di espressione genica
L’uso di un procedimento Bayesiano per l’analisi dei dati generati con microarray, illustrato nel precedente capitolo, non considera la struttura dell’esperimento, cosicché gli effetti del trattamento in esame possono essere confusi con alcune delle sorgenti di variabilità tipicamente presenti su questo tipo di dati.
Dal momento in cui queste fonti di variabilità vengono identificate, si può pensare di misurare il loro effetto sul dato o, come si dice in termini statistici, cercare di quantificare la varianza “spiegata” da tali sorgenti, in relazione alla varianza totale dell’insieme di dati, nel tentativo di eliminarla.
Questo può essere ottenuto disegnando in maniera opportuna l’esperimento, in modo da raccogliere un’adeguata quantità di misure e contribuire a monitorare gli effetti di alcune delle fonti di variabilità. Ad un disegno sperimentale idoneo si deve aggiungere la capacità di realizzare un modello descrittivo dei dati, che renda possibile la diversificazione dell’effetto del trattamento di interesse da quelli legati a fonti di variabilità indesiderate.
Kerr e Churchill (Kerr et al., 2001a, Kerr & Churchill, 2001b e Kerr et al., 2002) sono stati i primi a studiare le potenziali sorgenti di variabilità in esperimenti di microarray e ad incorporarle in un modello additivo attraverso il metodo statistico di analisi della varianza a più fattori ANOVA (ANalysis Of VAriance).
In questo capitolo verranno illustrate le principali fonti di variabilità dei dati, per poi metterle in relazione con diverse tipologie di disegno sperimentale e con diversi modelli proposti per la loro quantificazione.
5.1 Fonti di variabilità sui dati di espressione genica
Le sorgenti di variabilità che si hanno per i dati di espressione genica possono includere sia fattori sperimentali sia rumore casuale o “random”; il metodo dell’analisi della varianza cerca di quantificare tale variabilità e di esaminare se sia statisticamente comparabile con quella attribuita alle sorgenti “random”.
Si supponga, per esempio, di trattare con un farmaco un gruppo di cavie e di confrontare mediante microarray i campioni ottenuti dopo il trattamento con quelli di un gruppo di controllo non trattato: l’analisi della varianza consente di esaminare le differenze rilevate fra i gruppi, dividendole in effetto del trattamento ed effetto dovuto ai fattori sperimentali che si abbattono sull’espressione differenziale.
Il processo è concettualmente simile alla normalizzazione, poiché si tratta di eliminare, anche in questo caso, gli errori sistematici che contribuiscono a corrompere il dato di espressione, ma, in più, l’analisi della varianza permette di rilevare direttamente l’espressione differenziale sui dati ripuliti.
Il tipo più semplice di esperimento microarray consiste nel cercare di misurare i cambiamenti nell’espressione genica in campioni che differiscono per un unico fattore, ad esempio la somministrazione di un farmaco.
Si indicano con il termine varietà tutte le categorie del fattore di interesse: nel caso della somministrazione del farmaco le due categorie saranno trattato e non-trattato (controllo).
Nel loro lavoro Kerr e Churchill (Kerr e Churchill ,2001b) hanno messo in evidenza che la variabilità può essere dovuta essenzialmente a quattro sorgenti principali:
Effetto Array (A);
Effetto Varietà o Trattamento (V o T); Effetto Gene (G).
Sotto il nome di effetto “Array” vengono classificate le variazioni di segnale fra array, mediate su tutti i geni, i fluorocromi e i trattamenti. Una problematica frequente che può condurre alla rilevazione di questo effetto può essere la non uniformità del processo di ibridazione del campione marcato.
L’effetto “Fluorocromo” o “Dye” misura le differenze intrinseche di emissione dei due fluorocromi. Nel caso di microarray “dual-color”, dove si fa uso di due cianine per marcare il campione, si può facilmente rilevare sin dalla fase di acquisizione dell’immagine che il fluorocromo Cy5 (rosso) ha un’efficienza di emissione più bassa rispetto al fluorocromo Cy3 (verde).
Questo comportamento è dovuto ad una differente sensibilità dei due fluorocromi rispetto all’eccitazione indotta con il laser e si ripercuote sul bilanciamento del segnale nei due canali.
L’effetto “Varietà” si riscontra quando le categorie del fattore di interesse presentano livelli di espressione diversi, dovuti a fattori non riconducibili al trattamento. Questo potrebbe verificarsi, nel caso della somministrazione del farmaco, se venisse preso come controllo un tessuto diverso da quello trattato: l’espressione differenziale sarebbe riconducibile anche alle differenze fra i due tessuti.
L’effetto “Gene” si può verificare quando alcuni geni mostrano una diversa risposta all’ibridazione; ciò si manifesta con la generazione di una variazione del segnale, di intensità indipendente dalla quantità di campione ibridizzato.
Gli effetti descritti sono soltanto i fattori principali. Con quattro fattori principali è possibile considerare 24=16 effetti complessivi ripartiti
in:
quattro effetti principali, sei interazioni a due fattori,
quattro interazioni a tre fattori, una interazione a quattro fattori.
Anche nel caso dei fattori di interazione è possibile identificare alcune cause alla base dell’insorgenza degli effetti combinati.
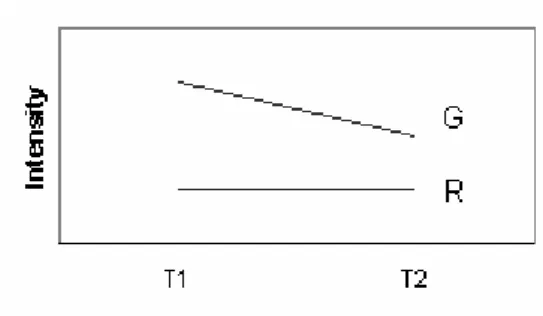
L’effetto combinato del fluorocromo e del trattamento (DV) si può ricondurre ad una differente efficienza di incorporazione del marcatore nei campioni di cDNA da analizzare. Si supponga, per esempio, che il fluorocromo verde presenti una differente efficienza di incorporazione rispetto a due diverse varietà, mentre il fluorocromo rosso si comporti in maniera equivalente con entrambe. Questa situazione è schematizzata nella figura 5.1, dove la linea orizzontale è indicativa del comportamento costante del fluorocromo rosso, mentre quella obliqua evidenzia la differenza di incorporazione del fluorocromo verde sui due campioni T1 e
T2.
Figura 5.1: Schematizzazione dell’effetto combinato DV
In un esperimento in cui venissero realizzate due ibridizzazioni su due microarray con marcatura invertita dei campioni, sarebbero rilevate delle differenze in espressione non imputabili ad un effetto del trattamento, ma attribuibili al comportamento non omogeneo del fluorocromo verde.
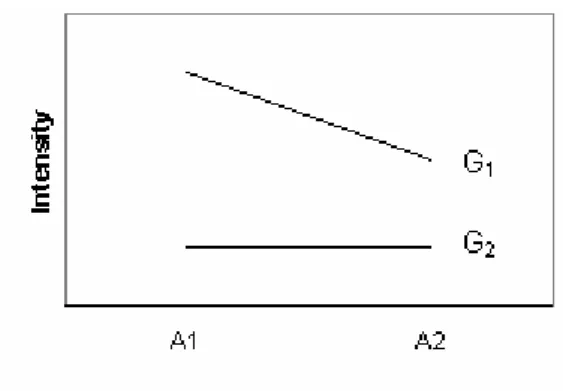
L’ effetto di interazione fra l’array e il gene (AG) si può verificare se lo stesso gene su diversi array è presente con una concentrazione diversa di sonde di cDNA disponibili per l’ibridazione. Questo effetto viene spesso denominato “Spot-effect” perché dipende fortemente dal processo di deposizione delle sonde sul microarray e, per eliminarlo, si possono seguire due strategie:
considerare ogni spot come un’unità a sé, anche se così facendo si perdono le informazioni globali sul gene (per esempio le repliche sperimentali);
cercare di ricostruire un modello statistico della densità dello spot o delle proprietà della punta di deposizione.
Figura 5.2: Schematizzazione dell’effetto combinato AG
Generalmente è difficile riuscire a modellare lo “Spot-effect”, per cui si tende a migliorare la misura relativa ad ogni spot aumentando il numero delle repliche sperimentali, in modo da avere più dati a disposizione per l’interpolazione del loro modello.
L’effetto Dye-Gene (DG) si realizza se ci sono interazioni gene-specifiche fra il gene e il fluorocromo; questo tipo di effetto è abbastanza raro e una sua schematizzazione è mostrata in figura 5.3.
Figura 5.3: Schematizzazione dell’effetto combinato DG
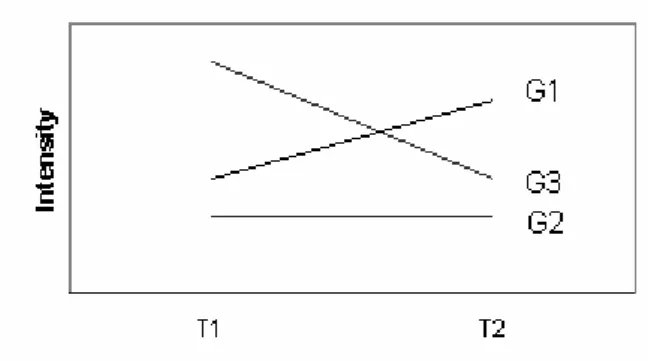
L’interazione fra il trattamento e il gene (VG) si realizza quando un gene mostra espressione differenziale nelle diverse varietà ibridizzate sul microarray e questa differenza è riconducibile proprio al trattamento. La quantificazione di questo effetto è l’obiettivo principale dell’esperimento e la sua schematizzazione è mostrata in figura 5.4.
Figura 5.4: Schematizzazione dell’effetto combinato VG
Le interazioni AD, AT e ADT non sono gene-specifiche ed è difficile connettere ognuno di questi effetti combinati ai processi che si realizzano sui microarray.
Le interazioni ADG, ATG, DTG e ADTG sono invece gene-specifiche. La presenza di tali interazioni dimostrerebbe che ci sono variazioni attribuibili a particolari coppie array-fluorocromo, array-trattamento, fluorocromo-trattamento o combinazioni di array-fluorocromo-trattamento in relazione ad un particolare gene. Queste interazioni di ordine superiore al secondo sono difficili da collegare a processi fisici o chimici che si realizzano nei microarray e generalmente si assume che non si verifichino.
Esistono diversi modelli per quantificare le fonti di variabilità illustrate; la possibilità di valutare tutti gli effetti che compaiono in essi è consentita, oltre che da un adeguato numero di dati sperimentali, anche da un’opportuna pianificazione del modello statistico e del disegno dell’esperimento.
5.2 Modelli additivi ANOVA per l’analisi dell’espressione
La formulazione di un modello dei dati di intensità è legata non soltanto all’identificazione delle sorgenti di variabilità, ma anche ad un’adeguata caratterizzazione statistica degli effetti.
E’ possibile distinguere fra effetti fissi, statisticamente modellabili con variabili aleatorie indipendenti ed identicamente distribuite, ed effetti “random”, che presentano le caratteristiche di variabili aleatorie generate da processi tipicamente utilizzati per descrivere l’errore non sistematico di misura o errore “random”.
Sulla base della descrizione statistica degli effetti, vengono definiti modelli “random”, in cui tutti gli effetti coinvolti vengono considerati casuali, modelli misti nei quali viene individuata una parziale componente sistematica, e modelli fissi in cui tutti gli effetti sono sistematici a meno dell’errore di misura.
Per poter utilizzare questi modelli è necessario operare delle trasformazioni sui dati in modo da renderli idonei per la successiva elaborazione.
La trasformazione più frequentemente utilizzata sui dati grezzi di intensità è quella logaritmica, al fine di generare un modello additivo piuttosto che moltiplicativo. Utilizzando questa scala si indica con yijkg il logaritmo dell’intensità della fluorescenza misurata per l’array i, il fluorocromo j, la varietà k e il gene g.
Assumendo che lo stesso insieme di geni sia depositato su ogni array dell’esperimento, si ha a disposizione un insieme completo di osservazioni per ogni combinazione di array, fluorocromo e varietà: in conseguenza di ciò l’effetto gene e le sue combinazioni sono ortogonali, ossia indipendenti, a tutti gli altri effetti e l’esperimento si dice bilanciato.
Questo porta a suddividere gli effetti in due gruppi: effetti globali, che coinvolgono solo gli effetti principali A, D e V, ed effetti gene-specifici, che coinvolgono G. L’effetto di interesse VG è, quindi, gene-specifico.
Se gli effetti non sono ortogonali, ossia la quantificazione di uno fornisce informazioni ridotte o complete anche sull’altro, si parla di confusione dell’informazione, ossia di mascheramento parziale o totale degli effetti.
5.2.1 Modelli additivi misti
La scelta più generale operabile quando non vi sono informazioni per caratterizzare gli effetti come variabili indipendenti ed identicamente distribuite è considerarli tutti come effetti casuali e generare un modello “random”. Malgrado questa scelta garantisca la completa generalizzabilità del modello, essa ha lo svantaggio di essere estremamente onerosa dal punto di vista della quantificazione di questi effetti.
Una valutazione più approfondita degli effetti e dei processi fisici che essi cercano di modellare può portare alla definizione di un modello misto, nel quale non tutte le sorgenti di variabilità vengono considerate casuali.
Questo tipo di modello per i dati di espressione genica è stato introdotto per la prima volta nel lavoro di Wolfinger (Wolfinger et al. (2001)) e si sviluppa in due stadi: un primo stadio di normalizzazione, cui segue un secondo di calcolo degli effetti gene-specifici.
La normalizzazione serve ad eliminare il contributo degli effetti principali globali ed essa viene realizzata attraverso la definizione di un sotto-modello per la loro stima, diverso a seconda degli effetti globali da stimare.
Un modello completo, in cui vengono presi in considerazione tutti gli effetti globali e gene-specifici, può avere la seguente forma:
y
ijkg= μ + A
i+ D
j+ V
k+ G
g+ (VG)
kg+ (AG)
ig+ (DG)
jg+ ε
ijkgdove:
il termine μ si riferisce all’intensità media totale calcolata su tutti i geni di tutti gli array;
il termine ε rappresenta l’errore “random”; questo è una quantità aleatoria che si distribuisce secondo una variabile di Fisher con media nulla e varianza σ2 e rappresenta tutta
l’informazione che non si riesce a modellare.
Supponendo di aver utilizzato un disegno dell’esperimento che mascheri l’effetto della varietà con quello combinato dell’array e del fluorocromo (AD), come accade quando si inverte la marcatura dei due
campioni confrontati nell’ibridizzazione su due vetrini, il modello parziale di normalizzazione ha la seguente forma:
y
ijkg= μ + A
i+ D
j+ AD
k+ x
ijkgdove:
xijkg rappresenta il termine dei residui del modello, che, per ipotesi, hanno distribuzione normale.
La stima degli effetti globali A, D e AD e della media totale μ serve a “centrare” la distribuzione dei dati rispetto a questi effetti e assolve, quindi, lo stesso compito della normalizzazione globale illustrata nel capitolo 2.
In questo modo i dati grezzi di intensità vengono “ripuliti” senza ricorrere a tecniche di normalizzazione, ma solo attraverso la quantificazione di queste sorgenti di variabilità.
Purtroppo, come evidenziato nel capitolo 2, spesso gli errori sistematici riscontrabili sui dati sono non lineari rispetto all’intensità, per cui è necessario fare una correzione che sia intensità-dipendente. A tale scopo, prima di effettuare la successiva elaborazione per identificare i geni differenzialmente espressi, è necessario operare una correzione del colore, realizzabile tramite una normalizzazione LO(W)ESS.
I residui xijkg del modello del primo stadio diventano i dati del
modello del secondo stadio, che è, invece, un modello gene-specifico, come deducibile dal pedice g di ogni effetto, e serve a generare la stima dell’effetto di interesse VG e degli altri effetti combinati gene-specifici. Questo avviene grazie all’interpolazione ai minimi quadrati della formula che schematizza il secondo stadio del modello:
x
ijkg= μ
g+ (VG)
kg+ (AG)
ig+ (DG)
jg+ ε
ijkgdove:
il termine μ g si riferisce all’intensità media totale calcolata
sul gene che si sta considerando.
Alcuni effetti possono essere considerati casuali in base alla considerazione che non vi è un’effettiva certezza che essi si abbattano in maniera sistematica su tutti i dati.
E’ questo il caso dell’effetto AG che, verosimilmente, potrebbe avere un’entità diversa sullo stesso gene in array differenti. Dal punto di vista statistico l’effetto AG viene, quindi, trattato come se fosse una variabile aleatoria con distribuzione normale a media nulla e il modello così definito viene detto misto.
5.2.2 Modelli additivi fissi
I modelli additivi fissi proposti da Kerr e Churchill ereditano, dal modello misto appena illustrato, la formulazione a due stadi; anche in questo caso, infatti, lo stadio gene-specifico di valutazione dell’effetto VG viene preceduto da quello di normalizzazione.
Un modello ANOVA semplice include solo i fattori principali e l’effetto VG e può essere schematizzato con la formula seguente:
Un modello più plausibile aggiunge le variazioni spot a spot, includendo l’effetto combinato AG, e la sua struttura è descritta dalla formula seguente:
y
ijkg= μ + A
i+ D
j+ V
k+ G
g+ (VG)
kg+ (AG)
ig+ ε
ijkgUn’altra possibilità è quella di aggiungere l’interazione fluorocromo-gene (DG), fluorocromo-generando così il modello completo già illustrato nel paragrafo precedente.
Questi modelli sono relativi a situazioni in cui ogni gene è presente in una sola copia per array. Se i geni sono depositati in r copie per ogni array, la varianza del termine di interesse VG decrementa di un fattore 1/r ed è possibile inserire un “effetto replica S” nel modello, per catturare le differenze fra gli spot duplicati all’interno dell’array, così come indicato nella formula:
y
ijkgr= μ + A
i+ D
j+ V
k+ G
g+ (VG)
kg+ (AG)
ig+ (DG)
jg+
S
r(ig)+ + ε
ijkgrPoiché si assume che tutti gli effetti presenti nei tre modelli siano variabili aleatorie indipendenti e identicamente distribuite con media nulla, a meno del termine di errore “random”, si parla di modello fisso.
Le stime degli effetti del modello sono realizzate attraverso un’interpolazione ai minimi quadrati, minimizzando la quantità:
2 r(ig) jg ig kg g k j i ijkgr
A
D
V
G
(VG)
(AG)
(DG)
S
]
[y
∑
−
−
−
−
−
−
−
−
−
ijkgrμ
con i vincoli che:
∑
=∑
=∑
=∑
=∑
=∑
=∑
=∑
= g ig i ig g kg kg g k j i D V G AG AG VG VG A ( ) ( ) ( ) ( ) k∑
=
∑
=
∑
=
=
g(
DG
)
jg j(
DG
)
jg rS
r(ig)0
L’effetto di interesse VGkg per ogni gene g e trattamento k è ottenuto
attraverso la stima ai minimi quadrati :
... . ... .. .. . ..
t
t
t
t
VG
kg=
kg−
k−
g+
dove t rappresenta il logaritmo delle intensità e ogni punto dei pedici identifica il termine sul quale è stata eseguita la media.
5.3 “Nested” F-test e determinazione dei geni
differenzialmente espressi
Una volta effettuata l’interpolazione ai minimi quadrati dei parametri del modello si può passare alla determinazione dei geni differenzialmente espressi.
Con questa tecnica di analisi dei dati di intensità, si decide se un gene è differenzialmente espresso realizzando un test delle ipotesi sul modello che è stato interpolato.
Seguendo lo schema classico del test delle ipotesi si definiscono:
ipotesi nulla o modello nullo: il trattamento non ha effetto sul gene e (VG)1g=…=(VG)kg=0 nel modello;
ipotesi alternativa o modello alternativo: il gene è differenzialmente espresso e vi è almeno un k per il quale il termine (VG)kg≠0 nel modello.
L’adeguatezza dei due modelli viene verificata attraverso un “nested” F-test.
Due modelli vengono dichiarati “nested” o “annidati” se il modello definito completo o alternativo contiene tutti i termini del modello definito parziale o nullo e almeno un termine addizionale diverso da zero.
Se si definisce il modello nullo secondo la classica formulazione statistica come:
( )
y
x
x
gx
gE
=
β
0+
β
1 1+
β
2 2…
+
β
dove E(y) rappresenta l’aspettazione dei dati y, allora il modello alternativo che lo contiene avrà la forma:
( )
y x x gxg g xg kxke dal punto di vista del test delle ipotesi, esse verranno definite come segue:
0
da
diverso
è
k
1,...
g
a
con
parametro
un
almeno
:
0
:
1 2 0+
=
=
=
=
=
+ + a a k g gH
H
β
β
β
β
…
Per testare queste ipotesi è possibile utilizzare un F-test in cui la classica statistica F viene sostituita con una che realizza il confronto fra i residui dei due modelli piuttosto che fra le varianze dei dati e che è definita come segue:
(
)
(
)
(
)
[
−
+
1
]
−
−
=
k
n
SSE
g
k
SSE
SSE
F
full full reduceddove SSE indica la somma degli errori quadratici dei residui per i due modelli secondo la definizione classica, k sono i gradi di libertà per il modello nullo, g quelli per il modello alternativo e n è il numero delle osservazioni.
Questa statistica si distribuisce ancora come una variabile F di Fisher con k−g gradi di libertà per il numeratore e n− k
(
+1)
gradi di libertàper il denominatore. La regola di rigetto dell’ipotesi nulla stabilisce che il modello nullo viene rifiutato se F >Fk−g,n−(k+1)., dove Fk-g,n-(k+1) è il valore critico della variabile tabulata.
Questo test è anche conosciuto con il nome di “F-test parziale” e per i dati di intensità ricavati dai geni si traduce in:
1 1 1 0 1 0
/
)
/(
)
(
df
rss
df
df
rss
rss
F
=
−
−
dove rss è l’equivalente di SSE e df sono i gradi di libertà del modello nullo (pedice 0) e alternativo (pedice 1). Questa statistica è gene-specifica, poiché vengono utilizzati i dati dell’interpolazione del modello gene-specifico.
In un procedimento di F-test è possibile utilizzare altre statistiche per la discriminazione dei geni differenzialmente espressi.
Se, per esempio, si vuole considerare una varianza dell’errore comune su tutti i geni di tutti gli array, la statistica F può essere definita come: 2 pool σ 2 1 0 1 0
)
/(
)
(
pooldf
df
rss
rss
F
σ
−
−
=
utilizzando un’informazione globale in supporto di quella gene-specifica espressa dal numeratore.
Una via di mezzo fra le due statistiche appena definite può venire dal considerare una combinazione di varianza globale e gene-specifica al denominatore della statistica da computare:
2
/
)
/
(
)
/(
)
(
2 1 1 1 0 1 0 pooldf
rss
df
df
rss
rss
F
σ
+
−
−
=
Le tre statistiche sono praticamente equivalenti e l’adozione di una di esse può dipendere dalle informazioni che si hanno sui dati e dalle ipotesi fatte su di essi.
Una volta stabilito il criterio da adottare per verificare le ipotesi, si può stabilire di effettuare delle permutazioni sui dati, senza o con sostituzione (bootstrap), per irrobustire il risultato del test statistico e acquisire un livello di confidenza opportuno.
A conclusione di tutto il procedimento è possibile realizzare un clustering dei risultati per raggruppare i geni differenzialmente espressi in base a profili di espressione comuni.
L’analisi dei residui del modello è utile non solo per determinare l’espressione differenziale dei geni, ma ha anche lo scopo di verificare l’adeguatezza del modello.
Infatti, dallo scatterplot dei residui è possibile rilevare la presenza di andamenti non casuali (o tendenze) sui dati dei residui; ciò indica l’inclusione di elementi di informazione in un elemento del modello che viene considerato “random” e, quindi, per definizione non informativo. Riscontrare una situazione del genere deve portare ad un’analisi più approfondita degli effetti da considerare nel modello che viene interpolato, per non rischiare di mantenere errori sistematici che corrompono i dati o di non quantificare effetti di interesse.
5.4 Disegno di esperimenti con microarray
Una corretta definizione del modello non può prescindere dal disegno sperimentale che è stato adottato, soprattutto, per determinare il mascheramento degli effetti e per massimizzare l’informazione che si può ottenere dall’esperimento stesso.
Gli esperimenti che utilizzano microarray per l’analisi di espressione genica generano un’insieme complesso e numeroso di dati multivariati. L’aspetto più impegnativo della bioinformatica nel settore dei microarray è la produzione di strumenti statistici e computazionali per l’analisi dei dati prodotti, ma un compito di importanza non secondaria è la valutazione di disegni sperimentali appropriati che rendano più efficiente e robusto il processo di generazione di queste informazioni.
Il problema del disegno sperimentale è particolarmente rilevante in esperimenti con microarray “two-color”, in cui è fondamentale stabilire quali campioni devono essere ibridizzati su ogni microarray per poter effettuare un’analisi di tipo comparativo fra le intensità dei due canali.
Su microarray di tipo Affymetrix il problema è meno stringente, per il fatto che ogni campione viene ibridizzato separatamente su un microarray dedicato e, poiché viene ricavato un valore assoluto dell’intensità, è possibile simulare, mediante software, qualunque disegno sperimentale accoppiando i campioni in fase di analisi.
Il principale compito del disegno sperimentale è rendere l’analisi dei dati e l’interpretazione dei risultati più semplice e potente possibile, in relazione alle domande cui l’esperimento deve dare una risposta e ai vincoli sul materiale che lo sperimentatore ha a disposizione.
Da un punto di vista pratico, i problemi che bisogna considerare quando si progetta un esperimento con microarray sono di diverso tipo:
Fissare l’obiettivo dell’esperimento:
- Domande biologiche alle quali si vorrebbe dare risposta; - Priorità da assegnare ad ogni questione;
Stabilire il tipo e le concentrazioni minime dei campioni che servono per realizzare l’analisi comparativa;
Decidere quali campioni devono essere messi a confronto sullo stesso microarray e, di conseguenza, quale marcatura va effettuata per ogni campione;
Determinare il numero di vetrini che servono per realizzare il disegno sperimentale adottato;
Definire un protocollo sperimentale per la preparazione, la marcatura e l’ibridizzazione dei campioni;
Valutare il costo dell’esperimento ed apportare eventuali modifiche in relazione al budget disponibile.
Prima di discutere gli elementi con i quali stabilire il disegno sperimentale più opportuno, è utile illustrare le convenzioni grafiche tipicamente utilizzate per una rappresentazione schematica (Yang and Speed, 2002).
Uno schema tipico di un esperimento con microarray si ottiene con l’utilizzo di un “multi-grafo orientato”. Ogni campione viene rappresentato con un quadrato e ogni array con una freccia fra i campioni: la punta della freccia indica il campione marcato in rosso, mentre l’altra estremità indica quello marcato in verde. La presenza di un numero posizionato sulla freccia indica il numero di array utilizzati per quel confronto, ossia il numero di repliche sperimentali.
Figura 5.5: Grafo del confronto diretto fra due campioni A e B
La struttura del grafo determina quali espressioni differenziali possono essere stimate e con che precisione: due campioni possono essere confrontati solo se nel disegno sperimentale esiste un “percorso”, cioè una sequenza di microarray con un campione in comune, che li unisce; la
precisione della stima è inversamente proporzionale alla lunghezza del percorso.
Si supponga di voler confrontare tre campioni A, B e C con un disegno sperimentale denominato “loop”, spiegato in seguito in dettaglio, osservabile in figura 5.6.
Figura 5.6: Grafo del disegno sperimentale a” loop”
Se si vogliono confrontare i campioni A e B esistono due percorsi per farlo: il percorso diretto fra A e B di lunghezza 1 e quello indiretto che passa da C di lunghezza 2. E’ facile comprendere come la stima del confronto fra i due campioni sia più precisa sul percorso più corto, piuttosto che su quello più lungo, dove si possono sommare errori dovuti al confronto indiretto di A e B con il campione C.
5.4.1 Criteri per la scelta del disegno sperimentale
La scelta di un adeguato disegno per l’esperimento che si vuole eseguire può dipendere da molteplici elementi.
Prima di tutto bisogna tenere in considerazione che su ogni microarray è possibile ibridizzare solo due campioni, per cui ogni vetrino è un blocco sperimentale di dimensione due. Se ci sono più di due varietà
del fattore di interesse non è possibile confrontarle sullo stesso array, per questo motivo si dice che il disegno sperimentale è a blocchi incompleti.
Chiarito questo aspetto è necessario decidere quali campioni devono essere ibridizzati su ogni vetrino e quali marcatori utilizzare per ogni campione.
La prima cosa da tenere in considerazione è lo scopo dell’esperimento, in modo da cogliere eventuali suggerimenti o disegni impliciti direttamente dalla modalità di trattamento dei campioni che si vogliono confrontare.
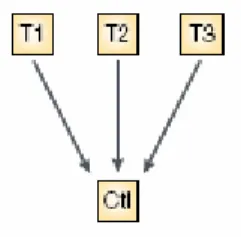
Si supponga, per esempio, di voler studiare l’effetto della somministrazione di diversi farmaci su un gruppo di cellule e che l’interesse principale di questo esperimento sia confrontare il loro mRNA con quello di un altro gruppo di cellule dello stesso tipo non sottoposto al trattamento. In questo caso un disegno appropriato deve considerare le cellule non trattate, o di controllo, come un riferimento de facto.
Figura 5.7: Grafo del confronto fra campioni trattati e campione di controllo
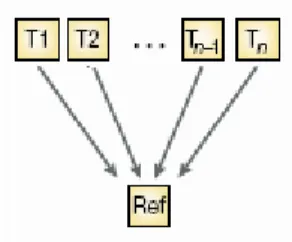
In un esperimento in cui si vogliano identificare sottogruppi di tumori della stessa famiglia sarà necessario mettere in relazione fra di loro i campioni tumorali provenienti dai diversi pazienti e ciò può essere ottenuto solo attraverso una base comune di confronto rappresentata da un RNA di riferimento. Se il numero di campioni è maggiore di tre, questo
disegno sperimentale offre il vantaggio di consentire il confronto fra tutti
gli n campioni in esame utilizzando n microarray piuttosto che ⎟⎟. ⎠ ⎞ ⎜⎜ ⎝ ⎛ 2 n
Figura 5.8: Grafo del disegno sperimentale con riferimento
Il disegno sperimentale, dunque, può essere suggerito direttamente dalla questione biologica alla quale si vuole dare una risposta; tuttavia, può succedere che disegni diversi descrivano ugualmente bene l’esperimento. E’ necessario, in questi casi, considerare altri vincoli per indirizzare la scelta sul modello migliore, ad esempio, decidere se realizzare un confronto diretto o indiretto fra i campioni.
5.4.2 Confronto diretto ed indiretto
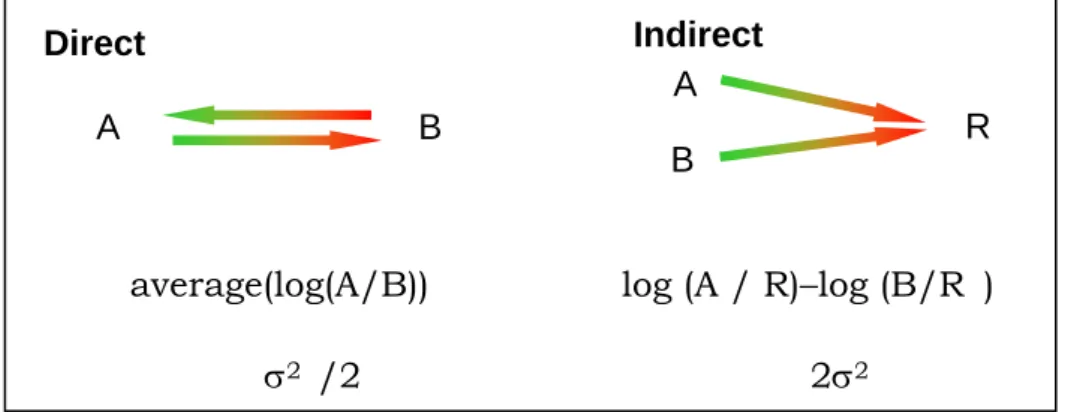
Si supponga di pianificare un esperimento in cui si ha a disposizione un microarray e la quantità di mRNA ricavata dai campioni non sia un fattore limitante. Per realizzare un confronto diretto fra un trattato e un controllo si può pensare di marcare il trattato con Cy3 e il controllo con Cy5 ed ibridizzare entrambi i campioni sullo stesso vetrino, come illustrato in figura 5.5. In questo caso, poiché il risultato dell’esperimento genera una sola osservazione del log(T/C) attraverso un confronto diretto, la varianza di questa misura sarà σ2.
Per migliorare la qualità dell’osservazione si può fare un confronto in “dye-swap”: con questo criterio gli stessi campioni vengono ibridizzati su due microarray diversi invertendo la marcatura con i fluorocromi.
Figura 5.9: Confronto diretto con scambio della marcatura
Se la varianza di ogni singola misura è ancora σ2, allora la varianza
della media delle due misure indipendenti è σ2/2 (figura 5.10).
La duplicazione della misura scambiando la marcatura è utile per ridurre l’errore sistematico dovuto alla diversa efficienza di emissione delle due cianine.
Se si fa uso di un riferimento comune o “reference”, denominato per esempio con R, allora le due ibridazioni disponibili per confrontare in maniera indiretta T e C saranno T rispetto ad R e C rispetto ad R. In questo caso il log(T/C) potrà essere ricavato dalle singole misure di T e C rispetto ad R. Essendo ancora la varianza di ogni singola misura pari a σ2,
ne segue che la varianza della differenza delle due misure è pari a 2σ2.
Le varianze relative ai due disegni sperimentali differiscono per un fattore quattro e questo può essere l’elemento critico che indirizza nella scelta di un metodo di confronto diretto piuttosto che di uno indiretto.
A B
A
B R
Direct Indirect
σ2 /2 2σ2
average(log(A/B)) log (A / R)–log (B/R )
Figura 5.10: Schema riassuntivo del confronto diretto ed indiretto
Il calcolo della varianza così come è stato eseguito considera i microarray come blocco sperimentale, senza entrare nel dettaglio dell’insieme di misure che sono state eseguite su tutti i geni o delle sorgenti di variabilità incluse nel modello dei dati.
Da questo punto di vista la scelta di un disegno sperimentale può essere influenzata dalla possibilità di quantificare tutti gli effetti presenti nel modello e ciò è strettamente legato ai gradi di libertà che è possibile riservare ad ognuno di essi e all’errore “random”.
Utilizzare un disegno con riferimento comporta di dover marcare con lo stesso fluorocromo, su tutti i vetrini, il campione di riferimento e con l’altro fluorocromo i campioni da analizzare: in questo modo le informazioni relative alle varietà e ai marcatori si sovrappongono, cioè i due effetti sono confusi o mascherati, e stimare l’uno o l’altro è completamente equivalente.
Una conseguenza di questo mascheramento è che anche gli effetti di interazione VG e DG si confondono, quindi, per utilizzare questo disegno bisogna assumere che non ci siano effetti gene-specifici nella marcatura o accettare che non possano essere quantificati.
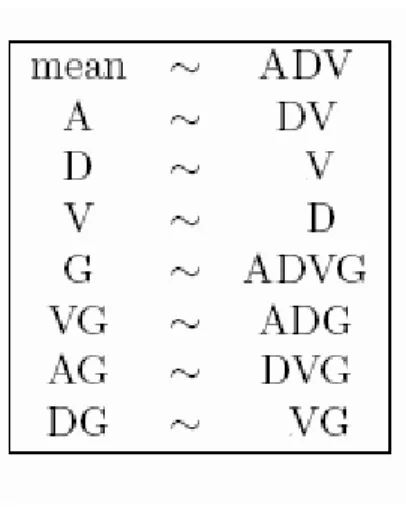
E’ possibile classificare numerosi altri effetti di mascheramento, riassunti, nel caso del disegno con riferimento, nella tabella 5.1. E’
interessante notare come l’effetto array è solo parzialmente confuso con l’effetto DV e questo sempre per il criterio con cui sono marcati i due campioni ibridizzati; se, infatti, si può identificare l’array sul quale è stato ibridizzato un campione quando si fissino i due indici di fluorocromo e di varietà per il campione da analizzare, lo stesso non avviene quando l’informazione riguarda il riferimento, per il quale la coppia di indici è uguale su tutti gli array.
Tabella 5.1: Mascheramento degli effetti nel disegno sperimentale con riferimento
Il disegno con riferimento per k varietà ed n geni produce 2kn osservazioni, quindi i gradi di libertà totali sull’insieme di dati sono 2kn-1. Se si calcolano i gradi di libertà per ogni effetto del modello si troverà che:
la media e gli effetti principali A, V e G coprono 2k+(n-1) gradi di libertà;
l’effetto combinato VG copre k(n-1) gradi di libertà; l’effetto AG copre (k-1)(n-1) gradi di libertà;
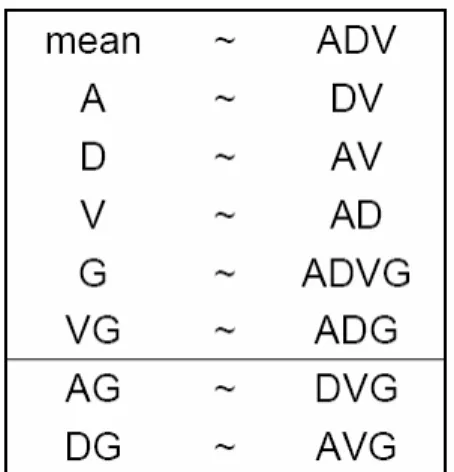
L’analisi del disegno che realizza una comparazione diretta con inversione della marcatura rivela che anche in questo caso vi sono mascheramenti di effetti. Si può facilmente verificare, infatti, che l’effetto array si confonde con l’effetto combinato della marcatura e della varietà, cioè una volta che viene fissata la coppia di indici che identifica il campione e il fluorocromo con cui esso è marcato si individua anche l’array su cui esso è stato ibridizzato.
Tabella 5.2: Mascheramento degli effetti nel confronto diretti con inversione della
marcatura
In questo caso, tuttavia, dal calcolo dei gradi di libertà si scopre che rimane un margine per stimare l’errore; infatti:
la media e gli effetti principali A, V e G coprono 2k+(n-1) gradi di libertà;
l’effetto combinato VG copre (k-1)(n-1) gradi di libertà; l’effetto AG copre (k-1)(n-1);
La scelta del disegno sperimentale più appropriato può avvenire, quindi, anche in base a quali effetti si desidera stimare o a quali si può ammettere di confondere con l’errore “random”.
Esiste un terzo disegno sperimentale che cerca di mettere insieme i pregi del disegno con riferimento e quelli del confronto diretto in “dye-swap”: il disegno a “loop”, di cui si è già accennato in precedenza (figura 5.6).
Questo tipo di disegno utilizza lo stesso numero di array del disegno con riferimento, ma supera il limite fondamentale di quest’ultimo, che consiste nel collezionare il maggior numero di misure sul campione di riferimento e non su quello di interesse.
Il disegno sperimentale a “loop” realizza il doppio delle misure sulle varietà di interesse e compie un bilanciamento fra i marcatori e le varietà, marcando ogni varietà una volta con un fluorocromo e una volta con l’altro su due array diversi.
Questo bilanciamento permette di separare gli effetti D e V e, di conseguenza, il loro effetto combinato; in questo modo è possibile rilevare sia la differenza intrinseca fra fluorocromi che un eventuale effetto gene-specifico della marcatura.
La stima dei gradi di libertà per questo disegno sperimentale è uguale a quella del confronto diretto in “dye-swap” e anche in questo caso rimangono n-1 gradi di libertà per stimare l’errore.
Un inconveniente pratico evidente di questo tipo di disegno è il fatto che bisogna realizzare il doppio delle reazioni di marcatura perché ogni campione deve essere marcato con entrambi i fluorocromi.