1
1. INTRODUZIONE
1.1 Gli amminociclitoli o amminocarbazuccheri
Gli amminociclitoli sono un gruppo di composti naturali di rilevanza significativa in chimica farmaceutica, perché sono componenti strutturali di molti antibiotici,1 di inibitori delle glicosidasi2-4 e di altre famiglie di composti biologicamente attive.5 Da un punto di vista strutturale, gli amminociclitoli sono cicloalcani che contengono almeno un gruppo amminico libero, o sostituito, e tre gruppi ossidrilici sull’anello.6 A causa della loro stretta relazione strutturale con gli zuccheri, gli amminociclitoli sono considerati amminocarbazuccheri.7
Gli amminociclitoli naturali sono metaboliti secondari che possono avere strutture semplici ma spesso costituiscono subunità strutturali presenti in strutture molto complesse come le validamicine, una famiglia di antibiotici isolati dalla fermentazione di colture di Streptomyces hygroscopicus.8
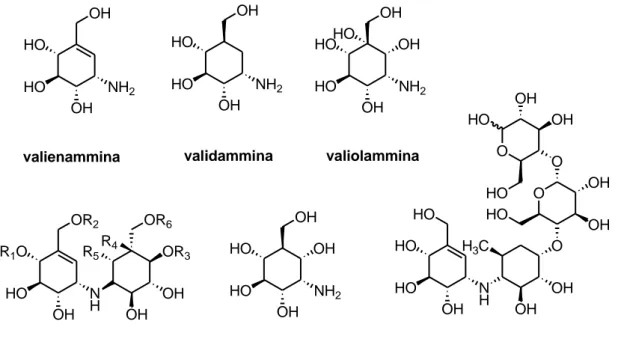
Una validamicina tipica è composta da una unità di valienamina, legata ad unità di validamina, valiolamina o idrossivalidamina. L’acarbosio è un altro prodotto naturale complesso contenente un’unità carbazuccherina ed è stato scoperto durante la ricerca di ceppi di vari generi di Actinomyceti. L’acarbosio è un inibitore dell’α-amilasi attualmente utilizzato in clinica per il trattamento del diabete insulino-dipendente di tipo II. Strutturalmente, l’acarbosio è un carba-tetrasaccaride, costituito dalla valienammina, da un desossiesoso e dal maltosio (Fig. 1.1).
2 HO OH NH2 HO OH valienammina HO OH NH2 HO OH validammina HO OH NH2 HO OH HO OH valiolammina N H OR2 R1O OH OH R5 OR6 HO OH OR3 R4 validamicine HO OH NH2 HO OH OH idrossivalidamina N H O O O O HO HO OH HO OH H3C OH OH HO OH OH OH HO HO acarbosio
Figura 1.1 C7N amminociclitoli isolati da fonti naturali.
Gli amminociclitoli sopra descritti, sono anche conosciuti come C7N amminociclitoli9 per la caratteristica catena idrossimetilica. Gli amminociclitoli più semplici del tipo amminocicloesitoli ed alcuni 1,3-diamminociclitoli (Fig. 1.2) sono stati trovati in diversi prodotti naturali con proprietà antibiotiche importanti per contrastare la resistenza degli agenti patogeni agli antibiotici non amminoglicosidici.10
HO HO OH H N OH HN NH2 HN NH2 streptidina HO HO OH NH2 H2N 2-deossistreptamina HO HO OH H N NH CH3 H3C spectinomicina O O O O X Y HO NH2 HO H2N NHR OH H2N HO HO kanamicina B tobramicina amicacina R X Y H H OH H NH2 NH2 OH OH NH2 OH O
3 Oltre alle proprietà antibiotiche degli 1,3-diamminociclitoli, molti amminociclitoli sono inibitori delle glicosidasi, per la loro capacità di comportarsi da mimetici dei carboidrati.11 Un gruppo importante di inibitori delle glicosidasi è costituito dagli amminociclopentitoli, rappresentati ad esempio dalla famiglia delle mannostatine, inibitori delle mannosidasi, dalla treazolina, inibitore della trealasi, e dalla allosamidina, inibitore della chitinasi (Fig. 1.3). Dato che le glicosidasi sono coinvolte in molti processi biologici differenti, gli inibitori delle glicosidasi sono importanti target farmaceutici per lo sviluppo di nuovi agenti terapeutici attivi contro infezioni virali, cancro, diabete ed altre patologie. In questo contesto alcuni inibitori delle glicosidasi sintetici, apparsi recentemente sul mercato, contengono il nucleo amminocicloesitolico, come nel caso del viglibosio (Fig. 1.3), inibitore dell’α-glicosidasi, utilizzato per abbassare i livelli di glicemia postprandiale in pazienti affetti da diabete mellito di tipo 2.
NH2 HO HO OH SMe mannostatina A NH2 HO HO OH SMe O mannostatina B O OH O N NH HO HO OH HO HO OH OH treazolina O O O HO HO NHAc HO OH OH NH Ac O HO OH N O NMe2 allosamidina H N OH OH HO OH HO OH OH viglibosio N H HO OH OH HO
N-ottil-valienamina (NOV)
4 Più recentemente, la capacità di alcuni amminociclitoli di interferire con il metabolismo degli sfingolipidi12 ha aperto la strada alla ricerca di nuove applicazioni terapeutiche per questa classe di composti. Questo è il caso dell’N-ottilvalienamina (NOV, Fig. 1.3), attualmente in studio come terapia complementare nel trattamento della malattia di Gaucher.13 Gli amminociclitoli inoltre sono candidati per lo sviluppo di agenti farmacologici coinvolti nel ciclo dell’inositolo fosfato e vari processi annessi.14
Dal punto di vista chimico i frammenti tipo 1,2-diamminoinositolo sono stati incorporati in catalizzatori asimmetrici tipo salen15-16 ed in nuovi agenti chelanti.17 Infine gli amminociclitoli rappesentano spesso intermedi chiave per la sintesi di alcuni prodotti naturali come ad esempio gli alcaloidi delle Amarilladaceae.18
1.2 Principali metodi sintetici per la sintesi di amminocarbazuccheri
1.2.1 Precursori per la sintesi di amminocarbazuccheri
La maggior parte delle sintesi degli amminocarbazuccheri utilizza come fonte principale di “starting material” un pool chirale. Diversi prodotti naturali contenenti scheletri carboniosi poliidrossicicloesanici, come il vibo-quercitolo, l’acido chinico o il ciclopellitolo (Fig. 1.4), sono stati utilizzati come starting material nella sintesi dei derivati amminociclitolici. Esempi classici si ritrovano nella sintesi della valiolammina19 e dei relativi amminocarbazuccheri20 a partire dall’acido chinico, nella sintesi di (-)-valiolammina e (-)-1-epi-valiolammina dal vibo-quercitolo,21 e nella sintesi della (+)validamicina B e (+)-validossilammina B da derivati ciclofellitolici.22
OH OH HO HO OH (-)-vibo-quercitolo OH OH OH HO O OH acido chinico OH HO HO O OH (+)-ciclofellitolo
Figura 1.4. Prodotti naturali contenenti scheletri polidrossicicloesanici.
I derivati dei carboidrati sono probabilmente i precursori più comuni degli amminocarbazuccheri grazie ai protocolli esistenti, ben definiti, che consentono la loro
5 conversione in carbacicli altamente funzionalizzati. Le strategie sintetiche che utilizzano tali precursori comprendono: a)reazioni di riarrangiamento come il classico riarriangiamento di Ferrier a partire dal metil-es-5-enopiranoside23 o il riarrangiamento di Claisen,24 b)ciclizzazioni radicaliche,25-27 c)reazioni di cicloaddizione,28 d)metatesi a chiusura d’anello29
Sono riportati anche esempi di sintesi enantioselettive di carbacicli. In questo contesto è da ricordare l’alchilazione allilica asimmetrica Pd-catalizzata per la desimmetrizzazione cinetica enantioselettiva di conduritoli achirali o racemici. Sono noti anche protocolli basati su approcci chemoenzimatici, per la sintesi di derivati ciclitolici, come nel caso della diidrossilazione enzimatica di sistemi aromatici con Pseudomonas putida, in cui il bromodiolo chirale inizialmente formato può comportarsi da precursore per diversi ciclitoli funzionalizzati30 (Schema 1.1).
Br Br OH OH Br O O HO OH Br OH OH Br O O Br O O O N R O O O O O O O O NHTs Ar NHR N3 OH Pseudomonas putida
Schema 1.1. Ciclitoli funzionalizzati a partire da diidrossilazione enzimatica di sistemi aromatici.
La risoluzione enzimatica del derivato racemico diacetossi-dibromo conduritolo B, ottenuto dal p-benzochinone è un altro esempio di approccio chemoenzimatico per ottenere un ciclitolo enantiopuro(Schema 1.2).31
6 O O OAc OAc Br Br risoluzione OH Br OAc Br OH Br Br OAc (racemo) (+)-diolo (-)-diacetato
Schema 1.2. Risoluzione enzimatica del diacetossi-dibromo conduritolo B racemo.
In un altro esempio la risoluzione enzimatica di un cicloesene meso diesterificato è stata usata per ottenere un monoestere chirale, che può essere utilizzato come precursore per una sequenza sintetica a più passaggi che conduce agli amminociclitoli (Schema 1.3).32 COOMe COOMe PLE 98% 96% e.e. COOH COOMe COO-tBu NHCOOMe amminociclitoli
Schema 1.3. Desimmetrizzazione enzimatica di un cicloesene diestere meso.
Di seguito verranno discussi alcuni approcci generali, emersi negli ultimi anni, per ottenere amminociclitoli, quali metodi chemoenzimatici non precedentemente riportati, ciclizzazioni intramolecolari di adatti precursori, reazioni fra derivati ciclitolici e nucleofili azotati e alcuni metodi basati su reazioni di riarrangiamento.
1.2.2 Approcci chemoenzimatici
Nonostante gli esempi sopra riportati riguardanti l’uso di enzimi nelle sintesi enantioselettive di amminociclitoli, la letteratura relativa alla formazione di amminociclitoli con stereochimica definita mediante metodi chemoenzimatici è ancora scarsa. Recentemente è stata riportata la desimmetrizzazione dei precursori 1.1 e 1.2 della 2,5-dideossistreptamina mediante acilazione enzimatica in solvente organico (Schema 1.4), che porta ai corrispondenti monoesteri 1.3 e 1.4 con un alto eccesso enantiomerico.33
7 HO OH N N 1.1 a AcO OH N N (-) 1.3 b AcO OH NH2 H2N HO OH N3 N3 a 1.2 HO OAc N3 N3 (+) 1.4 HO OAc NH2 H2N b
a) Candida rugosa lipase / vinil acetato b) H2 Pd/C, MeOH
Schema 1.4. Processo di desimmetrizzazione per fornire precursori della 2,5-dideossistreptamina.
Un altro approccio interessante riguarda la ciclizzazione intramolecolare tipo nitroaldolica tra la 3-idrossi-4-nitrobutiraldeide mascherata e il didrossiacetonefosfato (DHAP) catalizzata dal fruttosio-1,6-bifosfatoaldolasi (RAMA, Schema 1.5).34 Questa trasformazione è stata applicata alla sintesi di miscele diasteroisomeriche di nitrociclitoli 1.5, separati e ridotti ai corrispondenti amminociclitoli 1.6, analoghi strutturali della valiolamina che è un inibitore delle glicosidasi.
O2N OH OEt OEt HO O OPO3 -DHAP Aldolasi RAMA HO OH NO2 OH HO HO H2 PtO2 HO OH NH2 OH HO HO 1.5 1.6
8
1.2.3. Ciclizzazioni intramolecolari
Carbociclizzazione degli zuccheri con metodo dell’1,3-ditiano
Partendo dalla D-glucosammina sono stati sintetizzati una serie di O-protetti building block con struttura generale 1.7 contenenti una funzionalità propano-1,3-ditioacetale, successivamente utilizzati nelle sintesi di imminozuccheri e amminociclitoli.35 Il trattamento di composti 1.7 con nBuLi a basse temperature conduce ai derivati amminociclitoli 1.8 O-protetti, utilizzati come intermedi sintetici per una vasta gamma di amminociclitoli (Schema 1.6).
R2O R3O OTs OR1 NHR S S S S R3O OR2 NHR OR1 1.7 1.8 O OH NH2 HO OH HO D-glucosammina BuLi -78°C
Schema 1.6. Ciclizzazione intramolecolare dell’1,3-ditiano.
In un recente studio questa carboclizzazione è anche stata utilizzata per la sintesi di C7N amminociclitoli e amminocicloeptitoli.36 L’ossidazione del gruppo ossidrilico primario di 1.9 ad aldeide1.10, seguita da epossidazione di Corey-Chaykovsky conduce a 1.11, la successiva carbociclizzazione intramolecolare porta ad una miscela di precursori amminociclitolici 1.12 e 1.13, come risultato della competizione fra 6-exo e 7-endociclizzazione (Schema 1.7).
E’ interessante sottolineare che l’andamento della ciclizzazione può essere modificato dalla scelta opportuna dei gruppi protettori sul precursore iniziale a catena aperta. Ad esempio il tri-O-benzil protetto 1.11 (R1 = R2 = R3 = Bn) conduce selettivamente all’amminociclitolo 1.12. Inoltre il fatto che l’aggiunta di LiBr aumenta considerevolmente la percentuale dell’amminociclitolo cicloesanico a discapito di quello cicloeptanico, sembra sia dovuto alla formazione di un intermedio Li-chelato nel processo di ciclizzazione.
9 R2O R3O OH OR1 NHR S S 1.9 R2O R3O O OR1 NHR S S 1.10 R2O R3O OR1 NHR S S 1.11 O epossidazione di Corey-Chaykovsky 1.11 BuLi BuLi, LiBr reagente di Dess_Martin NHR OR2 R3O OR1 HO S S S S HO R3O R2O NHR R1O 1.12 1.13 6-exo ciclizzazione 7-endo ciclizzazione
Schema 1.7. Ciclizzazione intramolecolare con metodo dell’1,3-ditiano per fornire C7N amminociclitoli (1.12) e precursori amminocicloeptitolici (1.13).
Ciclizzazioni radicaliche di eteri ossima e reazioni di pinacol coupling
Il coupling riduttivo, in presenza di tributilstagno idruro (Bu3SnH) o ioduro di samario (SmI2), di ossime eteree poliossidrilate funzionalizzate nelle loro posizioni terminali con gruppi bromuro, esteri α,β-insaturi, aldeidi o chetoni possono condurre a amminociclopentitoli (n=1, Schema 1.8) e amminocicloesitoli (n=2, Schema 1.8) con buone rese e con alta stereoselettività in condizioni di reazioni blande.37-38
10 NOBn Br R1O R1O OR1 n SmI2 SmI2 SmI2 SmI2 SmI2 SmI2 R1O R1O OR1 NHOBn n R1O R1O OR1 NH2 n NOBn R1O R1O OR1 R1O R1O OR1 NHOBn COOEt R1O R1O OR1 NH2 COOEt NOBn O R1O R1O OR1 R3 n R1O R1O OR1 NHOBn OH R3 n R 1O R1O OR1 NH2 OH R3 n H2O H2O H2O COOEt
Schema 1.8. Ciclizzazione radicalica intramolecolare di eteri ossime.
L’applicazione di questo protocollo a substrati funzionalizzati, derivati dai carboidrati, permette di avere accesso ad una varietà di amminociclitoli con definita regio- e stereochimica. Inoltre il risultante intermedio, O-benzilidrossilamminico, può esser ulteriormente ridotto in situ con eccesso di SmI2 in presenza di acqua per fornire l’amminoalcol corrispondente con ottime rese (Schema 1.8). Gli amminociclipentitoli polidrossilati risultanti sono unità strutturali presenti in diversi prodotti naturali come la mannostatina A, la treazolina e l’allosamidina, inibitori delle glicosidasi e nucleosidi carbociclici come l’aristeromicina.
In un approccio sintetico correlato al precedente, il building block 1.14, derivato dalla D-glucosammina, è stato usato in una reazione di coupling pinacolico intramolecolare promosso ancora da SmI2 per ottenere miscele diasterosimeriche di nuclei amminociclopentitoli 1.15, presenti in prodotti naturali della famiglia degli opanoidi39 e strutturalmente correlati alla treazolina che è un inibitore della trealasi (Schema 1.9).
11 Il coupling pinacolico promosso da SmI2 può essere eseguito in un unico passaggio in situ dopo l’ossidazione di Swern dell’amminodiolo 1.14 iniziale, permettendo la sua trasformazione diretta nell’amminociclitolo corrispondente.
OBn OH OBn 2R1RN BnO OH 1) Swern 2) SmI2 OH OH BnO BnO NR2R1 OBn OH OH HO HO NH3+X -OH 1) H2 Pd/C 2) Dowex 1.14 1.15
Schema 1.9. Coupling pinacolico intramolecolare.
Cicloaddizione intramolecolare tra ossido di nitrile ed alchene (INOC) e cicloaddizione intramolecolare tra nitrone ed alchene (INAC)
Queste ciclizzazioni intramolecolari sono state usate per sintetizzare amminociclitoli funzionalizzati con diversa grandezza d’anello a partire dagli zuccheri.40 Sia l’ossido di nitrile che il nitrone possono essere generati da un precursore aldeidico comune (Schema 1.10). L’ossido di nitrile solitamente si forma per trattamento dell’aldeide iniziale con NH2OH, seguito dall’ossidazione dell’ossima risultante con un reagente clorurato (NCS, NaOCl o clorammina-T), e poi dall’eliminazione base-indotta di HCl dall’intermedio idrossimminolcloruro.41
La cicloaddizione [1,3] dipolare dell’intermedio ossido di nitrile porta all’isossazolina biciclica, un precursore avanzato dell’amminociclitolo che si ottiene grazie alla riduzione del legame C=N e alla contemporanea scissione riduttiva del legame N-O. Per quanto riguarda invece la ciclizzazione del tipo INAC, il trattamento dell’aldeide iniziale con ossime N-sostituite, seguito da trattamento in condizioni basiche del risultante nitrone, porta a sistemi isossazolidinici fusi che, conducono ad amminociclitoli grazie alla scissione riduttiva del legame N-O.
Applicazioni rilevanti di entrambe queste metodologie di sintesi degli amminociclitoli sono state riportate.
12 n N HO ossima n N O ossido di nitrile (b) N O n isossazolina n NH2 HO amminociclitolo (c) (a) n O aldeide (d) n N R O nitrone N O R isossazolidina (c) INAC INOC n
(a): NH2OH·HCl/ base (c): scissione riduttiva N-O (b): NaOCl/ Et3N (d): R-NHOH·HCl/ base
Schema 1.10. Approcci generali basati su metodologie INOC e INAC.
Ad esempio l’approccio INOC è stato utilizzato per sintetizzare nuovi inibitori della glicosidasi, aventi la struttura amminociclopentitolica.42 Come si può osservare nello Schema 1.11, nonostante la ciclizzazione intramolecolare fornisca miscele diasteroisomeriche di isossazoline, ciascuna di loro conduce ad un singolo diasteroisomero per riduzione dell’immina che si ottiene. L’alta diasteroselettività che si osserva nello step riduttivo in questo tipo di sistemi si pensa sia dovuta alla configurazione del centro stereogenico in α al legame C=N e alla natura dei sostituenti presenti su C(α).43
13 pool chirale O OMe BnO Br OBn OBn BnO OBn OBn O HONH2.HCl Py BnO OBn OBn N OH NaOCl Bu4NHSO4 N O BnO OBn OBn N O BnO OBn OBn 1)LiAlH4 /Et2O 2) H2 Pd/C MeOH-HBr HO OH OH NH2 HO HO OH OH NH2 HO +
Schema 1.11. Applicazione dell’approccio INOC alla sintesi di amminociclopentitoli.
Anche le cicloaddizioni intramolecolari tra il nitrone e l’alchene (INAC) sono state impiegate con successo per sintetizzare amminociclitoli a partire da precursori chirali.44 Come mostrato nello Schema 1.12 sottostante il trattamento di un alcol allilico protetto derivato dal D-glucosio con TFA/H2O porta ad un emiacetale intermedio che viene trattato direttamente con N-benzilidrossilammina cloridrata e NaHCO3 per dare una isossazolidina biciclica come singolo diasteroisomero. Questo risultato è stato interpretato sulla base della formazione “in situ” di un N-benzil nitrone e una concomitante regio- e stereoselettiva reazione di tipo INAC che fornisce il prodotto osservato. L’andamento stereochimico di questo processo dipende dall’orientazione dei sostituenti alcossilici presenti sull’anello dello zucchero.
14 O O O BnO BnO TFA /H2O O OH BnO BnO OH BnO OH OH OBn O NaHCO3 BnO OH OH OBn NBn O Pd/C HCOONH4 HO OH OH OH NH2 HO NHBn(OH).HCl
Schema 1.12. Sintesi di amminocicloesitoli basati sull’approccio INAC
Reazioni aldoliche intramolecolari
Le reazioni tipo aldoliche sono tra i metodi di sintesi più importanti nella formazione dei legami C-C. Nella chimica degli amminociclitoli ci sono diversi esempi di questa metodologia generale. Nella reazione aldolica intramolecolare di Mukaiyama, si ha enolizzazione del gruppo lattamico dell’aldeide 1.16 in presenza di TBSOTf e DIPEA, e quindi carbociclizzazione sililativa per fornire il sistema biciclico 1.17.45 Questo processo porta ad un sistema trans-configurato con un totale stereocontrollo (Schema 1.13). Questo intermedio è stato ulteriormente elaborato a dare un isomero della validamina. N Bn H OTBS O TesO N H O Bn OTBS O TesO TBSOTf DIPEA N O Bn OTBS OTBS TesO 1) Na /NH3 2) Boc2O /DMAP 3) LiOH, THF TesO NHBoc OTBS COOH TBSO HO NH2 OH HO OH 1.16 1.17 isomero della validamina O
15
Metatesi a chiusura d’anello
La reazione di ring-closing metatesi (RCM) si è rapidamente sviluppata come un metodo versatile ed efficace per sintetizzare carbacicli a partire da carboidrati.29 Come riportato da J. H. Van Boom e coll. la RCM dell’amminodiene 1.18 (Schema 1.14), ottenuto con diversi passaggi a partire da un opportuno furanoside protetto, conduce alla conduramina 1.19, un intermedio chiave nella sintesi di diamminociclitoli correlati alla 2-deossistreptamina, antibiotico amminoglicosidico.46,47
In un approccio simile, l’aldeide di Garner, ottenuta a partire dall’L-serina attraverso una sequenza multistep,48 è stata usata come precursore per la formazione dei diamminodieni 1.20 e 1.21 (Schema 1.15). Questi dieni sono stati utilizzati nelle reazioni RCM per fornire i diamminocicloalchenoli 1.22 e 1.23,49 poi ulteriormente funzionalizzati mediante epossidazione diastereoselettiva dell’olefina o diidrossilazione per fornire i desiderati amminociclitoli.
D-ribosio O OMe I O O (a) O O NRR1 RCM O O NRR1 NRR1 NRR1 OR1 R2O R3O 1.18 1.19
a) Zn (eccesso), bromuro di allile, benzilammina, THF, sonicazione
16 OH OH HO NH2 H2N HO N PMB Cbz BocHN N NHBoc HO PMB Cbz RCM O N CHO Boc HO NHBoc N PMB Cbz 1.23 1.21 NHBoc N PMB Cbz 1.20 N BocHN PMB Cbz H2N H2N 1.22 OH OH RCM Aldeide di Garner
Schema 1.15. Sintesi di amminociclitoli attraverso RCM di diamminodieni.
Ciclizzazioni N-alchilative
La ciclizzazione intramolecolare di carbammati o tiocarbammati ciclitolici con conseguente apertura dell’anello dell’intermedio ossazolidinone è stata usata per l’introduzione di gruppi amminici negli amminociclitoli. In condizioni basiche, possono essere generate specie nucleofile azotate che possono prendere parte alla formazione di un legame C-N attraverso una reazione di sostituzione nucleofila intramolecolare stereocontrollata.
In un approccio di questo tipo il trattamento del diolo 1.24 con benzil isocianato e successivamente con anidride triflica (Tf2O), seguita da trattamento del triflato 1.25 grezzo col bis(trimetilsilil)ammide, porta all’ossazolidinone 1.26, precursore dell’amminociclitolo 1.27(Schema 1.16).50
17 HO OCOPh HO OCOPh 1.24 Ph N C O Et3N Tf2O TfO OCOPh O OCOPh N H O Ph KN(TMS)2 OCOPh OCOPh 1.25 N O O Ph OH OH H2N HO O O 1.26 1.27
Schema 1.16. Ciclizzazione intramolecolare di un carbammato ciclitolico.
In un approccio simile, la iodociclizzazione del composto 1.29, ottenuto dalla condensazione del sale di potassio 1.28 con p-metossibenzil isotiocianato, seguita da “quenching” con CH3I, porta all’ossazolidinone 1.30 dopo trattamento con iodio e work up con solfito di sodio acquoso. Questo intermedio è stato poi elaborato per dare la valienammina (Schema 1.17).51
Schema 1.17. Iodociclizzazione del composto 1.29 per la sintesi di un intermedio della valienammina. BnO BnO O-K+ BnO 1.28 Ph N C S MeI BnO BnO O BnO SMe N CH2Ph 1.29 I2 Na2SO3 O N BnO I BnO O Ph BnO 1.30 HO OH NH2 HO HO valienammina
18 Un approccio concettualmente simile è stato utlilizzato nell’elaborazione di un composto amminociclopentitolico, utilizzato per la sintesi della (+)-treazolina, inibitore della glicosidasi (Schema 1.18).52 Il trattamento dell’alcool 1.31 con p-toluensolfonil isocianato e successiva iodociclizzazione conduce al composto 1.32, un precursore dell’amminociclitolo 1.33 ottenuto dopo l’idrolisi del carbammato ed opportuna manipolazione dei gruppi funzionali.
OTBS OH 1)TsNCO 2) I2/K2CO3 OTBS N O I Ts O 1.32 1.31 DBU benzene OTBS N O Ts O LiAlH4 MeOH OTBS NH OH Ts Na NH3 OTBS NH2 OH 1.33
Schema 1.18. Applicazione della reazione di iodociclizzazione nella sintesi di amminociclopentitoli.
Infine un approccio più sofisticato è stato descritto da Trost e coll. nella sintesi della (±) valienammina.53
In quest’esempio la specie elettrofila è il sistema 𝜋-allilico 1.35, ottenuto dal trattamento del vinil epossido(±) 1.34 con Pd come catalizzatore in presenza del tosil isocianato. E’ da sottolineare il controllo stereochimico del processo poichè la trasformazione complessiva prevede l’apertura dell’epossido sulla posizione allilica con ritenzione di configurazione (Schema 1.19).
19 O OR RO COOEt Pd cat TsNCO O OR COOEt RO Pd+(L)2 O -OR COOEt RO Pd+(L)2 O NTs OR RO COOEt O NTs O HO OH NH2 HO HO (R= TBDMS) 1.34 1.35 valienammina
Schema 1.19. Sintesi della valienammina attraverso una ciclizzazione intramolecolare sul sistema Pd π-allilico.
Ciclizzazione trans anulare
J. H. van Boom e coll. hanno riportato un metodo interessante per ottenere 2,3-diammino condutiroli basato sulla reazione di Ramberg-Backlund del 4,5-diazidotiepane 1,1 diossido (Schema 1.20).54 I gruppi azidici sono introdotti per spostamento nucleofilo, con inversione di configurazione, sul bis-mesilossi intermedio che si ottiene dal D-sorbitolo.
La stereochimica del sistema eterociclico dipende dalla natura dell’esoso utilizzato per elaborare l’anello tiepanico,55
quindi variando lo zucchero di partenza si possono ottenere con questa metodologia una buona varietà di diammino conduritoli configurazionalmente correlati.
20 D-sorbitolo S O2 OMe OMs MeO MsO NaN3 DMSO S O2 OMe N3 MeO N3 KOH CCl4 OMe N3 OMe N3 HS(CH2)3SH ET3N/ MeOH OMe NH2 OMe NH2
Schema 1.20. Sintesi di condurammine attraverso la reazione di Ramberg-Bӓcklund.
1.2.4 Trattamento dei derivati ciclitolici con nucleofili azotati
Apertura dell’anello di epossidi, aziridine, solfiti e solfati ciclici di tipo ciclitolico
Gli epossidi cicloesanici sono un gruppo abbondante di prodotti naturali per i quali sono state sviluppate diverse strategie sintetiche.56 In questo contesto l’apertura di epossidi ciclitolici con nucleofili azotati è stata ampiamente riportata in letteratura. In generale le condizioni di reazione richiedono l’utilizzo di eccesso di nucleofilo in presenza di solventi polari e ad alte temperature. Sebbene la stereoselettività dell’apertura dell’epossido sia dettata dalle regole di Fűrts-Platner,57
che prevedono l’apertura trans-diassiale della conformazione a sedia più stabile, la regiochimica dell’apertura dell’epossido può essere controllata in diversi modi. Ad esempio, in una sintesi della fortammina,32 la porzione 1,4-diamminociclitolica presente nell’antibiotico amminoglicosidico fortimicina A, le restrizioni stereochimiche imposte dal precursore iniziale, sono risultate cruciali per dare un risultato completamente regioselettivo (Schema 1.21).
21 N O MeOOC O Me C O N O MeOOC Me C O N O MeOOC Me C O OE OE E / N3 O NMe CO2Me N3 HO O OH NHMe OMe H2N HO fortammina (non osservato) N3 N3 OH
Schema 1.21. Sintesi della fortammina attraverso l’apertura dell’epossido in un sistema conformazionalmente rigido
La formazione del prodotto può anche essere controllata dalla giusta scelta delle condizioni di reazione. Un lavoro preliminare di Crotti e coll. sull’amminolisi e sull’azidolisi, controllata da processi di chelazione, di 1,2-epossiciclosani portanti un gruppo polare in posizione remota,58 ha ispirato l’uso di Yb(OTf)359 o LiClO460-62 per permettere l’apertura regioselettiva del derivato completamente O-protetto dell’epossi conduritolo B (1.36, Schema 1.22) attraverso un processo controllato dalla chelazione per dare amminociclitoli 1.37. E’ interessante sottolineare che gli addotti regioisomerici 1.38 sono stati ottenuti in condizioni non chelanti.61 Questa metodologia è risultata molto efficace ed è stata applicata con successo alla sintesi su larga scala (multi-gram) di una serie di building block utilizzati in chimica combinatoriale per la realizzazione di librerie di sistemi amminociclitolici.63
22 O OH BnO BnO OBn 1.36 OH BnO BnO OBn OH N3 OH HO HO OH OH NHR OH HO HO OH NHR OH 1.38 1.37 BnO H BnO H OBn H H OH O Li+ Li+ Nu LiClO4, 2N NaN3 / NH4Cl MeOH / H2O 1) RNH2 2) deprotezione apertura sul C2 apertura sul C1 1 2 1 2
Schema 1.22. Sintesi di amminociclitoli mediante apertura regio- e stereoselettiva dell’epossido del conduritolo B.
Le condizioni classiche non chelanti sono state ampiamente utilizzate per aperture selettive trans-diassiali di epossidi ciclitolici con diversi nucleofili.64-66
La reattività degli epossidi è anche stata sfruttata per la sintesi di analoghi amminociclitolici a cinque termini contenenti un gruppo amminico esociclico funzionalizzato con sostituenti alchilici, come nel caso dell’impiego di epossidi ciclopentanolici per la sintesi stereocontrollata della porzione amminociclitolica della (+)-treazolina (Schema 1.23).67 O PMBO OMOM OMOM OMOM NaN3 NH4Cl HO PMBO OMOM OMOM OMOM N3 AcO AcO OAc OAc OAc AcHN 1.39
Schema 1.23. Sintesi di amminociclopentitoli attraverso l’apertura di un derivato del ciclopentene ossido
23 In un altro approccio molto elegante, la catena laterale idrossimetilica della (+)-valiennammina è stata introdotta attraverso una ciclizzazione intramolecolare N-alchilativa a partire da un precursore carbammato con simultanea sostituzione allilica di uno spiroepossido (Schema 1.24).68
O O PMBO PMBO O NHBn KHMDS 18-crown-6 PMBO PMBO OH O NBn O 1) Na/ NH3-THF 2) LiOH 3) Ac2O/ Py RO RO OR OR NHR
Schema 1.24. Apertura dell’epossido con ciclizzazione N-alchilativa intramolecolare
La reattività delle aziridine, come quella degli epossidi, è stata usata per ottenere trans-1,2-diamminociclitoli con totale controllo stereochimico. Le aziridine possono essere ottenute facilmente per sostituzione intramolecolare del trans-1-ammino-2-mesilato vicinale originato dalla riduzione del corrispondente azido trans-1-ammino-2-mesilato (Schema 1.25). NH BnO OBn BnO OBn 1 2 BnO OBn BnO OBn BnO OBn BnO OBn N3 NH2 NH2 N3 apertura sul C1 apertura sul C2 BnO OBn N3 BnO OBn OMs BnO OBn OMs BnO OBn N3 LiAlH4 / THF
Schema 1.25. Sintesi di una aziridina derivata dal conduritolo e accesso stereocontrollato ai diamminociclitoli
24 Relativamente alla reattività delle aziridine l’apertura catalizzata dal TBAF dell’N-tosil-aziridina 1.4019 con p-toluensolfonnammide è stata applicata nella sintesi di derivati diammino inositolici(Schema 1.26).69
Br O O TsN Br p-TsNH TBAF O O Br NHTs TsHN AlBN n-Bu3SnH O O NHTs TsHN 1) OsO4 2) DMP O O NHTs TsHN O O 2) HCl / MeOH OH OH NH3+ +H 3N OH HO Cl- Cl -1.40 1) Na / NH3
Schema 1.26. Sintesi di diamminociclitoli attraverso l’apertura di N-tosil-aziridine.
I solfati ciclici ottenuti da dioli vicinali possono essere considerati equivalenti degli epossidi ma con maggiore reattività.70 Le loro proprietà, così come quelle dei solfiti ciclici,71 sono state sfruttate in un approccio sintetico per ottenere la (+)-valienammina72 e la validammina.73 Sia i solfiti ciclici 1.41 che i solfati ciclici 1.42 subiscono apertura dell’anello regio- e stereospecifica con lo ione azide per fornire i corrispondenti azido ciclitoli con buone rese (Schema 1.27).
O S O OBn O OBn OBn 1.41 LiN3 DMF OBn OBn OBn OH N3 OH OH OH OH H2N valienammina O S O OBn OBn OBn O O LiN3 DMF OBn OBn OBn OH N3 OH OH OH OH H2N validammina 1.42
25
Reazioni di sostituzione nucleofila
La sostituzione nucleofila con un nucleofilo azotato di un derivato inositolico attivato è un metodo ampiamente utilizzato nella sintesi di amminociclitoli. In un approccio molto diretto l’1,2-diamminoinositolo racemo è stato ottenuto da una sostituzione nucleofila SN2 a partire da un inositolo bismesilato protetto.74 Un approccio simile è stato utilizzato nella sintesi di amminocarbazuccheri a partire da precursori ottenuti da reazioni di riarrangiamento di derivati norbonilici,75 analoghi della 2-deossistreptamina,76 ed altri mono- diazido- ed ammino inositoli.77 Sono stati descritti anche esempi di sostituzione nucleofila SN2’, come nella sintesi della valienammina peracetilata riportata da Ogawa a partire dal cloruro allilico 1.43 e sodio azide (Schema 1.28).78 OAc OAc OAc Cl 1.43 AcO NaN3 / DMF OAc OAc OAc OAc N3 1) riduzione 2) acetilazione OAc OAc OAc OAc AcHN
Schema 1.28. Formazione di azidociclitolo mediante sostituzione SN2’ di un cloruro allilico
Il decorso stereochimico della sostituzione nucleofila può essere fortemente influenzato dalla partecipazione del gruppo vicinale come nel caso di un trans acetatovicinale. Così nella sintesi degli analoghi della valienammina,79 l’amminolisi del bromoacetato 1.44 con ammine primarie conduce all’ammino alcool 1.45 con una netta ritenzione di configurazione. L’andamento stereoselettivo della reazione può essere interpretato sulla base della formazione dello ione intermedio acetossonio che si forma grazie alla partecipazione del gruppo vicinale trans acetil. Inoltre nella reazione si ottiene un singolo regioisomero in seguito all’attacco nucleofilo sulla posizione allilica dello ione acetossonio (Schema 1.29). Lo stesso meccanismo è stato osservato nella sintesi della (-)-1-epi-valiolammina a partire da un precursore tosilato che presenta un gruppo trans-α-acetossilico partecipante.80
26 OAc Br O O O Me Me O Me 1.44 OAc O O O Me O Me + RNH2 -Br -OAc NHR OH O O Me Me 1.45 - RNHAc
Schema 1.29. Reazioni di sostituzione nucleofila in presenza di gruppi vicinali partecipanti. Sono state riportate anche reazioni interessanti tra alcool e nucleofili azotati nelle condizioni di Mitsunobu. Questo approccio è stato utilizzato in molte sintesi della valienammina a partire da precursori inositolici. L’esempio mostrato nello schema 1.30 è basato sull’utilizzo del classico sistema DEAD/Ph3P per attivare il gruppo ossidrilico e la ftalimmide come nucleofilo.81 Un approccio sintetico equivalente consiste nella conversione diretta dell’alcool in azide per trattamento con acido idrazoico in presenza di Ph3P/DEAD82 o, alternativamente, per trattamento con difenilfosforilazide (DPPA)83 e NaN3, come descrive Chang e coll. in una efficiente sintesi della valienammina.84 In ogni caso, si verifica una netta inversione di stereochimica che conduce al desiderato precursore amminociclitolico (Schema 1.30).
BnO OBn BnO OBn OBn 1) DPPA, DBU toluene, 0°C 2) NaN3 40°C BnO N3 BnO OBn OBn ftalimmide Ph3P/DEAD BnO N BnO OBn OBn O O valienammina
Schema 1.30. Sintesi di precursori della valienammina a partire da un ciclitolo nelle condizioni di Mitsunobu
27 E’ stata riportata anche l’addizione nucleofila tipo Michael catalizzata da acido di Lewis della ftalimmide a precursore ciclitolico protetto.85 In questo caso l’andamento stereochimico della reazione è influenzato dall’ingombro sterico del gruppo acetonide in assiale che dirige l’attacco nucleofilo sulla faccia meno ingombrata del sistema inositolico (Schema 1.31). O O O O MPMO CO2CH3 ftalimmide / iPrMgBr MgBr2.OEt /THF O O O O MPMO NPhth CO2CH3 O O O O H3CO2C MPMO NPhth NPhth Approccio favorito
Schema 1.31. Addizione di Michael in presenza di nucleofili azotati per ottenere amminociclitoli
E’ stata riportata anche una reazione tipo Michael sulla nitro-olefina 1.46. In questo caso, l’ammoniaca liquida è la specie nucleofila che permette l’addizione coniugata per fornire l’acetammide 1.47 dopo acetilazione in situ. Il gruppo nitro è stato poi rimosso per riduzione in condizioni radicaliche (Schema 1.32).86
28 BnO OAc AcO NO2 OBz 1.46 1) (liq) NH3 2) Ac2O AcO OAc NHAc NO2 BnO OBz nBu3Sn AIBN AcO OAc NHAc BnO OBz 1.47
Schema 1.32. Addizione nucleofila di Michael su nitro-olefina derivata da un ciclitolo per ottenere amminociclitoli.
E’ stata effettuata anche la sintesi di vari tipi di azido- e amminociclitoli mediante amminazione allilica catalizzata dal palladio.87 Per esempio il trattamento del diacetato 1.48 con la benzilammina in presenza di quantità catalitiche di Pd(PPh3)4 e Ph3P in acetonitrile a riflusso fornisce l’ammina 1.49 (Schema 1.33), un precursore immediato della valienammina. La configurazione risultante deriva dal meccanismo di doppia inversione, che complessivamente porta a ritenzione di configurazione, come atteso.88 OBn OBn OAc AcO OBn cat (Ph3P)4 Pd/ Ph3P RNH2 / CH3CN refl. OBn OBn OAc RHN OBn 1.48 1.49
Schema 1.33. Amminazione allilica catalizzata da Palladio per fornire amminociclitoli
In un processo correlato la reazione catalizzata dal palladio della sodio azide con un opportuno acetato allilico fornisce l’azide allilica 1.50. Questo composto è un precursore avanzato della 1-acetammido-6-epi-valienammina (Schema 1.34).89
29 NHAc OBn OBn AcO 1.49 cat (Ph3P)4Pd NaN3 NHAc OBn OBn N3 OBn 1.50 NHAc OH OH H2N OH 1-acetammido-6-epi-valienammina OBn
Schema 1.34. Amminazione allilica catalizzata da Palladio per fornire azidociclitoli
In un approccio simile il carbonato ciclico 1.51 e l’N-benzilnosilammide sono stati utilizzati per ottenere derivati 4,6-diamminocicloesenici 1.52, come precursori del nucleo amminociclitolico presenti in antibiotici amminoglicosidici tipo 2-deossistreptamina (Schema 1.35). O O N O Bn Ns 1.51 NsNHBn Pd2(dba)3.CHCl3 / Ph3P HO N N Ns Bn Bn Ns 1.52
Schema 1.35. Reazione di un carbonato ciclico catalizzata dal palladio per fornire precursori degli amminociclitoli
Un altro approccio interessante descritto da Trost per la sintesi di derivati amminociclitolici è basato sulla desimmetrizzazione catalizzata dal palladio del conduritolo A dicarbonato 1.53 (Schema 1.36).50 L’utilizzo del catalizzatore palladio complessato a ligandi C2 simmetrici permette la distinzione tra due centri prochirali presenti nel composto iniziale meso. Lo step enantiodiscriminante di questo processo si basa sull’abilità del catalizzatore palladio di reagire con uno dei due esteri allilici enantiotopici, in relazione alla chiralità del ligando. Il trattamento del dicarbonato 1.53 con l’azide, in queste condizioni, seguito da un’idrolisi alcalina del grezzo di reazione con simultaneo riarrangiamento sigmatropico [3,3] dell’azide allilica formatasi inizialmente, porta al’azidociclitolo 1.54 con una resa del 70% e con 93% di e.e. (Schema 1.36).
30 O O OCO2CH3 OCO2CH3 1.53 O NH PPh2 NH O Ph2P (S,S) (dba)3Pd2. CHCl 3 TMSN3 O O OCO2CH3 N3 K2CO3 MeOH O O OCO2CH3 N3 1.54
Schema 1.36. Desimmetrizzazione del condutirolo A dicarbonato catalizzata dal palladio.
Amminazione riduttiva
L’amminazione riduttiva dei cicloesanoni rappresenta un classico approccio per arrivare agli amminocicloesani e amminociclitoli. In una delle sintesi della valiolammina e di altri derivati N- sostituiti,81 l’amminazione riduttiva di derivati del cicloesanone funzionalizzati 1.55 in presenza di NaBH3CN permette l’introduzione di una catena laterale amminoalchilica con completo stereocontrollo grazie all’attacco dell’agente riducente sulla faccia meno ingombrata dell’intermedio costituito dalla base di Schiff. Inoltre la formazione della chetossima a partire dal cicloesanone 1.55, seguito dall’idrogenazione catalitica, porta ad una miscela di amminociclitoli epimeri in un rapporto α/β 16:1 (Schema 1.37).
31 O OBn BnO HO BnO OBn OBn BnO HO BnO OBn N OH OH 1.55 OBn BnO HO BnO OBn NOH OH HO HO HO OH NH2 HO OH NH2 NaBH3CN NH2OH.HCl NaOAc 1) Ni raney/ MeoH 2) Pd/C, acido formico MeOH
epimeri della valiolammina (
Schema 1.37. Amminociclitoli da amminazione riduttiva di cicloesanoni e riduzione della chetossima
Anche le amminazioni riduttive che conducono a C6, C7, C8 amminociclitoli sono state riportate in letteratura nell’ottica dell’ottenimento di nuovi inibitori delle glicosidasi.90-92
1.2.5 Reazioni di riarrangiamento
Nella chimica degli amminociclitoli sono state applicate con successo alcune reazioni di riarrangiamento. Come detto in precedenza, i carboidrati sono stati ampiamente utilizzati come precursori nella conversione a carbocicli funzionalizzati. 29-93
Il riarrangiamento di Ferrier è considerato un approccio classico.23 Quando viene applicato ad un appropriato amminozucchero protetto, questa trasformazione fornisce precursori amminocicloesenonici che sono stati utilizzati come composti di partenza nella sintesi di derivati amminociclitolici. Un esempio di questo approccio è dato dalla sintesi di pseudo-disaccaridi correlati all’allosamidina.94 In questo caso, il gruppo β-ossidrilico del β-idrossicicloesanone che si forma inizialmente è utilizzato per elaborare la porzione ossazolidinica caratteristica dei prodotti naturali di questo tipo (Schema 1.38).
32 O PMBO N OMe OBn Cbz Bn PMBO N OH O OBn Bn Cbz PMBO N OH HO OBn Bn Cbz PMBO HO OBn N O Bn O analoghi del-l'allosamidina HgSO4/ H2SO4 diossano, H2O NaBH4 / AcOH NaH/ DMF
Shema 1.38. Amminociclitoli attraverso riarrangiamenti di Ferrier di derivati della glucosammina.
Un esempio recente dell’uso del riarrangiamento di Ferrier può essere trovato nella sintesi del derivato 1-acetammido della 6-epi-valienammina, un inibitore di diverse β-N-acetilglucosamminidasi.89
Nonostante la reazione di riarrangiamento proceda velocemente a partire dal precursore 2-acetammido, i problemi associati alla reattività del risultante acetammidocicloesenone con il reagente di Grignard richiesto per l’introduzione della catena benzilossimetilica ha portato gli autori ad usare i corrispondenti precursori 2-azido 1.56, che risultano essere buoni substrati per questa reazione di riarrangiamento (Schema 1.39). L’intermedio avanzato 1.57 è stato usato come materiale di partenza per la sintesi di un precursore dell’1-acetammido-6-epi-valienammina (vedi anche Schema 1.34).
O BnO N3 OMe OBn 1.56 BnO N3 O OBn BnO N3 OBn OBn HO BnO NHAc OBn OBn
AcO (vedi schema 1.34)
1) Hg(CF3COO)2 acetone / H2O 2) MsCl /Et3N Mg / BnOCH2Cl HgCl2 1.57
Schema 1.39. Esempio di riarrangiamento di Ferrier nella sintesi di analoghi della valienammina.
33 Infine il riarrangiamento sigmatropico degli immidati allilici, conosciuto come riarrangiamento di aza-Claisen o di Claisen-immidati e oggi chiamato anche riarrangiamento di Overman,95 è stato usato per fornire derivati amminociclitolici da opportuni precursori. Il processo può essere effettuato termicamente o in presenza di catalizzatori come HgII o PdII. Metha e coll. hanno utilizzato questo riarrangiamento per convertire il tricloroacetimmidato 1.58 nel tricloacetammide 1.59 in condizioni termiche in prospettiva dell’ottenimento di analoghi degli amminocarbazzuccheri (Schema 1.40).75 OH OH OH O O O NH CCl3 O O N H O Cl3C OH OH ClH.H2N HO OH 1) Me2CO, Amberlyst 15 2) CCl3CN / DBU K2CO3 / p-xilene 1) OsO4 / NMMO 2) 1N HCl 1.58 1.59 riflusso