of TEs makes them responsible for chromosomal rearrangements via homologous recombination. When inserting in or near a gene they can provide
new regulatory elements, altering the expression of the genes (Green et al., 1994; Martienssen et al., 1989; Masson et al., 1987). Moreover chromosome breakage, and sequence amplification are identified outcomes of the transpositional and/or recombinational potential of all of the transposon types (Bennetzen, 2000).
Transposable Elements can promote mutations which can be deleterious, however, they can also increase the genetic diversity of a species. For example gene inactivation via insertion of TE sequences within a gene is common with low-copy-number TEs and can serve as raw material for the evolution of new gene functions.
Methods for the detection of repetitive sequences in the genomes
The ability to identify and recognize repetitive elements is a suitable starting point for the assembly of a genome, especially in genomes containing very high proportions of repeats.
The similarity-based methods search for TEs by comparing genome sequences to a library of TE reference sequences. The library can be a public database such as REPBASE (Jurka et al., 2005) or can be defined by the user. Typically the program RepeatMasker (Smit et al., 1996-2010) is used to perform a similarity search using the library as reference. This method is fast and accurate but can’t allow the discovery of new TE families. Anyways, this methods is still a good starting point to explore new genome, particularly if TE sequences from closely related species are available.
The signature-based methods use particular structural features (such as nucleotide or protein motif) of known TE classes to determine their occurrence in a genome sequence. This approach can identify new elements of a given class but can’t be used to discover new classes of elements. Moreover, these methods will only discover nearly complete and potentially active copies and miss degraded ones. Thus, such an approach can be complemented using a library based method once complete reference elements have been discovered based on their structure. Another important aspect is the level of knowledge available for a given class and the possibility of determining fixed and shared features among several families of the same class.
The de novo methods allow to search for new types of elements without a priori knowledge of the sequence itself taking advantage of the repetitive nature of TEs. These methods are particularly suitable when sequencing the genomes of species for which no close relatives are currently annotated. However they are particularly sensitive to genomic coverage and to the quality of sequence assembly and TE families containing very few copies will not be detected.
In a de novo approach the genome sequence is first compared against itself to locate all the repeated sequences; the repeated sequences that are located are then grouped into clusters of similar sequences. A consensus sequence is then built for each cluster and used in a library based approach to retrieve all occurrences within the genome.
In another de novo approach, the occurrence of multiple small words known as k-mers is searched within the genome sequence. The k-mer can then be extended to obtain longer sequence.
The project olea
The Italian project OLEA is an initiative dedicated to the development of genomic resources of olive, and aimed to identify, isolate and determine the function of genes that are associated with both vegetative and reproductive phenotype. The first part of this project is devoted the sequencing of the whole genome.
The olive genome is being sequenced using a combination of Next Generation Sequencing (NGS) technologies and a combination of assembly approaches, using the cultivar Leccino as the genotype to be sequenced. The Whole Genome Shotgun approach to assembling the genome is being pursued using Illumina and 454 sequencing with a combination of long single reads, paired end reads and mate pairs until a coverage of at least 40 genome equivalents is reached. The assembly is being performed using Abyss and CLC assemblers. A BAC pooling approach is being used to sequence random pools of 384 BACs using Illumina paired end reads. A BAC coverage of approximately 3-4 genome equivalents is going to be sequenced, with each BAC clone sequenced on average at a 50X coverage. The advantages of the BAC approach are of two types: on one hand each BAC pool is much smaller in size than the total genome size, reducing the assembly complexity, on the other hand within each BAC pool the problem posed by sequence heterozygosity among maternal and paternal-derived genomes, that strongly affects WGS approaches, should not be faced. The advantage of the WGS approach is the much more complete and homogeneous coverage of the genome. The two assemblies, the WGS and the pooled BAC assembly, will therefore be combined using a proprietary algorithm (GAM) to produce a consensus assembly. The consensus assembly will finally be anchored to the genetic map through the use of high throughput genotyping technologies.
Aim of the thesis
In the frame of the project OLEA aiming to obtain the complete sequence of the olive genome we performed a deep analysis of the repetitive component of this genome, using NGS techniques (454-Roche and Illumina). In this work, we used different computational procedures with the aim to isolate and characterize olive repeated sequences. These data were used to determine the structure of the genome and the composition of its repetitive fraction and will be used in the annotation of the genome when the complete sequence will be available.
Alagna F, D’agostino N, Torchia L, Servili M, Rao R, Pietrella M, Giuliano G, Chiusano ML, Baldo-ni L, Perrotta G (2009) Comparative 454 pyro-sequencing of transcripts from two olive genot-ypes during fruit development. BMC Genomics 10:349–353
Alkan, C., Sajjadian, S., & Eichler, E. E. (2011). Li-mitations of next-generation genome sequence assembly. Nature methods, 8(1), 61-65.
Angiolillo A, Mencuccini M, Baldoni L (1999) Olive genetic diversity assessed using amplified frag-ment length polymorphisms. Theor Appl Genet 98:411–421
Baldoni L, Belaj A (2009) Olive. In: Vollmann J, Rajean I (eds) Oil crops. Handbook of plant breeding, vol 4. Springer Science Business Me-dia, New York, pp 397–421
Baldoni L, Tosti N, Ricciolini C, Belaj A, Arcioni S, Pannelli G, Germana MA, Mulas M, Porceddu A (2006) Genetic structure of wild and cultivated olives in the central Mediterranean basin. Ann Bot 98:935–942
Baldoni L, Cultrera NG, Mariotti R, Riccioloni C, Ar-cioni S, Vendramin GG, Buonamici A, Porceddu A, Sarri V, Ojeda MA, Trujillo I, Rallo L, Belaj A, Perri E, Salimonti A, Muzzalupo I, Casagrande A, Lain O, Messina R, Testolin R (2009) A con-sensus list of microsatellites markers for olive genotyping. Mol Breed 24:213–231
Bartolini G (2008) Olive germplasm (Olea
euro-paea L.): cultivars, synonyms, cultivation area, collections, descriptors. http://www.oleadb.it Belaj A, Rallo L, Trujillo I, Baldoni L (2004) Using
RAPD and AFLP markers to distinguish indivi-duals obtained by clonal selection of ‘Arbequi-na’ and ‘Manzanilla de Sevilla’ olive. HortScien-ce 39:1566–1570
Bellini E. Genetic variability and heritability of some characters in cross-bred olive seedlings. Olivae. 1993;49:21–34
Bennetzen JL: The contributions of retroelements to plant genome organization, function and evo-lution. Trends Microbiol 4: 347–353 (1993) Bennetzen JL (2000). Transposable element
con-tributions to plant gene and genome evolution. Plant molecular biology, 42(1), 251-269
Besnard G, Baali-Cherif D (2009) Coexistence of diploids and triploids in a Saharan relict olive: evidence from nuclear microsatellite and flow cytometry analyses. CR Biol 332:1115–1120 Besnard G, Baradat PH, Chevalier D, Tagmount
A, Bervillé A (2001) Genetic differentiation in the olive complex (Olea europaea) revealed by RAPDs and RFLPs in the rRNA genes. Genet Resour Crop Evol 48:165–182
Besnard G, Henry P, Wille L, Cooke D, Chapuis E
(2007a) On the origin of the invasive olives (Olea
europaea L., Oleaceae). Heredity 99:608–619 Biémont C (2010). A brief history of the status of
transposable elements: from junk DNA to major players in evolution. Genetics, 186(4), 1085-1093
Biémont C & Vieira C (2006). Genetics: junk DNA as an evolutionary force. Nature, 443(7111), 521-524
Bitonti MB, Cozza R, Chiappetta A, Contento A, Minelli S, Ceccarelli M, Gelati MT, Maggini F, Baldoni L and Cionini PG (1999) Amount and organization of the heterochromatin in Olea
eu-ropaea and related species. Heredity 83, 188– 195
Bogani P, Cavalieri D, Petruccelli R, Roselli G (1994) Identification of olive tree cultivars by using random amplified polymorphic DNA. Acta Hortic 356:98–101
Bolger M E, Weisshaar B, Scholz U, Stein N, Usa-del B, & Mayer K F (2014). Plant genome se-quencing—applications for crop improvement. Current Opinion in Biotechnology, 26, 31-37. Bureau TE, Wessler SE: Mobile inverted-repeat
elements of the Tourist family are associated with the genes ofmany plant genomes. Proc Natl Acad Sci USA 91: 1411–1415 (1994) Cabezas JA, Ibáñez J, Lijavetzky D, Vélez D,
Bra-vo G, Rodrı´guez V, Carrenõ I, Jermakow AM, Carrenõ J, Ruiz-García L et al.: A 48 snp set for grapevine cultivar identification. BMC Plant Biol 2011, 11:153.
Cappello J, Handelsman K & Lodish H. Sequen-ce of Dictyostelium DIRS-1: an apparent retro-transposon with inverted terminal repeats and an internal circle junction sequence. Cell 43, 105–115 (1985)
Capy P, Bazin C, Higuet D & Langin T. (eds) Dyna-mics and evolution of transposable elements (Library of Congress, Austin, 1998)
Chandler M & Mahillon J. in Mobile DNA II (eds Craig, N., Craigie, R., Gellert, M. & Lambowitz, A.) (ASM, Washington D.C., 2002)
Contento A, Ceccarelli M, Gelati MT, Maggini F, Baldoni L and Cionini PG (2002) Diversity of Olea genotypes and the origin of cultivated oli-ves. Theor. Appl. Genet. 104, 1229–1238 De la Rosa R, James CM, Tobutt KH (2004) Using
microsatellites for paternity testing in olive pro-genies. HortScience 39:351–354
De la Torre F, Bautista R, Canovas FM, Claros MG (2004) Isolation of DNA from olive oil and oil sediments: application in oil fingerprinting. J Food Agric Environ 2:84–86
Dewannieux M, Esnault C and Heidmann T (2003). LINE-mediated retrotransposition of marked Alu
sequences. Nat. Genet. 35: 41–48
Dowsett AP & Young MW (1982). Differing levels of dispersed repetitive DNA among closely re-lated species of Drosophila. Proceedings of the National Academy of Sciences, 79(15), 4570-4574
Eickbush TH & Malik HS. in Mobile DNA II (eds Craig, N. L., Craigie, R., Gellert, M. & Lam-bowitz, A. M.) 1111–1146 (ASM, Herndon, 2002)
Eickbush T & Furano A. Fruit flies and humans respond differently to retrotransposons. Curr. Opin. Genet. Dev. 12, 669–674 (2002)
Evgen’ev M et al. Penelope, a new family of transposable elements and its possible role in hybrid dysgenesis in Drosophila virilis. Proc. Natl Acad. Sci. USA 94, 196–201 (1997)
Evgen’ev M & Arkhipova I. Penelope-like elements — a new class of retroelements: distribution, function and possible evolutionary significance. Cytogenet. Genome Res. 110, 510–521 (2005) Galla G, Barcaccia G, Ramina A, Collani S, Ala-gna F, Baldoni L, Cultrera NG, Martinelli F, Se-bastiani L, Tonutti P (2009) Computational an-notation of genes differentially espresse along fruit development. BMC Plant Biol 9:128–144 Gemas VJ, Rijo-Johansen MJ, Tenreiro R,
Feve-reiro P (2000) Inter and intra-varietal analysis of three Olea europaea L. cultivars using the RAPD techniques. J Hortic Sci Biotechnol 75:312–319 Goodwin T & Poulter R. A new group of tyrosine
recombinase-encoding retrotransposons. Mol. Biol. Evol. 21, 746–759 (2004)
Grandbastien M-A: Retroelements in higher plants. Trends Genet 8: 103–108 (1992)
Green PS. 2004. Oleaceae. In: Kubitzki K, Kade-reit JW. eds. The families and genera of vascu-lar plants. Vol. VII: Flowering plants, dicotyle-dons. New York: Springer, 296–306
Green B, Walko R, Hake S: Mutator insertions in an intron of the maize knotted-1 gene result in dominant suppressible mutations. Genetics 138: 1275–1285 (1994)
Greenblatt IM & Brink RA. Twin mutations in me-dium variegated pericarp maize. Genetics 47, 489–501 (1962)
Hamman-Khalifa AM, Castro AJ, Jìmenez-Lòpez JC, Rodrìguez-Garcìa MI, de Dios Alche´ J (2008) Olive cultivar origin is a major cause of polymorphism for Ole e 1 pollen allergen. BMC Plant Biol 8:10–18
Hess J, Kadereit W, Vargas P (2000) The coloni-zation history of Oleae uropea L. in Macarone-sia based on internal transcribed spacer 1 (ITS-1) sequences, randomly amplified polymorphic DNAs (RAPD) and intersimple sequence repe-ats (ISSR). Mol Ecol 9:857–868
Johnson LAS. 1957. A review of the family Olea-ceae. Contributions from the New South Wales National Herbarium 2: 397–418
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O & Walichiewicz J (2005). Repba-se Update, a databaRepba-se of eukaryotic repetitive elements. Cytogenetic and genome research, 110(1-4), 462-467
Kajikawa M and Okada N (2002). LINEs mobilize SINEs in the eel through a shared 39 sequence. Cell 111: 433–444
Kapitonov VV & Jurka J. Rolling-circle transpo-sons in eukaryotes. Proc. Natl Acad. Sci. USA 98, 8714–8719 (2001)
Kapitonov VV & Jurka J. (2008). A universal clas-sification of eukaryotic transposable elements implemented in Repbase. Nature Reviews Ge-netics, 9(5), 411-412
Katsiotis A, Hagidimitriou M, Douka A and Hatzo-poulos P (1998) Genomic organization, sequen-ce interrelationship, and physical localization using in situ hybridization of two tandemly repe-ated DNA sequences in the genus Olea. Geno-me 41, 527–534
Keys A (1995) Mediterranean diet and public health: personal reflections. Am J Clin Nutr 61:1321S–1323S
Kidwell MG & Lisch DR (2000). Transposable ele-ments and host genome evolution. Trends in ecology & evolution, 15(3), 95-99
Kim JM, Vanguri S, Boeke JD, Gabriel A & Voytas DF (1998). Transposable elements and geno-me organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Research, 8(5), 464-478
Kramerov D & Vassetzky N. Short retroposons in eukaryotic genomes. Int. Rev. Cytol. 247, 165– 221(2005).
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, & Grafham D. (2001). Initial se-quencing and analysis of the human genome. Nature, 409(6822), 860-921
León L, De la Rosa R, Gracia A, Barranco D, Rallo L Fatty acid composition of advanced olive se-lections obtained by crossbreeding. J. Sci. Food Agric.2008;88:1921–6.
Lorite, P., Garcia, M.F., Carrillo, J.A. and Palome-que, T. (2001) A new repetitive DNA sequence family in the olive (Olea europaea L.). Hereditas 134, 73-78
Loukas M, Krimbas CB (1983) History of olive cul-tivars based on their genetic distances. J Hortic Sci 58:121–127
Loureiro J, Rodriguez E, Costa A and Santos C (2007) Nuclear DNA content estimations in wild olive (Olea europaea L. ssp. europaea var.
syl-vestris Brot.) and portuguese cultivars of O.
eu-ropaea using flow cytometry. Genet. Res. Crop Evol. 54, 21-25
Maraia RJ, & Sarrowa, J. (1995). Alu-family SINE RNA: interacting proteins and pathways of ex-pression. The Impact of Short Interspersed
Ele-ments (SINEs) on the Host Genome. RG Lan-des compagny, TX, 163-196
Mariotti R, Cultrera NGM, Muñoz Diez C, Bal-doni L, Rubini A (2010) Identification of new polymorphic regions and differentiation of cul-tivated olives (Olea europaea L.) through pla-tome sequence comparison. BMC Plant Biol 10:211
Martienssen RA, Barkan A, Taylor WC, Freeling M: Somatically heritable switches in the DNA modification of Mu transposable elements mo-nitored with a suppressible mutant in maize. Genes Dev 4: 331–343 (1989)
Martins-Lopes P, Gomes S, Santos E, Guedes-Pinto H (2008) DNA markers for Portugue-se olive oil fingerprinting. J Agric Food Chem 56:11786–11791
Masson P, Surovsky R, Kingsbury J, Fedoroff NV: Genetic and molecular analysis of the Spm-dependent a-m2 alleles of the maize a locus. Genetics 177: 117–137 (1987)
Minelli S, Maggini F, Gelati MT, Angiolillo A and Cionini PG (2000) The chromosome comple-ment of Olea europaea L.: characterization by differential staining of the chromatin and in-situ hybridization of highly repeated DNA sequen-ces. Chromosome Res. 8, 615-619
Moreno-Alias I, Rapoport HF, Lopez R, Leon L, De la Rosa R Optimizing early flowering and pre-selection for short juvenile period in olive seedlings. HortScience. 2010;45:519–22
Muñoz-Mérida A, González-Plaza JJ, Cañada A, Blanco AM, del Carmen García-López M, Ro-dríguez JM, Pedrola L, Sicardo MD, Hernández ML, De la Rosa R, Belaj A, Gil-Borja M, Luque F, Martínez-Rivas JM, Pisano DG, Trelles O, Valpuesta V, Beuzón CR (2013) De novo as-sembly and functional annotation of the olive (Olea europaea) transcriptome. DNA Res 20: 93-108
Muzzalupo I, Perri E (2002) Recovery and cha-racterisation of DNA from virgin olive oil. Eur Food Res Technol 214:528–531
Nassif N, Penney J, Pal S, Engels W & Gloor G. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol. Cell. Biol. 14, 1613–1625 (1994) Natali L, Giordani T, Buti M and Cavallini A. (2007)
Isolation of Ty1-Copia putative LTR sequences and their use as a tool to analyse genetic diver-sity in Olea europaea. Mol. Breed. 19, 255–265 Ostertag EM & Kazazian HH. Genetics: LINEs in
mind. Nature 435, 890–891 (2005)
Pasqualone A, Caponio F, Blanco A (2001) Inter-simple sequence repeat DNA markers for iden-tification of drupes from different Olea europaea L. cultivars. Eur Food Res Technol 213:240–243 Pérez-Jiménez F, Ruano J, Pérez-Martinez P,
Lopez-Segura F, Lopez-Miranda J (2007) The influence of olive oil on human health: not
a question of fat alone. Mol Nutr Food Res 51:1199–1208
Petrov DA & Hartl DL. High rate of DNA loss in the Drosophila melanogaster and Drosophila virili-se species groups. Mol. Biol. Evol. 15, 293–302 (1998)
Pevzner PA, Tang H & Waterman MS (2001). An Eulerian path approach to DNA fragment as-sembly. Proceedings of the National Academy of Sciences, 98(17), 9748-9753
Phillippy, AM, Schatz MC & Pop M (2008). Geno-me assembly forensics: finding the elusive mis-assembly. Genome Biol, 9(3), R55
Rallo L, Barranco D, De La Rosa R, León L ‘Chi-quitita’ olive. HortScience.2008;43:529–31 Rasmussen DA & Noor MA. (2009). What can you
do with 0.1× genome coverage? A case study based on a genome survey of the scuttle fly Megaselia scalaris (Phoridae). Bmc Genomics, 10(1), 382
Reale S, Doveri S, Dıaz A, Angiolillo A, Lucentini L, Pilla F, Martin A, Donini P, Lee D (2006) SNP-based markers for discriminating olive (Olea
eu-ropaea L.) cultivars. Genome 49:1193–1205 Rodrıguez R, Villalba M, Batanero E, Gonzales
EM, Monsalve RI, Huecas S, Tejera ML, Lede-sma A (2002) Allergenic diversity of the olive pollen. Allergy S71:6–16
Rohwer JG. 1996. Die Frucht- und Samenstruktu-ren der Oleaceae. Bibliotheca Botanica 148: 1–177
Rowold DJ & Herrara RJ (2000) Alu elements and thehuman genome. Genetica 108, 57–72 Rugini E et al. “Olea.” Wild Crop Relatives:
Ge-nomic and Breeding Resources. Springer Berlin Heidelberg, 2011. 79-117
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, & Cordes M. (2009). The B73 maize genome: complexity, diversity, and dyna-mics. science, 326(5956), 1112-1115.
Servili M, Selvaggini R, Esposto S, Taticchi A, Montedoro G, Morozzi G (2004) Health and sensory properties of virgin olive oil hydrophilic phenols: agronomic and technological aspects of production that affect their occurrence in the oil. J Chromatogr A 1054:113–127
Simola, D. F., & Kim, J. (2011). Sniper: improved SNP discovery by multiply mapping deep se-quenced reads. Genome Biol, 12(6), R55 Smit AFA, Hubley R & Green P 1996–2010.
Re-peatMasker Open-3.0.URL: http://www. repeat-masker. org
Stergiou G, Katsiotis A, Hagidimitriou M and Loukas M (2002). Genomic and chromosomal organization of Ty1-Copia-like sequences in
Olea europaea and evolutionary relationships of Olea retroelements. Theor Appl Genet 104, 926-933
Straub SC, Fishbein M, Livshultz T, Foster Z, Par-ks M, Weitemier K, & Liston, A. (2011). Building
a model: developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC genomics, 12(1), 211
Swaminathan, K., Varala, K. and Hudson, M.E. (2007) Global repeat discovery and estimation of genomic copy number in a large, complex genome using a high-throughput 454 sequence survey. BMC Genomics 8, 132
Treangen TJ and Salzberg SL (2012) Repetitive DNA and next-generation sequencing: compu-tational challenges and solutions. Nature Rev. Genet. 13, 36-46
Wallander E, Albert VA. 2000. Phylogeny and classification of Oleaceae based on rps16 and trnL-F sequence data. American Journal of
Bo-tany 87:1827–1841
Wessler SR, Bureau TE, White SE: LTR-retrotran-sposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev 5: 814–821 (1995)
Wetzel J, Kingsford C & Pop M (2011). Assessing the benefits of using mate-pairs to resolve re-peats in de novo short-read prokaryotic assem-blies. BMC bioinformatics, 12(1), 95
Wicker T, Sabot F, Hua-Van A et al. (2007) A uni-fied classification system for eukaryotic transpo-sable elements. Nature Rev. Genet. 8, 973-982 Wu S, Collins G, Sedgley M (2004) A molecular
linkage map of olive (Olea europaea L.) based on RAPD, microsatellite, and SCAR markers. Genome 47:26–35
The peculiar landscape of repetitive sequences in the olive
(Olea europaea L.) genome
Elena Barghini1, Lucia Natali1, Rosa Maria Cossu1,2, Tommaso Giordani1, Massimo Pindo3, Federica
Cattonaro4, Simone Scalabrin4, Riccardo Velasco3, Michele Morgante4,5, Andrea Cavallini1*
1Dept. of Agricultural, Food, and Environmental
Sciences, University of Pisa, Pisa, Italy
2Institute of Life Sciences, Scuola Superiore Sant’Anna,
Pisa, Italy
3Research and Innovation Center, Fondazione E. Mach
di San Michele all’Adige, Italy
4Institute of Applied Genomics, Udine, Italy
5Dept. of Crop and Environmental Sciences, University
of Udine, Udine, Italy
*Author for Correspondence: Andrea Cavallini, Dept. of Agricultural, Food, and Environmental Sciences, University of Pisa, Via del Borghetto 80, 56124 Pisa, Italy, tel. +390502216663, E-mail: andrea.cavallini@ unipi.it.
All whole-genome-shotgun sequences described are available upon request or on NCBI Sequence Read Archive under SRA Project numbers SRX465835 and SRX474079. The OLEAREP database (sequences and annotations) is available at the repository sequence page of the Department of Agriculture, Food, and Environment of the University of Pisa (http://www.agr. unipi.it/Sequence-Repository.358.0.html).
Abstract
Analysing genome structure in different species allows to gain an insight into the evolution of the plant genome size. Olive (Olea europaea L.) has a medium sized haploid genome of 1.4 Gbp, whose structure is largely uncharacterized, despite the growing importance of this tree as oil crop. Next generation sequencing technologies and different computational procedures have been used to study the composition of the olive genome and its repetitive fraction. A total of 2.03 and 2.3 genome equivalents of Illumina and 454 reads from genomic DNA, respectively, were assembled following different procedures, which produced more than 200,000 differently redundant contigs, with mean length higher than 1,000 nt. Mapping
Illumina reads onto the assembled sequences was used to estimate their redundancy. The genome dataset was subdivided into highly and medium redundant and non-redundant contigs. By combining identification and mapping of repeated sequences, it was established that tandem repeats represent a very large portion of the olive genome (about 31% of the whole genome), consisting of six main families of different length, two of which were firstly discovered in these experiments. The other large redundant class in the olive genome is represented by transposable elements (especially LTR-retrotransposons). On the whole, the results of our analyses show the peculiar landscape of the olive genome, related to the massive amplification of tandem repeats, more than that reported for any other sequenced plant genome.
Key words: genome landscape, Olea
europaea, repetitive DNA, tandem repeats, retrotransposons, assembly of NGS reads
Introduction
Large genomes are filled with repetitive sequences, especially in plants (Morgante et al. 2007). Although some repeats appear to be non functional, others can have played a role in the evolution of a species (see for example Britten 2010),
acting as independent, ‘selfish’ sequence elements (Hua-Van et al. 2011), or creating novel functions (Morgante et al. 2005), or modelling the regulatory patterns of genes that result in phenotypic variation (Knight 2004).
Repeats arise from a variety of biological mechanisms that result in extra copies of a sequence being produced and inserted into the genome. They can be widely interspersed repeats, tandem repeats or nested repeats, and occur even in millions of copies, ranging in size from 1–2 bases to thousands of bases. In some cases, only a few repeat families account for the majority of genomic DNA (in the human genome, for example, the family of Alu repeat elements cover approximately 11% of the genome, Rowold and Herrara 2000); in other large genomes no prominent repeat families are found but many low redundant repeat classes account for the majority of genomic DNA (Cavallini et al. 2010). Generally, the most redundant sequences in plants are transposable elements, especially retrotransposons belonging to Gypsy and
Copia superfamilies, which transpose via a copy-and-paste mechanism (Wicker et al. 2007). For example, they cover about 80% of the maize genome (Schnable et al. 2009). In certain cases, as in the short-lived fish Nothobranchius furzeri, another class of sequences, the tandem repeats, are prominent (21%) in the genome (Reichwald et al. 2009).
Tandem repeats are commonly known as satellite DNAs (Schmidt and Heslop-Harrison, 1998), because they were initially isolated from satellite bands in
experiments with gradient centrifugation, due to the difference in A+T content from the rest of genomic DNA (Szybalski, 1968). Such sequences are arranged in tandem repeating units, where individual copies lye adjacently to each other. They are found preferentially at specific positions of the chromosomes, such as the pericentromeric, subtelomeric, telomeric or intercalary regions (Kubis et al. 1998). Tandem repeats from Secale
cereale were among the first satellite DNAs isolated, representing more than 6% of the rye genome (Bedbrook et al., 1980).
Families of tandem repeats show different homology, redundancy, and distribution pattern between related species of a plant genus or family, exhibiting species-, genome- and even chromosome-specificity (Wang et al. 1995). For example, within the plant genus Cucurbita, one satellite was detected differing in copy number among species, and another was present in a similar number of copies (King et al. 1995). By contrast, dramatic variation in copy number is reported for all satellites in all species within the tribe Triticeae (Vershinin and Heslop-Harrison 1998). In three species of the genus Chironomus, beside copy number variation, also chromosomal localization of the same tandem repeat is detected (Ross et al. 1997). Within a species, a satellite DNA shows sequence variability that depends on the ratio between the mutation and homogenization rates (Dover 1986). In this sense, each satellite DNA can be regarded as an independent evolutionary
unit (Ugarkovic and Plohl 2002).
Many basic questions about the evolution of plant genomes remain unanswered, especially regarding the occurrence of similar patterns of evolution among species. Genome evolution is based on the equilibrium between genome size increase by polyploidy and retrotransposon amplification and decrease by retrotransposon-mediated DNA loss (Morgante et al. 2007; Proost et al. 2011). The role of satellite DNAs in this respect is still largely unknown. Next generation sequencing (NGS) procedures can be conveniently used to study such dynamics by performing a global survey of the genome also in species whose genome has not been sequenced yet (Swaminathan et al. 2007; Treangen and Salzberg 2012).
The olive genome is largely uncharacterized, despite the growing importance of this tree as oil crop. Olive (Olea europaea L.) has a medium sized haploid genome of 1.4-1.5 Gbp (Loureiro et al. 2007). Concerning repeated sequences, the best characterized are four tandem repeats, isolated from genomic libraries and, in some instances, localized by cytological hybridization on olive chromosomes (Katsiotis et al. 1998; Bitonti et al. 1999; Minelli et al. 2000; Lorite et al. 2001; Contento et al. 2002). Also putative retrotransposon fragments have been isolated and sequenced (Stergiou et al. 2002; Natali et al. 2007), but a comprehensive picture of repeat elements landscape in the olive genome is still lacking.
In the frame of a project aiming to
obtain the complete sequence of the olive genome, we performed a deep analysis of the repetitive component of this genome, using NGS techniques (454-Roche and Illumina). In this work, we used different computational procedures to isolate and characterize olive repeated sequences. These data were used to determine the structure of the genome and the composition of its repetitive fraction. The results indicated that olive genome structure is peculiar among plant genomes, with a very large percentage of satellite DNA, related to a few tandem repeat families.
Materials and Methods
Illumina and 454 sequencing
Genomic DNA was extracted starting from young leaves of Olea europaea cv ‘Leccino’ following the nuclei extraction protocol of Zhang et al. (1995), modified for small volumes.
Paired-end libraries were prepared as recommended by Illumina (Illumina Inc., San Diego, CA, USA) with minor modifications. Illumina reads were pre-processed to remove Illumina adapters using Cutadapt (Martin 2011) with default parameters but -O 10 -n 2 -m 50. An internally developed Perl script was used to remove unpaired reads. In order to trim low quality regions, reads were further processed with ERNE-FILTER [erne. sourceforge.net] using default parameters but --min-size 50.
For 454 sequencing, two random shotgun ‘genomic’ libraries were
generated via fragmentation of 500ng each of genomic DNA employing the GS FLX+ Series XLR70 and XL+ Rapid Library preparation kit following the manufacturer’s recommendations (Roche, Indianapolis, IN, USA).
Low quality bases, empty reads, and adapter sequences were removed using CLC-BIO Genomic Workbench, version 5.1 (CLC-BIO) and ERNE-FILTER (Del Fabbro et al. 2013).
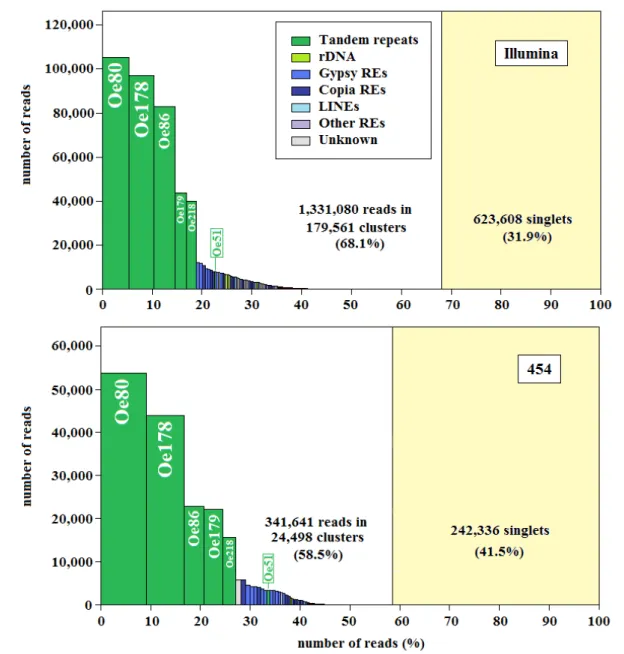
With Illumina technology, we obtained 157,049,970 paired-end reads, with mean read length, after trimming for base quality, of 98.6 nt. From these reads we produced two sets of sequences. The first set included 28,875,848 paired-end reads, corresponding to 2,847,904,818 nt and 2.03 genome equivalents and was used for assembly. The second set included 151,945,027 paired-end reads, that were trimmed at 75 nt in length, corresponding to 11,395,877,025 nt and 8.1 genome equivalents, and was used for mapping-based estimation of sequence redundancy. With 454 technology, we obtained 8,079,610 single reads, with mean read length of 407 nt, corresponding to a total of 3,275,110,538 nt and a 2.3 genome equivalents.
Graph-based clustering of sequences
The graph-based clustering method (Novák et al. 2010) was not feasible on the full data set because of computational requirements. Two reduced sets of randomly selected genomic Illumina and 454 reads (1x coverage for each set) were separately subjected to clustering
using RepeatExplorer website (http:// repeatexplorer.umbr.cas.cz/). The output of RepeatExplorer was also used to prepare two in-house libraries of olive repetitive sequences, the first containing tandem repeat sequences (characterized by sequence similarity search and structural analysis using DOTTER, Sonnhammer and Durbin 1995) and the second containing all contigs belonging to clusters identified by RepeatExplorer as retrotransposons, DNA transposons, rDNA, by similarity search against internal databases of known repeats (RepeatMasker, protein domains). These libraries were used for the annotation of assembled sequences (see next section).
Sequence assembly procedure
Illumina (2.03 genome equivalents) and 454 (2.3 genome equivalents) reads were assembled using CLC-BIO Genomics Workbench, version 5.1 (CLC-BIO). Initially, we performed a simple assembly of Illumina reads and obtained 1,788,026 contigs that were further assembled using Minimus 2 assembler (Sommer et al. 2007) using an overlap length cut-off of 40bp and an overlap identity cut-off of 90%. Alternatively, the pool of Illumina reads was split into 16, 64, 256, or 512 subpackages and assembled by CLC-BIO separately into contigs, based on unambiguous overlapping (indicated as split 0, 16, 64, 256, and 512, respectively); for each splitting, the resulting contigs were assembled on their turn using Minimus 2 assembler with an overlap length cut-off of 40bp and an overlap identity cut-off of
80%.
Also 454 reads were assembled using CLC-BIO. Possible contaminants resembling organellar sequences were removed by all assemblies masking contigs against an in-house olive organellar sequence database using RepeatMasker (Jurka 2000; http://www.repeatmasker. org/). A conservative threshold of 1% similarity was used for excluding any contamination of organellar sequences in the nuclear dataset.
Finally, all Illumina supercontigs and single contigs were assembled with 454 contigs to produce a whole genome set of assembled sequences (WGSAS) in which also single contig longer than 1,000 nt were included.
Estimation of sequence abundance
Redundancy of each supercontig or individual contig in the WGSAS was estimated by mapping a large Illumina sequence read set (total coverage 8.1 x) onto the WGSAS. To obtain uniformly long reads, all bases exceeding 75 nt were cut. Mapping was performed using CLC-BIO, which distributes multi-reads randomly; hence the number of mapped reads to a single sequence is only an indication of its redundancy. On the other hand, if all sequences of a sequence family or class are taken together, the total number of mapped reads (in respect to total genomic reads) reveals the effective redundancy of that family or class.
To establish the mapping parameters, 16 olive DNA sequences were selected whose copy numbers per haploid genome
were reported in the literature or were established by slot blot and hybridization experiments previously performed in our lab (Supplementary Materials S1). Mapping on sequences with known redundancy was performed using CLC-BIO with diverse parameters (mismatch cost, deletion cost, insertion cost, length fraction, similarity) and the significance of the correlation between copy number (as determined by slot blot) and average coverage for all 16 sequences was calculated for each set of parameters (see Supplementary Materials S2).
The parameters determining the largest correlation were selected to be used in the subsequent mapping of the WGSAS. After mapping, the WGSAS was subdivided into two classes of redundancy, redundant contigs (RC) and non-redundant contigs (NRC), using an arbitrary threshold of 16.2. RC were further subdivided into highly and medium redundant (HR and MR, respectively) according to their average coverage (> 1,620 and comprised between 16.2 and 1,620, respectively).
Annotation of redundant contigs
Annotation of supercontigs and individual contigs of the WGSAS was performed in two steps. In the first, sequences were masked by RepeatMasker (using as parameters –s –x –no_is –nolow) against the two libraries produced by the graph-based clustering method (see above) and against the RepBase database (Jurka 2000). In the second step, the remaining supercontigs and individual contigs were searched for homologies using the NCBI
BLAST with an e value cut-off of 1E-6.
In rare cases of ambiguity (i.e. supercontigs containing both tandem repeats and transposons fragments) the supercontigs were removed.
Preparation, sequencing and annotation of a small insert library
Five micrograms of olive genomic DNA were sheared by Hydroshear (Genomics Solutions) in fragments between 1.5-3 Kbp and the inserts cloned using pPCR-Script Amp SK(+) (Stratagene) according to the manufacturer’s instructions. One microliter of the ligation mix was electroporated into
E. coli ElectroMAX DH10B Cells (mcrA, mcrB, mcrC, mrr; Invitrogen), using the BioRad GenePulser II electroporator, in a 0.1 cm cuvette at the conditions of 2.0 kV, 200 W, 25 mF. The average insert size after cloning was about 2 Kb and insert from 3213 clones were selected for sequencing from both directions.
DNA for sequencing was prepared from selected transformants using the Montage Plasmid Miniprep Kit (Millipore). DNA sequencing was performed using an Applied Biosystems 3730 DNA Analyzer, the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and standard M13 forward and M13 reverse primers.
All sequences were annotated as above. Then, all sequences were compared to each other to detect additional repetitive sequences that did not show significant similarity to known repeated sequences but did overlap to each other by using the CAP3 sequence assembler (Huang and
Madan 1999) using parameter settings of 90% sequence identity and 40 bp minimum overlap.
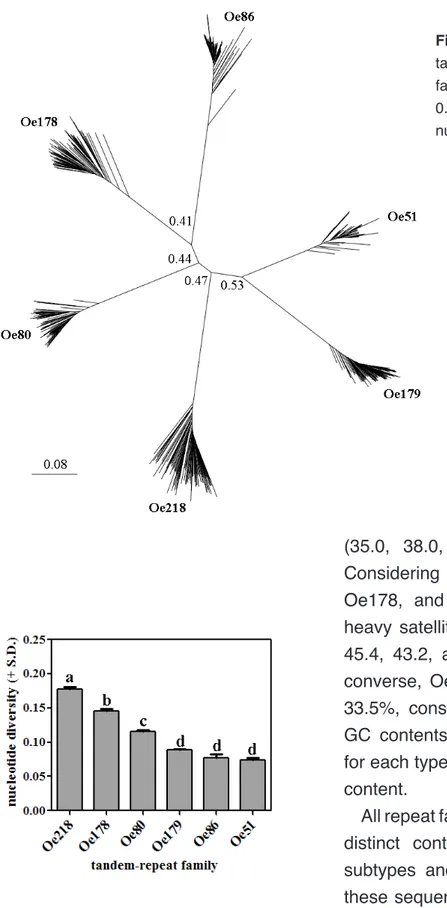
Sequence analyses of tandem repeats
One hundred sequences for each of the five main repeat families plus the Oe51 family were selected from 454 reads as follows: a preliminary consensus sequence of each tandem repeat type was deduced by dot plot analysis of the contigs assembled by RepeatExplorer and the subsequent alignment of the repeat units using CLC-BIO with default parameters. Then, a large set of 454 reads (1.0 genome-equivalent) were subjected to BLASTN (with an E-value
cut-off of 10-10) against the consensus
and the 100 most similar reads (i.e., “real” sequences) were selected for each type. Whenever more than one repeat was found within a read, only the most similar to the consensus was selected, i.e. each selected unit belonged to a different read, that represent a different locus.
Selected sequences were aligned using CLUSTALX 2.1 (Thompson et al. 1997) with default parameters. Then, 100 versions of the original multi-alignment were generated and a distance tree was produced by neighbour joining analysis. The tree was visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Alignments were also used to perform statistics of intraspecific polymorphism within tandem repeat type, using the DnaSP program version 3.51 (Rozas and Rozas 1999). Nucleotide diversity, i.e. the average number of nucleotide differences