3
M
ATERIALI E METODI
3.1 POLIETILENE
Il polimero con il quale è stato scelto di miscelare l’idrolizzato proteico è il polietilene a bassa densità, un materiale termoplastico molto diffuso, di basso costo, trasparente tendente al bianco traslucido, con una struttura a catena ramificata. Inoltre è dotato di buona flessibilità, resistenza alla corrosione e bassa permeabilità al vapore d’acqua.
Nella prima parte del presente studio sono stati utilizzati due tipi di polietilene a bassa densità, sotto forma di pellets denominati PE1 e PE2.
Il PE1 presenta le proprietà riportate in Tabella 3.1.1:
Nome commerciale Basell Lupolen 2426 F LDPE
Densità (g/cm3) 0,924
Assorbimento di acqua 0,01%
Temperatura di fusione (°C) 111
Tabella 3.1.1: Proprietà del PE1
Il PE2 presenta le proprietà riportate in Tabella 3.1.2:
Nome commerciale Basell Lupolen 3020 H LDPE
Densità (g/cm3) 0,927
MFR - 2.16kg/190°C (g/10min) 2,0
Temperatura di fusione (°C) 118
Nella seconda parte della sperimentazione i due tipi di idrolizzato proteico sono stati miscelati con un polietilene lineare a bassa densità disponibile in polvere, denominato PE, le cui proprietà sono state riportate in Tabella 3.1.3
Nome commerciale Orene 4-3545
Densità (g/cm3) 0,935
MFR - 2.16kg/190°C (g/10min) 4,5
Temperatura di fusione (°C) 120
Tabella 3.1.3: Proprietà del PE
3.2 PREPARAZIONE FILM
3.2.1
Mescole in brabender
Le mescole polimeriche sono state preparate a partire dal materiale fuso in un miscelatore Brabender, uno strumento progettato e utilizzato per miscelare polimeri termoplastici ed elastomeri, eventualmente con additivi e cariche, ed equipaggiato con un sistema riscaldante dotato di un adeguato controllo di temperatura.
Il Brabender è costituito da una camera di miscelazione riscaldata avente sezione “ad otto” e contenente una coppia di viti controrotanti che possono ruotare fino ad una velocità massima di 200rpm con un gioco ridotto tra i rotori e la parete della camera in modo da favorire il mescolamento dei componenti .
La temperatura di lavoro e la velocità di rotazione vengono scelte in funzione dei componenti, generalmente si lavora ad una temperatura non eccessivamente superiore alla temperatura di fusione del materiale per evitare fenomeni di termodegradazione. Le miscele oggetto del presente studio sono state ottenute utilizzando uno strumento Brabender Plastigraph, operando a 130°C per 5 minuti a una velocità di rotazione di 50rpm.
I materiali, prima di essere processati, sono stati essiccati in stufa a 40°C sottovuoto per una notte, ed in seguito sono stati mantenuti in essiccatore.
3.2.2
Film tramite termopressa
I film sono stati ottenuti utilizzando una termopressa idraulica, uno strumento dotato di due piastre riscaldate di cui la superiore è fissa e quella mobile inferiore viene premuta contro la piastra superiore con un pistone idraulico garantendo una forza di compressione massima di 125kN.
La pressa è provvista anche di un sistema di raffreddamento a cassette, grazie al quale vengono posizionate sul film caldo contenuto tra due piastre di acciaio due cassette in alluminio, all’interno delle quali scorre acqua di rubinetto che raffredda il film polimerico e isolate termicamente nella zona di contatto con le piastre calde, permettendo di mantenere le piastre riscaldanti alla temperatura desiderata per un successivo riutilizzo.
I film del materiale in esame sono stati realizzati usando una termopressa Collin P200E (Figura 3.2.2.1) operando a pressioni di 100bar e temperature pari a 130°C per 5minuti e raffreddando per 4minuti.
3.3 METODI D’ANALISI
Per determinare le proprietà chimico-fisiche nonché la morfologia di un materiale è possibile ricorrere a vari metodi d’analisi. Le tecniche utilizzate nel presente studio sono le seguenti:
• prove meccaniche: curve sforzo-deformazione, • microscopia elettronica a scansione (SEM), • analisi termogravimetrica (TGA),
• analisi calorimetria differenziale a scansione (DSC), • analisi termica dinamico meccanica (DMTA), • spettrometria infrarossa (IR),
• valutazione della biodegradabilità, • termodegradazione,
• valutazione del contenuto di umidità.
3.3.1
Prove meccaniche: curve sforzo-deformazione
Le prove meccaniche permettono di determinare le proprietà di tensione e deformazione del materiale in esame, dopo aver definito le caratteristiche geometriche del provino e il tipo di forza applicata.
Lo strumento utilizzato per eseguire le prove meccaniche è costituito da un basamento su cui è posta una pinza fissa e due colonne sulle quali scorre la traversa mobile nella quale è situata l’altra. Tali pinze hanno potere autobloccante, cioè aumentano la presa al procedere della deformazione per azione di un comando pneumatico. Esse possono avere una superficie rigata, dentellata o liscia, in quest’ultimo caso, per migliorare la presa, il campione può essere posizionato sullo strumento interponendo tra esso e la superficie delle pinze un materiale abrasivo.
Il campione deve essere allineato con la direzione in cui è applicato lo sforzo in modo che non ci siano movimenti rotatori che possano indurre scorrimento.
Durante la prova viene fatta variare la distanza tra le pinze e tramite una cella di carico, posta tra la pinza superiore e la guida, viene registrato lo sforzo che il provino offre in termini di resistenza.
Il software che gestisce lo strumento permette di impostare la velocità di deformazione ed i vari criteri per il calcolo a posteriori delle caratteristiche meccaniche. Per effettuare la prova è necessario caratterizzare il provino dal punto di vista geometrico, poiché queste informazioni vengono usate dal software per standardizzare i valori di ritorno dello strumento e fornire le caratteristiche meccaniche relative all’unità di sezione. Le proprietà tensili di maggior interesse (Figura 3.3.1.1) sono:
• carico di rottura: sollecitazione alla quale avviene la rottura;
• carico di snervamento: punto di massimo della curva sforzo-deformazione che incorre alla fine del tratto di curva a comportamento elastico;
• modulo di Young: rapporto tra il carico nominale (calcolato in riferimento alla sezione iniziale) e la deformazione misurati nel tratto lineare della curva sforzo-deformazione;
• deformazione a rottura: rapporto tra l’allungamento a rottura e la lunghezza iniziale; • deformazione a snervamento: deformazione nel punto di snervamento.
Figura 3.3.1.1: Curva sforzo-deformazione per un materiale polimerico
Le proprietà tensili variano al variare della preparazione del provino, della velocità di deformazione adottata e delle condizioni ambientali perciò, dovendo effettuare comparazioni è necessario tener conto anche di queste variabili.
Figura 3.3.1.2: Strumento utilizzato per le prove meccaniche
L’analisi è stata eseguita utilizzando il modello Instron 5564 con velocità di deformazione pari a 10 mm/min, testando dodici campioni per ogni tipo di materiale. La prova è stata effettuata nel rispetto delle normative ASTM D638M e ASTM D882 relative ai metodi per i test di proprietà tensili di film sottili.
3.3.2
Microscopia elettronica a scansione (SEM)
Il microscopio elettronico a scansione (SEM) è uno strumento operante in condizioni di vuoto spinto nel quale il campione viene fatto interagire con un fascio di elettroni ad elevata energia (elettroni primari). Dalle modificazioni provocate dagli elettroni nella struttura atomica del preparato vengono originati e raccolti numerosi segnali, utilizzabili per la formazione d’immagini della struttura morfologica del campione e della sua composizione chimico-fisica.
Per l’esecuzione dell’analisi chimica il principio di funzionamento del SEM si basa sull’interazione tra un fascio di elettroni che bombarda il campione ed il campione stesso. Per effetto di questa interazione il campione emette una serie di segnali che sono caratteristici della sua composizione chimica. Infatti, quando elettroni veloci bombardano un materiale, entrano in esso e lo ionizzano causando l’emissione di un elettrone dagli orbitali interni; l’atomo, che è quindi energeticamente instabile, decade tramite passaggio di un elettrone appartenente ad un orbitale più esterno il quale va ad occupare la lacuna formatasi: il salto energetico effettuato si traduce nell’emissione di un fotone X di energia pari alla differenza di energia tra gli orbitali interessati. Questo processo determina una lacuna in un orbitale ancora superiore, per cui si ha un’ulteriore transizione a questo livello con emissione di un nuovo fotone X di energia pari a questo nuovo salto.
Queste transizioni tra livelli atomici danno dunque luogo ad un insieme di raggi X distribuiti secondo uno spettro discreto di energie dette “righe caratteristiche” univocamente attribuite a quell’elemento essendo così possibile rilevare la presenza dell’elemento stesso in un campione.
Il modello di SEM da noi utilizzato è JEOL 5600LV che utilizza come sorgente elettronica quella ottenuta dal riscaldamento di un filamento di tungsteno. La temperatura a cui viene portato il filamento al fine di ottenere un’emissione costante è dell’ordine di 2500-2600K. Il filamento riscaldato emette elettroni in ogni direzione: l’indirizzamento verso una particolare direzione ed il controllo delle emissioni viene effettuato mediante un dispositivo metallico che lo circonda detto “cilindro di Wehnelt”. I segnali emessi dal campione possono essere di vario tipo (Figura 3.3.2.1), tra i più importanti si possono citare:
• i raggi X, fondamentali per l’analisi chimica.
Figura 3.3.2.1: Segnali generati dall’interazione tra elettroni e campione
Volendo indagare la morfologia di superficie di un campione, il segnale più frequentemente utilizzato è quello prodotto dagli elettroni secondari (SE). L’interazione del fascio elettronico primario con gli elettroni degli orbitali esterni del preparato provoca, a seguito di trasferimento di energia cinetica, l’allontanamento degli stessi elettroni di valenza. L’elettrone espulso, denominato elettrone secondario, possiede un’energia non superiore a 50 eV.
Il segnale, originato a seguito dell’interazione, viene raccolto da un opportuno rilevatore e trasferito alla griglia di controllo di un oscilloscopio a raggi catodici (CRT). La modulazione prodotta permette di regolare l’intensità del fascio elettronico dell’oscilloscopio stesso in funzione della quantità di segnale ricevuto, ottenendo un’immagine corrispondente sullo schermo del CRT. Esiste una perfetta corrispondenza tra il segnale proveniente dal campione e l’immagine ottenuta sullo schermo poiché il trasferimento sequenziale del pennello elettronico sul preparato viene prodotto da un generatore di scansione che contemporaneamente agisce in modo sincrono sull'avvolgimento di deflessione del fascio elettronico dell’oscilloscopio.
Il sistema che genera e trasferisce il fascio elettronico primario ed il campione stesso devono essere contenuti in un ambiente ad un elevato grado di vuoto, in modo da ridurre l’interazione tra elettroni e molecole di gas.
E’ possibile in definitiva considerare il SEM composto da diversi sistemi: • un sistema di illuminazione del campione;
• un sistema di rilevazione e trasferimento del segnale; • un sistema di produzione e registrazione dell’immagine; • un sistema del vuoto.
Si riporta in Figura 3.3.2.2 uno schema a blocchi di un microscopio a scansione elettronica.
3.3.3
Analisi termiche
Con il termine termoanalisi si intende quel gruppo di tecniche analitiche aventi in comune il seguente principio operativo: registrare una o più proprietà chimico fisiche in funzione della temperatura, quando il campione viene riscaldato o raffreddato secondo un determinato programma, o del tempo se il campione viene mantenuto a temperatura costante.
Le tecniche più comunemente impiegate devono soddisfare tre principi base:
a) la proprietà fisica di interesse e la temperatura del campione devono essere misurate in maniera continua;
b) detti parametri devono essere registrati in maniera automatica;
c) la temperatura del campione deve aumentare o diminuire con velocità nota e uniforme entro certi intervalli di temperatura.
L’interpretazione di una curva termoanalitaca consiste nel mettere in relazione un certo andamento della particolare proprietà chimico fisica di interesse con possibili trasformazioni che hanno luogo nel campione, quali reazioni chimiche o processi fisici come passaggi di stato.
Le tecniche termoanalitiche possono essere classificate in tre gruppi a seconda della modalità di registrazione del parametro di interesse:
a) registrazione del valore assoluto dalla proprietà misurata (TGA);
b) registrazione della differenza di una qualche proprietà tra il campione e una sostanza di riferimento, in tal modo si effettua una misura differenziale (DSC, DTA);
c) registrazione della velocità con cui un dato parametro sta cambiando in funzione della temperatura o del tempo, in tal modo si effettua una misurazione derivativa (DTG).
3.3.3.1
Analisi termogravimetrica (TGA)
Un parametro fisico particolarmente adatto allo studio dei cambiamenti chimici in un materiale è la variazione di peso che si verifica quando un materiale interagisce con l’atmosfera circostante evolvendo o assorbendo gas. Nell’analisi termogravimetrica (TGA) viene infatti misurata la variazione di peso che si registra in conseguenza del riscaldamento o raffreddamento di un certo campione secondo un determinato programma di temperatura.
Il campione del materiale in esame viene posto in un ambiente a temperatura controllata su un apposito sistema di pesatura in cui le variazioni di temperatura e di peso sono registrate in continuo. L’apparecchiatura necessaria deve quindi essere dotata di una bilancia di precisione e di un forno riscaldato elettricamente la cui temperatura possa essere controllata con sufficiente precisione.
Negli strumenti moderni il forno è alimentato elettricamente attraverso un riscaldamento resistivo, le resistenze sono posizionate attorno ad un supporto tubolare, isolato elettricamente, che è un buon conduttore termico all’interno del quale viene posto il campione.
L’esterno della fornace deve essere isolato, inoltre di norma sono presenti specifici sistemi di raffreddamento ad aria (ventole) e/o termostatazione a circolazione di un opportuno fluido refrigerato (termostatato).
L’atmosfera intorno al campione può essere condizionata mediante l’immissione controllata di un gas con l’ausilio di opportune valvole e di un sistema di vuoto, in tal modo è possibile condurre prove sotto vuoto, in atmosfera statica o sotto flusso di gas che, a seconda delle esigenze, può essere inerte, riducente o ossidante e per certe particolari applicazioni anche corrosivo. Il principale svantaggio che si ha utilizzando un’atmosfera statica deriva dalla possibile condensazione dei prodotti di reazione sulle parti più fredde dello strumento, ciò può causare corrosione nei meccanismi della bilancia o errori di pesatura se essi si depositano sugli elementi della bilancia. Inoltre in questo caso si possono avere reazioni secondarie tra i prodotti e il campione residuo. La granulometria del campione influenza la perdita di peso dal momento che ha effetto sulla diffusione dei volatili e il trasferimento di calore attraverso il campione (Figura 3.3.3.1.1).
Figura 3.3.3.1.1: Dipendenza della curva TG dalla granulometria del campione
I campioni possono essere in forma di solido polverizzato (compresso sotto forma di pellet o sparso finemente sopra la superficie del crogiolo), di film sottile o di liquido. Generalmente sono preferiti i liquidi e le polveri fini, queste ultime non dovranno essere però eccessivamente fini poiché altrimenti potrebbero essere asportate dal flusso di gas.
Il peso del campione, come la sua forma, può influenzare la curva TG incidendo sulla diffusione, e sul trasferimento di calore (Figura 3.3.3.1.2).
Quando si opera con campioni di peso intorno a pochi milligrammi la spinta idrostatica del gas sul campione o la presenza di correnti convettive possono manifestarsi come disturbi nella curva termogravimetrica o apparenti variazioni di peso. In particolare scaldando un portacrogiolo vuoto la spinta idrostatica causa un apparente aumento di peso pari al peso del gas spostato dal portacrogiolo. Poiché la temperatura cambia, anche la densità del gas cambia e ciò provoca una variazione della spinta idrostatica (galleggiamento) che si traduce in una apparente variazione di peso anche se di norma trascurabile. Per questo motivo per analisi accurate è necessario effettuare delle prove in bianco nelle stesse condizioni di analisi e sottrarre dalla curva dell’analisi quella della prova in bianco.
Una curva termogravimetrica di norma mostra, in un graficoin cui si riporta la perdita di peso in funzione della temperatura, una serie di scalini più o meno accentuati che possono essere separati da tratti ad andamento costante del peso; alcuni processi però possono avvenire in un intervallo di temperatura più ampio tale da sovrapporsi ad altri fenomeni così da rendere non risolti i vari stadi di perdita di peso. In questi casi risulta utile ricorrere alla termogravimetria derivativa (DTG) grazie alla quale è possibile individuare una serie di picchi che corrispondono ai vari stadi della perdita di peso e i cui massimi corrispondono ai punti di flesso della curva TG (Figura 3.3.3.1.3). Inoltre l’area dei picchi è proporzionale alla perdita di peso di ogni singolo stadio.
Figura 3.3.3.1.3: Curva TG ideale, curva TG reale, curva DTG
Il punto in cui la curva TG devia da un andamento orizzontale è definito come punto di inizio di una reazione, quello in cui la curva ritorna ad avere un andamento orizzontale è il punto di fine reazione. Tali punti sono di norma difficili da determinare e quindi si ricorre ad elaborazioni via software estrapolando gli andamenti rettilinei prima e dopo tale punto e tenendo conto della loro intersezione.
L’aspetto della curva termogravimetrica dipende da alcuni parametri operativi quali velocità di riscaldamento, tipo di crogioli impiegati, caratteristiche del campione, tipo di atmosfera ecc.
Un riscaldamento veloce aumenta la temperatura alla quale ha luogo una reazione e alla quale la velocità di perdita di peso raggiunge il massimo (Figura 3.3.3.1.4), inoltre aumenta l’intervallo nel quale è osservata la perdita di peso; quest’ultimo effetto si verifica anche nel caso di bassa velocità di riscaldamento, in tal caso però diminuisce la temperatura alla quale appare la reazione.
Figura 3.3.3.1.4: Dipendenza della curva TG dalla velocità di riscaldamento
Tra i possibili impieghi della termogravimetria appare interessante lo studio del comportamento termico dei materiali in relazione alla loro degradazione. La degradazione termica provoca infatti lo sviluppo di sostanza volatili che si allontanano dal campione in fase gassosa, pertanto la termogravimetria si presta particolarmente bene per seguire l’evolversi delle materie volatili mentre un dato campione viene sottoposto a riscaldamento. Ciò permette di valutare la temperatura al di sopra della quale un dato materiale perde le sue originali caratteristiche e quindi permette anche di definire il campo di impiego entro certi intervalli di temperatura.
Le analisi termogravimetriche sono state effettuate utilizzando uno strumento Netzsch STA409; i campioni, di peso intorno ai 20mg, sono stati riscaldati in atmosfera di azoto, per evitare l’ossidazione, da temperatura ambiente a 700°C con velocità di riscaldamento di 10°C/min ed è stato utilizzato un crogiolo di Pt di circa 1cm di
Lo strumento consente di registrare la variazione del peso del campione in funzione della temperatura, in tal modo si ricavano le curve termogravimetriche (TG); per derivazione via software di queste ultime si ottengono le curve DTG dette profili di perdita di peso.
3.3.3.2
Calorimetria differenziale a scansione (DSC)
Un gruppo particolarmente utile di tecniche termoanalitiche è basato sulla rilevazione delle variazioni di entalpia o calore specifico di un campione in funzione della temperatura.
Fornendo energia al campione aumenta la sua entalpia e di conseguenza anche la sua temperatura dipendente dal suo calore specifico. Il calore specifico di un materiale cambia lentamente con la temperatura in un particolare stato fisico ma varia discontinuamente in un passaggio di stato.
All’aumentare della temperatura l’energia termica fornita può indurre processi chimici o fisici nel campione accompagnati da variazioni di entalpia (calore latente di fusione,ecc.), tale variazione di entalpia può essere rilevata attraverso un’analisi termica e messa in relazione con il processo che sta avvenendo nel campione. In tal modo è possibile ottenere informazioni sia sulla transizione vetrosa, associata alla componente disordinata presente nel materiale, che su tutte le transizioni di fase che interessano la componente ordinata.
Le più importanti tecniche basate su cambiamenti di entalpia sono: l’analisi termica differenziale (DTA) e la calorimetria differenziale a scansione (DSC);in entrambi i casi il sistema è costituito da un forno riscaldante, due portacrogioli (uno per il campione e uno per il materiale di riferimento) posizionati simmetricamente intorno all’asse della fornace cilindrica e una serie di termocoppie per la registrazione della temperatura del campione e del materiale di riferimento.
Nella DTA il campione e il materiale di riferimento sono riscaldati in uno stesso ambiente dal medesimo forno, la sostanza di riferimento non subisce eventi termici nel range di temperature studiato quindi la sua temperatura è esattamente quella impostata dal programmatore, mentre quella del campione aumenta o diminuisce rispetto a quella programmata a seconda che questo subisca un evento rispettivamente esotermico o
endotermico ; in questo caso si registra la differenza di temperatura tra campione e riferimento.
Nel caso della DSC invece il campione e la sostanza di riferimento sono mantenuti alla stessa temperatura da due riscaldatori separati (Figura 3.3.3.2.1), ciò significa che in corrispondenza di determinati eventi termici il sistema reagisce fornendo più o meno energia al campione rispetto al riferimento in modo da compensare istantaneamente l’effetto endotermico o esotermico verificatosi.
Figura 3.3.3.2.1: Schematizzazione di un calorimetro differenziale a scansione (DSC)
In questo caso si registra la differenza di energia elettrica fornita tra campione e riferimento e tale parametro sperimentale figura come ordinata della curva DSC; l’area dei picchi che rappresentano le transizioni è proporzionale alla variazione di entalpia (Figura 3.3.3.2.2). Effettuando una misura con i crogioli vuoti è possibile ottenere la linea di base; tale linea teoricamente dovrebbe risultare orizzontale ma nella pratica non lo è poiché riflette le lievi differenze costruttive esistenti tra la parte dello strumento in cui si trova il campione e quella in cui si trova il riferimento. La presenza di un campione provoca uno spostamento della linea di base proporzionale al suo calore specifico inoltre un cambiamento del calore specifico con la temperatura dà una linea di base inclinata. Questa può risultare inclinata anche se esiste una considerevole differenza di emissività tra il campione e il riferimento, questo effetto è particolarmente marcato alla alte temperature ma può essere eliminato usando crogioli muniti di coperchio con un piccolo foro al centro.
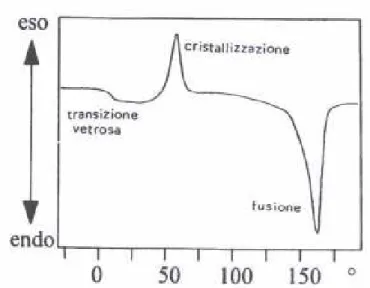
Figura 3.3.3.2.2: Segno dei picchi eso/endotermici per un generico materiale
La forma e la posizione dei picchi eso- ed endotermici è influenzata da vari fattori, tra questi figurano la massa, la granulometria, l’impaccamento del campione e la velocità di riscaldamento; è opportuno usare piccole quantità e strati sottili di materiale per sfavorire gradienti di temperatura ed avere un buon contatto termico. Inoltre poichè l’energia fornita o sottratta è correlata alla percentuale di sostanza coinvolta nella transizione, il valore del calore di fusione/cristallizzazione cresce al crescere della percentuale di cristallinità del campione.
Una particolare applicazione della DSC consiste nella determinazione della temperatura a cui ha luogo la transizione vetrosa. I parametri che caratterizzano la transizione vetrosa sono: la temperatura di transizione vetrosa (Tg) e l’aumento di calore specifico ∆cp. Dal valore sperimentale di ∆cp, se è noto l’incremento di calore specifico del campione totalmente amorfo ∆cpa , è possibile risalire al grado di cristallinità Wc
attraverso la relazione:
Wc=1-(∆cp/∆cpa)
Nel caso di transizioni di fase i parametri caratteristici sono: la temperatura del processo esaminato, normalmente ricavata dalla temperatura corrispondente all’inizio del picco eso- o endotermico, e la variazione di entalpia ∆H associata alla transizione. Facendo riferimento, per semplicità, al fenomeno della fusione in un polimero parzialmente cristallino, i risultati DSC vengono normalmente utilizzati per ottenere una stima del grado di cristallinità confrontando il valore di ∆Hf misurato sperimentalmente con
quello di un campione dello stesso polimero a cristallinità nota o con quello del polimero totalmente cristallino ∆Hfo
Wc=∆Hf/∆Hfo
Inoltre ogni apparecchiatura presenta delle costanti strumentali che è necessario conoscere per programmare in maniera corretta la prova DSC, esse si riferiscono al tempo di ritardo (τlag) cioè al ritardo con cui il campione ed il riferimento raggiungono
la temperatura programmata, e all’inerzia termica (τsignal) che fornisce una misura del
tempo necessario perché la curva DSC ritorni alla linea di base una volta completato il processo termico osservato.
Per lo studio delle proprietà termiche è stato usato un calorimetro differenziale a scansione (DSC 7 Perkin Elmer), i campioni di peso intorno a 3-5 mg sono stati sottoposti prima ad un riscaldamento da 50°C a 200°C in atmosfera di azoto con velocità di riscaldamento di 10°C/min e poi ad un raffreddamento fino a 50°C.
3.3.3.3
Analisi termico dinamico meccanica (DMTA)
L’analisi termica dinamico meccanica (DMTA) è una tecnica di indagine non distruttiva che permette di determinare le proprietà dinamico meccaniche di materiali che hanno un comportamento viscoelastico.
Questi materiali hanno sia le caratteristiche dei fluidi viscosi, cioè la capacità di dissipare energia e non immagazzinarla, che quelle dei solidi elastici i quali immagazzinano energia senza dissiparla. Quando un materiale polimerico viene deformato parte dell’energia viene immagazzinata come energia potenziale e parte è dissipata come calore, quest’ultima si manifesta come uno smorzamento meccanico (attrito interno) ed è alta nei polimeri viscoelastici amorfi.
Durante l’analisi il materiale viene sottoposto ad una sollecitazione e viene misurata la risposta (deformazione) al variare della temperatura e/o della frequenza, facendo attenzione a non superare la deformazione critica, rimanendo cioè nella regione in cui la risposta è lineare, e ad operare in un range di temperatura in cui il campione si mantiene
Le proprietà determinate sono:
• il modulo dinamico o modulo di immagazzinamento (E’); • il modulo di perdita (E”);
• lo smorzamento o attrito interno o tangente di perdita (tanδ=E”/E’).
La sollecitazione applicata può essere di vario tipo, in questo studio i campioni sono stati sottoposti ad una deformazione che varia sinusoidalmente con il tempo ottenendo così una forza che varia anch’essa con il tempo ma non è in fase (Figura 3.3.3.3.1).
ε = εosin(ωt)
σ = σosin(ωt+δ)
σ = σosin(ωt)cosδ+σocos(ωt)sinδ
con ω = frequenza di sollecitazione δ = angolo di sfasamento
Figura 3.3.3.3.1: Relazione tra i parametri usati per esprimere i risultati delle prove dinamico meccaniche
La forza applicata viene separata in due componenti, una in fase con la deformazione (σocosδ) e l’altra fuori fase di 90° (σosinδ) quindi è possibile valutare la componente del
modulo in fase (E’) e fuori fase (E”): σ=εoE’sin(ωt)+εoE”cos(ωt)
E ’= (σo/εo)cosδ
E” = (σo/εo)sinδ
Inoltre è possibile esprimere il modulo E attraverso un numero immaginario in cui la parte reale è costituita dalla componente in fase (E’) mentre la parte immaginaria è rappresentata dalla componente fuori fase (E”):
ε = εoexp(iωt)
σ = σoexp[i(ωt+δ)]
La parte reale E’ è chiamata modulo di immagazzinamento perché è legato all’energia immagazzinata come energia elastica durante la deformazione e liberata durante il rilascio; E’ è quindi una misura della rigidità del materiale.
La parte immaginaria E” è chiamata modulo di perdita ed è associata alla dissipazione di energia come calore quando il materiale è deformato.
Il rapporto tra l’energia dissipata e quella immagazzinata in ogni ciclo prende il nome tangente di perdita o smorzamento o attrito interno (tanδ = E”/E’).
Tali proprietà variano all’aumentare della temperatura: mentre E’ diminuisce, E” e tanδ mostrano dei picchi, corrispondenti ai punti di flesso della curva E’, che indicano cambiamenti delle proprietà del materiale; il materiale infatti subisce alcune transizioni durante le quali dissipa l’energia assorbita.
Per transizione si intende un rilassamento, un riarrangiamento interno in corrispondenza del quale l’energia in eccesso è dissipata come calore, poiché un segmento della catena che è libero di muoversi può immagazzinare meno energia rispetto ad un segmento congelato.
Al variare della temperatura nella curva tanδ è possibile individuare alcune transizioni (Figura 3.3.3.3.2):
• il picco α rappresenta la transizione che si trova alla temperatura più alta ed è dovuta a piccoli moti microbrowniani delle catene molecolari alla regione di interfaccia tra la parte amorfa e quella cristallina ed è perciò più grande nei polimeri amorfi rispetto a quelli semicristallini, perché questi ultimi impongono restrizioni ai segmenti di catena;
• il picco β, che si trova ad una temperatura di circa 0,75Tα, è associato a rilassamenti
della fase amorfa, movimento di gruppi laterali o di un piccolo numero di monomeri attorno alla catena principale;
• il picco γ è legato al movimento di piccoli gruppi laterali (es. metili) nella fase amorfa e nella fase cristallina, alla rotazione di gruppi terminali, movimenti di piccoli segmenti o gruppi della catena principale, difetti cristallini e separazione di fase di impurezze.
In alcuni casi può essere osservata un’altra transizione (picco αc) situata tra il picco α e
Figura 3.3.3.3.2: Proprietà dinamico meccaniche di un polimero amorfo
Un parametro importante nella caratterizzazione di un materiale è la transizione vetrosa (Tg), un intervallo di temperature in corrispondenza del quale il coefficiente di
espansione termica subisce un improvviso cambiamento (Figura 3.3.3.3.3), per cui il polimero amorfo passa da uno stato rigido vetroso ad uno gommoso, anche se non ancora liquido. Questa transizione avviene grazie all’energia termica entrata nel sistema che permette alle catene polimeriche della regione amorfa di muoversi e rotare intorno alla catena principale.
Figura 3.3.3.3.3: Andamento del volume specifico in funzione della temperatura per vari tipi di materiali
Le analisi termiche dinamico meccaniche sono state effettuate con uno strumento DMTA Rheometric Scientific V su campioni rettangolari aumentando la temperatura da -150°C a 120°C con velocità di riscaldamento di 4°C/min, frequenza costante pari a 1Hz e applicando una deformazione iniziale εo di 0,015.
3.3.4
Spettroscopia infrarossa (IR)
La spettroscopia infrarossa (IR) è una tecnica che si basa sull’interazione delle molecole con l’energia elettromagnetica nel campo delle radiazioni infrarosse comprese tra la regione del visibile e la regione delle microonde. La porzione di maggior interesse pratico per il chimico organico è quella limitata tra 4000 e 400 cm-1, poiché in tale regione cadono i più comuni moti vibrazionali delle molecole.
Un composto irradiato da una radiazione elettromagnetica assorbe energia a determinate lunghezze d’onda, corrispondenti a determinate vibrazioni di legami chimici, così irradiando il composto con l’energia associata a molte lunghezze d’onda e determinando quali vengono assorbite e quali vengono trasmesse è possibile ottenere lo spettro di assorbimento di quel composto organico.
Poiché un legame chimico irradiato con luce infrarossa assorbe energia per mettersi a vibrare solo se la frequenza della luce e la frequenza della vibrazione sono uguali,è possibile identificare vari tipi di legami presenti in un materiale confrontando picco per picco lo spettro del composto sconosciuto con i dati presenti nelle tabelle delle frequenze dei vari gruppi funzionali e quindi risalire alla struttura della molecola.
Lo spettro IR in realtà non è uno spettro a righe, come risulterebbe dai soli salti quantici dei livelli vibrazionali, bensì uno spettro a bande in quanto alle transizioni vibrazionali si sovrappongono quelle rotazionali di minor energia ma in numero molto maggiore creando un numero di righe elevato che dà appunto luogo alla banda IR.
Le intensità delle bande possono essere espresse sia come trasmittanza (T) che come assorbanza (A), la trasmittanza è il rapporto tra la potenza radiante trasmessa da un campione (I)e la potenza radiante incidente sul campione(Io), l’assorbanza è il logaritmo
in base 10 del reciproco dalla trasmittanza.
T=I/Io A=Log 1/T
La frequenza o la lunghezza d’onda dell’assorbimento dipendono dalle masse relative degli atomi, dalle costanti di forza dei legami e dalla geometria degli atomi. Le posizioni delle bande nello spettro IR sono indicate come numeri d’onda (
ν
) la cui unità di misura è il reciproco del cm (cm-1), questa unità è proporzionale all’energia vibrazionale, i moderni strumenti forniscono risultati lineari in cm-1. La lunghezzad’onda (λ) era espressa nella letteratura in tempi passati come micrometri. I numeri d’onda sono inversamente proporzionali alla lunghezza d’onda
λ
ν = 1 cm-1=104µm
Le vibrazioni dei legami indotte dalle radiazioni IR si possono classificare in:
• stretching (stiramento): movimento ritmico lungo l’asse di legame con conseguente aumento o diminuzione della distanza interatomica; si dice simmetrico se i legami si allungano o si accorciano contemporaneamente, asimmetrico se un legame si allunga mentre l’altro si accorcia (Figura 3.3.4.1);
stretching simmetrico stretching asimmetrico
Figura 3.3.4.1: Vibrazioni di stretching
• bending (piegamento): variazione di angolo nei legami con un atomo in comune o movimento di un gruppo di atomi rispetto al resto della molecola senza che gli atomi nel gruppo si muovano uno rispetto all’altro; tali vibrazioni possono avvenire in uno stesso piano e allora si distinguono in scissoring (simmetrico) o rocking (asimmetrico), altrimenti se avvengono fuori dal piano possiamo essere nel caso del wagging o del twisting (Figura 3.3.4.2).
wagging twisting Figura 3.3.4.2: Vibrazioni di bending
Una molecola ha tanti gradi di libertà quanti sono i gradi di libertà dei suoi singoli atomi, ogni atomo ha tre gradi di libertà corrispondenti alle coordinate cartesiane (x, y, z) necessarie per descrivere la sua posizione relativa agli altri atomi nella molecola. Una molecola di n atomi perciò ha 3n gradi di libertà; per le molecole non planari tre gradi di libertà descrivono la rotazione, altri tre descrivono la traslazione e i rimanenti 3n-6 gradi di libertà sono gradi di libertà vibrazionale anche detti vibrazioni fondamentali. Le molecole lineari hanno invece 3n-5 gradi di libertà essendo solo due i gradi di libertà richiesti per descrivere la rotazione.
Vengono osservate nell’IR solo quelle vibrazioni che portano ad un cambiamento ritmico del momento dipolare della molecola.
Il numero teorico di vibrazioni fondamentali (frequenze di assorbimento) sarà osservato raramente poiché gli overtone (multipli di una data frequenza) e i toni di combinazione (somma di altre due vibrazioni) aumentano il numero di bande mentre altri fenomeni lo riducono.
Il numero teorico delle bande può essere ridotto dai seguenti fenomeni: • frequenze fondamentali che cadono fuori della zona 4000-400 cm-1; • frequenze fondamentali che sono troppo deboli per essere osservate; • vibrazioni fondamentali che sono così vicine da confondersi;
• il manifestarsi di una banda degenere provocata da diversi assorbimenti della stessa frequenza in molecole molto simmetriche;
• l’incapacità di alcune vibrazioni fondamentali di apparire nell’IR dovuta alla mancanza di variazione del dipolo molecolare.
I calcoli delle frequenze di stretching possono essere approssimati applicando la legge di Hooke, in base alla quale due atomi ed il legame di connessione sono considerati come un semplice oscillatore armonico rappresentato da due masse legate da una molla. La seguente equazione, derivante dalla legge di Hooke, stabilisce la relazione tra la frequenza di oscillazione, le masse atomiche e la costante di forza del legame.
) ( * ) ( 2 1
M
M
M
M
y x y x f c + =π
ν
ν
= frequenza di vibrazione (cm-1)c = velocità della luce (cm/s)
f = costante di forza del legame (dine/cm) Mx = massa dell’atomo x
My = massa dell’atomo y
Il valore di f è approssimativamente di 5x105dine/cm per i legami singoli e circa il doppio ed il triplo di tale valore per i legami doppi e tripli.
Lo strumento utilizzato per la registrazione dello spettro è uno spettrofotometro infrarosso a trasformata di Fourier (FT IR). La radiazione emessa, contenente tutte le lunghezze d’onda IR, viene divisa in due raggi: il raggio di riferimento, che percorre una distanza fissa, e il raggio che passa attraverso il campione e percorre una distanza variabile. Se viene colpita una molecola vibrante con luce IR, la molecola stessa assorbirà le frequenze della luce che si combinano esattamente con le frequenze dei diversi oscillatori armonici che la formano. La luce residua, non assorbita da alcun oscillatore all’interno della molecola, viene trasmessa ad un rilevatore, che misura la differenza di intensità dei due raggi per ciascuna lunghezza d’onda, ed analizzata da un computer. Se la frequenza di una vibrazione della molecola della sostanza cade nell’intervallo percepito dallo strumento, la molecola stessa assorbirà energia di tale frequenza dal raggio di luce.
Figura 3.3.4.3: Principio di funzionamento dell’analisi FT IR
La spettrometria infrarossa a trasformata di Fourier presenta numerosi vantaggi: la trasformata di Fourier permette di convertire l’interferogramma ottenuto da grafico nel dominio tempo al familiare spettro nel dominio della frequenza, l’intero intervallo di radiazione viene fatto passare attraverso il campione simultaneamente con un notevole risparmio di tempo, inoltre poiché i dati sono sottoposti ad una conversione dall’analogico al digitale possono essere elaborati facilmente, i segnali di campioni puri o di solventi possono essere sottratti dallo spettro delle miscele e gli spettri possono essere stampati in numeri d’onda o in lunghezze d’onda a partire dallo stesso set di dati. Gli spettri infrarossi possono essere ottenuti da composti allo stato gassoso, liquido o solido.
Gli spettri dei gas o dei liquidi basso bollenti possono essere ottenuti facendo espandere il campione all’interno di una cella sotto vuoto.
I liquidi possono essere analizzati puri o in soluzione: nel primo caso il campione liquido viene analizzato disponendolo tra due pastiglie piane di cloruro di sodio (la compressione produce un film poiché le pastiglie vengono tenute insieme per capillarità); nel caso di soluzioni lo spettro che si ottiene è quello del soluto dato che una cella di riferimento contenente il solvente puro viene interposta nel cammino del raggio di riferimento così da eliminare i segnali del solvente.
I solidi sono generalmente analizzati come dispersione in olio, come pastiglia pressata o come film trasparente depositato.
La tecnica del pellet (disco pressato) si basa sul fatto che il bromuro di potassio anidro in polvere può essere compresso sotto vuoto per formare dei dischi trasparenti; il campione (0,5-1,0 mg) viene intimamente miscelato con 100mg di KBr anidro in polvere e la miscelazione può essere fatta attraverso un’accurata macinazione in un mortaio d’agata o con un piccolo mulino a palle; la miscela viene poi compressa con una pressa per dare un disco trasparente. La qualità dello spettro dipende dalla omogeneità e dalla finezza della miscelazione.
Queste tecniche basate sull’impiego del KBr possono essere semplificate attraverso l’uso di una Mini Press, procedendo come segue: la miscela campione e KBr viene collocata all’interno della cavità di un dado cieco e il campione viene compresso avvitando il bullone. La successiva rimozione del bullone consente di recuperare dalla cavità del dado il disco che verrà utilizzato per registrare lo spettro.
La tecnica del film depositato è utile solo quando il campione può essere cristallizzato da una soluzione o ottenuto, per raffreddamento del composto fuso, in forma microcristallina o di film vetroso.
Inoltre è stata messa a punto una tecnica denominata spettroscopia a riflessione totale attenuata (ATR) che consente di ottenere spettri di solidi prescindendo dallo spessore del campione. La tecnica si basa sul fatto che un raggio di luce, che è riflesso internamente da una superficie di un mezzo trasparente, attraversa una breve distanza oltre il limite di riflessione e ritorna al mezzo trasmittente come una parte del processo di riflessione. Se una sostanza avente indice di rifrazione inferiore rispetto al mezzo trasmittente viene posta a contatto con la superficie riflettente, la radiazione penetra nel campione per una profondità di pochi micrometri producendo uno spettro di assorbimento. Un’estensione di questa tecnica si avvale di riflessioni interne multiple lungo la superficie del campione. La tecnica di riflessione interna multipla produce spettri di intensità confrontabili agli spettri di trasmittanza.
I campioni oggetto del presente studio sono stati analizzati secondo la tecnica di riflessione attenuata ATR; successivamente sono stati ottenuti film abbastanza sottili e quindi è stato possibile realizzare l’IR in trasmittanza. Essi hanno comunque uno spessore ancora troppo grande per cui in alcuni casi certe bande vanno in saturazione. La spettrometria infrarossa sui due tipi di idrolizzato puro è stata effettuata secondo la
3.3.5
Valutazione della biodegradabilità
3.3.5.1
Introduzione
In questo periodo si sta compiendo un notevole sforzo per standardizzare metodi facilmente riproducibili atti a valutare quantitativamente la biodegradabilità dei polimeri.
Devono, però, essere considerati una grande varietà di parametri, non solo attinenti all’ambiente in cui avviene il processo ma anche al campione da biodegradare; quindi, il problema si presenta molto complesso.
Metodi standard sono stati pubblicati dalla ASTM e riguardano in particolare:
• biodegradazione aerobica ed anaerobica di materiali plastici mediante inoculo prelevato da fanghi attivati provenienti da impianti per la depurazione di acque reflue;
• biodegradazione aerobica di plastiche ad opera di microrganismi specifici; • biodegradazione aerobica di materiali plastici in condizioni controllate di
compostaggio.
In genere questi test consistono in misure indirette di biodegradazione e spesso conducono a risultati difficilmente riproducibili da laboratorio a laboratorio.
Considerando queste problematiche i campioni preparati sono stati sottoposti ad una prova di biodegradazione in condizioni aerobiche seguendo la procedura elaborata e sperimenta da E. Chiellini et al. [25]. Anche in tale metodo la misura è indiretta, però è utilizzato un terreno naturale, senza inoculo di nessun microrganismo da selezionare o testare, in tal modo è facilmente riproducibile.
Bisogna inoltre osservare che l’eventuale fallimento di un test di biodegradazione non esclude la possibilità che tale sostanza sia biodegradabile, indica soltanto che in quelle condizioni ambientali la biodegradazione non si è sviluppata. Saranno quindi necessari ulteriori prove, condotte in altre condizioni, prima di confermare la non biodegradabilità.
3.3.5.2
Descrizione metodo
Seguendo la metodologia citata [25], inizialmente deve essere preparata una miscela costituita di terreno, adeguatamente selezionato e setacciato, e di perlite; il ruolo di questo strato è quello di assicurare la presenza di microrganismi presenti nel terreno e garantire loro soddisfacenti condizioni di incubazione. I provini sono stati collocati all’interno di questa miscela, che a sua volta è stata posta tra due strati ciascuno con 10g di perlite umidificata con 15ml di acqua distillata.
Il ruolo di questo materiale è quello di garantire un’ottima aerazione e una fonte di umidità.
Tutto ciò è stato realizzato all’interno di barattoli cilindrici (vedi Figura 3.3.5.2.1) ciascuno dei quali contenenti uno specifico campione. Inoltre ogni tipo di mescola è stato ripetuto in tre recipienti diversi in modo da poter effettuare una media ed ottenere poi un risultato totale che sia statisticamente valido. Tutti i contenitori sono stati chiusi, posti al buio e mantenuti a temperatura ambiente.
Per quanto riguarda la quantità di campione da sottoporre alla prova, è stata rispettata la seguente proporzione: 25mg di materiale per grammo di miscela terreno e perlite.
Per catturare la CO2 sviluppata dai processi biodegradativi, in ciascun contenitore è
stato inserito un beaker contenente 40ml di una soluzione di KOH con normalità tra 0,05 e 0,1.
La soluzione basica serve per assorbire chimicamente la CO2 prodotta, perciò dovrà
essere sostituita e titolata secondo scadenze (normalmente ogni 7-14 giorni) da stabilire in funzione della produzione giornaliera di CO2 o del massimo contenuto di O2
Figura 3.3.5.2.1: Sistema adottato per i test di biodegradazione
Il bianco e la carta
La CO2 che risulta assorbita dalla soluzione basica non è prodotta solo ed
esclusivamente dai campioni per effetto dell’azione biodegradativa dei microrganismi presenti nel terreno, ma anche questo ultimo contribuisce alla produzione di CO2 dato
che rappresenta un ottimo substrato per gli stessi organismi.
Per risolvere questo problema sono stati preparati tre barattoli con all’interno soltanto la miscela con la perlite senza alcun campione, in questo modo, sempre attraverso le titolazioni, è possibile risalire alla quantità di CO2 prodotta solo da esso. Questi tre
contenitori sono stati denominati con la sigla bianco.
Questa informazione è molto importante dal momento che la CO2 realmente prodotta
per biodegradazione si ricava per differenza, cioè:
[CO2]biod, y = [CO2]recipiente, y - [CO2]bianco
Dove: [CO2]biod, y sono i mg CO2 prodotta per biodegradazione del campione y;
[CO2]barattolo, y sono i mg di CO2 prodotta nel recipiente, campione y e terreno;
[CO2]bianco sono i mg di CO2 ottenuta dal bianco (terreno).
Infine sono stati preparati tre ulteriori barattoli con dei filtri di carta inseriti all’interno del terreno che hanno la funzione di controllo positivo; come è ben noto, la carta è un materiale biodegradabile e quindi questa operazione è stata effettuata per verificare che in questi barattoli i microrganismi siano in grado di attuare processi degradativi.
terreno
perlite
campione
soluzione di KOH
Qualora non fosse misurata alcuna produzione di CO2 in questi recipienti sarebbe
evidente che sono stati commessi errori nella preparazione della prova. Nel caso contrario si avrebbe la certezza che la sperimentazione sia stata realizzata correttamente, perciò la mancata produzione di CO2 nei barattoli con i campioni di interesse
indicherebbe che essi non sono stati ancora attaccati dai microrganismi presenti.
La titolazione
All’interno dei contenitori in cui è stata realizzata la prova è stata posta una soluzione di KOH a concentrazione nota, così la CO2 sviluppata neutralizza parte della KOH
presente; una successiva titolazione per determinare la KOH residua permette di risalire alla quantità di carbonio metabolizzato e quindi di CO2 prodotta. Le reazioni che hanno
luogo sono di seguito riportate:
CO2 + H2O ↔ H2CO3
H2CO3 + 2KOH → K2CO3 + 2 H2O
La CO2 prodotta dai campioni si scioglie in soluzione acquosa dando acido carbonico
che reagisce immediatamente con la KOH.
La KOH residua, che non ha reagito con la CO2 , è valutata in base alla quantità di
titolante (HCl) necessario per neutralizzarla secondo la reazione di titolazione:
KOH + HCl → KCl + H2O
È quindi possibile determinare l’ammontare della KOH consumata che corrisponde alla quantità di CO2 prodotta dal campione.
Risulta necessario introdurre nella soluzione anche il BaCl2 poiché esso permette di
precipitare sotto forma di BaCO3 insolubile i carbonati formatisi che altrimenti
porterebbero falsare il risultato dell’analisi reagendo essi stessi con l’HCl:
I carbonati infatti, per effetto tampone, potrebbero portare alla produzione di altri ioni OH- che verrebbe anch’esso titolato dall’HCl, si avrebbe cioè:
K2CO3 ↔ 2K+ + CO3
-CO3- - + H2O ↔ HCO3- + OH
-HCO3- + H2O ↔ H2CO3 + OH
-Le titolazioni sono state ripetute a scadenze temporali stabilite tenendo conto della produzione giornaliera stimata di CO2 e del contenuto di O2 all’interno del contenitore.
Attraverso questo procedimento si ottiene la massa di CO2 che si è sviluppata; essa deve
essere ricondotta al campione tramite differenza con la produzione di anidride carbonica del terreno come già descritto precedentemente.
3.3.6
DETERMINAZIONE DEL CONTENUTO DI UMIDITA’
Poiché l’idrolizzato proteico è un composto altamente idrofilo è apparso interessante valutare il contenuto di acqua all’interno dei campioni analizzati e nelle polveri di idrolizzato puro da sottoporre a lavorazioni.
I campioni e le polveri sono stati introdotti in stufa, mantenuti a 105°C e, dopo il loro raffreddamento fino a temperatura ambiente in essiccatore, sono stati pesati ad intervalli di tempo crescenti fino a che il peso non è risultato pressoché costante.
Questa analisi permette di capire quali siano le tecniche migliori da adottare nelle fasi di trasporto, stoccaggio e lavorazione per evitare che il materiale assorba una eccessiva quantità di acqua.