TYROSINE KINASE RECEPTORS (RTKs)
Protein tyrosine kinases represent a wide family of homologous enzymes, both transmembranous and cytoplasmic, that catalyze the transfer of the γ-phosphate group from ATP to a hydroxy group of selected tyrosine residues in target protein substrates. Under physiological conditions, tyrosine phosphorylation represent a fundamental signal transduction mechanism that proceeds hierarchically in a ordered sequence of protein interactions, ensuring cross-talk between cells and regulating key aspects of cell life such as proliferation, differentiation, metabolism, and apoptosis.1
Approximately 90 TK receptors have been identified, 58 of which are transmembrane receptor type (eg: EGFR, Epidermal Growth Factor Receptor) and 32 the cytoplasmic “non receptor” type.
Transmembrane RTKs are floating protein in the double layer phospholipid membrane, all of which share a similar structure characterized by three regions: a cytosolic, an extracellular, an intramembrane portion. For their different structural features of domains approximately 20 RTK classes have been identified (Figure 1). The best characterized are:2
RTK class I (Epidermal Growth Factor (EGF) receptor family) RTK class II (Insulin receptor family)
RTK class III (Platelet-derived Growth factor (PDGF) receptor family) RTK class IV (Fibroblast Growth Factor (FGF) receptor family)
RTK class V (Vascular Endothelial Growth Factor (VEGF) receptors family) RTK class VI (Hepatocyte Growth Factor (HGF) receptor family)
RTK class VII (Tropomyosin-receptor-kinase (Trk) receptor family) RTK class XI (Angiopoeitin (TIE) receptor family)
Figure 1: Human Receptors Tyrosine Kinase2
The extracellular region exposes N-terminal domain to the interstitial fluid and presides over the recognition and interaction with the ligand.
Hydrophilic intracellular portion, C-terminal, is equipped with sites that govern signal transduction after ligand-receptor interaction. This portion is characterized by tyrosine residues in specific positions for any receptor and after interaction with the ligand undergo autophosphorylation. It’s the specific arrangement that influence the interaction with the cytoplasmic proteins and its phosphorylation, because those exposed on their surface domains enable to recognize specific phosphorylated aminoacid sequences of the catalytic domain of intracellular portion of the receptor.3
On the contrary, cytoplasmic receptors are devoid of the extracellular portion and generally allow the signal transduction within the cell only after the activation of transmembrane receptors.
Normally, TK receptors are encoded by protoncogenes, and when they undergo mutations or are overexpressed, become oncogenes and activate enzymes prematurely, leading to the appearance of tumors.
The basis of many cancers are anomalies involving two important classes of genes:
• tumour suppressor genes, that normally inhibit cell growth, may undergo mutations that are usually recessive with loss of function;
• protoncogenes, that stimulate the cell to advance its cell cycle, are affected by dominant mutations with gain of function, which leads to increased or uncontrolled activity of the product, or oncogenes.
So inactivated tumour suppressor genes lead to a lack of controller proteins preventing the excessive cell proliferation, while protoncogenes mutation leads to production of excessive amounts or of an excessively active form of the protein growth promoters.
In both cases the result is the same: an uncontrolled cell growth that degenerates into a tumour. The difference between oncogenic and normal genes may also be related to the replacement of a single amino acid, which often results in a radical change in protein function.
While normal cytoplasmic receptors encoded by protoncogenes are activated by transmembrane receptors, in turn activated by their ligand, those resulting from the respective oncogenes are constitutively endowed with enzymatic activity.
Under physiological conditions the kinase activity is very low, while it’s increased in tumor cells: in fact, the increase in intracellular phosphotyrosine, associated with increased cell proliferation, represents a real tumor marker status.
In the last years some RTK have become the targets of anti-tumor drugs, which, inhibiting the TK activity, could block the proliferation of cancer resulting from signal transduction.3
DESIGN OF TYROSINE KINASE INHIBITORS
The literature describes a number of heterocyclic derivatives as effective inhibitors of protein tyrosine kinases. They work by replacing the ATP molecule in connection with the specific receptor site thereby blocking, reversibly or irreversibly, the kinase activity of proteins. These molecules show chemically heterogeneous, especially for what concerns the compounds of natural origin. In contrast, inhibitors of synthetic origin are generally represented by derivatives of bicyclic nitrogen. Tyrosine kinase inhibitors that act at the intracellular domain are small molecules able to across cell membranes, that block or compete with the binding of ATP on the catalytic site, preventing the phosphorylation of tyrosine
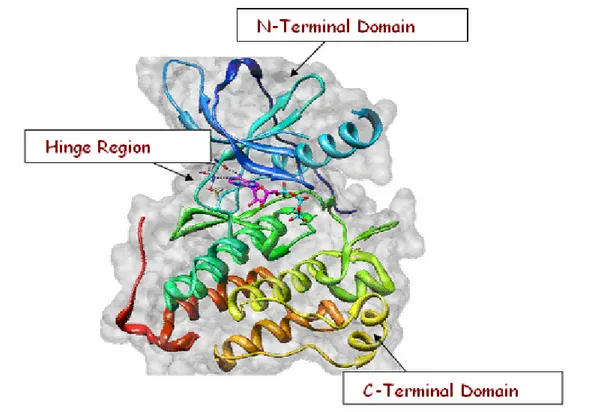
residues. The catalytic site consists of two kinase domains separated by a deep cleft, where the molecule of ATP is located. The smaller N-terminal domain contains the binding site of. The largest C-terminal domain is the region that contains the thyrosine residues that are able to realize autophosphorilation in order to make the receptor attivation possible (Figure 2).
Figure 2. Catalytic site of protein tyrosine kinase.
Conceptually, the binding site of ATP can be divided into five regions (Figure 3):
Adenine region: An hydrophobic region that accomodates the purine ring of ATP, which joins the lobes through the formation of two hydrogen bonds with the polypeptide chain, in which are involved the nitrogen atom N1 and the amino
group in the 6 position, which behave as donor and acceptor of hydrogen. This region is also called the "hinge" region. In addition to these polar interactions, the purine ring also forms non-polar interactions with hydrophobic residues located at the N- and C-terminal lobes. The region of adenine’s binding is not characterized
by large amino acid variability and therefore the interaction with this site doesn’t provide many useful elements for selectivity towards specific protein kinases.
Sugar Region: Where the riboside portion of ATP is located, whose 2'-OH group forms a hydrogen bond with a polar residue located at the beginning of the C-terminal lobe.
Phosphate region: This region accepts the triphosphate group and is mainly composed of a glycine-rich flexible loop, and an alpha helix structure that properly orients the phosphate group of ATP for catalysis. In most crystal structures of ATP-kinase, a hydrogen bond between the α and β phosphate groups of ATP and a residue of lysine has been found. The γ-phosphate group instead interacts with an arginine residue.
Buried region and solvent accessible region: ATP does not occupy these two regions. They constitute the main source of structural and sequence diversity among the members of the kinase superfamily. The buried region consists in a lipophilic pocket variable in shape and dimension, opposite to the sugar region.
The shape of solvent accessible region depends, however, on the presence or absence of a glycine residue, which can cause a conformational change of the protein between the hinge region and the initial portion of the C-terminal tail.15,16
Inside the ATP binding site, typical amino acid residues stand out as important structural elements, some of which are common features of the entire RTK family.17 Hydrogen bonds between the 6-NH2 and the backbone carbonyl of Gln
767 and between N1 and the backbone amide of Met 769 fix the purine onto the
extended coil stretch, that connects the N-terminal lobe with the C-terminal lobe of the enzyme. A third hydrogen bond between N7 of adenine and the hydroxyl
group of Thr 830 located on strand 8 preceding the activation loop may be less important because it is not conserved in all protein kinases.
Non-polar interactions occur between the purine and amino acids Val 702, Ala 719 and Leu 768, which form part of the conserved glycine-rich flap. The ribose and the triphosphate moieties extend towards the opening of the cleft where phosphorylation occurs. The ribose 2'-OH may form a hydrogen bond with Cys 773, either directly or indirectly through water. Since the early studies, the design of new ATP-competitive inhibitors selective towards one or a group of protein kinases turned out as a difficult goal, because of the similarities of the binding site inside the kinases family. Despite this, several potent inhibitors with an acceptable degree of selectivity have been reported in the literature.
INDIRECT AND DIRECT TARGETING OF ANGIOGENESIS PATHWAYS
The development of a vascular supply is essential not only for organ development and differentiation during embryogenesis, but also for wound healing and reproductive functions in the adult. Angiogenesis is also implicated in the pathogenesis of a variety of disorders: proliferative retinopathies, age-related macular degeneration, rheumatoid arthritis, and psoriasis and demonstrated to be a fundamental event in the process of tumour growth and metastatic dissemination. Hence, the molecular basis of tumour angiogenesis has been of keen interest in the field of cancer research. The vascular endothelial growth factor (VEGF) pathway is well established as one of the key regulators of this process.31
VASCULAR ENDOTHELIAL GROWTH FACTORS AND RECEPTORS VEGF/VEGFR
The VEGF family and its receptors are key regulators of angiogenesis and are highly specific for endothelial cells.32
VEGF was isolated and cloned in 1989 by Ferrara, Plouet and co-workers33; the predominant member of this family is VEGF-A, a 45 kDa glycoprotein composed of 165 amino acids34 that interacts with two transmembrane tyrosine kinase receptors: VEGFR-1 and VEGFR-2. VEGF-A also exists in other isoforms characterized by a different number of amino acids.
The VEGF-B protein trough its binding to VEGFR-1 exert a mitogenic effect on endothelial cells and moreover may link VEGF-A, increasing its action. VEGF-C and VEGF-D are structurally similar and may be considered as members of a subfamily (Figure 4).
Figure 4. VEFG structure
These proteins, interacting with VEGFR-2, induce a slight mitogenic effect on endothelial cells, while binding VEGFR-3 regulate lymphangiogenesis. At last,
VEGF-E, the most recently identified factor, is specific only for endothelial cells of endocrine glands.
Human VEGFR-1 and VEGFR-2 consist of 1338 and 1356 amino acids, respectively. They are structurally similar and formed by four region: the extracellular ligand domain, a transmembrane domain, an intracellular kinase domain that is comprised of a regulary domain and a catalytic domain tethered togheter by a hinge region, and downstream carboxy-terminal region. The kinase domains of VEGFR-1 and VEGFR-2 are the most conserved regions, with about 70% homology, while the carboxy-terminus rapresents the most differentiated region.35,36 Both VEGFR-1 and VEGFR-2 are located on surface of vascular endothelial cells, and they bind VEGF with high affinity. More recently, a third receptor has been discovered, VEGFR-3, which is closely related to VEGFR-1 and -2 and activated by VEGF, but limited in action to lymphangiogenesis; its expression is associated with the dissemination of tumor cells to regional limph nodes.37
VEGFR SIGNALLING39
The mechanism by which VEGFR-1 and VEGFR-2 receptors trasduce signal have been exstensively studied at the molecular level. The binding of VEGF to two proximal VEGFR-2 receptors causes their dimerization, following activation and autophosphorylation of the intracellular kinase domain, which catalyzed the phosphorylation of cytosolic substrate proteins and downstream cellular events (Figure 5).
Figure 5. Representation of vascular growth factors and their receptor interaction38
Among several tyrosine residues that have been shown to be phosphorylated, two major VEGF-dependant autophosphorylation sites have been identified in VEGFR-2; however, only the autophosphorylation of Tyr 1175 has been identified as crucial for VEGF-dependant endothelial cell proliferation.40
Activation of the MAPK pathway in response to VEGF has been observed in many types of endothelial cells. The PLC-γ-PKT pathway has also been implicated in the mitogen action of VEGF. VEGFR-1 interacts with the PLC-γ SH2 domain, inducing the phosphorylation and activation of PLC-γ leading to the hydrolysis of phosphatidylinositol-4,5-bisphosphate to diacylglycerol and inositol-1,4,5-trisphosphate. Inositol-1,4,5-trisphosphate is likely responsible for the increase in intracellular Ca2+ after VEGF stimulation, whereas diacyl-glycerol, in turn, activates PKC isoforms expressed in the target cells.
VEGF also activate PI3-K that activates Akt, a serine kinase involved in antiapoptotic signaling. Akt has also been reported to directly activate endothelian
nitric oxide synthase, suggesting that Akt may regulate the increased production of nitric oxide in response to VEGF stimulation. STATs are latent cytoplasmic transcription factors. STAT activation by the VEGFRs has been studied in transient transfection assay. All three receptors were shown to be strong activators of STAT3 and STAT5, whereas STAT1 was not activated by the VEGFRs. However, the role of this pathway in endothelian cell biology is unknown(Figure 6).
Figure 6. VEGFR pathway
Although very little is known about the specific signal transduction of VEGFR-3 in the lymphatic endothelium, mutations in VEGFR-VEGFR-3 have been linked with hereditary lymphedema, an autosomal dominant disorder of the lymphatic system.
Endothelial cell proliferation and survival in response to VEGF may require the association of VEGFR-2 with cell surface adhesive proteins. Activated VEGFR-2 was found in a complex with integrin αvβ3, an adhesion molecule specifically expressed on angiogenic endothelium; αvβ3 has been shown to be involved in the regulation of the cell cycle and the survival of endothelial cells. VE-cadherin, an endothelium-specific cell-adhesion protein, has also been implicated in molecular interactions with the VEGF.
VEGF/VEGFR IN ANGIOGENESIS41
Angiogenesis is the process that leads to the formation of new capillaries sprouting or splitting from pre-existing vessels, in fact, in order to grow beyond a size of 1-2 mm, tumors need new blood capillaries to create their own nutrient supply, to remove metabolic waste and to encourage metastasis formation.42 Hypoxic conditions in the centre of tumor mass stimulate the release of proangiogenic factors from tumor cells that are localized in the proximity of pre-existing blood vessels, and lead to the formation of new capillaries around the tumor mass.15
VEGF and its receptors have a central role in promoting cancer and a strategic position in the regulation of angiogenesis; although the precise role of VEGFR-1 function is still emerging, in recent years several studies have suggested that this receptor may play both negative and positive role in angiogenesis. In fact, VEGFR-1 has negative function, probably trapping VEGF-A in the embryo, while showing a positive role in adulthood in a tyrosine kinase-dependent manner. VEGFR-1 acts primarily as a decoy receptor, modulating the availability of VEGF for VEGFR-2, which is the principal receptor for VEGF signalling.43
Elevated VEGF expression, due to abnormal and inefficient tumor vasculature, with many blind ends, and it is difficult for convetional drug molecules to gain access to the tumor tissue; moreover, drug penetration is diminished by the high interstitial pressure of many tumor.44 Anti-VEGF therapy may produce vascular
normalization, resulting in a decrease in vascular volume and interstitial pressure, leading to enhanced delivery of cytotoxic therapeutic agents.
So blockade of angiogenesis is one of the more attractive approaches for the treatment of both solid tumors and haematological malignancies.45
In human disease, VEGFR-2 was shown to have a major role in tumor angiogenesis and diabetic retinopathy, while VEGFR-1-dependent signalling was shown to play a role in angiogenesis of certain tumors, the progression of rheumatoid arthritis, and aterosclerosis.46,47
ANTI-VEGFR AGENTS
Therapeutic strategies targeted at blocking VEGF or its receptor signalling systems are an attractive approach for the treatment of different diseases, primarily tumors. Therapies molecularly targeted at angiogenesis are supposed to be less toxic for chronic administration, in comparison with conventional treatments such as chemotherapy, but it is thought that anti-VEGF agents alone will be not sufficient for cancer monotherapy; in fact, such molecules are often evaluated in clinical trials in combination with chemotherapy.48
Agents currently in development against VEGF or its receptors include neutralizing monoclonal antibodies (such as bevacizumab), and small molecules TK-inhibitors.
MONOCLONAL ANTIBODY
The most advanced approach to VEGF-dependent angiogenesis inhibition is the humanized monoclonal antibody directed against VEGF-A, bevacizumab, by Genentech.34,49 Bevacizumab prevents the binding of all VEGF isoforms to all VEGFRs; in preclinical animal model, it blocks the growth of a variety of human
tumor xenografts. The results obtained in clinical trials on different malignancies such as: combination with 5-fluorouracil- and irinotecan-based chemotherapy in metastatic colorectal cancer, and association with paclitaxel in metastatic breast cancer, led to its approval by the FDA as a first-line treatment in combination with chemotherapy for metastatic colorectal cancer.
This antibody is generally well tolerated, even though toxicities such as gastrointestinal perforation and tromboembolic complications have been observed in a small percentage of cases.50
A different anti-VEGF-A monoclonal antibody, ranibizumab, has been developed by Genentech for the treatment of the age-related macular degeneration (AMD), charaterized by neovascularisation; this new agent maintains or improves vision and was approved by the FDA in June 2006.51
Aptamers are nucleic acids that recognize their target with high affinity and specificity.
Macugen® is a modified RNA developed by Eyetech Pharmaceuticals and Pfizer that inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF-A with extremely high affinity (Kd = 50 pM), and was approved
by USA (United States) FDA in December 2004 for the treatment of AMD.52 AngiozymeTM, by Chiron Corporation/Rybozyme Pharmaceuticals, is a stabilized rybozime against the pre-mRNA of the VEGF-1 gene that reduces the expression of VEGFR-1 through targeted cleavage of VEGFR-1 mRNA in cells, and the truncated soluble forms in plasma.53 Recent phase I clinical trials showed that Angiozyme is well tolerated, with minimal toxicities and good bioavailability. Phase II trials for specific tumor types are ongoing to further asses the biological and clinical activity of Angiozyme.54
A different anti-angiogenic approach is based on the use of a soluble recombinant decoy receptor, called VEGF Trap, a protein constructed from the VEGF receptor-binding domain linked to an immunoglobulin. It posses a high affinity for VEGF and demonstrated marked efficacy in inhibiting angiogenesis and the growth of tumors in preclinical animal models. Currently, it is being studied in phase I clinical trials in humans with advanced solid malignancies55 and for treatment of patients with intraocular neovascularisation due to AMD.56
The negative aspects of both anti-VEGF and anti-VEGFR based therapies are the high cost of manufacturing and the necessity for parental application.48
SMALL MOLECULES TK INHIBITORS
Another important class of angiogenesis inhibitors currently in development is that of the small-molecule tyrosine kinase inhibitors. Generally these compounds have dual activity inhibiting both VEGFR-1 and VEGFR-2 in vascular endothelial cells.
Among ATP site-directed inhibitors of VEGFR, indolin-2-ones, identified in 1993, represent an important class of anti-angiogenesis agents.57,58 The oxindole derivative sunitinib (SU11248) is an inhibitor of VEGFR-1, VEGFR-2, VEGFR-3 (IC50 values 15, 38, 30 nM respectively), but also inhibits PDGFRs
(Platelet-Derived Growth Factor Receptors), Kit, Flt3, RET (REarranged during Transfection), and CSF-1R (Colony Stimulating Factor 1 Receptor) with similar efficacy, and is approved multinationally for the treatment of advanced renal cell carcinoma (RCC) and imatinib-resistant or imatinib-intolerant gastrointestinal stromal tumour. In January 2006, it was approved by FDA for the treatment of gastrointestinal and kidney cancer.
F N H O N H O NH NEt 2 Sunitinib
Sunitinib is characterized by a 5-fluoro-substituted indolinone structure, bearing a diethylaminoethyl group that confer good solubility. The co-crystal X-ray structure of the catalytic domain of the FGF (Fibroblast Growth Factor)
receptor with several oxindoles suggests that the compound bind in the ATP pocket with the indolin-2-one core participating in key H-bond donor/acceptor capacities with the carbonyl of Glu 915 and the NH Cys 917, typical residues of the hinge region of VEGFR-2. Other key SAR features include the preference of a Z methylidene geometry that can be enforced by a heteroaromatic ring capable of participating in intramolecular H-bond interaction with the indolin-2-one core.57,58 Another important class of angiogenic inhibitors is that of anilinophthalazines disclosed by Novartis. These compounds were selective for human VEGFR.
The anilinophtalazine derivative valatanib52 (PTK787/ZK222584) is one of the most potent and selective first-generation VEGFR kinase inhibitors, with IC50
values of 110, 43, 195 nM against VEGFR-1, VEGFR-2, VEGFR-3, respectively. It also inhibits other kinases including PDGFR-β and c-Kit, but at higher concentrations. This compound is currently in phase III clinical trials for metastatic colorectal cancer.
Valatanib was docked in a model of ATP binding site of VEGFR-2 constructed using the available X-ray structures of the domain of FGF receptor 1.
According to Authors’ hypothesis, valatanib does not form direct hydrogen bonds with the peptide backbone of the hinge region as does ATP and many reported kinase inhibitors, but rather occupies the hydrophobic regions of the binding site. N N HN Cl N COOH COOH Valatanib
The aniline moiety is located in a hydrophobic pocket, while the phthalazine bicycle also makes hydrophobic contacts with other amino acids. Although none direct hydrogen bond with the hinge region is established, the aniline NH group forms water-mediated hydrogen bonds with Glu 915 and Cys 917 of the hinge region of VEGFR, and the inhibitor pyridil nitrogen is assumed to form a hydrogen bond with Lys 1060, a residue of the kinase activation loop.52
In the context of the same structural class of gefitinib (anilinoquinazolines) appropriate modifications of the substituents have led to the identification of vandetanib, (AstraZeneca) a novel drug, that joins two actions with a dual pharmacological profile, is an orally active tyrosine kinase inhibitor that exhibits potent agonist activity toward VEGFR-2 and, to a lesser extent, to EGFR.
N N NH Br F O O N CH3 Vandetanib
The clinical development of vandetanib is being progressed, as in vivo tests have demonstrated the ability to inhibit, in a dose-dependent way, the growth of a wide range of cancers. Its administration in mice leads to inhibition of VEGF signal, of angiogenesis, of neuro-vascularization induced by the tumour, and the tumour growth. Vandetanib is undergoing phase I clinical testing in patients who have advanced solid tumours.39,40
Pazopanib
Pazopanib (5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methyl-amino]-2-pyrimidinyl] amino]-2-methyl-benzensulfonamide) is an orally bioavailable inhibitor of VEGFR, PDGFR, c-Kit tyrosine kinases. Its chemical structure is shown in the above picture. It inhibits angiogenesis in mouse corneal micropocket and matrigel plug assays; murine models demonstrate antitumor acitivity against a variety of tumor xenograft models including lung, renal, melanoma and prostate.