CAPITOLO 2
RISULTATI E DISCUSSIONE
2.1. Reazioni di reagenti di organoalluminio con aziridine
ariliche
Nel laboratorio dove ho svolto la presente tesi, uno dei progetti in corso di studio riguardava l’apertura di aziridine, tra cui quelle ariliche, usando come reagenti alchil- e alchinilorganoalluminio per ottenere i corrispondenti prodotti di apertura, cioè ammine sostituite in maniera regioselettiva. Inizialmente i risultati delle prime reazioni dell’N-tosil fenilaziridina racema, 2.1a, con Me3Al, effettuate in solventi come etere dietilico e THF,
furono un pò scoraggianti in quanto si ottenevano solo tracce dei prodotti alchilati con basse conversioni (<10%), e non cambiava la situazione anche se si prolungava il tempo di reazione. La chiave di volta è stata quella di utilizzare il diclorometano come solvente di reazione tanto da ottenere una completa conversione in un’ora a 0 °C. Quindi questo tipo di reazione è risultata essere fortemente solvente dipendente. Probabilmente nei solventi eterei utilizzati all’inizio non si avevano buoni risultati perchè l’alluminio ha la proprietà di essere ossigenofilico, quindi andava a complessarsi fortemente con l’atomo di ossigeno delle molecole del solvente e così veniva persa parte della reattività del reagente stesso, con una diminuzione notevole nella velocità di metilazione. Facendo quindi reagire il substrato 2.1a con Me3Al (2 eq.) usando CH2Cl2 è stato ottenuto nel tempo e alla
temperatura precedentemente descritti, il prodotto di apertura con l’attacco completamente selettivo del frammento metilico in posizione benzilica (2.2a, Tabella 2.1.1).
Quando il substrato in questione è stato fatto reagire con Et3Al (2 eq.), è stato ottenuto il
corrispondente prodotto di apertura 2.3a, anche se con una regioselettività più ridotta. Poi abbiamo preso in considerazione come materiale di partenza anche la N-Cbz fenilaziridina 2.1b per farla reagire con il Me3Al (2 eq.) ed è stato recuperato come
prodotto il corrispondente addotto 2.2b nonostante le rese siano state un pò più basse, dovute al fatto che, probabilmente, il gruppo tosile sull’azoto è un miglior attivante rispetto ad un gruppo carbobenzilossi24.
Tabella 2.1.1.
Reazioni di apertura di fenil aziridine con alchilalaniESEMPIO AZIRIDINA AlR3/
CONDIZIONI REGIOSELETTIVITA' RESA DEI PRODOTTI 1 Ph Ts N 2.1a AlMe3 1h, 0°C >95/<5 Ph Me NHTs 2.2a, 92% 2 Ph Ts N 2.1a AlEt3 1h, 0°C 88/12 Ph NHTs 2.3a, 70% Et 3 Ph Cbz N 2.1b AlMe3 1h, 0°C >95/<5 Ph Me NHCbz 2.2b, 58%
Quando queste reazioni sono state realizzate usando l’aziridina enantiomericamente pura (R)-2.1a (>98% di ee), i corrispondenti prodotti 2.2a e 2.3a sono stati ottenuti con una bassa enantioselettività (14% di ee). Stessa cosa anche quando è stata usata l’aziridina enantiomericamente pura (R)-2.1b, per la quale è stato ottenuto solo il 25% di ee; in entrambi i casi viene quindi osservata una estesa racemizzazione, con livelli molto bassi di eccesso enantiomerico.
Questo fenomeno di racemizzazione che è stato osservato, insieme con l’attacco sulla posizione benzilica, sono caratteristiche attribuibili ad una reazione di apertura dell’aziridina che avviene sotto controllo elettronico. Probabilmente l’alta affinità dell’alluminio per l’ossigeno fa sì che ci sia una coordinazione del reagente AlR3 ai
lone-pairs dell’ossigeno del gruppo protettivo dell’azoto, in maniera da formare un complesso più reattivo che è in grado di trasferire il metile in modo intramolecolare sulla posizione benzilica cationica; questo però avviene con una sostanziale perdita dell’integrità della stereochimica dell’aziridina di partenza (57% di inversione, 43% di ritenzione, come determinato da analisi HPLC), come mostrato nello Schema 2.1.1.
Ph N S O O Ar Al Me Me Me Ph N S Ar O O Al Me Me Me
Schema 2.1.1
Meccanismo di apertura delle fenilaziridine sotto controllo elettronicoInoltre, tali substrati sono stati fatti reagire con alchinilalani in maniera da introdurre frammenti aventi ibridazione sp. Tali reagenti sono preparati di fresco nel nostro laboratorio dai corrispondenti alchinillitio e dal Me2AlCl. Ad esempio, l’esino ed il
fenilacetilene, sono stati trattati con n-BuLi per ottenere i corrispondenti alchinillitio; quest’ultimi sono stati fatti reagire con Me2AlCl in maniera da ottenere i corrispondenti
organoalani in seguito alla reazione di scambio (Schema 2.1.2). I reattivi sintetizzati 2.1m e 2.1n sono stati impiegati quindi nelle nostre reazioni.
CH2Cl2, 0°C n-BuLi Li Me2AlCl 0°C per 40 min. e poi t.a. per 20 min. AlMe2 Ph Ph Li Me2AlCl 0°C per 40 min. e poi t.a. per 20 min.
Ph AlMe2 10-15 min. CH2Cl2, 0°C n-BuLi 10-15 min. H H 2.1m 2.1n
Schema 2.1.2
Reazione di scambio per ottenere alchinilalaniQuesti reagenti hanno una reattività molto più alta rispetto ai normali alchilalluminio, visti precedentemente; infatti se vengono fatte reagire le aziridine N-protette 2.1c e 2.1d con Me3Al sono ottenute solo tracce dei prodotti di alchilazione, invece gli alchinilalani nella
quantità di 3 equivalenti reagiscono con le stesse aziridine a 0 °C in CH2Cl2 per dare le
omopropargilammine 2.2c e 2.2d in buone rese. Questo è riportato nello Schema sottostante, 2.1.3 dove è stato preso in considerazione l’alchinilalano derivante dall’esino.
AlMe2 PG N 2.1c, PG= Ts 2.1d, PG= Cbz CH2Cl2 0°C, 1-3h NHPG 2.2c (PG= Ts, 80%) 2.2d (PG= Cbz, 70%)
Schema 2.1.3
Reazioni di alchinilazione di aziridine monosostituiteOppure la stessa reazione può essere effettuata con l’alchinilalano derivante dal fenilacetilene, Schema 2.1.4, e anche in questo caso sono stati ottenuti i composti 2.3c e
2.3d in buone rese. Ph AlMe2 PG N 2.1c, PG= Ts 2.1d, PG= Cbz CH2Cl2 0°C, 1-3h Ph PGHN 2.3c (PG= Ts, 66%) 2.3d (PG= Cbz, 60%)
Schema 2.1.4
Reazioni di alchinilazione di aziridine monosostituiteNonostante la deprotonazione dell’alchino potrebbe essere condotta anche in un solvente diverso dal diclorometano, quest’ultimo è stato invece necessario per ottenere elevate rese dei prodotti alchinilati e non è stata osservata nessuna reazione competitiva di deprotonazione del diclorometano stesso. La facilità con cui viene trasferito facilmente il gruppo alchinilico da parte dell’alchinilalano è attribuibile al fatto che nei reagenti misti viene trasferita la porzione che presenta un maggiore carattere di ibridazione s.13
Quindi sono state fatte reagire le arilaziridine con vari tipi di alchinilalani per verificare la loro reattività. Ad accezione della reazione in cui è stato utilizzato l’organoalano derivante dall’eptino, tutte le altre reazioni sono andate a completezza in un’ora a 0 °C dopo l’addizione dell’alchinilalano all’aziridina. La regioselettività dell’apertura dell’anello è risultata eccellente in tutti i casi analizzati, utilizzando come substrati le fenilaziridine 2.1a e 2.1b, come mostrato nella Tabella 2.1.2, dove si vede che l’alchino è sempre trasferito in posizione benzilica.24
Tabella 2.1.2
Reazioni di apertura di fenilaziridine con alchinilalaniESEMPIO AZIRIDINA R AlMe2 E CONDIZIONI
REGIOSELETTIVITA' RESA DEI PRODOTTI 1 Ph Ts N 2.1a R=Ph 1h, 0°C >95/<5 Ph Ph NHTs 2.4a, 85% 2 Ph Ts N 2.1a R=C5H11 18h, t.a. >95/<5 Ph NHTs C5H11 2.5a, (70%) Ph Ts N 2.1a R=C3H5 1h, 0°C >95/<5 Ph NHTs 2.6a, (88%) 4 Ph Cbz N 2.1b R=Ph 1h, 0°C >95/<5 Ph Ph NHCbz 2.3b, (60%) 3
Quindi partendo da fenilaziridine, con questo tipo di reazioni è stato possibile ottenere
ammine β-fenil-β-sostituite, in particolar modo sono state ottenute ammine
omopropargiliche che possono essere utilizzate successivamente in reazioni di idroamminazione intramolecolare per fornire derivati pirrolici.
Successivamente è stata considerata anche la fenilaziridina 2.1e, che presenta un gruppo difenilfosfinoil come gruppo protettivo, e tale substrato è stato fatto reagire con l’alchinilalano derivante dal fenilacetilene. Anche in questo caso è stato ottenuto un unico prodotto che deriva dall’attacco dell’alchino in posizione benzilica (resa NMR circa del 60%), dimostrando ancora una volta la notevole regioselettività di questi reagenti, come riportato nello Schema 2.1.5.
Ph N P O Ph Ph 2.1e Ph AlMe2 CH2Cl2, 0°C, 1h Ph Ph NH P O Ph Ph 2.2e
Schema 2.1.5
L’unica reazione che non ha dato un buon esito pur usando uno di questi reattivi di alchinil alluminio, è stata quella dell’aziridina alifatica 2.1f, N-Cbz protetta; questa, se fatta reagire nelle medesime condizioni usate con gli altri substrati, ha portato a livelli di conversione estremamente bassi, recuperando essenzialmente l’aziridina stessa immodificata mentre il prodotto di apertura è stato ottenuto solo in tracce. Questo substrato presenta un gruppo poco attivante sull’azoto che potrebbe spiegare il motivo di questo comportamento; però la medesima reazione, in cui è stato utilizzato come substrato l’aziridina 2.1d, la quale presenta sempre lo stesso gruppo carbobenzilossi sull’azoto, è andata a completa conversione. Quindi probabilmente il motivo della scarsa reattività è attribuibile al fatto che l’aziridina 2.1f è disostituita a differenza di 2.1d che è monosostituita, come appare chiaro dal confronto tra i due substrati. Nell’insieme quindi il substrato 2.1f risulta essere poco reattivo verso l’apertura dell’anello, come riportato nello Schema 2.1.6. NCbz C4H9 AlMe2 CH2Cl2 da 0°C a t.a. 2.1f
solo tracce del prodotto alchinilato
2.2. Reazioni di reagenti di organoalluminio con aziridine
viniliche
Oltre a considerare le fenilaziridine sono state prese in considerazione anche aziridine viniliche, in particolar modo le aziridine derivanti dall’1,3-cicloesadiene 2.1g o dal ciclopentadiene 2.1h. Anche in questo caso era presente una posizione allilica dell’aziridina in grado di reagire preferenzialmente in quanto in grado di fornire un carbocatione stabile. Sono stati quindi testati questi tipi di substrato in reazioni che utilizzano i reagenti analizzati nel caso delle fenilaziridine, quali alchil- e alchinilalani. L’addizione del Me3Al (2 eq.) al 7-tosile-7-azabiciclo[4.1.0]ept-2-ene (2.1g), senza utilizzo
di catalizzatori è avvenuta con completa conversione in un’ora a 0 °C e con una buona regioselettività. Infatti è prevalsa la modalità di attacco SN2, che ha portato all’addotto 1,2
rispetto alla modalità di attacco SN2’, che invece avrebbe portato all’addotto 1,4 con un
rapporto SN2/SN2’= 93/7. Sfortunatamente il prodotto principale è risultato essere costituito
da una miscela di diastereoisomeri, cis-2.2g (60%) e trans-2.3g (40%), che inoltre non è stato possibile separare tramite purificazione cromatografica (Schema 2.2.1).
L’addizione ancora non catalizzata del medesimo reagente, Me3Al (2 eq.), all’analoga
aziridina vinilica a 5 membri 2.1h, è avvenuta sempre in modo prevalente nella posizione allilica (rapporto SN2/SN2’= 91/9). Però in questo caso la reazione ha fornito
principalmente l’addotto cis-2.2h (92%) che quindi è stato ottenuto con una buona stereo- e regioselettività e con una buona resa isolata dopo avere effettuato una TLC preparativa (Schema 2.2.1). La stereochimica del prodotto 2.2h è stata confermata da esperimenti 1H NMR, in cui si è osservata una costante di accoppiamento di circa 7 Hz tra H1 e H2, e dal
confronto con i dati di letteratura del corrispondente addotto trans.25 L’apertura di tale aziridina che porta ad un addotto 1,2-cis è un tipo di reazione raramente osservato con achil carbanioni ma può essere razionalizzato considerando la coordinazione dell’organoalano al gruppo protettore dell’aziridina con il successivo trasferimento intramolecolare del gruppo alchilico dalla stessa parte in maniera da ottenere addotti del tipo cis.24
NTs 2.1g Me NHTs 2.2g,2.3g miscela inseparabile 2.1h AlMe3 1h, 0°C Me NHTs 2.2h, (62%) NTs H1 H2
Schema 2.2.1
Reazionidi apertura di vinilaziridine con alchilalaniLe reazioni delle vinil aziridine con reagenti organometallici alcune volte vengono promosse da sali di rame come catalizzatori e questo spesso porta a reazioni anti-SN2’
selettive anche se motivi sterici possono portare a reazioni syn-stereoselettive.26 A questo proposito sono state condotte le reazioni precedenti utilizzando sempre le aziridine viniliche e il Me3Al ma in più è stato aggiunto il Cu(OTf)2 (1.5 mol%) come catalizzatore
e come ligante una fosforoammidite chirale L1 del tipo (R,R,R), lasciando immodificato il solvente. Nel caso dell’aziridina 2.1h è stato ottenuto lo stesso prodotto 2.2h che era stato ottenuto con le reazioni non catalizzate, anche se però abbiamo assistito ad un leggero aumento del prodotto derivante dall’attacco di tipo SN2’, infatti il rapporto tra i due
prodotti SN2/ SN2’ in questo caso è risultato essere 75:25. Considerando invece l’aziridina
2.1g, nel caso in cui è stato utilizzato il catalizzatore di rame più il ligante, è stato ottenuto
un prodotto diverso da quello ottenuto senza l’utilizzo del catalizzatore; confrontando gli spettri NMR della reazione catalizzata e di quella non catalizzata è stato osservato che i doppietti a 0.9 ppm risultano diversi così come è evidente il multipletto intorno a 3.8 ppm, relativo al segnale CH-N, invece del ddd a 3.5 ppm presente nella reazione non catalizzata in cui si forma il prodotto di tipo 1,2.7 Da questo è stato dedotto che probabilmente il composto principale ottenuto è l’addotto 1,4-trans, derivante da un meccanismo quindi di tipo SN2’ che il rame è in grado di esaltare (Schema 2.2.2).
NTs 2.1g Me3Al (2 eq.) Cu(OTf)2: 1.5 mol% L1: 1.5 mol% CH2Cl2, 0°C, 1h Me NHTs prodotto principale O O P N Ph Me Ph Me
Schema 2.2.2
Reazione di apertura Cu(II) catalizzata della vinilaziridina con Me3AlSuccessivamente, come per le fenilaziridine, ci siamo occupati di far reagire questi substrati con alchinilalani. Nel caso delle aziridine 2.1g e 2.1h, facendole reagire con alchinilalani, si osserva che il gruppo alchinile del reagente ha una significativa influenza sulla regio- e stereoselettività nella reazione di apertura dell’anello che dipende dal tipo di substrato usato. Infatti la reazione dell’alchinilalano che deriva dal fenilacetilene con l’aziridina derivante dal cicloesadiene 2.1g è avvenuta con un’alta regioselettività di tipo SN2; l’addizione di tipo 1,2 non è risultata completamente stereoselettiva (circa 9/1
trans/cis), ma il maggior prodotto che è stato isolato in seguito a cromatografia risulta essere la trans-ammina omopropargilica 2.5g, come mostrato nella Schema 2.2.3. Con nostra sorpresa invece la reazione dello stesso alchinilalano con l’aziridina vinilica a cinque membri 2.1h ha mostrato una inusuale stereoselettività cis-SN2’. La relazione
cis-1,4 tra i due sostituenti è stata dedotta dalla grande differenza di chemical shift (0.90 ppm) tra i due protoni metilenici dell’anello e dalle loro caratteristiche costanti di accoppiamento.24
7/ 93 (SN2'/SN2) 1h, 0°C NTs 2.1g Ph NHTs 2.5g, (60%) NTs 89/11 (SN2'/SN2) NHTs Ph 2.1h 2.3h, (45%) Ph AlMe2
Schema 2.2.3
Reazioni di apertura di vinilaziridine con alchinilalaniQuesto può essere spiegato considerando che l’anione derivante dall’alchinilalano risulta avere un carattere più “soft” e così risulta favorito l’attacco SN2’ dell’alchino sul doppio
legame. Invece il trimetilalluminio ha un carattere più “hard” e in questo caso l’interazione favorita è quella tra il metile e il carbocatione, che si forma in seguito alla rottura del legame C-N dell’aziridina, dopo la complessazione tra l’atomo di alluminio e l’ossigeno del gruppo tosile presente sull’azoto; al contrario l’aziridina a sei membri, proprio per la sua struttura si dispone in modo tale che sia favorita la classica apertura trans-diassale sia con un alchinilalano, sia con il trimetilalluminio (Schema 2.2.4).
H NH S O O Ph H Al R Me Me
stato di transizione del tipo syn-SN2'
VS
stato di transizione del tipo anti-SN2
N H H S O O Ph R AlMe2
Schema 2.2.4
Razionalizzazione della differenza di reattività tra le due aziridine vinilicheLa stereoselettività ottenuta nell’apertura dell’aziridina a cinque termini 2.1h è complementare al protocollo sintetico sviluppato recentemente in cui si ottenevano ciclopenteni 1,4-trans disostituiti partendo dalla desimmetrizzazione di idrazine bicicliche.27
Inoltre, tali aziridine viniliche, sia 2.1g che 2.1h sono state fatte reagire con altri tipi di alchinilalani derivanti dall’etossiacetilene nelle medesime condizioni. È noto che l’utilizzo di alchinilalani derivanti dall’etossiacetilene è in grado di esaltare i processi di tipo SN2’ secondo quanto riportato da Somfai.28 Però in questo caso sia per l’aziridina
derivante dal ciclopentadiene, sia per quella derivante dal cicloesadiene sono state ottenute miscele complesse. Nel caso dell’aziridina a sei termini non è stata osservata neppure completa conversione e il recupero in peso del grezzo di reazione è risultato sempre assai scarso (Schema 2.2.5). NTs 2.1g NTs 2.1h 1h, 0°C AlMe2 EtO miscele complesse
Schema 2.2.5
Utilizzo di alchinilalani derivanti dall’etossiacetileneSono state prese poi in considerazione vinil aziridine di natura alifatica, quali l’aziridina 2.1i e 2.1l, ottenute con due processi sintetici diversi (Schema 2.2.6).29-30
PhI N-Ts CH3CN Cu(Acac)2 (10 mol%) 0 °C / Ts N 2.1l Ph NTs S Ts N Ph 2.1i CH3CN KOH (1.2 eq.) H
miscela cis - trans
(68/32)
13% dell'altro regioisomero Br
Per ottenere l’aziridina 2.1i è stato inizialmente sintetizzato il sale di 1-allil tetraidrotiofene bromuro, facendo reagire il tetraidrotiofene con il bromuro di allile in acetone; una volta ottenuto questo composto come un solido altamente igroscopico, è stato solubilizzato in CH3CN e a questa soluzione è stata aggiunta la fenil-N-tosilmetanimmina sotto argon e il
KOH a temperatura ambiente. L’attacco dell’ilide dello zolfo fornisce un grezzo che è stato sottoposto a purificazione ottenendo come prodotto 2.1i che risulta costituito da entrambe le aziridine, cis- trans, in rapporto 68/32 che viene usato come tale nelle reazioni successive. Invece per sintetizzare il composto 2.1l è stata preparata una soluzione di 1,3-pentadiene in CH3CN a cui sono stati aggiunti 10 mol% di Cu(acac)2 (rame
acetilacetonato); successivamente dopo aver raffreddato a 0 °C è stato aggiunto l’N-tosil fenil immino iodinano in tre porzioni. Come grezzo è stato ottenuto il prodotto 2.1l costituito dall’aziridina principale derivante dall’attacco sul doppio legame meno sostituito e per il 13 % anche dal suo regioisomero; tale grezzo non può essere sottoposto a purificazione e viene utilizzato come tale.
È stata quindi testata la reattività di tali substrati verso reagenti di alchilalluminio come il trimetilalluminio sia verso alchinilalani derivanti dal fenilacetilene. In tutti i casi sono state ottenute delle rese non molto alte e in certi casi non è stata ottenuta una conversione completa neppure dopo 18 ore a temperatura ambiente. In entrambi i casi il Me3Al ha permesso di ottenere addotti 1,2 in seguito ad un meccanismo SN2; nel caso
dell’alchinilalano invece con l’aziridina 2.1i è stato ottenuto l’attacco dell’alchino mediante un meccanismo coniugato per ottenere l’addotto di apertura 1,4. Questo è chiaramente visibile perchè analizzando lo spettro 1H NMR non si vede più il segnale del doppio legame terminale. Quando sempre lo stesso alchinilalano è stato fatto reagire con l’aziridina 2.1l il prodotto che si è formato è stato invece di tipo 1,2 (Tabella 2.2.1).
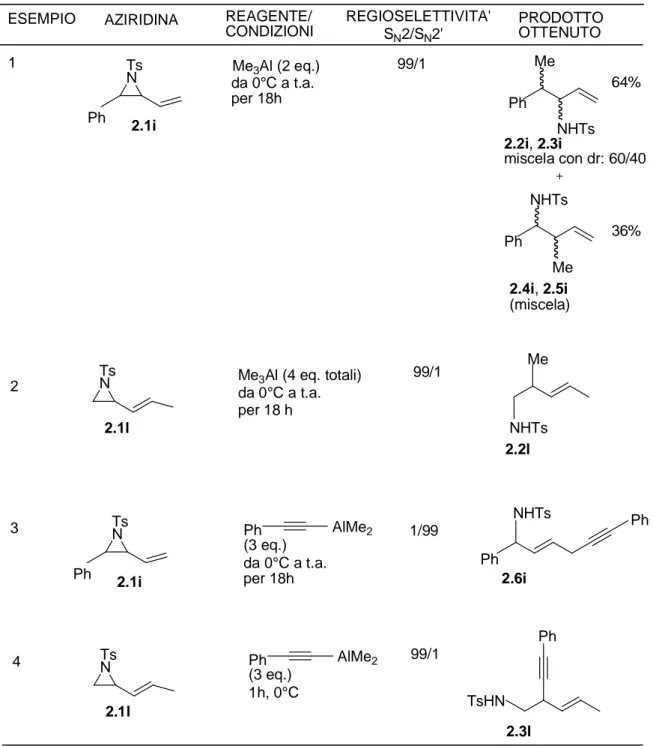
L’aziridina 2.1i fatta reagire con Me3Al ha mostrato una conversione del 90% dopo 18 ore
a temperatura ambiente; sono stati ottenuti principalmente 2 prodotti, a seconda che il metile abbia attaccato in posizione benzilica oppure sull’altro carbonio dell’aziridina. Per i fattori elettronici è prevalso l’attacco sulla posizione benzilica rispetto a quella allilica con un rapporto di 64/36 e in entrambi gli attacchi sono state ottenute due coppie di diastereoisomeri. Queste due coppie di prodotti però sono state ottenute in miscela e non è stato possibile separarle neppure effettuando una separazione tramite lastra preparativa. La resa della miscela dei prodotti di apertura 2.2i-2.3i e 2.4i-2.5i, come accennato in precedenza, è risultata non molto alta, circa del 50%.
L’aziridina 2.1l, con 2 equivalenti di Me3Al per 18 ore a temperatura ambiente, non ha
mostrato nessun tipo di conversione; aggiungendo ancora 2 equivalenti, dopo 2 ore dall’ulteriore aggiunta la reazione è andata a completezza per ottenere selettivamente l’attacco del metile sulla posizione allilica dell’aziridina con una resa di circa 58%; è stato quindi ottenuto il prodotto 2.2l del quale non è stato necessario effettuare una purificazione cromatografica. Trattando l’aziridina 2.1i con l’alchinilalano, dopo 18 ore a temperatura ambiente, è stata ottenuta una conversione del 70%; in seguito a separazione tramite lastra preparativa è stato ottenuto il prodotto di apertura 2.6i derivante da un’addizione coniugata dell’alchino su tale substrato per dare il prodotto di apertura 1,4 con una resa isolata solo del 35% . Infine la reazione dell’aziridina 2.1l con l’alchinilalano è l’unica che è avvenuta in un’ora a 0 °C con una conversione completa e con l’attacco selettivo sulla posizione allilica. Dopo purificazione tramite lastra preparativa è stato ottenuto il prodotto 2.3l con una resa che è comunque bassa , circa del 40%. Questi due tipi di aziridine hanno evidenziato un comportamento un pò particolare, certe volte simile alle aziridine viniliche e a quelle ariliche e certe volte del tutto opposto. Nel caso del substrato 2.1i è evidente una certa competizione tra la posizione benzilica e tra quella allilica, infatti dopo essere stato fatto reagire con il trimetilalluminio, il metile ha attaccato entrambe le posizioni mediante un meccanismo SN2, con prevalenza di attacco in posizione benzilica. Invece quando la
reazione è stata condotta usando l’alchinilalano è stato ottenuto esclusivamente l’attacco sul doppio legame secondo un meccanismo SN2’ per ottenere l’addotto 1,4, comportandosi
come l’aziridina vinilica a cinque membri (Schema 2.2.7). L’aziridina derivante dall’ 1,3-pentadiene 2.1l invece fatta reagire con il trimetilalluminio ha portato all’ottenimento di prodotti di tipo 1,2 con l’attacco esclusivo del metile in posizione allilica. In questo caso anche l’alchinilalano ha portato a prodotti di tipo 1,2, con l’attacco dell’alchino sempre in posizione allilica, reagendo in una modalità simile all’aziridina vinilica a sei termini. Non è stato quindi osservato nessun tipo di attacco con modalità SN2’ forse a causa del diverso
grado di sostituzione del doppio legame (Schema 2.2.7).
Ts N Ph Ph AlMe2 Me3Al Me3Al Ts N Me3Al Ph AlMe2 VS
Tabella 2.2.1
Apertura di vinilaziridine di natura alifatica con alchil- e alchinilalaniESEMPIO AZIRIDINA REAGENTE/ CONDIZIONI REGIOSELETTIVITA' SN2/SN2' PRODOTTO OTTENUTO 1 Ph Ts N 2.1i Me3Al (2 eq.) da 0°C a t.a. per 18h 99/1 Ph Me NHTs 2.2i, 2.3i miscela con dr: 60/40 Ph NHTs Me 2.4i, 2.5i (miscela) 2 Ts N 2.1l Me3Al (4 eq. totali) da 0°C a t.a. per 18 h 99/1 NHTs Me 2.2l 3 Ph Ts N 2.1i Ph AlMe2 da 0°C a t.a. per 18h (3 eq.) Ph Ph NHTs 2.6i 4 TsN 2.1l Ph AlMe2 1h, 0°C (3 eq.) 99/1 TsHN 2.3l 64% 36% 1/99 Ph
2.3 Reazioni di reagenti di organoalluminio con cicloaddotti
acilnitroso
Durante il periodo di laboratorio, uno degli obiettivi è stato quello di riuscire ad ottenere composti funzionalizzati, ad esempio derivati cicloesanici diversamente sostituiti considerati come “building blocks” da utilizzare in processi sintetici di vario tipo. A questo scopo sono stati anche impiegati sistemi biciclici contenenti azoto e ossigeno, cioè cicloaddotti acilnitroso in reazioni di apertura con reagenti di organoalluminio.
Di particolare interesse è risultato il biciclo 3-aza-2-ossabiciclo-[2.2.2]oct-5-ene 2.7, con un gruppo t-butossicarbonilico (Boc) sull’atomo di azoto come gruppo protettivo.
Per ottenere questo substrato è stato necessario sintetizzare l’acido idrossammico corrispondente, e quì sono stati trovati un pò di problemi perchè inizialmente è stato utilizzato un metodo descritto che è risultato non molto efficace. Secondo tale processo ad una soluzione di idrossilammina cloridrato in acqua e CH2Cl2 a 0 °C doveva essere
aggiunto in più porzioni NaHCO3, e dopo 10 minuti, il di-terz-butil dicarbonato (l’anidride
del Boc) sempre a 0 °C e la reazione doveva essere lasciata ad agitare per 3 ore (Schema 2.3.1).31 Però anche dopo avere effettuato una cristallizzazione con esano è stato ottenuto l’acido idrossammico con molte impurezze e quindi la successiva reazione con l’1,3-cicloesadiene non ha portato all’ottenimento dell’addotto biciclico desiderato.
Il tentativo successivo ha previsto l’utilizzo di un altro protocollo sintetico.32 In questo caso è stata preparata una sospensione di idrossilammina cloridrato in Et2O a cui è stato
aggiunto Na2CO3 e H2O e tale sospensione è stata lasciata in agitazione per un’ora. Dopo,
a 0 °C è stata aggiunta goccia a goccia una soluzione di di-terz-butil dicarbonato in Et2O e
la sospensione è stata agitata a temperatura ambiente per tutta la notte. In questo caso in seguito alla cristallizzazione con esano è stato ottenuto l’acido idrossammico desiderato allo stato di purezza, come un solido bianco (Schema 2.3.1).
NH2OHHCl H2O/CH2Cl2 NaHCO3 Boc2O, 0 °C molte impurezze O O NHOH NH2OHHCl Et2O/H2O NaCO3, Boc2O 0 °C t.a.
A questo punto il t-butilidrossicarbammato è stato fatto regire con l’1,3-cicloesadiene e NaIO4 in una miscela di MeOH/H2O (3/1); la specie acilnitroso che si viene a formare per
trattamento dell’acido idrossammico con periodato di sodio viene intrappolata in situ con l’1,3-cicloesadiene in una reazione di etero Diels-Alder che ha portato direttamente alla formazione del biciclo desiderato. Questo è stato poi purificato tramite cromatografia per dare il composto 2.7 allo stato di purezza come un olio giallino (Schema 2.3.2).15
O O NHOH NaIO4, MeOH/H2O (3/1)0-5°C O NBoc 2.7
Schema 2.3.2
Sintesi del 3-aza-2-ossabiciclo-[2.2.2]oct-5-enePrecedentemente nel laboratorio dove è stata svolta la presente tesi, erano già stati utilizzati dei cicloaddotti acilnitroso presentanti un diverso gruppo protettore sull’azoto, in particolare il biciclo 3-aza-2-ossabiciclo-[2.2.2]oct-5-ene, con un gruppo carbobenzilossi sull’azoto 2.8b. Su questo substrato era stata valutata la reattività in reazioni di apertura con reagenti organometallici in condizioni di risoluzione cinetica per ottenere derivati cicloesanici funzionalizzati enantioarricchiti di tipo 1,2-trans 2.9b e 2.10b. Il substrato
2.8b nonostante si differenzi dall’omologo inferiore 2.8a solo per la presenza di un –CH2-
aggiuntivo sul ponte, è risultato caratterizzato da una reattività marcatamente inferiore rispetto all’ossazabiciclo [2.2.1].33 Mentre l’omologo inferiore 2.8a aveva reagito con conversione completa dopo 3 ore con 1.5 equivalenti di dietilzinco, 1.5 mol% di Cu(OTf)2
e 3 mol% di ligante chirale L1 del tipo (S,S,S) per dare il prodotto di apertura 1,2-trans (Schema 2.3.3), nel caso dell’ossazabiciclo [2.2.2] con 3 equivalenti dello stesso reattivo la conversione era solo del 20%. Per cui era stato usato il trietilalluminio, sempre nella quantità di 3 equivalenti, in toluene e in questo modo è stata ottenuta una conversione completa dopo 5 ore a 0 °C per dare il prodotto di apertura 1,2-trans (Schema 2.3.3). Con 1.8 equivalenti di questo reattivo, dopo 24 ore, si era osservata una conversione del 40% con un 58% di eccesso enantiomerico del substrato non reagito; con dimetilzinco (3 eq.) in diclorometano, dopo 24 ore la conversione era del 67% con un 70% di ee del substrato non reagito. Utilizzando infine come agente alchilante il trimetilalluminio, 3 equivalenti, dopo
1 ora a 0 °C è stata ottenuta una conversione completa con l’ottenimento però di un grezzo di reazione molto complesso che non era stato ulteriormente analizzato.
O N "RM"/ solvente Cu(OTf)2 (1.5 mol%) L1-(S,S,S) (3.0 mol%) R N OH O O Ph R=Me, 2.9b R=Et, 2.10b 2.8b O N O Ph O Ph 2.8a Et2Zn/ solvente Cu(OTf)2 (1.5 mol%) L1-(S,S,S) (3.0 mol%) N O O OH Ph O Et 2.9a
Schema 2.3.3
Esperimenti effettuati precedentemente su cicloaddotti acilnitroso con reagenti organometallici
Quindi lo scopo del lavoro è stato di utilizzare cicloaddotti acilnitroso [2.2.2] con un gruppo Boc sull’azoto per farli reagire con organoalluminio per trasferire gruppi alchilici o fenilici nella reazione di apertura e ottenere così derivati cicloesanici funzionalizzati.
La prima reazione che è stata eseguita sul composto 2.7 è stata quella che ha sfruttato come reagente il fenildimetil alluminio, PhAlMe2, cioè l’organoalluminio
derivante dal fenillitio e dal Me2AlCl; anche questo reattivo come gli alchinilalani visti in
precedenza, è stato preparato di fresco nel laboratorio al momento dell’uso. In questo caso la sintesi è partita dal PhLi a cui a 0 °C è stato aggiunto 1 equivalente di Me2AlCl sotto
argon; la miscela di reazione è stata lasciata in agitazione per circa 40 minuti a 0 °C e poi per 20 minuti a temperatura ambiente per poter far avvenire lo scambio (Schema 2.3.4).
PhLi
Me2AlCl
CH2Cl2 0°C/40 min.
e poi t.a./ 20 min.
PhAlMe2
Schema 2.3.4
Reazione di scambio per la formazione di organoalani mistiQuesto tipo di reazione nel caso del substrato 2.7, così come tutte le altre reazioni che sono state eseguite, non è stata in grado di avvenire in assenza di un catalizzatore, quale
Cu(OTf)2; infatti sono state eseguite contemporaneamente due prove, una senza
catalizzatore e una che ha previsto il suo utilizzo. Nella prova in bianco la reazione è rimasta ferma e anche dopo circa 20 ore a temperatura ambiente, la conversione è risultata pari a zero recuperando solo il materiale di partenza non reagito. Invece, usando sempre 3.0 equivalenti di reagente, ma aggiungendo 5 mol% di Cu(OTf)2 e 6 mol% di (±)-BINAP
dopo circa 16 ore a temperatura ambiente la reazione è risultata terminata ed è stato ottenuto un prodotto di apertura; il procedere della reazione viene osservato anche visivamente perchè da giallo chiaro la reazione è diventata verde chiaro.
Una volta isolato il prodotto principale tramite cromatografia usando come miscela eluente esano/acetato di etile (8/2), con una resa del 60%, dallo spettro 1H NMR e dal 2D-COSY è stato osservato un risultato diverso rispetto a quello aspettato; infatti il prodotto di apertura principale ottenuto non è stato quello in cui si è trasferito il fenile, come si pensava inizialmente ma l’addotto ottenuto è risultato essere quello in cui si è trasferito il metile; in particolar modo è stato ottenuto l’addotto 1,2-trans, 2.7a (Schema 2.3.5).
O NBoc 2.7 PhAlMe2 (3 eq.) Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% 0°C, CH2Cl2 Me ()-2.7a 60 % di resa N Boc H1 H2 OH
Schema 2.3.5
Reazione di apertura del biciclo [2.2.2] con PhAlMe2, ottenuto daPhLi
Il prodotto ottenuto 2.7a è di tipo 1,2 e questo è stato osservato dallo spettro 2D-COSY; la stereochimica trans è stata dimostrata facendo esperimenti di disaccoppiamento protonico. Per cui disaccoppiando il protone H2 dai protoni del metile, era stato osservato un
doppietto di tripletti a = 2.63 con J= 2.5 Hz e 10.3 Hz. Il valore di J pari a 10.3 Hz dimostra che c’è una relazione trans-diassale tra i due protoni H1 e H2.
Il fatto che venga trasferito il metile invece del fenile da un reagente di questo tipo è insolito. Infatti è noto che nei reagenti misti del tipo ArAlR2 viene trasferita
principalmente la porzione che presenta un maggiore carattere di ibridazione s.12 Per esempio, molto recentemente, utilizzando reagenti misti di questo tipo PhAlMe2 in reazioni
di apertura con un substrato quale un cicloesadienone prochirale, è stato ottenuto il trasferimento del fenile nell’addizione di tipo 1,4 e non è stata osservata traccia del
prodotto derivante dal trasferimento del metile.34 Diversamente da questo risultato, più
recentemente Alexakis, nel suo studio sulla desimmetrizzazione degli
ossabenzonorbornadieni, ha effettuato reazioni di apertura sfruttando anche questi reagenti misti come il PhAlEt2, in presenza di rame tiofene-2-carbossilato (CuTF) e liganti della
famiglia SimplePhos. In questi casi veniva osservato l’attacco competitivo dell’etile rispetto al fenile ottenendo un rapporto di 40:60 tra il prodotto di apertura in cui è stato trasferito il fenile e quello con l’etile, con prevalenza dell’attacco del gruppo alchilico. Anche la scelta successiva di usare il fenildiisobutile alluminio, PhAl(i-Bu)2, più
ingombrato, non ha migliorato la situazione perchè in questo caso il rapporto è diventato addirittura di 35:65, con prevalenza di attacco da parte dell’isobutile, senza ottenere nessun miglioramento nel trasferimento del fenile.35 Nel nostro caso, facendo reagire il cicloaddotto acilnitroso di nostro interesse 2.7 con il fenilalluminio dimetile, è stato ottenuto per l’86% il prodotto di apertura in cui è stato trasferito il metile e dallo spettro 1H NMR si vedono tracce del prodotto in cui è stato trasferito il fenile (14%) ma non è stato isolato in seguito alla cromatografia(Schema 2.3.5).
È degno di nota il fatto che cambiando reagente e utilizzando il PhAlEt2, preparato
sempre a partire dal fenillitio ma fatto reagire con il Et2AlCl, invece del PhAlMe2, non è
stato ottenuto nessun risultato; la reazione è stata condotta sempre in presenza di Cu(OTf)2
e (±)-BINAP, e dopo 60 ore, lasciandola andare a temperatura ambiente, non è stato osservato nessun tipo di conversione ed è stato recuperato solo il materiale di partenza (Schema 2.3.6). O NBoc 2.7 PhAlEt2 (3 eq.) Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% 0°C, CH2Cl2 materiale di partenza
Schema 2.3.6
Tentativo di apertura del biciclo [2.2.2] con PhAlEt2 ottenuto dalPhLi
Allo scopo di verificare se il Me2AlCl fosse in grado da solo di trasferire il metile, è
stata eseguita una reazione utilizzando solo questo reagente; anche in questo caso la reazione è stata catalizzata dal rame e come solvente è stato usato sempre il diclorometano. Dopo 16 ore dalla TLC analitica la reazione è sembrata terminata, ma il successivo spettro
1
una miscela complessa (Schema 2.3.7). Sono state eseguite anche due reazioni con reagenti di alchilalluminio; in primo luogo è stato usato il Me3Al per provare se fosse stato
possibile anche in questo caso il trasferimento del metile ed in caso positivo per verificare se il prodotto ottenuto fosse stato uguale a quello ottenuto con il PhAlMe2. E poi è stato
usato il Et3Al per verificare se poteva essere trasferito un etile a questi substrati, visto che
la reazione con il PhAlEt2 non aveva portato a nessun risultato. Nel caso del
trimetilalluminio ne sono stati utilizzati 2 equivalenti, senza variare le condizioni di reazione usate in precedenza e dopo 2 ore e mezzo la reazione è risultata terminata ma si sono ottenute delle miscele troppo complesse di prodotti ed è risultato perciò difficile effettuare una cromatografia. Stessa cosa anche con il trietilalluminio, di cui ne sono stati usati 3 equivalenti e in questo caso la reazione risulta terminata dopo 30 minuti, per dare però ancora delle miscele di prodotti troppo complesse (schema 2.3.7).
Me2AlCl(3 eq.) Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% 0°C, CH2Cl2 degradazione del cicloaddotto iniziale O NBoc 2.7 Me3Al (2 eq.) Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% 0°C, CH2Cl2 miscele complesse Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% Et3Al (3 eq.) 0°C, CH2Cl2 miscele complesse
Schema 2.3.7
Tentativi di alchilazione del cicloaddotto acilnitroso
Quindi a questo punto era evidente che per poter trasferire un gruppo metilico a questi sistemi, era necessario utilizzare un reagente misto ottenuto dallo scambio tra un organolitio e il dimetilalluminio cloruro. Tra le reazioni analizzate, l’unica che aveva dato un buon esito era stata quella che aveva permesso l’introduzione di un gruppo metilico sul sistema; era necessario però trovare un modo per poter trasferire anche un gruppo fenilico su tali sistemi, visto che questo era il nostro scopo originario.
Abbiamo quindi provato a variare il reagente organometallico primario per effettuare lo scambio, usando questa volta un reagente di Grignard invece dell’organolitio (Schema 2.3.8).
PhBr Mg Et2O anidro PhMgBr
Me2AlCl
CH2Cl2 0°C/40 min.
e poi t.a./ 20 min.
Schema 2.3.8
Reazione di scambio per la formazione di organoalani misti dal GrignardDa un esame della letteratura sembra che questo tipo di scambio non abbia precedenti. Solo in un caso è documentato l’uso di reagenti di Grignard in combinazione con Et2AlCl
utilizzato come acido di Lewis senza menzionare un’eventuale reazione di scambio.36 Con questa combinazione di reagenti, dopo 16 ore, lasciando andare a temperatura ambiente la reazione, è stata ottenuta una conversione completa, e anche in questo caso il procedere della reazione è stato osservato visivamente, la reazione infatti da gialla è diventata marrone scuro e poi rosso violacea. Ovviamente la reazione è sempre rame catalizzata e l’aggiunta del reattivo è stata effettuata a 0 °C, mentre poi la reazione è stata lasciata andare a temperatura ambiente per 16 ore. Dallo spettro 1H NMR del grezzo della reazione sono stati individuati due prodotti; però in seguito alla separazione cromatografica è stato possibile isolare un solo prodotto, quello principale, che risulta essere il composto arilato
2.7b senza il gruppo t-butossicarbonilico sull’azoto, con una resa del 55% (Schema 2.3.9).
O NBoc 2.7 Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% PhAlMe2 (3 eq.) 0°C, CH2Cl2 Ph N H ()-2.7b 55% OH H1 H2
Schema 2.3.9
Reazione di apertura del biciclo [2.2.2] con PhAlMe2 ottenuto dalGrignard
Anche la deprotezione del gruppo Boc risulta inattesa in queste condizioni di reazione. Infatti la deprotezione selettiva di ammidi e carbammati N-Boc protetti è possibile con 0.2 eq. di Mg(ClO4)2 in CH3CN a 50 °C, ma in queste condizioni le N-Boc ammine sono
inerti.37
Quindi sorprendentemente con questa combinazione di reagenti è stato possibile introdurre un fenile sul cicloaddotto per ottenere prodotti di apertura 1,2-trans. Il prodotto ottenuto
2.7b è di tipo 1,2 e questo è stato osservato dallo spettro 2D- COSY; la stereochimica trans è stata dimostrata dai valori delle costanti di accoppiamento; infatti per quanto riguarda H1,
allo spettro 1H NMR è presente un doppio doppio doppietto a =3.02 con J= 2.8 Hz, 8.0 Hz e 10.7 Hz, dimostrando così che sono presenti due correlazioni trans-diassali con i protoni vicinali quali H2 e H5. Per quanto riguarda il protone H2 è stato osservato un
doppio doppio doppietto a =3.45 con J= 2.8 Hz, 5.5 Hz e 8.0 Hz, confermando la relazione trans-diassale tra questo protone e H1.
Sulla scia di questi risultati interessanti è stata provata l’analoga reazione che però ha utilizzato il Et2AlCl (Schema 2.3.10).
PhBr Mg Et2O anidro PhMgBr
Et2AlCl
CH2Cl2 0°C/40 min.
e poi t.a./ 20 min.
Schema 2.3.10
Reazione di scambio per la formazione di organoalani misti dal GrignardIn questo caso con 3 equivalenti di reattivo è stato ottenuto solo il 50% di conversione dopo 16 ore a temperatura ambiente partendo da 0 °C, temperatura alla quale è stata effettuata l’addizione del reagente. Quindi è stata ripetuta la reazione usando 6 equivalenti di organoalluminio, sempre in presenza di Cu(OTf)2, e dopo 16 ore è stata raggiunta
completa conversione. È stata ottenuta una miscela di prodotti dove è stato possibile individuare con certezza l’addotto 1,2-trans 2.7b, senza il gruppo t-butossicarbonilico, derivante dal trasferimento del fenile (Schema 2.3.11).
O NBoc 2.7 Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% PhAlEt2 (6 eq.) 0°C, CH2Cl2 Ph N H ()-2.7b OH
Schema 2.3.11
Reazione di apertura del biciclo [2.2.2] con PhAlEt2 ottenuto dalGrignard
A questo punto è stato pensato di provare la reazione del cicloaddotto acilnitroso direttamente con PhMgBr, utilizzandone sempre 3 equivalenti e di impiegare ugualmente il diclorometano come solvente, in presenza di Cu(OTf)2 come catalizzatore. Dopo circa 16
ore la reazione ha una conversione completa; anche qui l’aggiunta del Grignard è stata eseguita a 0 °C e poi si è lasciata andare a temperatura ambiente. Tutti i tentativi effettuati
di separazione cromatografica hanno condotto a miscele di prodotti non chiaramente identificabili. L’esame 1H NMR mostra comunque la presenza di un prodotto arilato di natura simile a quello ottenuto come prodotto secondario mediante l’uso dell’eventuale reagente misto PhAlMe2 ottenuto da PhMgBr (Schema 2.3.9). Comunque la presenza di
altri segnali non ci permette un’attribuizione definitiva della struttura del composto principale ottenuto, anche se si può dire che il trasferimento del fenile sul cicloaddotto acilnitroso è stato in parte ottenuto. E’ stato poi provato un altro solvente oltre al diclorometano, quale il THF, lasciando invariate le altre condizioni di reazione, però anche dopo 60 ore la conversione è risultata circa del 20%.
L’attenzione è stata poi rivolta ad un altro reagente di Grignard quale il MeMgBr per verificare se poteva avvenire il trasferimento del metile; ma la reazione, svolta nelle medesime condizioni di quella che utilizza il PhMgBr, non ha avuto conversione; dopo 60 ore quello che si è recuperato è stato solo il cicloaddotto di partenza immodificato, con conversione pari a zero.
Un’ altra prova che è stata eseguita è stata quella che ha previsto l’utilizzo di un particolare reagente di organoalluminio, il trifenil(tetraidrofurano)alluminio, Ph3Al[THF];
anche questo particolare tipo di reagente è stato preparato di fresco in laboratorio seguendo una procedura di letteratura.38 Partendo dalla soluzione di PhMgBr in THF anidro è stato aggiunto AlCl3 precedentemente sublimato e solubilizzato anch’esso in THF anidro;
l’aggiunta è stata eseguita a 0 °C e la reazione è stata lasciata andare sotto agitazione per 12 ore a temperatura ambiente; successivamente, è stato portato a secco, sono stati aggiunti 7 ml di toluene anidro e si è filtrato su Buchner per ottenere una soluzione di Ph3Al[THF]
in toluene con molarità pari a 0.25. Quindi sono state eseguite due reazioni in parallelo, una senza Cu(OTf)2 e una con il rame, utilizzando 3 equivalenti di questo reattivo. In
entrambi i casi come solvente è stato usato CH2Cl2 e l’aggiunta è stata fatta a 0 °C. Però
entrambe le due reazioni non hanno portato a buoni risultati perchè dopo 60 ore circa, a temperatura ambiente la conversione è risultata pari a zero ed è stato recuperato solo il cicloaddotto iniziale 2.7, senza traccia del prodotto di apertura, neanche nel caso della reazione catalizzata dal rame (Schema 2.3.12).
O NBoc 2.7 materiale di partenza materiale di partenza Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% Ph3AlTHF3 eq.) 0°C, CH2Cl2 0°C, CH2Cl2 Ph3AlTHF3 eq.)
Schema 2.3.12
Tentativo di reazione di apertura con Ph3Al[THF]Sono stati anche effettuati tentativi di alchinilazione usando reagenti misti del tipo RCCAlMe2; in particolare è stato utilizzato l’alchinilalano derivante dal fenilacetilene
nella quantità di tre equivalenti e la reazione è stata eseguita in presenza di Cu(OTf)2. Però
non è stata osservata nessuna conversione e anche lasciando andare la reazione per 48 ore a temperatura ambiente non è stato ricavato il prodotto di apertura ma solo il cicloaddotto
2.7 immodificato (Schema 2.3.13). O NBoc 2.7 Cu(OTf)2, 5 mol% ()-BINAP, 6 mol% 0°C, CH2Cl2 Ph AlMe2(3 eq.) è stato recuperato solo il materiale di partenza
Schema 2.3.13
Tentativo di reazione di apertura con alchinilalani2.4 Reazioni di trasposizione di cicloaddotti acilnitroso
Nel nostro laboratorio sono state studiate, infine, le trasposizioni dei cicloaddotti acilnitroso, partendo inizialmente dal composto 3-aza-2-ossabiciclo[2.2.1]ept-5-ene, per arrivare a considerare la trasposizione del composto omologo superiore, analizzato già in precedenza nelle reazioni con gli organoalluminio, entrambi con un gruppo t-butossicarbonilico sull’azoto.
Partendo dall’osservazione che il composto 2,3-diazabiciclo-[2.2.1]ept-5-ene 2.11, in presenza di un acido di Lewis come Cu(OTf)2 nella quantità di 3 mol% e in presenza di 6
mol% di (±)-BINAP, dava il prodotto di trasposizione 2.11a mediante un meccanismo formalmente di tipo [3,4]-sigmatropico (Schema 2.4.1), si è pensato che anche il cicloaddotto acilnitroso derivante dal ciclopentadiene 2.12 potesse dare un analogo tipo di trasposizione in quelle condizioni.39
Per cui il biciclo in questione è stato trattato con 5 mol% di Cu(OTf)2 e con 6 mol% di
(±)-BINAP usando come solvente CH2Cl2 e in meno di un’ora è stato possibile ottenere il
prodotto di trasposizione 2.12a che si presenta come un solido giallo. Tale prodotto è risultato quindi un idrossammato 5,5-biciclico ottenuto mediante un meccanismo formalmente di tipo 3,4-sigmatropico, innescato dall’acido di Lewis che si coordina all’ossigeno, seguito poi dall’eliminazione dell’isobutene per dare l’idrossammato 2.12a, quale prodotto di trasposizione (Schema 2.4.1).
Cu(OTf)2 : 5 mol% ()-BINAP:6 mol% N O OH O H O N OH O 2.12a N N 2.11 Boc O O N O 2.12 O O THF anidro N O NHBoc O H O N H N O 2.11a Cu(OTf)2 : 5 mol% ()-BINAP:6 mol% CH2Cl2 Boc
Schema 2.4.1
Reazioni di trasposizione dei bicicli diaza e ossaza [2.2.1]Una volta ottenuto il riarrangiamento 2.12a in CH2Cl2, cambiando il solvente e usando
THF anidro, la reazione ha proceduto ugualmente bene per dare il medesimo prodotto di trasposizione 2.12a però è stata osservata la polimerizzazione del THF a differenza di quanto è stato riscontrato per il prodotto 2.11a. Quindi probabilmente questo processo non è del tutto concertato e non si può parlare di trasposizione sigmatropica. Questo tipo di reazione è stata comunque condotta sotto argon, in quanto l’acido di Lewis usato è molto igroscopico e quindi è stato più conveniente lavorare sempre sotto argon, sia per formare il complesso rame-BINAP, sia per effettuare la trasposizione stessa, in modo da avere delle condizioni ottimali.
Durante lo svolgimento del lavoro sperimentale, l’osservazione della letteratura ha evidenziato come Miller abbia riportato, all’inizio di questo anno, un analogo lavoro in cui ha ottenuto lo stesso prodotto di trasposizione usando quantità catalitiche di acido di Brønsted. In questo caso viene ipotizzato un meccanismo di tipo carbocationico innescato dall’acido usato, che risulta essere l’unico reattivo di questa reazione di trasposizione (Schema 2.4.2).22
O N O OtBu 2.12 H+ O N O OtBu H 2.13 N OH O OtBu 2.14 O N O OH H A 2.15 O N O OH 2.12a H+ O N O OtBu H + 2.16
Schema 2.4.2
Meccanismo di trasposizione di tipo carbocationico del biciclo [2.2.1]Successivamente sono state eseguite delle reazioni con il tentativo di elaborare l’idrossammato ottenuto 2.12a, il quale infatti risulta essere di natura allilica e quindi suscettibile di sostituzione allilica.
Era interessante per esempio farlo reagire con un Grignard come il MeMgBr, in presenza di Cu(OTf)2, come era stato fatto precedentemente nel caso del carbazato 2.11a;27 però la
reazione condotta a zero gradi in THF anidro ha portato a delle miscele troppo complesse (Schema 2.4.3) e anche cambiando solvente e usando diclorometano la situazione non è migliorata molto. O N OH O 2.12a MeMgBr (3 eq.) Cu(OTf)2 5 mol%
()-BINAP 6 mol% miscela complessa THF/0°C
Schema 2.4.3
Tentativo di apertura dell’idrossammato 5,5-biciclico con MeMgBrPer cui è stata fatta una prova “one pot” con il Grignard, partendo dal cicloaddotto 2.12, facendolo riarrangiare e poi aggiungendo 2 equivalenti di MeMgBr a -78°C; anche in questo caso si sono ottenute delle miscele troppo complesse e soprattutto non si è osservata
una conversione completa. Anche la reazione analoga, sempre “one pot”, con l’aggiunta però a 0 °C di vinilmagnesio bromuro non ha dato buoni risultati; dallo spettro 1H NMR del grezzo si sono intravisti dei segnali di un possibile attacco del Grignard, come il doppietto con =2.2, ma è impossibile ottenere prodotti puri mediante una separazione cromatografica. È stato provato così ad ottenere il cloridrato ma si è ottenuto un precipitato marrone il cui spettro 1H NMR non è risultato positivo (Schema 2.4.4).
Cu(OTf)2 5 mol% ()-BINAP 6 mol% CH2Cl2 MeMgBr (2 eq.) -78°C miscela complessa MgBr (3 eq.) 0°C miscela complessa 2.12 O NBoc
Schema 2.4.4
Tentativo di apertura “one pot” dell’idrossammato 5,5-biciclico con GrignardProbabilmente in una struttura come quella dell’idrossammato che è stato ottenuto, 2.12a, l’attacco del Grignard presenta molti inconvenienti, per la presenza dell’OH libero legato all’azoto con cui può reagire; quindi è stata tentata la trasformazione dell’ossidrile in un gruppo uscente migliore facendolo reagire con il pivaloil cloruro, ottenendo così un estere molto ingombrato scarsamente suscettibile di attacco da parte del Grignard. Sfortunatamente non è stato ottenuto il prodotto derivatizzato di nostro interesse (Schema 2.4.5). O N OH 2.12a C O Cl (1.5 eq.) piridina, 0 °C miscela complessa O
Schema 2.4.5
Tentativo di derivatizzazione dell’ossidrile dell’idrossammato 5,5-biciclicoQuindi il riarrangiamento che deriva dal cicloaddotto acilnitroso [2.2.1] è stato ottenuto facilmente, in condizioni facilmente riproducibili, ma per il momento, non è stato possibile un suo utilizzo successivo perchè i tentativi di alchilazione allilica con reagenti di Grignard hanno portato a miscele troppo complesse, soprattutto non purificabili; in alcuni casi si sono intravisti dei segnali di alchilazione dallo spettro 1H NMR, quali dei doppietti con intorno a 1 (nel caso del MeMgBr) e intorno a 2.2 (nel caso del vinilmagnesio bromuro), però è risultato impossibile ottenere prodotti puri mediante una separazione cromatografica. Anche una prova “one pot” usando il Me3Al non ha portato a nessun buon
risultato; infatti inizialmente è stata ottenuta una miscela complessa dove però dallo spettro
1
H NMR erano stati osservati segnali buoni come il doppietto a 1.0 ppm derivante dall’attacco del metile. Provando a derivatizzare la funzionalità ossidrilica libera con il TBDMSCl non è migliorata molto la situazione, anzi sono aumentate le impurezze andando a creare una miscela complessa di difficile purificazione (Schema 2.4.6).
Cu(OTf)2 5 mol% ()-BINAP 6 mol% CH2Cl2, 1h 2.12 Me3Al (3 eq.) 0 °C miscela complessa O NBoc
Schema 2.4.6
Tentativo di apertura “one pot” dell’idrossammato 5,5-biciclico con Me3AlL’attenzione poi è stata spostata sulla reazione di trasposizione del 3-aza-2-ossabiciclo[2.2.2]oct-5-ene. Sono state eseguite numerose prove per riuscire ad individuare la combinazione ottimale tra acido di Lewis e ligante e per scegliere il tipo di solvente più adatto e la temperatura più idonea. Inizialmente il prodotto di trasposizione aspettato era quello derivante da una trasposizione formalmente di tipo 3,4-sigmatropico come nel caso del biciclo omologo inferiore e quindi il nostro obiettivo era quello di ottenere un idrossammato 5,6-biciclico.
La prima reazione effettuata è stata quella in cui è stato usato come acido di Lewis Cu(OTf)2 ( 3 mol%) e come ligante il (±)-BINAP (6 mol%) in THF anidro; dopo 60
ore a temperatura ambiente non è stata osservata nessun tipo di conversione. Quindi, dopo aver aggiunto ancora 9 mol% di acido di Lewis e 6 mol% di ligante, la reazione è stata messa a riflusso a 60-65 °C e dopo 24 ore è stato osservato un cambiamento in seguito alla
formazione di un precipitato nero. É stato poi eseguito il trattamento della reazione, consistente nell’aggiunta di una soluzione acquosa satura di cloruro d’ammonio goccia a goccia e nell’estrazione con etere dietilico; dallo spettro 1H NMR del grezzo però non è stata osservata traccia del prodotto di trasposizione ma solo polimerizzazione del THF, che comunque poteva essere un indice di un’eventuale reazione coinvolgente carbocationi (Schema 2.4.7). Quindi sono state fatte altre prove ed è stato usato il diossano come solvente aumentando la quantità di sistema catalitico [(12 mol% di Cu(OTf)2 e 12 mol% di
(±)-BINAP)]; tale reazione è stata scaldata a 105 °C per 16 ore ed è stata ottenuta la completa degradazione del cicloaddotto iniziale. Invece effettuando la reazione in toluene e lasciando invariata la combinazione acido di Lewis-ligante, è stato recuperato solo il cicloaddotto immodificato sia a temperatura ambiente che a riflusso a 105 °C (Schema 2.4.7). O NBoc Cu(OTf)2 (3 mol%) )-BINAP(6 mol%) t.a./ 60 h THF anidro materiale di partenza Cu(OTf)2 (9 mol%) )-BINAP(6 mol%) 60-66 °C/ 24h no reazione, solo polimerizzazione del THF Cu(OTf)2 (12 mol%) )-BINAP(12 mol%) diossano 102-103 °C/ 16 h 2.7 degradazione Cu(OTf)2 (12 mol%) )-BINAP(12 mol%) toluene t.a./ 16 h materiale di partenza 105 °C/ 6 h e poi t.a./ 16 h materiale di partenza
Schema 2.4.7
Tentativi di reazione di trasposizione del cicloaddotto acilnitroso [2.2.2]Dopo questi tentativi deludenti, è stato deciso di utilizzare un equivalente di Cu(OTf)2 e di
usare come ligante la trifenilfosfina (2 equivalenti); il solvente scelto è stato il toluene e la reazione è stata lasciata andare a temperatura ambiente. Tale reazione è stata monitorata tramite TLC e dopo 16 ore è stato effettuato un prelievo, sul quale è stato eseguito un
è stato osservato un 46% di conversione con i segnali di un probabile prodotto di trasposizione 2.17 a = 3.75-3.94.(m, 1H), 4.67-4.79 (m, 1H), 5.83-5.94 (m, 1H), 6.15-6.24 (m, 1H) (Schema 2.4.8). N O O O O N OH O 2.7 Cu(OTf)2 ( 1 eq.) Ph3P ( 2 eq.) toluene t.a./16h 2.17 trattamento acquoso 46% di conversione
Schema 2.4.8
Reazione di trasposizione del biciclo [2.2.2] in presenza di Cu(OTf)2 ePh3P
Nel tentativo di aumentare la conversione è stato prolungato il tempo di reazione (72 ore) lasciando immodificati il solvente e la temperatura; è stata così ottenuta una conversione del 70%, con la formazione dell’idrossammato 5,6-biciclico 2.17. Sempre con lo scopo di aumentare la conversione le due reazioni precedenti sono state scaldate a riflusso a 100 °C per 5 ore ma è stata ottenuta solo completa degradazione del prodotto. Per ottenere una conversione completa è stato necessario condurre tale reazione a 70-80 °C per due ore ma insieme al composto 2.17 era ottenuto un altro composto incognito risultato non isolabile per cromatografia su gel di silice. Quindi è stato effettuato uno “screening” di solventi per migliorare la conversione a temperatura ambiente, migliorare la solubilità del complesso acido di Lewis-ligante che appare poco solubile e cercare di ottenere solo il prodotto di trasposizione e non una miscela di prodotti. In diclorometano dopo 60 ore a temperatura ambiente è stata ottenuta una conversione completa anche se il complesso del rame (II) con il ligante non è risultato essere molto solubile e venivano ottenuti due prodotti in rapporto 58/42; con il benzene è stata osservata una solubilità migliore del complesso rame-ligante e dopo 18 ore a temperatura ambiente è stato osservato solo il 27% di conversione però con l’ottenimento del solo prodotto di nostro interesse 2.17. Anche in questo caso nel tentativo di aumentare la conversione la reazione è stata scaldata a 80-85 °C per 18 ore causando però completa degradazione del prodotto.
Sono state tentate inoltre delle prove in cui non è stato utilizzato nessun tipo di ligante ma solo 1 equivalente di Cu(OTf)2 sia in benzene che toluene scaldando a 105 °C
stata ottenuta solo degradazione del materiale di partenza; anche scaldare a 40 °C per 18 ore in benzene ha causato totale degradazione e stessa cosa anche dimezzando la quantità di rame e scaldando a 80-85 °C per 2 ore. È stato anche provato un ligante diverso quale la tributil fosfina nella stessa quantità (due equivalenti) e dopo aver scaldato a 80-90 °C per 4 ore in toluene sono state ottenute solo miscele troppo complesse. Stessa cosa anche con il trietilfosfito dopo 36 ore a temperatura ambiente in benzene e anche con il (±)-BINAP (1 equivalente) dopo 18 ore a temperatura ambiente in benzene o diclorometano. É stato tentato poi un altro acido di Lewis, quale Sc(OTf)3 (1 equivalente) senza utilizzare nessun
ligante e dopo 60 ore a temperatura ambiente in diclorometano è stata ottenuta una miscela complessa; utilizzando il Cu(OTf) in toluene a temperatura ambiente dopo 72 ore la reazione è risultata ferma e scaldando a 100 °C è stata ottenuta completa degradazione. Quindi effettuare la reazione in assenza di ligante, solo con l’acido di Lewis, causa completa degradazione del cicloaddotto iniziale, e anche liganti diversi dalla trifenilfosfina così come acidi di Lewis diversi dal Cu(OTf)2 non hanno permesso di ottenere il prodotto
desiderato ma solo delle miscele complesse. Importante era da considerare anche che temperature troppo alte potevano danneggiare la reazione con degradazione del prodotto.
Arrivati a questo livello la combinazione migliore era risultata quella in cui il reagente era costituito da un equivalente di Cu(OTf)2 e da due equivalenti di trifenilfosfina,
utilizzando come solvente il benzene. In questo caso era stato osservato che il complesso rame-ligante inizialmente risultava essere di colore arancione scuro e non appena veniva introdotto il cicloaddotto 2.7, la soluzione diventava più chiara. Per quanto riguarda la temperatura, quella ottimale è risultata essere 40 °C con un tempo di reazione di circa 24 ore per ottenere una conversione del 100% (Schema 2.4.9).
O NBoc 2.7 Cu(OTf)2 ( 1 eq.) Ph3P ( 2 eq.) benzene 40 °C/ 24h O N OH O 2.17 (non isolato) conversione del 100%
Schema 2.4.9
Reazione di trasposizione del biciclo [2.2.2] in presenza di Cu(OTf)2 ePh3P
Dopo 24 ore è stato effettuato il trattamento della reazione consistente nell’aggiunta di una soluzione acquosa satura di cloruro d’ammonio goccia a goccia e nell’estrazione con etere,
diclorometano e acetato di etile, essendo il composto molto polare; è stato ottenuto il probabile prodotto di trasposizione quale l’idrossammato biciclico 2.17, confermato da analisi NMR e anche dall’analisi dell’infrarosso in cui si vede un picco a 1768 cm-1 e uno a 3504 cm-1. Dalla TLC analitica eseguita era evidente che nel corso della reazione si era formata trifenilfosfina ossido, TPPO, e anche dallo spettro 1H NMR erano presenti molti segnali a livello aromatico. Tale ossido si è formato in seguito all’ossidazione della trifenilfosfina da parte del Cu(OTf)2; infatti effettuando una prova in bianco utilizzando
solo Ph3P e Cu(OTf)2 in rapporto 2/1 in benzene è stata recuperata TPPO. Inoltre, da
un’analisi accurata dello spettro protonico è stato osservato che in realtà, anche usando il benzene, il composto di trasposizione di nostro interesse non è risultato il solo prodotto ottenuto ma si è formato per il 20% anche un altro prodotto con = 5.61-5.72 (m, 1H), 5.95-6.09 (m, 1H), che comunque non viene mai isolato.
Quindi successivamente è stato fatto il tentativo di purificare tale composto 2.17, in modo da poterne avere una resa isolata e poterlo così caratterizzare; però in seguito a separazione cromatografica utilizzando il normale gel di silice, con miscela eluente costituita da esano/acetato di etile (6/4), il composto è stato totalmente degradato e al termine della cromatografia non è stato recuperato niente se non TPPO. Consultando la letteratura è risultato chiaro come la silice normalmente usata per la cromatografia abbia la tendenza a trattenere prodotti analoghi a quello ottenuto nel nostro caso, cioè idrossammati liberi, per la presenza di tracce di ferro contenute in essa, impedendone il recupero; per questo motivo è risultato utile preparare un gel di silice libero da tracce di ferro, seguendo una procedura descritta in letteratura.40 Secondo questo procedimento è stata aggiunta la silice in una soluzione al 10% di metanolo in diclorometano, poi è stato filtrato su Buchner e questo gel è stato lavato con una soluzione di 3-idrossi-2-metil-4-pirone in metanolo, il quale risulta in grado di complessarsi al ferro per dare una colorazione arancione e infine di nuovo il gel è stato lavato ancora con la soluzione al 10% di metanolo in diclorometano per far eluire il complesso con il ferro. Per essere sicuri che tale complesso non fosse più presente è stato necessario che la soluzione eluente diventasse trasparente ed è stata anche fatta una TLC analitica per conferma e solo quando non è stata più osservata la sua presenza allora la silice è stata considerata libera dal ferro. É stata poi tirata a secco sotto vuoto per 18 ore e alla fine è stata ottenuta la silice di nostro interesse, completamente libera dal ferro in grado da essere utilizzata. Utilizzando quindi tale silice per la purificazione, il prodotto non è stato degradato ed è stato recuperato ma purtroppo sempre insieme alla TPPO e mai completamente puro.
Questo ha indotto a pensare che probabilmente la trifenilfosfina ossido formatasi rimanga sempre legata al prodotto ottenuto tramite un forte legame ad idrogeno con l’ossidrile libero dell’idrossammato, come mostrato nello Schema 2.4.10. Sono presenti numerosi esempi in letteratura che mostrano come la TPPO possa dare dei legami ad idrogeno molto forti, fungendo da accettore e interagendo con un donatore.41-42
O N O O 2.17 P O Ph Ph Ph H OPPh3
Schema 2.4.10
Complesso tra l’idrossammato 5,6-biciclico e la trifenilfosfina ossidoÉ stato anche effettuato uno spettro del 31P dal quale è stato ottenuto un segnale a -0.1, mentre invece la trifenilfosfina libera dà un picco a -5 e la trifenilfosfina ossido dà un picco a +27, dimostrando in questo modo che non sono presenti nè la trifenilfosfina nè la TPPO come tali nel prodotto; questo effetto di schermo può spiegare che la TPPO risulta complessata con il composto ottenuto riducendo le caratteristiche del doppio legame fosforo-ossigeno. Anche dall’analisi dell’infrarosso è stata ulteriormente validata questa ipotesi perchè lo spettro infrarosso della trifenilfosfina ossido libera presenta un segnale a 1190 cm-1 relativo allo stretching del legame P-O, mentre quello del prodotto ottenuto ci ha dato un segnale a 1165 cm-1; in questo modo può essere spiegata la presenza del legame tra TPPO e il composto 2.17 dal momento che ci sono degli spostamenti batocromici significativi nei segnali rispetto a quando la TPPO è libera, in accordo con quanto riportato in letteratura.43 Anche dallo spettro di massa ESI-MS otteniamo un picco a 589.07 che può essere spiegato ammettendo una combinazione tra il prodotto 2.17 e la TPPO che quindi in un certo modo può ulteriormente spiegare la possibilità di un complesso come indicato nello Schema 2.4.10.
Per poter ottenere il prodotto di trasposizione puro 2.17, libero dalla trifenilfosfina ossido è stato deciso di usare la trifenilfosfina supportata su materiale polimerico, quale un copolimero di stirene e divinilbenzene, effettuando così tale reazione in fase eterogenea piuttosto che in fase omogenea e lasciando le altre condizioni invariate. Dallo spettro 1H