CAPITOLO 1
INTRODUZIONE
1.1. Reazioni di aziridine con reagenti organometallici.
Durante il mio periodo di tesi sperimentale, uno degli obiettivi è stato quello di effettuare reazioni di apertura di aziridine viniliche e ariliche, opportunamente attivate con gruppi protettivi sull’azoto, con alte rese e in modo regioselettivo utilizzando organoalani come nucleofili in grado di trasferire a tali substrati dei gruppi alchilici o alchinilici.
Le aziridine sono ampiamente usate come versatili intermedi sintetici in chimica organica. Le reazioni che le coinvolgono sono dominate dal carattere elettrofilo di tali eterocicli e generalmente riguardano l'apertura dell'anello che essendo a tre termini risulta tensionato. Sweeney definisce le aziridine come i “cattivi cugini” degli epossidi analizzandone le loro proprietà fisiche e la loro reattività, considerando diversi tipi di sostituenti sull’azoto e la regiochimica delle loro reazioni di apertura.1 C’è da sottolineare che molti composti naturali contengono nel loro nucleo aziridine, le quali sono essenziali per la loro attività antitumorale e antibiotica. Le loro reazioni di apertura includono una larga gamma di nucleofili (Schema 1.1.1).
N X R1CO R2OC NHX R N3 R NHX Hal NHX R R' R NHX R'S R NHX R'O R NHX NHX P(OR)2 O= R R3P CO2R R NHX R
Schema 1.1.1
I reagenti più spesso usati sono O-, S-, N- nucleofili, e meno esempi riguardano C-nucleofili come Grignard, organolitio, reagenti di Wittig, cuprati e malonati nonostante l'apertura del ciclo con questi ultimi porti a trasformazioni importanti poiché si viene a formare un nuovo legame C-C. Nel caso di aziridine non simmetriche in genere l’attacco avviene sul carbonio meno sostituito anche se fattori elettronici possono modificare questo trend. Ad esempio nelle 2-aril aziridine l’attacco nucleofilo avviene in maniera prevalente sul carbonio secondario benzilico. Inoltre a differenza degli epossidi, i quali per la presenza dell’ossigeno si comportano da basi di Lewis, e in presenza di acidi di Lewis la velocità delle loro reazioni di apertura aumenta, le reazioni delle aziridine sono molto meno dominate dall’utilizzo degli acidi di Lewis. Solo quando c’è un sostituente non ossigenato sull’azoto, H o gruppi alchilici, si può avere un’interazione diretta con tali acidi; se invece c’è un sostituente polare sull’azoto del tipo COR o SO2R, tale interazione è resa
più difficile e la reazione di apertura non è attivata in modo diretto come quando invece ci sono gruppi alchilici.1 È importante sottolineare quindi che se c’è un sostituente polare ossigenato sull’azoto può verificarsi un’interazione indiretta tra i lone-pairs dell’ossigeno e l’acido di Lewis come rappresentato nella figura di seguito, attivando comunque tali composti per le reazioni di apertura (Schema 1.1.2).
N R R1 LA R1= H, alchile N R X O= R1 LA
interazione diretta con acidi di Lewis
interazione indiretta con acidi di Lewis
N R O=X R1 LA sfavorito favorito
Schema 1.1.2
Possono esserci diversi tipi di sostituenti ossigenati sull’azoto che attivano le aziridine per le reazioni di apertura, quali sulfonile, sulfinile, fosforile, fosfinoile, carbonile; a seguito dell’interazione di risonanza tra il doppietto dell’azoto e il legame X=O, aumenta la
N O=X R1 R N X R O R1
Schema 1.1.3
Le aziridine con gruppi sulfonilici sull’azoto risultano essere quelle più attivate verso reazioni di apertura. Quando altri sostituenti sono presenti sull’aziridina il problema della regiochimica può essere superato da giuste scelte di protezione. L’aziridina N-tosile che in posizione 2 aveva un estere, riportata nello Schema 1.1.4, portava ad una miscela di prodotti di apertura e il nucleofilo andava anche ad attaccare il carbonile dell’estere; tale problema è stato superato idrolizzando l’estere ad acido carbossilico, ottenendo così un solo prodotto in seguito alla reazione di apertura che utilizza cuprati di ordine superiore;1 la medesima reazione invece fatta con l’aziridina derivante dall’ (S)-treonina aveva un più basso livello di regiocontrollo (Schema 1.1.4).
Ts N RO2C Ts N HO2C R2CuCNLi2 54-68% resa R CO2H TsHN Ts N HO2C R2CuCNLi2 TsHN R CO2H R CO2H NHTs 1:3
Schema 1.1.4
Il controllo della regioselettività è più problematico quando vengono considerate aziridine non simmetriche in cui i due sostituenti sono dello stesso tipo; Tanner e Somfai hanno risolto questo problema usando direttamente un ossidrile non protetto nell’aziridina che deriva dal butene 1,4 diolo e l’attacco nucleofilo con organo cuprati avviene così solo sul carbonio adiacente al gruppo dirigente (Schema 1.1.5).1
HO TsN OR Et2CuLi HO NHTs OR
Schema 1.1.5
Per quanto riguarda le N-acil aziridine lo stato di ossidazione dell’acile e quindi l’elettrofilicità del carbonile, gioca un ruolo cruciale; quando sono presenti semplici gruppi acilici non osserviamo l’apertura dell’anello aziridinico ma piuttosto la deacilazione; questo dipende molto anche dal tipo di nucleofili utilizzati, perchè nucleofili “soft” permettono di dare il cleavage dell’anello meglio di nucleofili “hard”, che invece deacetilano l’aziridina e quindi non portano al prodotto di apertura aspettato.
Tra le N-alchil aziridine abbiamo le N-benzil aziridine che subiscono le reazioni con organo cuprati in presenza di acidi di Lewis contenenti il boro, ma ci sono pochi esempi di reazioni di apertura di questo tipo di aziridine. Anche gruppi contenenti il fosforo possono essere in grado di attivare le aziridine; le N-dialchilfosforil aziridine danno reazioni di apertura con nucleofili rapresentativi tra cui gli anioni derivanti da composti 1,3-dicarbonilici e anche le N-difenilfosfinoil aziridine subiscono reazioni di apertura da parte di C- e di altri tipi di etero nucleofili; questo gruppo protettivo richiede condizioni più blande di quelle usualmente richieste per la desolfonilazione, la quale avviene in condizioni fortemente acide o riduttive.
Uno dei primi lavori sull’utilizzo sistematico di reagenti organometallici risale al 1978 quando Kozikowski e i suoi collaboratori dell’Università di Pittsburgh, nel loro studio sull’uso dei piccoli anelli eterociclici per la sintesi di sistemi alcaloidei, iniziarono a valutare l’abilità di questi reagenti per effettuare reazioni di cleavage alchilativo di aziridine N-sostituite.2 Il loro studio era mirato a definire il carattere organometallico richiesto per far avvenire tali trasformazioni; inoltre si concentrarono sulla regiochimica di queste reazioni per capire se l’attacco avveniva in posizione benzilica oppure se era possibile anche sull’altro carbonio.
L’attenzione era rivolta principalmente a derivati N-sostituiti di 2-fenil aziridine (composti
N N N
1.1a 1.1b 1.1c
EtO2C Ts H3C
Schema 1.1.6
Principalmente vennero utilizzati reagenti di litio, magnesio e rame. Inizialmente gli studi rivelarono che gli organolitio davano risultati insoddisfacenti, poichè decomponevano le aziridine o spiazzavano il gruppo protettore sull’azoto; comunque il cleavage dell’anello si otteneva in buone rese con gli altri due reagenti.
L’ N-tosil aziridina 1.1b risultò essere un substrato più reattivo dell’aziridina 1.1a, mentre
l’aziridina 1.1c non era molto adatta per questi studi a causa della sua instabilità. Le reazioni di 1.1a e 1.1b con omocuprati procedevano con buone rese (esempi 5,6,14,
Tabella 1.1.1); solo il meno reattivo litio divinil cuprato non era in grado di dare un prodotto isolabile (esempio 7). Di particolare interesse era il fatto che la reazione di 1.1a con MeCu·BF3 (esempio 8) procedeva in condizioni più blande di quelle osservate con i
corrispondenti omocuprati. Questo perchè la più alta acidità di Lewis di questo reagente assiste la rottura del legame attraverso l’interazione del complesso con il doppietto elettronico presente sull’azoto. I Grignard reagivano con le aziridine 1.1a e 1.1b per dare feniletilammine, anche se con rese più basse rispetto a quelle ottenute con gli omocuprati. Nel caso dell’ esempio 13 il Grignard era stato ottenuto dal corrispondente litio derivato dell’allilossi carbanione facendolo reagire con MgBr2 anidro; al contrario solo basse rese si
ottenevano quando si utilizzava il corrispondente reagente di rame e si utilizzava questo per il cleavage dell’anello.2
Tabella 1.1.1 Reazioni di aziridine con organometallici
ESEMPIO AZIRIDINA REAGENTE CONDIZIONI
DI REAZIONE PRODOTTI RESE ISOLATE (%)
1 1.1a CH3Li 0°C/2h, poi t.a./2h/THF Ph H N 85 2 1.1a H2C=C=C(OMe)Li -20°C/7h/ THF recuperato solo materiale di partenza 3 1.1a CH 3MgBr 0°C/3h, poi t.a./3h/Et2O PhCH(CH3)CH2NH COOEt 48 4 1.1a CH2=CHCH2MgBr t.a./6h/ Et2O-THF (1/1) PhCHBrCH2NH COOEt 75 5 1.1a (CH3)2CuLi 0°C/6h/ Et2O PhCH(CH3)CH2NH COOEt 98
6 1.1a (n-Bu)2CuLi 0°C/4h/
Et2O PhCH(n-Bu)CH2NH COOEt + PhCH(NHCOOEt) CH2-nBu (3:1) 70
7 1.1a (CH2=CH)2CuLi -70°C/3h, poi
0°C/3h/ Et2O decomposizione 8 1.1a CH3CuBF3 -70°C0°C, poi t.a./1h/ Et2O PhCH(CH3)CH2NH COOEt 95 9 1.1b CH3Li t.a./10h/THF decomposizione 10 1.1b CH3MgBr (1 eq.) 0°C/3h, poi t.a./3h/ Et2 O-THF (1:2) PhCH(NHTs)CH2CH3 + PhCH(CH3)CH2NHTs (1:3.5) 41 11 1.1b CH3MgBr (4 eq.) t.a./10h/Et2 O-THF (1:2) PhCH(CH3)CH2NHTs 46 12 1.1b CH2=CHCH2MgBr (4 eq.) t.a./24h/ THF PhCH(CH2CH=CH2)CH2 NHTs 94 13 1.1b CH2=C CH H3C O(THP) Li MgBr2 (4 eq.) -70°C/3h/Et2 O-THF (2:3.5) PhCHCH2NHTs HC C(CH3)=CH2 O(THP) 56 14 1.1b (CH3)2CuLi -78°C/1h/Et2O PhCH(CHPhCH(NHTs)CH3)CH2NHTs + 2CH3 (1:2) 70 15 1.1c (CH3)2CuLi -70°C/3h, poi 0°C/3h/Et O decomposizione
In tutti i casi che sono stati esaminati la regiochimica di queste aperture dell’anello avviene con un meccanismo a volte di tipo A2 e a volte di tipo A1; secondo il meccanismo A1, il
trasferimento di un gruppo alchilico avviene sul carbonio che è più in grado di sopportare una carica positiva. Uno stato di transizione del tipo A2 certamente richiede l’attacco sul
carbonio meno ingombrato. Inoltre le quantità di reagente usate in questi tipi di reazione hanno un importante effetto sulla regiochimica del processo; quando l’aziridina 1.1b è trattata con 4 equivalenti di Grignard piuttosto che con 1 equivalente, l’eccesso di reagente fa sì che l’apertura diventa completamente regiospecifica senza influenzare la resa isolata (esempi 10 e 11, Tabella 1.1.1). Il meccanismo push-pull spiega questo risultato in quanto l’eccesso di Grignard funzionando da acido di Lewis si coordina sull’azoto e aumenta la facilità della rottura del legame C-N nello stato di transizione, come riportato nello Schema 1.1.7.
N Ts R Mg2+ MgX Ph
Schema 1.1.7
È da sottolineare come l’aumento del potere di ionizzazione del solvente giochi un ruolo importante; un solvente polare favorisce il meccanismo di tipo A1 in quanto stabilizza
meglio la carica positiva, mentre invece solventi apolari favoriscono il meccanismo di tipo A2.
É poi da considerare l’apertura di aziridine simmetriche con reagenti organometallici proposta nel 1990 da David Tanner insieme al suo gruppo di ricerca dell’università di Uppsala.3 I substrati da lui usati erano i seguenti:
Ts N CO2Et EtO2C N Ts CO2Et EtO2C 1.2b 1.2a
Schema 1.1.8
Tali aziridine erano facilmente ottenibili dall’ (+)- e (-)-acido tartarico, rispettivamente. La simmetria di tali molecole evitava il problema della regiochimica nelle reazioni di apertura, e l’attacco nucleofilo previsto avveniva così con inversione, conservando la purezza enantiomerica. Inoltre i due gruppi esterei e il tosile sull’azoto attivavano queste aziridine verso l’attacco nucleofilo. Il suo gruppo di ricerca dimostrò che la coppia enantiomerica di aziridine 1.2a e 1.2b, rappresentava un metodo sintetico utile per ottenere i derivati N-tosil dietilici dell’acido L- o D-aspartico.
Tabella 1.1.2
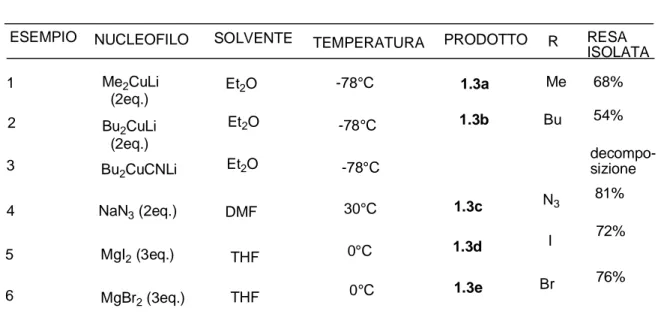
N Ts CO2Et EtO2C 1.2a NHTs R CO2Et EtO2C 1.3a-eESEMPIO NUCLEOFILO SOLVENTE TEMPERATURA PRODOTTO R RESA
ISOLATA 1 Me2CuLi (2eq.) Et2O -78°C 1.3a Me 68% 2 Bu2CuLi (2eq.) Et2O -78°C 1.3b Bu 54% 3 Bu 2CuCNLi Et2O -78°C decompo-sizione 4 NaN3 (2eq.) DMF 30°C 1.3c N3 81% 5 MgI2 (3eq.) THF 0°C 1.3d I 72% 6 MgBr2 (3eq.) THF 0°C 1.3e Br 76%
I primi nucleofili testati (esempi 1 e 2, Tabella 1.1.2) erano i cuprati di Gilman, e si vide che il composto 1.2a reagiva in modo estremamente veloce (meno di due minuti) con Me2CuLi in una soluzione eterea per dare il prodotto 1.3a in rese isolate accettabili. Il
dibutil cuprato dava delle rese più basse, a causa della formazione concomitante del prodotto di riduzione dell’ N-tosil dietil aspartato. L’uso del cianocuprato di più alto ordine di Lipshutz (esempio 3) causava solo decomposizione3. Il composto 1.2a (Schema
un pò più alte, -20 °C (Schema 1.1.9). Inoltre, è stato studiato il comportamento dell’aziridina 1.6 utilizzando sempre lo stesso reattivo, come mostrato nello Schema 1.1.9. Tale aziridina presenta il gruppo tosilico sull’azoto con potere attivante e il gruppo estereo come sostituente, con potere dirigente, in modo tale che la reazione avviene a -78°C in sole 3 ore, anche se è presente un gruppo etereo non coordinante, a differenza del composto 1.5 che presenta come sostituenti due eteri benzilici.
N Ts BnO OBn Me2CuLi, Et2O, -20°C 6h, 82% CH3 OBn BnO NHTs 1.5 1.5a N Ts MeO2C OSitBuMe2 Me2CuLi, Et2O,-78°C 3h,84% MeO2C Me OSitBuMe 2 1.6 1.6a NHTs O CO2Et EtO2C Me2CuLi, Et2O, -20°C 4h, 64% OH EtO2C CO2Et 1.4 1.4a Me
Schema 1.1.9
La rispettiva serie enantiomerica di prodotti era ottenuta dalle analoghe reazioni utilizzando l’aziridina 1.2b.3
Di notevole interesse risulta il lavoro del gruppo di ricerca di Bergmeier che nel 1997 esaminò la reattività dell’aziridina 1.7 con diversi reagenti organometallici in maniera che si formasse il prodotto di apertura come intermedio, il quale poi riciclizzava per dare l’aziridina (R)-1.7b come indicato di seguito nello Schema 1.1.10 (via b).4
N R' LG H a b a b N R' R H R LG NR' R N R' R H 1.7 (S)-1.7a (R)-1.7b
Schema 1.1.10
Trattando l’aziridina 1.8 con n-Bu2CuLi si otteneva direttamente l’aziridina 1.8a attraverso
un singolo step: apertura/riciclizzazione dell’aziridina (Schema 1.1.11).
NTs TsO R2CuLi N Ts R (R)-1.8a, R=n-Bu, 74% 1.8 (R)-1.8b, R=Me, 60% (R)-1.8c, R=CH2Si(CH3)3, 69% (R)-1.8d, R=n-Hexyl, 68% (R)-1.8e, R= SiMe3
Schema 1.1.11
Oppure l’aziridina 1.8a si poteva ottenere in una maniera alternativa partendo dall’aziridina 1.9 dalla quale era ottenuto il prodotto di apertura derivante dall’attacco del cuprato sul carbonio meno sostituito. Il corrispondente prodotto di apertura era poi riciclizzato, dopo la deprotezione, mediante la reazione di Mitsunobu, come riportato nello Schema 1.1.12.
N Ts TBSO n-Bu2CuLi, 86% RO NHTs R=TBS R=H n-Bu4NF,98 % Ph3P, DEAD, 88% TsN 1.9 (R)-1.8a
Schema 1.1.12
In entrambi i casi comunque si otteneva la stessa stereochimica del prodotto 1.8a. Considerando lo Schema 1.1.11, si notava che nè n-BuLi, nè n-BuMgBr davano il prodotto, si aveva solo decomposizione. I cuprati derivanti da metillitio, esillitio e (trimetilsilil)metillitio (commerciali) davano i composti 1.8b-1.8d. Venne usato anche il reattivo non commerciale, ottenibile in “situ”, la cui sintesi sfruttava il 6-iodio-1-(trimetilsilil)-2-esene più t-BuLi e poi CuI, per dare un cuprato insolubile, ottenendo così
1.8e con basse rese; invece addizionando n-Bu3P alla reazione si otteneva 1.8e con rese del
75%. Inoltre i cuprati derivanti da fenillitio, vinillitio e vinil magnesio bromuro portavano alla decomposizione di 1.8.4 Quindi con questi studi era stata sfruttata la reazione di apertura di un’aziridina N-tosile con organocuprati primari per ottenere intermedi da utilizzare successivamente per ottenere aziridine monosostituite, pure dal punto di vista enantiomerico.
David Tanner e il suo gruppo di ricerca, sempre nello stesso anno, studiarono la regioselettività dell’apertura di aziridine derivanti da alcoli omoallilici usando reagenti come LiAlH4, Red-Al, DIBAL e organocuprati di Gilman e di Lipshutz.5
N Ts R OH 1.10a R=Et 1.10b R=C6H11 1.10c R=Ph N Ts R O M Nu 1.10' M=metallo Nu=nucleofilo attacco C-3 OH Nu R NHTs 1.11a (1,4-isomero) 1 2 3 4 attacco C-4 OH R NHTs Nu 1.11b (1,3-isomero)
Schema 1.1.13
Tali reazioni di apertura passavano attraverso uno stato di transizione ciclico, a 6 o 7 membri a seconda che l’attacco fosse su C-3 o su C-4 rispettivamente, con l’aspettativa che avvenisse principalmente su C-3 (Schema 1.1.13). Vennero considerati substrati con R scelti in modo da verificare sia gli effetti sterici che quelli elettronici.
Da considerare con attenzione opportuna è soprattutto la reazione dell’aziridina 1.10c con i cuprati, sia quello di Lipshutz, sia quello di Gilman. La presenza del gruppo fenilico rendeva più favorito dal punto di vista elettronico l’attacco sul C-4, dal momento che una carica positiva in posizione benzilica è più stabilizzata e questo si contrapponeva con l’effetto di coordinazione che avrebbe favorito invece l’attacco sul C-3. Facendo reagire questa aziridina con il cianocuprato, Me2CuCNLi2 (3 eq.), in etere dietilico, si aveva quindi
una notevole regioselettività, con attacco principale sul C-4 e con un rapporto tra l’isomero 1,4 e quello 1,3 di 30:70; tale rapporto aumentava a 20:80 facendo uso del cuprato di Gilman Me2CuLi, sempre nelle stesse condizioni. Invece considerando gli altri tipi di
aziridine, non presentanti un gruppo fenilico, ma un gruppo alchilico, la regioselettività era del tutto opposta e l’attacco avveniva sul carbonio meno ingombrato, cioè sul C-3.
Da considerare con attenzione è lo sviluppo di un protocollo catalitico per la desimmetrizzazione di meso aziridine, N-tosil protette, proposto nel 1999 da Paul Müller.6 Questo è uno dei pochi esempi di desimmetrizzazione catalitica asimmetrica di aziridine con reagenti organometallici.
NTs 1.12 MeMgX CuL* NHTs X 1.12a X=Me 1.12b X=Br
Schema 1.1.14
Il composto 1.12 reagiva con MeMgBr in THF (0 °C, 6h), ma si otteneva il trans-2-bromo derivato (1.12b) e non l’aspettato trans-metil-N-p-toluensolfonilcicloesanammina (1.12a); se invece la reazione era condotta in presenza di Cu(acac)2 (10 mol%), l’apertura avveniva
più velocemente (0 °C, 1h) e si otteneva 1.12a con 85% di resa (Schema 1.1.14).
Successivamente una serie di catalizzatori chirali a base di rame (CuL*) furono testati in questo processo, non evidenziando una chiara relazione tra la struttura del catalizzatore e la sua efficacia. In generale le rese delle reazioni variavano dal 60 all’89% e l’attacco competitivo dell’alogenuro era la causa delle rese basse. Una bassa enantioselettività era osservata con il catalizzatore Cu-semicorrin, con un complesso di base di Schiff e salicilaldeide, con un complesso di base di Schiff derivante dall’1,2-diamminocicloesano e con il catalizzatore Cu-pybox. La più alta enantioselettività invece si otteneva con ammiditi derivanti dal BINOL e con complessi di basi di Schiff, derivanti dalla condensazione della salicilaldeide con amminoacidi. In questo caso, cambiando l’amminoacido si osservava un considerevole effetto sulla enantioselettività, ma comunque non era possibile stabilire un comportamento univoco.
Il MeMgBr era superiore al MeMgI in THF ma meno efficiente in etere; aumentando la quantità di catalizzatore la resa delle reazioni diminuiva, poichè ad una più alta concentrazione di catalizzatore predominava l’attacco del bromuro; aggiungendo il reattivo lentamente (in 10 minuti) alla miscela di reazione e non goccia a goccia in 5 minuti si avevano delle rese accettabili. Con 30% di catalizzatore si otteneva 1.12a con 91% di ee e con resa del 52%, ma con 40% dello stesso catalizzatore la resa scendeva e questo perchè aumentando la concentrazione del catalizzatore la presenza di una specie elettrofila quale il Cu (II) catalizzava l’attacco del bromuro. L’addizione di un eccesso di ligante alla reazione con il 10% di catalizzatore in THF non dava effetti benefici. Un’alta enantioselettività (83%) è stata ottenuta in Bu2O con 10% di catalizzatore. Come
applicazioni successive poi il composto 1.12a poteva subire il cleavage per ottenere l’ammina corrispondente con una precisa stereochimica.6
Un’altro importante esempio da riportare viene dal gruppo di ricerca in cui è stata svolta la presente tesi.7 In questo caso era stato individuato un protocollo di risoluzione cinetica di vinil aziridine con reagenti organometallici
NCbz rac- 1.13 "RCu" R NHCbz R NHCbz NHCbz R trans -addotto 1.14a, R=Me 1.14b, R=Et cis -addotto 1.15a, R=Me 1.15b, R=Et -addotto 1.16a, R=Me 1.16b, R=Et
Schema 1.1.15
La reazione dell’aziridina 1.13 con Me2CuLi in Et2O anidro non risulta essere SN
2’-regioselettiva a differenza delle aziridine alifatiche N-difenilfosfinoil viniliche, e si ottiene una miscela di prodotti tra cui il 1.16a è il principale prodotto di alchilazione. Quando CuCN (20 mol%) catalizza l’addizione di EtMgBr si ottiene una miscela complessa di prodotti; interessante è che l’addizione di Et2Zn (1.5 eq.) in presenza di Cu(OTf)2 (0.03
eq.) porta in 3 ore all’ottenimento di una miscela equimolare di ammine alliliche trans e
cis, 1.14b e 1.15b con una conversione > del 95% (Schema 1.1.15). L’ottenimento di una
notevole quantità di prodotti di syn addizione è in contrasto netto con quello che si ottiene dal relativo epossido, per il quale si ha un’elevata anti-stereoselettività nelle reazioni con reagenti di organorame. Generalmente una alchilazione allilica catalizzata dal rame avviene secondo una modalità anti ma si può avere un processo syn-stereoselettivo qualora si abbia la coordinazione del gruppo uscente con il reagente; e questo è proprio quello che succede in questo caso, in cui si ha la complessazione del rame con l’atomo di azoto o con il gruppo protettivo della vinil aziridina. L’addizione pseudoassiale del reagente organometallico probabilmente dà un complesso –allil rame (B) e la successiva eliminazione riduttiva porta all’addotto cis 1.15, come mostrato nello Schema 1.1.16.7
CbzN Cu R HN Cbz Cu R A B HN Cbz R 1.15
Schema 1.1.16
La syn stereoselettività viene drasticamente soppressa usando un ligante (chirale) esterno per il rame (la cui struttura è riportata nello Schema 1.1.17); usando ad esempio quantità catalitiche di rame complessate con un ligante racemico (R,S,S) (S,R,R)-L1 si ottiene una stereoselettività anti-syn di 95:5. La natura del gruppo protettore su queste aziridine esercita una grossa influenza sulla loro reattività. Mentre il Cbz risulta essere ottimo considerando la stabilità e la reattività, le aziridine benzil protette non sono sufficientemente reattive in queste condizioni, anche usando un eccesso di Et2Zn e
aumentando il tempo di reazione. Usando 0.75 eq. di Et2Zn in tempi più brevi, 2 h,
seguendo un protocollo di risoluzione cinetica si hanno le allil ammine corrispondenti 1.14
b con un alto livello di diastereoselettività e una moderata enantioselettività (61% ee). Una
più alta enantioselettività si ottiene con il ligante L2 a più bassi valori di conversione e quando una simile reazione è condotta in THF si osserva la stessa velocità e la stessa diastereoselettività ma una bassa enantioselettività. L’uso di un ligante come L3 che non presenta una simmetria C2 permette di ottenere il prodotto 1.14b con un’eccellente diastereoselettività e una moderata enantioselettività.7
O O P N (S,R,R)-L1 O O P N (S,S,S)-L2 O O P N Ph (S,R)-L3 O O P N (S)-L4 Ph Ph Ph Ph Ph Ph
Con il substrato 1.13, non è possibile effettuare una risoluzione cinetica regiodivergente (RKR), al contrario dei corrispondenti sistemi 2-alchenil ossiranici. Infatti quando la reazione è condotta in presenza di un complesso chirale di rame con L2 con un eccesso di Et2Zn è ottenuto pincipalmente l’addotto γ (8% ee) e solo poche tracce di addotto addotto
α (1.16b, <5%); il solo effetto visibile è l’aumento dell’addotto syn (1.15b). Usando invece un eccesso di Me2Zn si ottiene una quantità maggiore di addotto α, (1.16a, 20%)
quando la reazione va a completezza ma entrambi gli addotti α e γ sono ottenuti come racemi; l’utilizzo di 0.4 eq. di Me2Zn porta al composto 1.14a con la più alta
enantioselettività ottenuta per questa reazione (83% ee al 48% di conversione).
NCbz 1.17 Cu(Otf)2, 3 mol% (S,R,R)-L1 6 mol% Et2Zn 0.75 eq.
toluene, -78°C fino a t.a. 18h, 44% di conversione Et NHCbz Et NHCbz 1.17a 52% ee 1.17b
Schema 1.1.18
Come riportato nello Schema 1.1.18 l’aziridina 1.17, portante una unità eso-metilidenica in posizione allilica rispetto all’anello aziridinico, è meno reattiva nelle reazioni considerate prima, comunque si può ottenere il prodotto di addizione γ, 1.17a , con il 52% di ee e poi in quantità più bassa l’addotto α (14%).
Sukbok Chang e il suo gruppo di ricerca invece nel 2004 hanno preso in considerazione reazioni di apertura di aziridine con un sostituente piridinsolfonammidico sull’azoto.8 Tali composti sono stati ottenuti facendo reagire la 5-metil-2-piridinsolfonammide con un ossidante quale PhI(OAc)2 per generare il precursore nitrene
che è trasferito cataliticamente all’alchene diversamente sostituito per ottenere i diversi tipi di aziridine. Mentre la reazione dell’ N-tosilfenilaziridina 1.18 con il metil cuprato generato in situ è caratterizzata da una più bassa selettività favorendo la posizione meno ingombrata (1.20a/1.20b, 1:2), si osserva una completa inversione della regioselettività usando le aziridine con un gruppo piridinsolfonammide sull’azoto, come mostrato nello Schema 1.1.19.
N Ph SO2 X 1.18 X=CH 1.19 X=N 2 3 MeLi, CuI Et2O, -78°C, 2h Ph ArSO2HN Ph NHSO2Ar
1.20a (Ar= 4-Me-Ph) 1.21a (Ar= 5-Me-Py)
1.20b 70% (1:2) 1.21b 79% (5:1)
Schema 1.1.19
L’apertura preferita sul C-2 di 1.19 viene attribuita dagli autori ad una pre-coordinazione del complesso del cuprato all’azoto piridinico seguita poi dal trasferimento del metile nella posizione benzilica più elettrofila in un modo intramolecolare (Schema 1.1.20).
N SO2 N 2 3 Cu Me Me Li Ph Ph ArSO2HN 2 3 N Ph SO2 N 2 3 1.19 1.21a
Schema 1.1.20
Una particolare elaborazione delle aziridine è stata sviluppata da Xue-Long Hou e dal suo gruppo di ricerca nel 2004, i quali hanno proposto l’alchinilazione di aziridine da parte di reagenti di alchinillitio in presenza di Cu(OTf) per ottenere i corrispondenti prodotti di apertura con alte rese (Schema 1.1.21).9
R H n-BuLi (2 eq.) Et2O, -78°C 30 min. NTs R1 R2 CU(OTf) (0.1 eq.) t.a. R1 R2 NHTs R R2 R1 NHTs R
Schema 1.1.21
L’acido di Lewis è essenziale per avere il processo di apertura con alte rese e sembra che il Cu(OTf) risulti il migliore. Anche la quantità di n-BuLi è importante, in quanto ne sono
necessari 2 eq.; anche per quanto riguarda il solvente l’etere etilico risulta migliore del THF. Sono state testate numerose aziridine e vari alchini, soprattutto aziridine con gruppi elettron attrattori sull’azoto, e in queste condizioni di reazioni di apertura Cu(OTf) catalizzate si hanno buone rese. La reattività di aziridine che derivano da cicloalcheni è più bassa di quelle che derivano da alcheni aciclici, ma in tempi più lunghi si ottengono comunque buone rese. Gli alchini aromatici sono più reattivi di quelli alifatici e tutte le reazioni condotte con aziridine bicicliche danno prodotti con stereochimica anti; per le aziridine che derivano da un alchene terminale vengono ottenuti solo i prodotti di attacco sul C terminale a causa di ragioni steriche. Per quanto riguarda le aziridine 2-fenil sostituite si ottengono due prodotti, uno derivante dall’attacco in posizione benzilica e l’altro dall’attacco sul carbonio meno ingombrato e tra i due il principale risulta essere il secondo.
Lo stesso gruppo di ricerca ha sviluppato un metodo alternativo per ottenere l’alchinilazione di aziridine. In particolare, si può avere un’efficiente alchinilazione di aziridine usando acetilene e fenilacetilene in presenza di basi senza l’ausilio di acidi di Lewis (Schema 1.1.22).10 N Ts R2 R1 Ph H base DMSO TsHN Ph R2 R1
Schema 1.1.22
Considerando le aziridine che derivano da alcheni ciclici, si è visto che tra le varie basi, NaH e CsOH sono le migliori, specie se vengono usate in combinazione con setacci molecolari. Come solventi l’etere dietilico, THF o toluene non sono adatti e anche DMF o acetonitrile non vanno bene perchè danno prodotti incogniti. NaH permette di ottenere alte rese quando l’aziridina deriva da alcheni ciclici, ma se deriva da alcheni aciclici la resa scende e invece risulta buona con una base come t-BuOK (1.5 eq.), con la quale anche le stesse aziridine derivanti da alcheni ciclici reagiscono in 30 min. – 2.5h, dando i prodotti di apertura con alte rese. Anche in questo caso la reazione è anti-stereoselettiva; una
terminali sia, soprattutto, per le fenil aziridine, dalle quali invece in presenza di Cu(OTf) si ottenevano due regioisomeri derivanti dall’attacco sulla posizione benzilica e sul carbonio terminale.9 La novità è soprattutto l’addizione di acetilene, in presenza di base, alle aziridine condotta in DMSO così da ottenere omopropargilamine. Uno dei vantaggi dell’utilizzo di acetilene è che i prodotti ottenuti presentano un gruppo acetilene terminale, il quale può essere convertito in altri prodotti. Un’interessante applicazione di queste reazioni di apertura riguarda la sintesi di diidropirroli partendo da un protocollo già sviluppato e modificandolo per cui alla iodo-ciclizzazione di omopropargilammine proposta da Knigh sono aggiunti 300 mol% di AgOAc per aumentarne la resa (Schema 1.1.23).10 R1 R2 NHTs R3 I2 (300 mol%)/K2CO3 (300 mol%) AgOAc (300 mol%) CH3CN, 0°C-20°C N R1 R2 I R3 Ts H H 83% di resa
Schema 1.1.23
Quindi facendo reagire numerose aziridine con una larga varietà di acetileni si ottengono omopropargilamine con alte rese e con alta regioselettività; l’utilizzo dell’acetilene rende queste reazioni ancora più utili; oltre a poter usare una più larga varietà di aziridine e ad ottenere una migliore regioselettività rispetto a quella ottenuta con CuOTf, si possono sintetizzare diidropirroli in una modalità one-pot.
Ancora altri esempi di apertura di aziridine sono stati condotti da Albert Padwa e dai suoi collaboratori.11 Anche loro presentano esempi di alchinilazione di aziridine usando reazioni rame catalizzate e usando acetiluri; ad esempio il litio fenilacetiluro determina apertura nucleofila dell’aziridina 1.12 per dare un cicloesil alchino in alte rese (1.22). Oppure il litio dimetil cianocuprato attacca l’aziridina 1.23 sul carbonio meno sostituito per dare un 4-metil-N-(ottano-3-il)benzensolfonammide, 1.24 (Schema 1.1.24).
NTs 1.12 Ph Li Cu(OTf) (10 mol%) Et2O, 25°C, 45h NHTs Ph 1.22 96% di resa Ts N 1.23 Li2CuCN(Me)2 Et2O da -78°C a 25°C 12h NHTs 1.24 63% di resa
Schema 1.1.24
1.2. Reagenti di organoalluminio
I composti di organoalluminio sono tra i più economici tra i reattivi contenenti metalli, non sono complessi da preparare e stanno per questo sostituendo gli altri organometalli come agenti riducenti e alchilanti. La facile accessibiltà di questi reattivi si combina con le loro proprietà chimiche particolari, tra cui quella più spiccata risulta essere il forte carattere di acido di Lewis, aspetto direttamente correlato alla tendenza dell’atomo di alluminio a completare l’ottetto di elettroni. Proprio per questo motivo l’alluminio ha una grande affinità per alcuni eteroatomi, in particolare i legami tra l’alluminio e atomi elettronegativi come l’ossigeno o gli alogeni sono molto forti, l’energia del legame Al-O risulta essere intorno a 138 Kcal mol-1.12 Viene così spiegato perchè tutti i reagenti di organoalluminio sono estremamente reattivi in presenza di ossigeno, soprattutto quelli con gruppi alchilici fino a 4 atomi di carbonio sono solitamente piroforici, ovvero danno combustione spontanea in aria, a temperatura ambiente; tutti gli alchilalluminio reagiscono inoltre violentemente con l’acqua. Per questo motivo le reazioni svolte in laboratorio, durante il periodo di tesi, in cui venivano usati alchilalluminio, come il trimetilalluminio, sono state condotte sempre sotto argon e l’aggiunta del reattivo è sempre avvenuta a 0 °C. Aumentando il peso molecolare di tali reagenti di organoalluminio, attraverso l’incremento degli atomi di carbonio del gruppo alchilico, o tramite la sostituzione di questi con alogeni, la reattività piroforica viene ridotta. E inoltre la complessazione con gruppi quali basi di Lewis può renderli stabili all’aria e più facilmente maneggevoli.
Il legame C-Al possiede poi delle importanti caratteristiche, tra cui l’abilità riducente nei confronti dei sali dei metalli di transizione, la tendenza a formare complessi a ponte contenenti altri metalli e la tendenza a reagire con le olefine in particolari condizioni. L’utilizzo della loro proprietà di eterogenofilicità e in particolar modo l’ossigenofilicità, in chimica organica permette semplici reazioni di questi reagenti con composti contenenti eteroatomi in particolare con quelli contenenti ossigeno o composti carbonilici. Comunque, in netto contrasto con i classici acidi di Lewis come BF3.Et2O, AlCl3, SnCl4, e
TiCl4, il loro carattere nucleofilo è latente ed emerge in seguito alla coordinazione con
gruppi funzionali portanti un eteroatomo. L’atomo di alluminio quindi ha la funzione principale di sito di coordinazione per il substrato, mentre il nucleofilo attaccato all’atomo di alluminio può essere attivato dalla formazione del complesso di attivazione (“ate complex”), che facilita l’attacco nucleofilo sul substrato.
L’esito delle reazioni che prevedono l’utilizzo di reagenti di organoalluminio dipende molto dal tipo di solvente utilizzato; rispetto agli organolitio o magnesio, in questo caso i solventi eterei possono ridurre la reattività dei composti di organoalluminio dal momento che, l’alluminio avendo un’elevata affinità per l’ossigeno, permette la formazione di complessi 1:1 anche con basi deboli come l’etere. In soluzioni eteree, l’atomo di ossigeno del solvente si coordina fortemente con l’atomo di alluminio e si forma il dialchilalluminio alcossido che è un dimero stabile e in questo modo la velocità di alchilazione è ridotta notevolmente. In solventi non eterei, la coordinazione dell’atomo di alluminio al solvente è minimizzata; così in questi solventi, come il diclorometano, può essere presente l’autoassociazione degli atomi di alluminio, la quale può essere significativa; ad esempio il trimetilalluminio in toluene a -55 °C mostra che per la maggior parte può avere una struttura dimerica, considerando che il rapporto tra il metile a ponte e il metile terminale è di 1:2 come mostrato nello Schema 1.2.1.12
Al H3C H3C CH3 CH3 Al CH3 CH3 metile a ponte metile terminale
Schema 1.2.1
Aumentando la temperatura, uno dei metili è rapidamente scambiato e questo è visibile dai segnali NMR dei due tipi di gruppi metilici. A più basse temperature, quindi, come -55 °C, l’orbitale vacante dell’alluminio può essere occupato dal gruppo metilico di altre molecole per dare questi dimeri per cui a queste temperature questi reagenti non sono adatti per fare avvenire la reazione. Mentre a più alte temperature si forma un trimetilalluminio monomerico reattivo e la reazione procede.
Una differenza importante tra gli organoalluminio e gli acidi di Lewis è attribuibile inoltre alla flessibilità strutturale dei reagenti di organoalluminio; la loro struttura è facilmente modificabile a seconda dei gruppi funzionali che sono legati.12
possono essere rimossi, si ottiene una miscela di alcoli come prodotto. Per far avvenire questo tipo di riarrangiamento, il reagente necessita di due siti, un sito che funzioni da acido di Lewis per coordinare l’ossigeno dell’epossido e un sito che funzioni da base di Lewis per rimuovere il protone dal sistema. Un organoalluminio ammide risulta il reagente di scelta per questo impiego, come riportato nello Schema 1.2.2. Con l’atomo di alluminio fortemente complessato all’ossigeno (“ate complex”), il lone pair di elettroni sull’atomo di azoto diventa essenziale per la rimozione del protone, e si ottiene uno stato di transizione ben definito. Con una struttura di transizione così rigida, la deprotonazione procede con un’elevata regioselettività.12 C O C C H Al N C C C O Al N H
Schema 1.2.2
I trialchil alani quali AlR3, con R=Me, Et, n-Pr, n-Bu, i-Bu, sono prodotti
commerciali di basso costo e sono prodotti su scala industriale. Reagenti più specifici come alchinilalani possono essere ottenuti facilmente in svariati modi, tra cui la deprotonazione di un alchino terminale seguita dalla transmetallazione del rispettivo cloruro di alluminio (in genere Et2AlCl o Me2AlCl). É da notare che la reattività dei composti di organorame
rispetto a questi reagenti è completamente diversa; infatti quando è attaccato al rame, il ligante alchinilico è stabilizzato dall’interazione con gli orbitali d del rame e non può essere trasferito facilmente; questa stabilizzazione non avviene con i reagenti di organoalluminio e il ligante alchinilico, essendo il meno basico e il più nucleofilo, viene trasferito di preferenza.13Questa differenza viene riportata nello Schema 1.2.3 in cui si nota che l’organorame non trasferisce l’alchino al substrato, a differenza dell’organoalluminio che invece trasferisce la porzione alchinilica all’epossido.
Cu Li O Si tBu O 80% O O Si tBu O (CH2)5CH3 98% H C C (CH2)5CH3 Et2AlC C
Schema 1.2.3
Interessante risulta poi la sintesi di organoalani attraverso l’inserzione di un legame C-C insaturo nei legami idrogeno (idroalluminazione) o nei legami alluminio-carbonio (carboalluminazione). La reazione di idroalluminazione con DIBAL-H (diisobutilalluminio idruro) con un alchino terminale in genere procede con cis-stereoselettività fornendo E-alchenilalani. Inoltre la carboalluminazione, ottenuta dall’addizione del trimetilalluminio ad un alchino assistita dallo zirconocene, ha dimostato la sua grande utilità in applicazioni sintetiche e in analoghe trasformazioni di alcheni può
essere ottenuto un alto enantiocontrollo per dare miscele di trialchilalani. 13 In letteratura ci sono esempi di addizione di reagenti organoalani a composti
carbonilici, soprattutto α, β-insaturi, dove si stabiliscono i livelli di selettività dei prodotti ottenuti, e soprattutto nel caso di addizioni 1,2-1,4 asimmetriche catalizzate da metalli si ha la possibilità di introdurre sulla molecola gruppi arilici, alchenilici e alchinilici in modo enantioselettivo.13
Uno dei progetti portato avanti nel laboratorio durante la mia tesi è stato l’utilizzo di questi reagenti organoalani, in particolare di alchil e alchinilalani, per effettuare reazioni di apertura di aziridine viniliche e ariliche in modo tale da trasferire gruppi alchilici e alchinilici a tali substrati, studiando quindi una tematica mai affrontata precedentemente.
1.3 Sintesi e reazioni di sistemi ossazabiciclici
Un altro argomento di cui ci siamo occupati durante la mia tesi, è stato quello di considerare i bicicli del tipo [2.2.1]ept-5-ene e 3-aza-2-ossabiciclo-[2.2.2]oct-5-ene, per osservare la loro reattività e le loro possibili reazioni di riarrangiamento, soprattutto quando il gruppo protettore sull’azoto era un t-butossi carbonile (Boc).
Il biciclo 3-aza-2-ossabiciclo-[2.2.1]ept-5-ene, 1.25, può essere facilmente ottenuto in scala multigrammo dalla rezione di etero Diels-Alder tra il ciclopentadiene e una specie acilnitroso transiente, generata in situ tramite ossidazione dal corrispondente acido idrossamico (Schema 1.3.1). R1 NHOH O O N O 1.25 O R1
Schema 1.3.1
È un substrato veramente interessante perchè può essere sottoposto a varie manipolazioni, che permettono di ottenere diversi composti, la maggior parte dei quali risultano essere molto interessanti come precursori di molecole biologicamente attive.
L’ossazabiciclo esaminato può condurre all’ottenimento di amminoalcoli (via A, Schema 1.3.2) attraverso la riduzione del legame N-O, a diacidi (via B, Schema 1.3.2) per rottura ossidativa del doppio legame C=C, a vari acidi idrossammici tramite reazione con nucleofili (vie C, D ed E) e a composti di alchilazione per reazione con reagenti oraganometallici (vie F, G, H e I). Tra i composti ottenibili, gli acidi idrossammici sono di particolare interesse in quanto precursori di composti dotati di attività biologica o farmaceutica, ma ciò che rende più interessante questo substrato è la possibilità di ottenere dei prodotti di apertura in cui si ha la nuova formazione di un legame carbonio-carbonio.
N O 1.25 O R1 N-O Redn via A HO NH O R1 C=C oxdn via B O N OH O HO O O R1 Pd(0), Nu via C N O R1 OH Nu HCl via D OH N O R1 OH R2OH via E N O R1 OH R2O R2M via F R2 O N O R1 H R2M via G N O R1 OH R2 R2M via H R2 O N O R1 H R2M via I N O R1 OH R2 R2M via L HN O O R1 R2
Schema 1.3.2
Gli 1,4-ammino ciclopentenoli (via A) sono intermedi molto importanti nella sintesi di nucleosidi, prostaglandine ed altri prodotti naturali, composti quindi caratterizzati da un’alta purezza ottica.
Miller e i suoi collaboratori, ad esempio, utilizzano l’amminoalcool 1.26 come intermedio per la sintesi del nucleoside carbociclico antivirale 5’- noraristeromicina 1.26a, dell’agente antivirale aristeromicina 1.26b e dell’antitumorale naturale neoplanocina A
NH2 HO O O O O HO N N N N NH2 O O HO n N N N N NH2 1.26 1.26a, n=0 (-)-5'-noraristeromicina 1.26b, n=1 aristeromicina 1.26c, neoplanocina A
Schema 1.3.3
Il metodo usato dagli autori per ottenere l’amminoalcol 1.26 in forma otticamente attiva prevede la generazione della specie acil-nitroso a partire da amminoacidi omochirali (Schema 1.3.4). D-Ala BocHN O NH OH ossidazione di Swern N O BocHN O 1.27 1.28 1) OsO4 2) (CH3)2C(OMe)2 3)NaBH4 HN O O O 1.29 H2, Pd/C NH2 HO O O 1.26
Schema 1.3.4
La sintesi del composto 1.26 parte dall’acido idrossamico 1.27 facilmente ottenibile dal corrispondente amminoacido, la D-alanina. L’intrappolamento della specie acilnitroso transiente, generata secondo le condizioni di Swern, con il ciclopentadiene porta all’ottenimento del cicloaddotto 1.28 come miscela 5.9:1 di diastereoisomeri facilmente separabili. La diidrossilazione del cicloaddotto porta ad un singolo diastereoisomero il quale viene protetto come acetonide 1.29. La rimozione tramite riduzione in condizioni blande dell’ausiliare chirale conduce infine all’ottenimento del desiderato amminoalcol,
1.3.4).14 È possibile quindi variare la chiralità del prodotto finale desiderato agendo sulla scelta dell’amminoacido naturale di partenza, usato come ausiliario chirale. La valenza di questa reazione di cicloaddizione è quindi, strettamente correlata alla possibilità di ottenere cicloaddotti chirali da utilizzare come building blocks.
1.3.1. Reazioni di apertura con nucleofili di diversa natura
Nel 1998 Miller e i suoi collaboratori presentano il primo esempio di apertura regio- e stereoselettiva del 3-aza-2-ossabiciclo-[2.2.1]ept-5-ene catalizzata da Pd(0) o da acidi di Lewis a dare i corrispondenti acidi idrossamici 1,4-anti e 1,4-syn (via C ed E, Schema 1.3.2).15 O N R1 Ph 2 O 3 4 FeLn 1 Fe(III) N O O Ph R1 1.30 Pd(0) O N R1 Ph 2 4 1 O Pd R2OH R2O N OH O Ph R1 1.30a: 1,4-syn R2OH N OH O Ph R1 R2O 1.30b: 1,4-anti
Schema 1.3.5
L’acido idrossammico è una funzionalità presente in molti composti biologicamente attivi. La sua presenza infatti influenza la capacità del composto di legare metalli come il ferro (III), il nichel (II) e lo zinco (II). Quindi le molecole che contengono questa funzionalità
ad attività biologica. Viene evidenziata la grande versatilità sintetica di questo metodo che consiste nella possibiltà di influenzare la formazione dell’addotto syn o anti agendo sul catalizzatore utilizzato.15 La chelazione operata infatti dal ferro trivalente, l’acido di Lewis che ha portato all’ottenimento dei risultati migliori, costringe il sistema a subire l’attacco sul carbonio 1, chelazione che non avviene nell’addotto che si ottiene in presenza di Pd(0), come mostrato nello Schema 1.3.5. Con lo scopo di ottenere il maggior numero possibile di informazioni sulla reattività di questo composto, il cicloaddotto 1.30 è stato anche messo a reagire con un acido di Brønsted, cioè in presenza di HCl 6N.
N O O Ph 1.31 HCl 6N N O OH Bn 1.32 N OH OH O Bn 1.33
Schema 1.3.6
Il composto che viene fatto reagire in queste condizioni di reazione è il fenacetil-derivato
1.31; gli autori osservano un chiaro riarrangiamento del cicloaddotto di partenza al sale 1.32, il quale in seguito idrolizza a ciclopentenolo idrossammato 1,2-disostituito 1.33
(addotto 1,2-syn) come riporta la via D (Schema 1.3.6).15
Studi successivi dell’autore mostrano come questa regio- e diastereoselettività di attacco possa anche essere influenzata modificando l’ingombro sterico del solvente nucleofilo utilizzato.16 All’aumentare infatti dell’ingombro sterico dell’alcol, da metanolo a t-butanolo, la quantità di prodotto 1,4-anti che si forma diminuisce e aumenta invece la quantità dell’addotto 1,4-syn. Dati sperimentali riportati nell’articolo mostrano che la quantità di prodotto 1,4-anti che si forma dalla reazione con nucleofili poco ingombrati può essere aumentata cambiando l’acido di Lewis utilizzato da Fe(III) a Cu(II). La selettività può inoltre essere spostata completamente verso la formazione dell’addotto
1,4-syn utilizzando l’alcol nella quantità di 4 equivalenti in un solvente non polare, come ad
esempio il toluene. La formazione preferenziale dell’addotto 1,4-syn rispetto all’addotto 1,4-anti viene incrementata anche dall’utilizzo di alcoli più ingrombati come nucleofili; infatti usando come nucleofilo il t-butanolo si osserva solo la formazione del prodotto
prodotto portano gli autori a proporre un meccanismo per spiegare le rezioni di apertura del cicloaddotto (Schema 1.3.7). O N R 1 M Ln O O N R 1 M Ln O O N R 1 M Ln O R2O H R2O N OH O R1 1.34a: 1,4 anti N O LnM R1 O coppia ionica intima N O M (OR2)n R2O R1 O R2O N OH O R1 1.34b: 1,4-syn A B
Schema 1.3.7
Un meccanismo plausibile suppone l’iniziale apertura del cicloaddotto mediata dall’acido di Lewis coinvolto nella reazione a dare una coppia ionica intima, il cui destino viene determinato dalle condizioni di reazione: se la reazione avviene nel solvente nucleofilo poco ingombrato, come il metanolo, il solvente può attaccare la coppia ionica intima dalla faccia opposta dell’idrossamamato a dare il prodotto 1,4-anti 1.34a (via A, Schema 1.3.7). Se invece il nucleofilo è troppo ingombrato o se la sua concentrazione è troppo bassa, avviene un rilascio intramolecolare del nucleofilo da parte del metallo a dare un attacco dalla stessa parte dell’idrossammato e quindi il composto 1,4-syn 1.34b (via B, Schema 1.3.7).16
Lo studio sulla reattività di questi cicloaddotti è proseguito da Miller stesso con il tentativo di utilizzo dei reagenti di Grignard come nucleofili.17 Il cicloaddotto offre vari siti per l’attacco nucleofilo del reagente organometallico: ci si può infatti aspettare che il
idrossammato 1.36b’. Alternativamente può essere anche possibile un attacco di tipo coniugato che sostituisca indirettamente l’ossigeno (via C, Schema 1.3.8) o l’azoto (via C’, Schema 1.3.8) per dare i prodotti 1,2-anti (1.36c e 1.36c’). Affinche la trasformazione sia sinteticamente utile è necessario, dicono gli autori, che una di queste vie prevalga sulle altre e che la via che prevale non sia l’A.
N O R1 O A B B' C' C 1.35 A B B' R2MgX O R1 R2 1.36a HN O R2 N O R1 OH R2 O N H O R1 1.36b 1.36b' C C' N O R1 OH R2 R2 O N H O 1.36c 1.36c' R2MgX R1
Schema 1.3.8
I risultati di questo studio, riportati nell’articolo, dimostrano come il cicloaddotto 1.35 possa essere fatto reagire con i reagenti di organomagnesio in presenza di quantità catalitiche di Cu(II), per dare acidi idrossammici fino al 98% di resa e con una selettività che può arrivare fino a 18:2:1 rispettivamente per i prodotti 1,2-anti:1,4-anti:1,4-syn. Le condizioni di reazione ottimali prevedono l’utilizzo di CuCl2 come catalizzatore e di un
reagente di Grignard, in particolar modo EtMgBr. Gli autori riguardo al ruolo del rame in questa reazione concludono che è più complesso di quello di un semplice acido di Lewis, come semplicemente supposto nei precedenti lavori, ma non danno molte spiegazioni al riguardo. Probabilmente si ha la formazione in situ di un cuprato che è poi la specie che effettivamente si addiziona al cicloaddotto.17 Viene poi in questo stesso lavoro riportato un esempio di applicazione di questo protocollo di reazione al fine di ottenere composti di interesse biologico. L’apertura Cu-catalizzata del cicloaddotto, infatti, si è detto che conduce ad analoghi di inibitori della 5-lipoossigenasi. Un esempio di una potente classe di
questi inibitori è quello degli acidi idrossammici 3,4-diidronaftilici, tra i quali il composto
1.37 che presenta un gruppo fenossi in posizione 5 sull’anello 3,4-diidronaftalenico si è
rivelato essere quello più potente (Schema 1.3.9).17
O N O OMe OH 1.37
Schema 1.3.9
Gli analoghi di questo composto derivati dall’ossazabiciclo possono essere sintetizzati in un solo passaggio. Infatti il bromuro alchilico può essere convertito in situ nel corrispondente reagente di organomagnesio ed usato per aprire il cicloaddotto in presenza di quantità catalitiche di CuCl2 a dare il composto desiderato 1.39 (Schema 1.3.10).
N O MeO O 1.38 O Br Mg, HgCl2, CuCl2, Et2O O N O OMe OH 1.39
Schema 1.3.10
Questo composto è stato ideato per poter interagire perfettamente con il sito dell’enzima come è noto interagirci il substrato naturale, cioè l’acido arachidonico: mentre l’acido idrossammico si lega al ferro (III), le porzioni aromatiche nelle due strutture vanno ad interagire con i siti lipofili dell’enzima.17
In un lavoro più recente lo stesso Miller evidenzia ulteriori applicazioni sintetiche che gli acidi idrossammici derivanti dal processo di apertura di questi ossazabicicli possono avere. Infatti alchilando il cicloaddotto 1.40 con reagenti di Grignard è possibile ottenere il β-ammino diestere 1.43 il quale è un importante intermedio che permette di
N O O R1O 1.40 MeMgBr, CuCl2 THF N OH O R1O 1.41 TiCl3, KOAc MeOH,H2O N O R1O H 1.42 O3, NaOH MeOH, CH2Cl2 O O MeO OMe NHG 1.43 N O H R2 CO2H 1.44
Schema 1.3.11
L’intermedio 1.43 viene ottenuto dal cicloaddotto 1.40 in soli tre passaggi (Schema 1.3.11): la sintesi messa a punto da Miller prevede l’iniziale trattamento del cicloaddotto con reagenti di Grignard in modo da ottenere l’acido idrossammico 1.41. Questo composto subisce una riduzione del legame N-O tramite trattamento con titanio tricloruro a dare il composto 1.42 che viene sottoposto a scissione ozonolitica del doppio legame in modo da ottenere il composto 1.43 con la stereochimica appropriata che consente l’ottenimento del carbapenem 1.44.
È sempre lo stesso Miller ad eseguire studi riguardo la reattività del sistema 3-aza-2-ossabiciclo-[2.2.2]oct-5-ene, omologo superiore rispetto al biciclo descritto fino a questo momento.19 Di questo substrato vengono riportate reazioni di apertura utilizzando come nucleofili gli alcoli a dare acidi idossammici 1,4-anti 1.45a, 1,4-syn 1.45b e 1,2-anti 1.45c.
O BnO 1.45 R1OH N O O OBn MLn O N acidi di Lewis R1O N OH O OBn 1.45a R1O N OH O OBn 1.45b N OH O OBn OR1 1.45c
Schema 1.3.12
Il cicloaddotto protetto sull’azoto con il gruppo carbobenzilossi (Cbz) viene fatto reagire in presenza di FeCl3 o CuCl2 fornendo i prodotti di apertura in buona resa, sebbene
mostrando una ridotta stereoselettività rispetto al biciclo [2.2.1], a favore del prodotto
1,2-anti 1.45c (Schema 1.3.12). La perdita della tensione d’anello nel cicloaddotto [2.2.2] determina infatti una diminuzione della reattività che rende necessario un aumento di temperatura necessario per indurre l’apertura dell’anello, il quale però può essere la causa della perdita nella stereoselettività osservata. Comunque il cicloaddotto può essere aperto con successo da alcoli in presenza di acidi di Lewis a fornire i corrispondenti acidi idrossammici senza che si osservi la perdita del gruppo protettivo. Inoltre, i prodotti vengono ottenuti con una regioselettività che, come gli autori osservano, non sembra essere dipendente da specifiche variazioni apportate. La causa di questa bassa e non prevedibile regio- e stereoselettività viene attribuita ad una bassa affinità di legame del Fe(III) e di Cu(II) ai cicloaddotti protetti sull’azoto come carbammati, avendo gli autori riscontrato le stesse problematiche anche usando come substrato il cicloaddotto N-Boc protetto.
Molto recentemente, nel 2008, sempre Miller continua lo studio sugli acidi idrossammici e sulla sintesi di molecole biologicamente rilevanti partendo dal substrato
1.46, sviluppando un metodo semplice e versatile per la riduzione del legame N-O.20 Come fonte di titanio (III) non usa più il titanio tricloruro ma il titanocene monocloruro (Cp2TiCl)
N-idrossicarbammati come composti chiave nella sintesi di nucleosidi carbociclici, antibitioci β-lattamici e benzodiazepine.
N O n O R 1.46 Cp2TiCl2 (2.5 eq.) Zn ( 5eq.) THF, MeOH -30°C, 45min HO n N O R H 1.47
Schema 1.3.13
Il cicloaddotto 1.46 con R=Ph e n=1 è aggiunto ad una soluzione fredda di Cp2TiCl2 e
zinco e in 45 minuti si forma l’aminociclopentenolo 1,4-syn come unico prodotto con resa del 95% e anche gli altri bicicli, con R= CH2Ph, OC(CH3)3, 4-NO2Ph e n=1, danno gli
stessi risultati anche se con rese più basse. Il titanio (III) è in grado di ridurre i gruppi nitro e si osserva che i sistemi ossazinici si riducono preferibilmente in presenza di un gruppo nitro aromatico, mentre il cicloaddotto che contiene un gruppo nitro aromatico, se sottoposto alla reazione con Cp2TiCl fornisce una miscela complessa e non si ottiene il
prodotto desiderato.
Anche i cicloaddotti con R= CH2Ph, OC(CH3)3 e n=2 danno come prodotti
amminocicloesenoli 1,4-syn con buone rese (Schema 1.3.13).20
Il meccanismo di riduzione proposto è che un singolo elettrone è trasferito dal Cp2TiCl a 1.46 e determina la rottura del legame N-O e genera il radicale intermedio 1.48; un
secondo equivalente di Cp2TiCl trasferisce un altro elettrone alla specie radicale per
formare il prodotto 1.49 che è protonato in presenza di MeOH/H2O per dare il prodotto 1.47 (Schema 1.3.14). N O n O R 1.46 Cp2TiCl N O R ClCp2TiO 1.48 Cp2TiCl N ClCp2TiO R OTiCp2Cl 1.49 MeOH H2O HO N O R H 1.47
Proprio in quest’anno William Tam e i suoi collaboratori si sono occupati di considerare le reazioni di apertura del composto 3-aza-2-ossabiciclo[2.2.1]ept-5-ene 1.50 con alcoli usando come catalizzatore il rutenio.21 Considerando il substrato 1.50, a differenza dei catalizzatori quali Pd(0), Pd/Cu(II) o acidi di Lewis come Fe(III) o Cu(II) che portano alla formazione di 1,4-ciclopenteni, con il rutenio si ottengono 1,2-ciclopenteni. In particolar modo con il catalizzatore a base di rutenio neutro Cp*RuCl(COD) si ottiene il ciclopentene 1,2-trans 1.51a mentre se si usa un catalizzatore a base di rutenio cationico [CpRu(CH3CN)]PF6 si forma preferibilmente il ciclopentene
1,2-cis 1.51b (Schema 1.3.15). N O O OtBu Ru-catalizzatore MeOH 60°C, 24h MeO N OH O OtBu 1.50 1.51a 1,2-trans MeO N OH O OtBu 1.51b 1,2-cis
Schema 1.3.15
Vengono poi considerati gli effetti della temperatura, del tempo di reazione e se si usano o meno cosolventi quando si utilizza il catalizzatore neutro; poca conversione si osserva a 25 °C, invece a 40 °C la reazione in 28h è completata e fornisce l’addotto trans come unico prodotto; aumentando a 60 °C si ha un decremento delle rese. Da notare è che se si tratta il substrato 1.50 sempre con lo stesso catalizzatore in THF o DCE a 40-60 °C in assenza di MeOH si osserva la decomposizione del prodotto; quando si usa un cosolvente come THF o DCE ma in rapporto 1:1 con il metanolo allora la reazione procede e fornisce dei risultati simili a quando si usa il metanolo da solo. Comunque aumentando il cosolvente la resa totale scende; usando il toluene la resa è praticamente zero e usando l’esano o l’1,4-diossano si forma anche un pò di addotto cis.
Ulteriori studi dimostrano che quando si usa il catalizzatore neutro Cp*RuCl(COD) (cat.A, Schema 1.3.16) con tutti gli alcoli nucleofili si ottengono esclusivamente i prodotti
decomposizione. Quando viene usato il catalizzatore cationico (cat.B, Schema 1.3.16) la reazione è ancora altamente regioselettiva e si ottiene il prodotto di apertura 1,2. Se come nuclefilo si usa il metanolo si ottiene solo il prodotto 1,2-cis, mentre con tutti gli altri alcoli si ha una miscela di isomeri trans/cis a favore dei cis comunque. In ogni caso gli isomeri
cis e trans sono facilmente separabili per cromatografia.21
N O O OtBu 1.50 Ru = cat. A Ru = cat. B cat.A N O O OtBu + Ru 1.54 N O Ru OtBu O MeOH MeO N OH O OtBu 1.51a 1,2-trans 1.55 cat.B N O O OtBu Ru + 1.52 N O O OtBu Ru + MeOH 1.53 MeO N OH O OtBu 1.51b 1,2-cis
Schema 1.3.16
Secondo il meccanismo proposto (Schema 1.3.16) il catalizzatore neutro preferisce coordinarsi all’ossigeno del biciclo e si ottiene l’intermedio 1.54 e l’inserzione del Ru sul legame C-O fornisce l’intermedio 1.55; il nucleofilo MeOH attacca poi la faccia meno ingombrata, cioè la faccia eso del frammento del biciclo e si ottiene l’addotto trans. Invece
l’intermedio 1.52; si forma un complesso π-allile seguito dall’addizione del metanolo dalla faccia endo per dare l’addotto 1,2-cis.
1.3.2. Reazioni di trasposizione
In questo stesso anno Miller si è occupato di reazioni di riarrangiamento ispirandosi al lavoro di Procter, il quale nel 1995 aveva individuato un procedimento molto utile dal punto di vista sintetico per quanto riguarda i riarrangiamenti di sistemi ossazabiciclici indotti da acidi.23 Procter era partito dall’effettuare cicloaddizioni asimmetriche di intermedi acilnitroso che portavano, come ausiliario chirale, una unità di acido mandelico per poter avere sotto controllo la stereochimica assoluta di queste cicloaddizioni; il trattamento di tali cicloaddotti, derivanti dal cicloesadiene, con acidi in condizioni anidre e blande, serviva per rimuovere l’ausiliario chirale, ma se era esposto il substrato 1.56 a queste stesse condizioni, si otteneva la totale decomposizione del biciclo. Per cui, a seguito dei risultati ottenuti, venne trattato il composto 1.56 con un acido diluito in soluzione acquosa e si ottenne un prodotto cristallino come singolo diastereoisomero 1.57 e in alta resa; la struttura e la stereochimica di tale prodotto venne dedotta dalle proprietà fisiche e chimiche, confermate dai raggi-X (Schema 1.3.17).
N O O Ph H HO 1.56 6N HCl/diossano (1:1) t.a., 96% NH2 HO O O OH Ph Cl 1.57
Schema 1.3.17
Lo Schema 1.3.18 spiega il meccanismo proposto con cui è stato ottenuto 1.57; il successo di questo procedimento sintetico è attribuibile alle condizioni acquose acide utilizzate, a differenza delle condizioni anidre che non permetterebbero di ottenere il prodotto desiderato e questo è ben comprensibile dall’osservazione dello Schema. Inoltre, è
Secondo il meccanismo proposto da Procter, l’aspetto fondamentale di questa sintesi era appunto la formazione dell’intermedio tetraedrico 1.56a che poi idrolizzava diventando
1.56b; e questo era possibile per la presenza dell’acqua. Presumibilmente gli equilibri
mostrati nello Schema 1.3.18 erano guidati dalla formazione irreversibile del cloridrato
1.57. Era evidente quindi che la metanolisi, condotta con metanolo anidro in acido
cloridrico, non portava al prodotto 1.57 o a relativi prodotti; infatti l’intermedio tetraedrico ottenuto con metanolo, analogo a 1.56a, in questo caso non era in grado di idrolizzarsi per dare il corrispondente estere, al contrario di quanto accade con la reazione condotta in presenza di acqua. N O O Ph H H HO N O O Ph H OH H N HO O Ph H HO N HO O Ph H HO H2O N HO O Ph H HO HO HO NH O O OH Ph HCl NH2 HO O O OH Ph Cl 1.57 1.56 1.56a 1.56b
Schema 1.3.18
Miller, partendo dall’esempio di Procter ha studiato un processo sintetico per poter effettuare reazioni di riarrangiamento di bicicli ossaza. Nel suo lavoro trattando il substrato
1.58 con 35 mol% di acido p-toluensolfonico in diclorometano a temperatura ambiente è
stato ottenuto l’idrossammato 1.58a con una bassa resa (Schema 1.3.19).22
O N Boc H+ O N O OH 1.58 1.58a resa del 20%
Schema 1.3.19
Con acidi più forti si sono ottenute maggiori rese dell’idrossammato 1.56a; rispetto all’acido p-toluensolfonico, quello triflico fornisce completa conversione e rese più alte in meno tempo; l’acido trifluoroacetico non è risultato buono per questo scopo; invece l’uso dell’acido triflico nella quantità di 2 mol% in THF a 0° C è risultato essere ottimo per ottenere il prodotto di riarrangiamento, con alta resa e purificato triturandolo con etere.
O N O OtBu 1.58 H+ O N O OtBu H 1.59 N OH O OtBu 1.60 O N O OH H A 1.61 O N O OH 1.58a H+ O N O OtBu H + 1.62
Schema 1.3.20
Lo Schema 1.3.20 illustra il meccanismo di questo tipo di reazione; avviene la protonazione del cicloaddotto 1.58 e si ottiene 1.59 o 1.62; quello che interessa è la formazione della specie 1.59 che subisce il cleavage del legame C-O per dare 1.60; quest’ultimo infine subisce una ciclizzazione intramolecolare e si ottiene 1.61. Come ultimo passaggio segue la perdita di isobutilene per dare l’idrossammato 1.58a e si rigenera il catalizzatore acido.
Se invece di N-Boc abbiamo N-Cbz, è più difficoltosa la perdita del gruppo benzile e il corrispondente idrossammato viene ottenuto con rese basse (Schema 1.3.21).
N Cbz
O N
O OH
TfOH (5 mol%) conversione incompleta +
Incoraggiati da questi risultati gli autori hanno provato queste reazioni su bicicli di maggiori dimensioni ma non è stata osservata nessun tipo di reazione recuperando solo il materiale di partenza. Gli autori spiegano questo considerando che in questi ultimi diminuisce molto la tensione dell’anello (Schema 1.3.22).
O
N Boc TfOH (5 mol%) CH2Cl2, t.a. n n=2, 1.63a n=4, 1.63b è recuperato solo il materiale di partenza
Schema 1.3.22
Gli autori poi hanno voluto studiare se, nel caso di un ossazabiciclo che presenta sull’azoto non un Boc ma un fenile o un benzile, si formi una specie nitrone invece di formarsi l’idrossammato come nel caso invece del cicloaddotto 1.58a.22
O N 1.64 (R=Ph) 1.31 (R=Bn) TfOH CH2Cl2, t.a. O N R O 1.64a (R=Ph) 1.31a (R=Bn) O R
Schema 1.3.23
Partendo dai cicloaddotti 1.64 e 1.31 e usando le condizioni precedenti non si osservano nitroni nella miscela di reazione ma aumentando gli equivalenti di acido triflico si forma
1.64a anche se in bassa resa mentre il cicloaddotto 1.31a si decompone. Questo
probabilmente è dovuto al fatto che il nitrone 1.64a è più stabile per la struttura di risonanza, come aveva proposto Procter nel caso dei bicicli derivanti dall’acido mandelico.23
Una buona parte di questa tesi sperimentale è stata dedicata allo sviluppo di nuove metodiche che consentono l’alchilazione con reagenti organometallici dei meno reattivi addotti acilnitroso [2.2.2]. Di questa classe di composti assai poco studiata sono state anche esaminate reazioni di trasposizione catalizzate da acidi di Lewis.