77 3. MATERIALI E METODI
Il materiale oggetto di studio è rappresentato da una miscela di semi di cultivar di lino (Linum usitatissimum L.) e dai panelli ottenuti dal processo di estrazione a freddo dell’olio.
I panelli sono stati analizzati a due diversi tempi di conservazione e in relazione a due diverse tipologie di packaging.
I tempi stabiliti sono 2:
T0: il panello appena prodotto;
T6: il panello dopo 6 mesi (conservazione a -20°C). I packaging utilizzati sono 2:
Carta: panello conservato in contenitori di carta;
Plastica: panello conservato in contenitori di plastica. Lo schema del disegno sperimentale è riportato in Figura 30.
OLIO SEME SPREMITURA A FREDDO PANELLO T0 T6 CARTA T6 PLASTICA
Figura 30. Schema del disegno sperimentale
I campioni sopracitati, prima dell’utilizzo, sono stati triturati con un macinino elettrico e successivamente polverizzati con azoto liquido fino ad ottenere una polvere omogenea. I campioni sono stati conservati in congelatore (-20°C) fino al momento delle analisi.
78 3.1 Determinazione del contenuto in carotenoidi mediante cromatografia liquida ad alta prestazione (HPLC)
Le procedure di preparazione ed estrazione dei campioni sono state eseguite al riparo dalla luce al fine di prevenire l’isomerizzazione e la foto-degradazione dei pigmenti. L’analisi dei carotenoidi è stata eseguita utilizzando per l’estrazione il metodo usato da Franke et al., 2010 (modificato).
A circa 1.5 g di campione sono stati aggiunti 0.15 g di carbonato di magnesio (per evitare la formazione di isomeri).
Il campione è stato poi combinato con 35 ml di una miscela di metanolo e THF (tetraidrofurano) 1:1 v/v contenente come antiossidante BHT (butilidrossitoluene) allo 0.1% (Figura 31).
Figura 31. Struttura del tetraidrofurano (THF) e del butilidrossitoluene (BHT)
Il campione è stato omogeneizzato per 5 minuti mediante Ultra-Turrax e filtrato sottovuoto con filtri Boro 3.3 (porosità 3 μm).
L’estrazione è stata eseguita per 2 volte e gli estratti sono stati raccolti in un pallone e ridotti a un volume di circa 0.5 ml mediante evaporatore rotante.
Il volume così ottenuto è stato controestratto con 1 ml di esano, filtrato con filtri idrofobici di 0.22 μm (Sartorius, Gӧttingen, Germany) e iniettato in HPLC (Spectra SYSTEM P4000 HPLC corredata di rilevatore UV 6000 LP [Thermo Fisher Scientific, Waltham, MA, USA]).
Il contenuto di carotenoidi (luteina e β-carotene) e clorofille (clorofilla a e clorofilla b) è stato determinato mediante HPLC, registrando i picchi in uscita alla lunghezza d’onda (λ) di 450 nm.
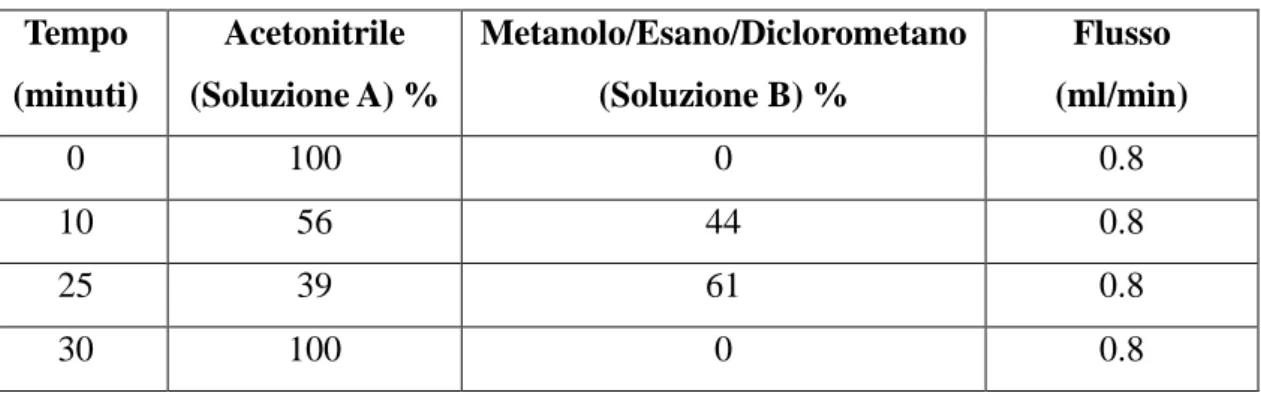
La separazione dei carotenoidi è stata condotta utilizzando una colonna Phenomenex Prodigy ODS LC-18 (4.6 mm x 250 mm, 5 μm) (Phenomenex srl, Castel Maggiore, Italy), ad un flusso di 0,8 ml/min utilizzando le due fasi mobili: Acetonitrile (CH3CN)
Butilidrossitoluene (BHT) Tetraidrofurano (THF)
79 (soluzione A) e una miscela 1:1:1 Metanolo/Esano/Diclorometano (soluzione B), secondo il gradiente riportato nella Tabella sottostante.
Tabella 8. Gradiente delle fasi mobili Tempo (minuti) Acetonitrile (Soluzione A) % Metanolo/Esano/Diclorometano (Soluzione B) % Flusso (ml/min) 0 100 0 0.8 10 56 44 0.8 25 39 61 0.8 30 100 0 0.8
L’elaborazione dei dati è stata effettuata tramite software ChromQuest versione 4.1 della Thermo Electron Corporation.
Le quantificazioni di luteina, β-carotene, clorofilla a e clorofilla b sono state effettuate mediante rette di taratura con standard commerciali a concentrazione nota (Figura 32).
y = 1E+06x R² = 0,9973 0,0E+00 5,0E+06 1,0E+07 1,5E+07 2,0E+07 0 5 10 15
Retta taratura luteina
A re a ppm y = 642518x R² = 0,9994 0,0E+00 5,0E+06 1,0E+07 1,5E+07 2,0E+07 0 10 20 30
Retta taratura β-carotene
A re a ppm y = 32403x R² = 0,9547 0,0E+00 1,0E+06 2,0E+06 3,0E+06 4,0E+06 5,0E+06 6,0E+06 0 50 100 150
Retta taratura clorofilla a
A re a ppm y = 774567x R² = 0,9899 0,0E+00 1,0E+07 2,0E+07 3,0E+07 4,0E+07 5,0E+07 6,0E+07 0 20 40 60 80
Retta taratura clorofilla b
A
re
a
ppm
80 3.2 Estrazione dei composti fenolici totali
L’estrazione dei composti fenolici totali è stata eseguita utilizzando il metodo usato da Terpinc et al., 2012 (modificato).
Ogni determinazione è stata eseguita in doppio.
L’estrazione è stata condotta pesando 0.5 g di campione e addizionando 5 ml di una soluzione di metanolo al 70% (v/v). La miscela è stata sonicata per 30 minuti e successivamente agitata per 30 minuti mediante l’impiego di un agitatore magnetico rotante. Il campione è stato quindi centrifugato per 30 minuti a 6000 giri a temperatura ambiente, per separare la parte liquida da quella solida.
Al termine della centrifugazione il surnatante è stato recuperato in un pallone da evaporatore rotante e conservato in frigorifero a 4°C, mentre il pellet è recuperato, risospeso in 5 ml di metanolo al 70% e riestratto senza sonicazione, ma tenendolo in agitazione per 60 minuti con un agitatore magnetico rotante, poi centrifugato e recuperato secondo la procedura riportata sopra.
Nella terza estrazione, il pellet è recuperato, risospeso in altri 5 ml di metanolo al 70% e riestratto per 30 minuti mediante l’impiego di un agitatore magnetico rotante, centrifugato e recuperato secondo la procedura riportata sopra.
I tre surnatanti riuniti nel pallone (per un volume complessivo di 15 ml) sono concentrati tramite evaporatore rotante fino al raggiungimento di un volume finale di 6 ml.
3.3 Dosaggio dei fenoli totali (Metodo di Folin-Ciocalteu)
I fenoli totali sono stati quantificati spettrofotometricamente tramite il metodo del Folin-Ciocalteu, adattato ai nostri campioni, in accordo con la metodica utilizzata da Barbolan et al., 2003.
Il reattivo di Folin-Ciocalteu è costituito da una miscela di acido fosfotungstico (H3PW12O40) e fosfomolibdico (H3PMo12O40) e si presenta come una soluzione gialla che grazie all’ossidazione dei fenoli si riduce in una miscela di ossidi di tungsteno (W8O23) e molibdeno (Mo8O23) caratterizzata da una colorazione blu.
L’analisi fornisce un dato che corrisponde al contenuto totale di fenoli in relazione alla variazione colorimetrica che viene misurata ad una lunghezza d’onda di 750 nm.
Per il dosaggio vengono aggiunti in una provetta di vetro:
25 μl di campione;
81
125 μl di reagente Folin-Ciocalteu.
La miscela preparata viene incubata a temperatura ambiente per 8 minuti, affinchè si realizzi la reazione di ossidoriduzione. Successivamente vengono aggiunti:
500 μl di una soluzione di sodio carbonato (NaCO3) al 20% (p/v);
600 μl di acqua Milli-Q per arrivare a un volume finale di 2.5 ml. Dopo 30 minuti di incubazione la reazione si è stabilizzata:
Na2WO4/Na2MoO4 (fenolo-MoW11O40)-4 Mo(VI) (giallo) + e- fenolo Mo(V) (blu)
Successivamente è stata misurata l’assorbanza della miscela a 750 nm in cuvette VIS con cammino ottico di 1 cm. Il contenuto in fenoli totali è espresso come mg equivalenti di acido gallico per grammo di peso secco, attraverso una retta di taratura ottenuta utilizzando lo standard di acido gallico a concentrazione nota.
La lettura dell’assorbanza a 750 nm è stata effettuata contro un bianco preparato utilizzando 25 μl di metanolo al 70% al posto del campione.
La retta di taratura dell’acido gallico è stata ottenuta utilizzando acido gallico puro, pesato e disciolto in metanolo al 70% in modo da ottenere una soluzione madre con concentrazione finale di 1000 ppm (mg/l).
Partendo dalla soluzione madre sono state preparate una serie di provette con diluizioni seriali che vanno dalla concentrazione di 500 ppm fino ad arrivare a 31.2 ppm di acido gallico.
La concentrazione di polifenoli in soluzione è calcolata sulla base della retta di taratura Y = 0.0012 X con coefficiente di correlazione, R2 pari a 0.9987 (Figura 33).
y = 0,0012x R² = 0,9987 0,0 0,2 0,4 0,6 0,8 0 100 200 300 400 500 600
Retta taratura acido gallico
y = 0,0012x R² = 0,9987 0 0,2 0,4 0,6 0,8 0 100 200 300 400 500 600
Retta taratura acido gallico
ppm
A
b
s
82 3.4 Dosaggio dei flavonoidi totali
I flavonoidi totali sono stati quantificati in accordo con il metodo di Kim et al., 2003. In una provetta si aggiungono:
100 μl di campione estratto;
60 μl di NaNO2 al 5% (p/v);
La miscela preparata viene incubata per 5 minuti.
40 μl di AlCl3 al 10% (p/v);
La miscela preparata viene nuovamente incubata per 5 minuti.
400 μl di NaOH 1M;
200 μl di acqua Milli-Q per arrivare a un volume finale di 800 μl.
Il contenuto di flavonoidi è stato misurato tramite lettura dell’assorbanza a 510 nm ed espresso come mg equivalenti di catechina su g di peso secco, attraverso una retta di taratura ottenuta utilizzando lo standard di catechina a concentrazione nota.
La lettura dell’assorbanza a 510 nm è stata effettuata contro un bianco preparato utilizzando 25 μl di metanolo al 70% al posto del campione.
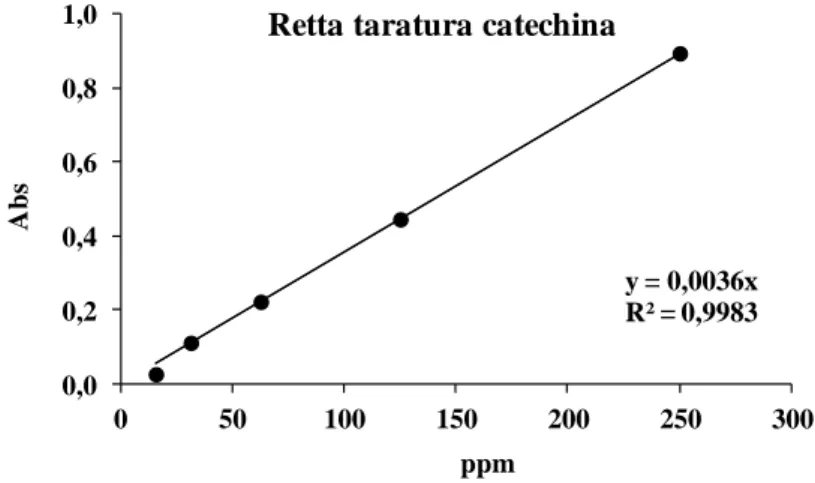
La retta di taratura della catechina è stata ottenuta utilizzando catechina pura, pesata e disciolta in metanolo al 70% in modo da ottenere una soluzione madre con concentrazione finale di 1000 ppm.
Partendo dalla soluzione madre sono state preparate una serie di provette con diluizioni seriali che vanno dalla concentrazione di 250 ppm fino ad arrivare a 15.64 ppm di catechina.
La concentrazione di flavonoidi in soluzione è calcolata sulla base della retta di taratura Y = 0.0036 X con coefficiente di correlazione, R2 pari a 0.9983 (Figura 32).
y = 0,0036x R² = 0,9983 0,0 0,2 0,4 0,6 0,8 1,0 0 50 100 150 200 250 300
Retta taratura catechina
ppm
A
b
s
83 3.5 Dosaggio dei flavonoli totali
Il contenuto dei flavonoli totali è stato quantificato in accordo al metodo proposto da Romani et al., 1996 (modificato).
I campioni sono stati utilizzati come tal quali e diluiti 1:2 con etanolo al 10% (v/v), successivamente in una provetta sono stati aggiunti:
25 μl di campione estratto (tal quale e diluito 1:2);
225 μl di etanolo al 10%;
250 μl di una soluzione di HCl 0,1% in etanolo al 95%;
1 ml di HCl al 2% in acqua per arrivare a un volume finale di 1.5 ml.
Il contenuto di flavonoli totali è stato misurato tramite lettura dell’assorbanza a 360 nm, ed espresso come mg equivalenti di quercetina su g di peso secco, attraverso una retta di taratura ottenuta utilizzando lo standard di quercetina a concentrazione nota.
La lettura dell’assorbanza a 360 nm è stata effettuata contro un bianco ottenuto utilizzando 25 μl di metanolo al 70% al posto del campione.
La retta di taratura della quercetina è stata ottenuta utilizzando quercetina pura, pesata e disciolta in metanolo al 70% in modo da ottenere una soluzione madre con concentrazione finale di 1000 ppm.
Partendo dalla soluzione madre sono state preparate una serie di provette con diluizioni seriali che vanno dalla concentrazione di 125.12 ppm fino ad arrivare a 7.82 ppm di quercetina (Figura 35).
La concentrazione di flavonoli in soluzione è calcolata sulla base della retta di taratura Y = 0.0132 X con coefficiente di correlazione, R2 pari a 0.9857.
y = 0,0132x R² = 0,9857 0,0 0,5 1,0 1,5 2,0 0 20 40 60 80 100 120 140
Retta taratura quercetina
ppm
A
b
s
84 3.6 Separazione e analisi quantitativa dei principali composti fenolici tramite HPLC
I principali acidi fenolici sono stati quantificati utilizzando gli estratti preparati come descritto precedentemente.
I campioni sono stati utilizzati tal quali, per la quantificazione delle forme libere, oppure previa idrolisi, per la quantificazione delle forme glicosilate (Krimer-Malešević et al., 2014, modificato).
3.7 Estrazione degli acidi fenolici liberi
L’estrazione è stata condotta aggiungendo in una provetta:
1 ml di campione estratto;
acido solforico (H2SO4) 2N, fino al raggiungimento di pH 2.
Questa operazione è stata seguita da una centrifugazione a 6000 giri per 10 minuti. Successivamente si è proceduto a controestrarre il campione aggiungendo:
1 ml di etilacetato puro.
La miscela è stata vortexata per alcuni minuti e centrifugata velocemente.
Il surnatante è stato raccolto in un pallone da evaporatore rotante e posto in frigorifero a 4°C.
La procedura di controestrazione è stata effettuata altre 2 volte, aggiungendo però al campione 0.5 ml di etilacetato puro.
Successivamente la miscela nel pallone è stata portata a secco mediante evaporatore rotante.
Il campione così ottenuto è stato risospeso con:
0.5 ml di metanolo puro.
3.8 Idrolisi degli acidi fenolici esterificati solubili
L’idrolisi degli acidi fenolici esterificati solubili è stata condotta sulla frazione rimanante dall’estrazione degli acidi fenolici liberi aggiungendo:
1 ml di NaOH 4N contenente l’1% di ASA (p/v) e 0.037 g di EDTA; ed incubando al buio per 4 ore. In seguito la miscela è stata acidificata con:
H2SO4 2N, fino al raggiungimento di pH 2.
Questa operazione è stata seguita da una centrifugazione a 6000 giri per 10 minuti. Si è poi proceduto a controestrarre il campione come per gli acidi fenolici liberi.
85 3.9 Idrolisi degli acidi fenolici esterificati insolubili
L’idrolisi degli acidi fenolici esterificati insolubili è stata condotta sul pellet ottenuto dall’estrazione dei composti fenolici totali, aggiungendo:
5 ml di NaOH con EDTA e ASA (precedentemente descritto).
La miscela è stata incubata al buio per 4 ore e successivamente acidificata con:
H2SO4 2N, fino al raggiungimento di pH 2.
Questa operazione è stata seguita da una centrifugazione a 6000 giri per 10 minuti. Successivamente si è proceduto a controestrarre il campione aggiungendo:
10 ml di etilacetato puro.
La miscela è stata vortexata per alcuni minuti e sottoposta a una breve centrifugazione. Il surnatante è stato raccolto in un pallone da evaporatore rotante e posto in frigorifero a 4°C.
La procedura di controestrazione è stata effettuata altre 2 volte, aggiungendo però al campione 5 ml di etilacetato puro.
Successivamente la miscela nel pallone è stata portata a secco mediante evaporatore rotante.
Il campione così ottenuto è stato risospeso con:
1 ml di metanolo puro.
3.10 Quantificazione mediante HPLC
I campioni derivanti dalle diverse procedure di estrazione e idrolisi, dopo filtrazione con filtri idrofilici (porosità 0.45 μm), sono stati analizzati mediante HPLC (Spectra System P4000 dotata di un detector a diodi) registrando le letture a due diverse lunghezze d’onda:
λ = 280 nm per visualizzare l’acido gallico;
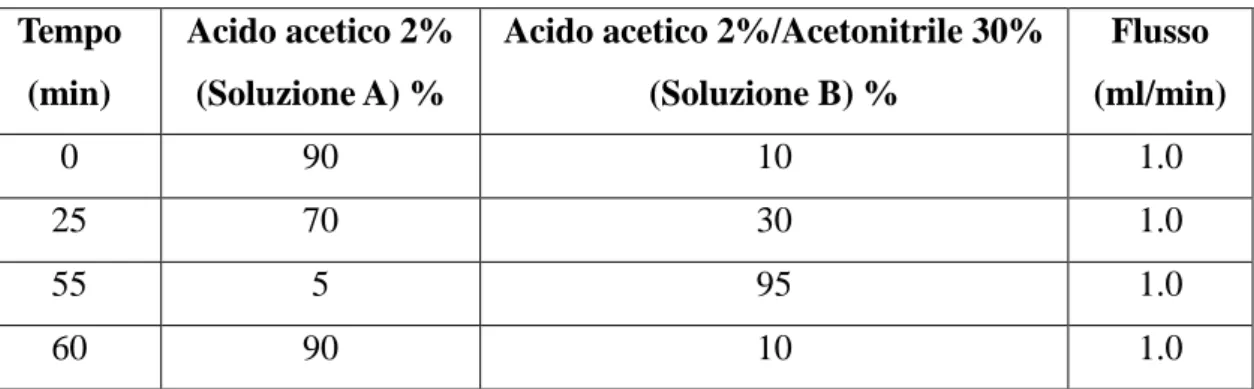
λ = 330 nm per visualizzare l’acido cumarico, l’acido caffeico e l’acido ferulico. La separazione cromatografica è stata effettuata utilizzando una colonna Phenomenex Prodigy ODS LC-18 (250 mm × 4.6 mm, 5 μm) e acido acetico al 2% (soluzione A) con acido acetico al 2% e acetonitrile al 30% (soluzione B) come fasi mobili. Durante tutta la fase di separazione il flusso delle soluzioni è mantenuto costante a 1 ml/min. Il programma di eluizione è riportato nella tabella sottostante.
86 Tabella 9. Gradiente delle fasi mobili
Tempo (min)
Acido acetico 2% (Soluzione A) %
Acido acetico 2%/Acetonitrile 30% (Soluzione B) % Flusso (ml/min) 0 90 10 1.0 25 70 30 1.0 55 5 95 1.0 60 90 10 1.0
Confrontando i risultati ottenuti, con le rette di taratura specifiche per gli acidi in esame, si ottengono le quantità dei composti fenolici presenti nel campione analizzato, espresse come μg del composto specifico su g di peso secco.
3.11 Determinazione dell’attività antiossidante totale (TEAC: Trolox Equivalent Antioxidant Capacity) mediante saggio ABTS
Questo metodo è stato messo a punto per la prima volta da Miller et al. (1993), e poi ulteriormente modificato da Re et al. (1999), Van den Berg et al. (1999) e Cano et al. (2000).
Il metodo si basa sull’utilizzo di una soluzione contenente una sostanza radicalica (ABTS•+: acido 2,2’-azinobis-3-etilbenzotiazolin-6-sulfonico), la cui assorbanza diminuisce in maniera proporzionale alla quantità di composti antiossidanti aggiunti alla soluzione stessa.
L’ABTS è una sostanza cromogena incolore che può essere convertita nella sua forma monocationica radicalica (ABTS•+), di colore blu-verde, se trattata con un agente ossidante, il persolfato di potassio (K2S2O8). L'aggiunta di sostanze antiossidanti, capaci di donare un elettrone o un atomo di idrogeno, determina una decolorazione della soluzione iniziale ed una diminuzione dell'assorbanza proporzionale alla quantità di antiossidante aggiunto (Figura 36).
ABTS•+(λmax= 734 nm) ABTS2-(incolore)
- K2S2O8 + Antiossidante
87 Preparazione del radicale ABTS•+
La soluzione cromogena (blu-verde) di ABTS•+ è stata preparata con:
5 ml di una soluzione 7 mM di ABTS in acqua;
88 μl di una soluzione 140 mM di persolfato di potassio.
La soluzione è poi mantenuta per 12-16 ore al buio e a temperatura ambiente (per far avvenire la reazione di ossidazione e produrre il catione radicalico ABTS) prima del saggio.
Al momento dell’analisi, la soluzione di ABTS•+ viene diluita con metanolo affinchè l’assorbanza a 734 nm (a 30°C) sia pari a 0.700 ± 0.020 e la soluzione sia quindi stabile. Dopodichè si misura l’assorbanza del bianco, preparato aggiungendo:
100 μl di metanolo al 70%;
1 ml della soluzione di ABTS•+.
I campioni, prima del saggio sono diluiti con metanolo al 70%, in modo tale da inibire tra il 20 e l’80% l’assorbanza della soluzione di ABTS•+.
In seguito si misura l’assorbanza dei campioni, preparati aggiungendo:
100 μl di campione opportunamente diluito;
1 ml della soluzione di ABTS•+.
La loro assorbanza è misurata dopo 4 minuti dall’aggiunta dell’ABTS•+. Sono state fatte 2 repliche per ogni diluizione del campione.
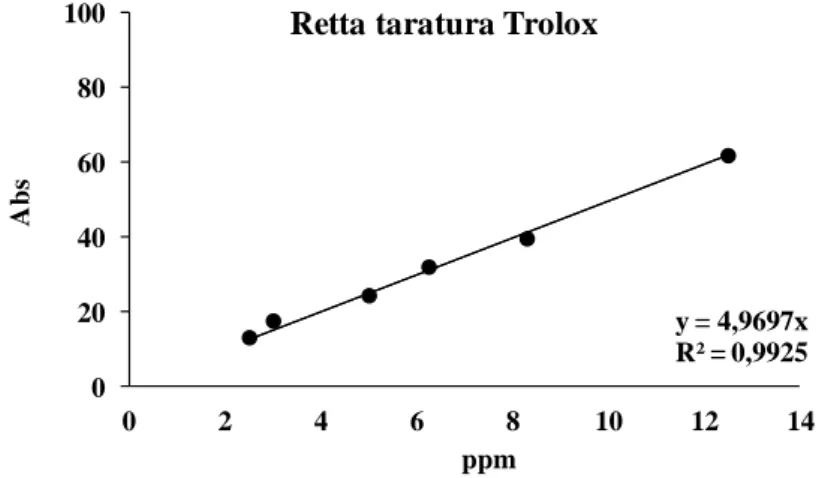
La capacità antiossidante dell’estratto è determinata utilizzando la retta di taratura del Trolox (acido 6-idrossi-2,5,7,8-tetrametilcromano-2-carbossilico), antiossidante sintetico omologo della vitamina E, e viene espressa come mmoli equivalenti di Trolox/g di peso secco di campione. La molecola del Trolox è riportata in Figura 37.
Figura 37. Molecola di Trolox (acido 6-idrossi-2,5,7,8-tetrametilcromano-2-carbossilico)
88 Retta di taratura del Trolox
La retta di taratura è preparata utilizzando concentrazioni di Trolox tra 0 e 12.5 μM, preparate a partire da una soluzione madre 2500 μM in metanolo al 70%.
Si costruisce la retta riportando in ascissa (y) la molarità delle soluzioni di Trolox e in ordinata (x) la % di inibizione dell’assorbanza, che si calcola con la seguente formula:
% inibizione = 1 – (AbsCampione/AbsBianco) * 100 Si ottiene quindi la retta nella figura sottostante:
y = 4,9697x R² = 0,9925 0 20 40 60 80 100 0 2 4 6 8 10 12 14
Retta taratura Trolox
ppm
A
b
s
Figura 38. Retta di taratura del Trolox per la determinazione dell’attività antiossidante totale
3.12 Determinazione del potere antiossidante totale mediante saggio DPPH
Il potere antiossidante totale è stato determinato in accordo al metodo proposto da Brand-Williams et al. (1995, modificato).
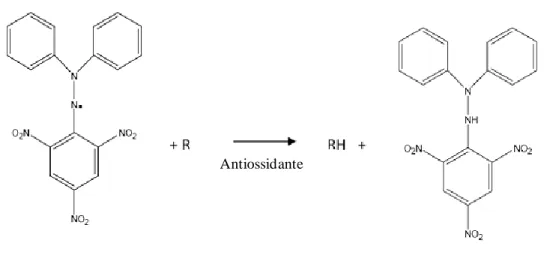
Il 2,2-difenil-1-picrilidrazile (DPPH•) è un radicale azotato molto stabile e caratterizzato da un'intensa colorazione rosso porpora, che decolora quando viene ridotto in presenza di una molecola dotata di capacità antiossidante (Figura 39).
89
Antiossidante
DPPH•(forma ossidata) DPPH (forma ridotta)
Figura 39. Riduzione del DPPH•
L’attività antiradicalica nei confronti del radicale stabile DPPH• è stata valutata misurando la variazione di assorbanza a 515 nm dopo 60 minuti (tempo necessario per raggiungere lo stadio stazionario) a 25°C.
Tutte le procedure sono state eseguite al buio.
Per la preparazione di 100 ml di soluzione madre di DPPH• 0.1 mM in metanolo sono stati pesati:
2.5 mg di DPPH•.
La soluzione madre, di colore viola scuro, è stata preparata e utilizzata il giorno stesso. Il saggio è stato condotto miscelando in cuvetta VIS:
975 μl della soluzione di DPPH•;
25 μl di quantità crescenti di estratto grezzo diluito in metanolo al 70% (tal quale, 1:2 e 1:3).
Le cuvette sono state immediatamente tappate (al fine di evitare l’evaporazione del solvente) ed agitate vigorosamente per alcuni secondi. L’assorbanza è stata registrata a 515 nm dopo 60 minuti di cinetica di reazione.
Il bianco è preparato con:
975 μl di DPPH•;
25 μl di metanolo al 70%.
90 La percentuale di DPPH inibito è stata calcolata utilizzando la formula:
% DPPH•inibito = (1- (DPPH•campione/DPPH•bianco))* 100 Dove:
DPPH•campione = Concentrazione di DPPH• in presenza di campione dopo 60 minuti; DPPH•bianco = Concentrazione di DPPH• del bianco dopo 60 minuti.
L’attività antiradicalica è stata espressa in IC50 (concentrazione efficiente), che
rappresenta la concentrazione di campione in cuvetta (mg di GAE/g di peso secco) necessaria a ridurre del 50% la quantità iniziale di DPPH radicalica. Di conseguenza, minore è il valore di IC50 e maggiore è l’attività antiradicalica (e dunque l’efficacia antiossidante) del campione testato. Il valore di IC50 è stato estrapolato da una curva dose-risposta costruita plottando i valori di % DPPH•inibito vs la concentrazione del campione.
3.13 Determinazione del potere riducente totale (FRAP: Ferric Reducing Antioxidant Power)
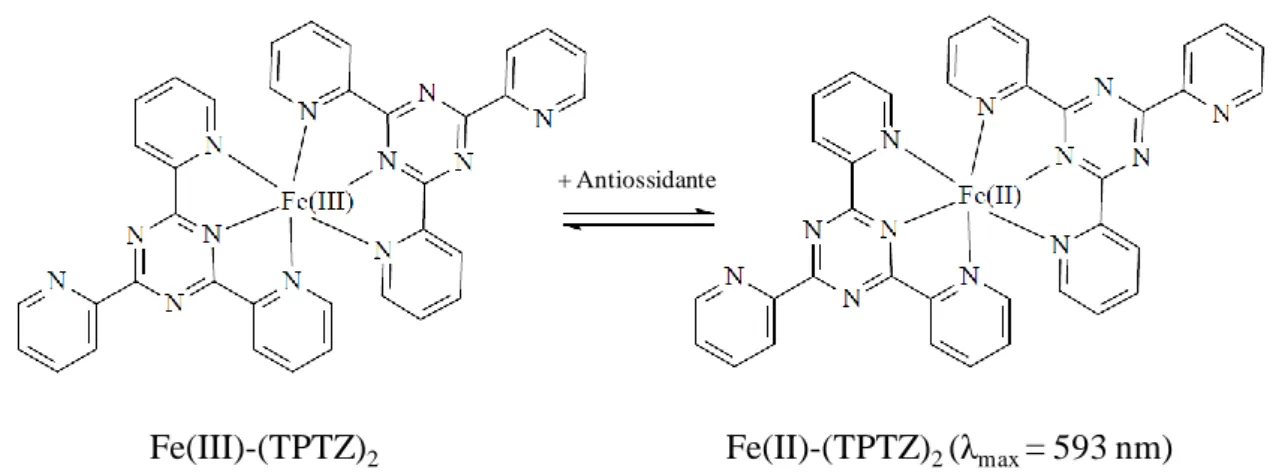
Il metodo FRAP rappresenta una misura diretta del potere riducente totale di una soluzione nei confronti degli ioni ferro. Si tratta di un metodo basato sul trasferimento di elettroni, in cui gli ioni ferro passano da Fe3+ a Fe2+. In determinate condizioni di pH (3.6) e in presenza di TPTZ (2,4,6-tris[2-piridil]-s-triazina), tali ioni formano dei complessi con caratteristiche diverse, in particolare il derivato ridotto (Fe2+-TPTZ) assume una colorazione blu intenso che presenta un assorbimento massimo a 593 nm misurabile per via spettrofotometrica (Figura 40).
Fe(III)-(TPTZ)2 Fe(II)-(TPTZ)2(λmax= 593 nm)
+ Antiossidante
- e
Figura 40. Riduzione del complesso ferrico Fe(III)-(TPTZ)2 al complesso ferroso Fe(II)-(TPTZ)2
91 La capacità riducente di una sostanza antiossidante può quindi essere misurata come variazione dell'assorbanza della soluzione contenente l'antiossidante alla lunghezza d'onda stabilita per confronto con la variazione relativa ad uno standard (acido ascorbico).
Il metodo utilizzato per la determinazione è quello descritto da Majer e Hideg (2012). Il reagente FRAP, preparato il giorno dell’utilizzo, è composto da:
25 ml di tampone acetato 0.3 M; pH 3.6;
2.5 ml TPTZ 10 mM in HCl 40 mM;
2.5 ml di FeCl3 x 6H2O 20 mM.
Un quantitativo pari a 1 ml di FRAP è stato mescolato con 40 μl di campione diluito 1:2, 1:4 e 1:8 in metanolo al 70% e agitato.
Dopo 30 minuti di incubazione a 37°C, è stata misurata l’assorbanza a 593 nm. Il bianco è stato preparato con:
1 ml di FRAP;
40 μl di metanolo al 70% (al posto del campione).
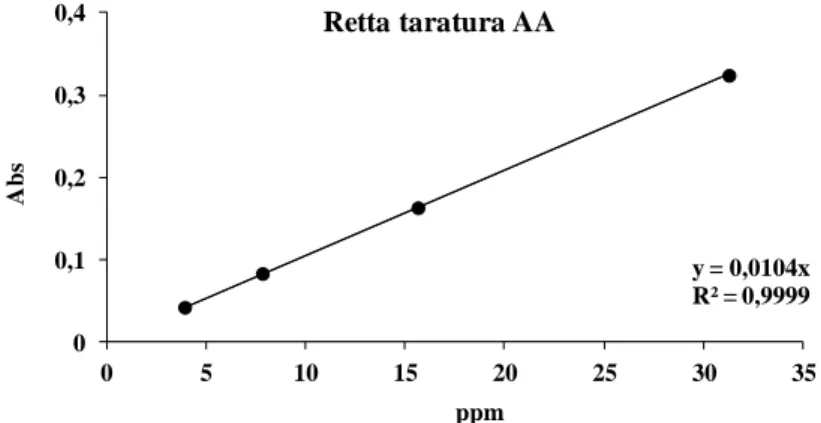
Il FRAP dei campioni, espresso come μg di acido ascorbico equivalenti/g di peso secco, è stato ricavato sulla base di una retta di taratura costruita utilizzando uno standard di acido ascorbico (AA) a concentrazioni crescenti (Figura 41).
y = 0,0104x R² = 0,9999 0 0,1 0,2 0,3 0,4 0 5 10 15 20 25 30 35 Retta taratura AA A b s ppm
Figura 41. Retta di taratura dell’acido ascorbico per la determinazione del potere riducente totale
3.14 Capacità chelante il ferro
La capacità chelante i metalli è una misura che valuta l’efficienza di un insieme di componenti o di un antiossidante puro di chelare stabilmente i metalli di transizione; sottraendoli al sistema, l’antiossidante metal-scavenger evita la formazione di radicali
92 liberi che accelerano e perpetuano la catena delle reazioni ossidative (Halliwell, 1991). Data l’alta reattività, il ferro è il più potente pro-ossidante tra i metalli di transizione (Halliwell et al., 1984).
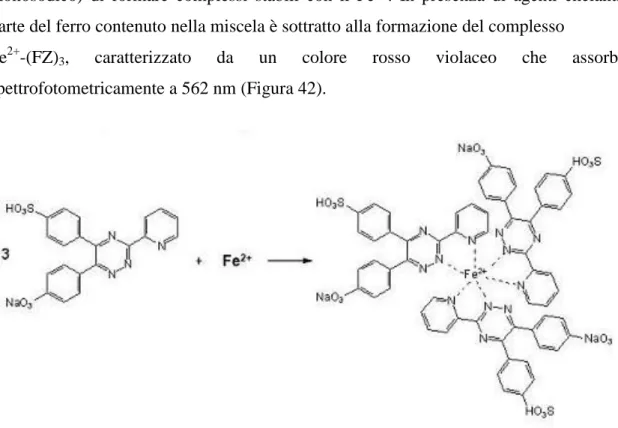
Il metodo più utilizzato per valutare l’efficienza di un antiossidante di chelare il Fe2+ è quello riportato da Decker e Welch (1990). Questo metodo sfrutta la capacità della ferrozina (FZ) (acido 3-(2-piridil)-5,6-difenil-1,2,4-triazina-p,p′-disulfonico sale idrato monosodico) di formare complessi stabili con il Fe2+. In presenza di agenti chelanti, parte del ferro contenuto nella miscela è sottratto alla formazione del complesso
Fe2+-(FZ)3, caratterizzato da un colore rosso violaceo che assorbe spettrofotometricamente a 562 nm (Figura 42).
Complesso Fe2+-(FZ)
3(λmax= 562 nm) FZ (ferrozina)
Figura 42. Chelazione della ferrozina (FZ)
Pertanto, maggiore è l’attività chelante di un campione, minore è l’assorbimento a 562 nm che si registra aggiungendo FZ ad una miscela contenente Fe2+ e antiossidante. La capacità chelante il ferro è stata misurata mediante il saggio di inibizione della formazione del complesso Fe2+-(FZ)3, utilizzando il metodo Decker e Welch (1990, modificato).
I campioni sono stati utilizzati tal quali e diluiti 1:2 e 1:4 con metanolo al 70%. In provette sono stati inseriti:
740 μl di metanolo al 70%;
200 μl di campione (tal quale, diluito 1:2 e 1:4);
93
40 μl di ferrozina 5 mM.
Il tutto è stato agitato e lasciato a riposo per 10 minuti.
Trascorso il tempo necessario è stata effettuata la lettura spettrofotometrica a 562 nm. Il bianco deve essere preparato impiegando soluzioni di campione + ferro, in maniera da tener conto della presenza nella miscela di reazione di eventuali complessi di chelazione cromofori, il cui assorbimento va sottratto nel calcolo utilizzato per determinare i risultati del saggio.
Il bianco è stato preparato aggiungendo nella cuvetta:
740 μl di metanolo al 70%;
200 μl di campione;
20 μl di FeCl2 2 mM;
40 μl di metanolo al 70% (al posto della ferrozina 5 mM). Il controllo è stato preparato aggiungendo nella cuvetta:
740 μl di metanolo al 70%;
200 μl di metanolo al 70% (al posto del campione);
20 μl di FeCl2 2 mM;
40 μl di ferrozina 5 mM.
La capacità chelante è stata calcolata in base alla seguente formula: Capacità chelante % = Abs1 – (Abs2 – Abs3) *100
Abs1 Dove:
Abs1 = assorbanza del complesso Fe2+-(FZ)3, privo di campione;
Abs2 = assorbanza del complesso Fe2+-(FZ)3, dopo l’aggiunta del campione; Abs3 = assorbanza del campione + Fe2+, senza FZ.
La capacità chelante il ferro è stata espressa in IC50 (concentrazione efficiente), che rappresenta la concentrazione di campione in cuvetta (mg di GAE/g di peso secco) necessaria a ridurre del 50% la quantità iniziale di FZ. Di conseguenza, minore è il valore di IC50, maggiore è la capacità chelante il ferro (e dunque l’efficacia chelante) del campione testato. Il valore di IC50 è stato estrapolato da una curva dose-risposta costruita plottando i valori di capacità chelante % vs la concentrazione del campione.
3.15 Composizione degli acidi grassi mediante esterificazione diretta
In accordo con il metodo Palmquist e Jenkins (2003) l’estrazione è stata condotta pesando 150 mg di campione, cui si aggiungono 0.5 mg circa di standard interno
94 (concentrazione madre 5 mg/ml). I campioni sono stati mantenuti sotto flusso di azoto per circa 1 minuto e quindi estratti con 3 ml di HCl al 10% metanolico a 50°C overnight. Una volta raffreddati, 1 ml di esano e 10 ml di K2CO3 al 6% sono stati aggiunti e, dopo agitazione su vortex, l’estratto è stato centrifugato per 10 minuti a 5000 giri a 4°C. La fase superiore è stata recuperata in una vial ambrata cui è stato aggiunto 1 g di sodio solfato anidro. Dopo centrifugazione per 10 minuti a 5000 giri sempre a 4°C, il surnatante è stato trasferito in una nuova vial e portato a secco sotto flusso di azoto, infine, diluito con 3 ml di esano e iniettato in gas-cromatografo.
3.16 Contenuto di dieni coniugati
Il campione (2-3 g) è stato omogeneizzato con Ultra-Turrax in presenza di 6 ml di acqua distillata. Successivamente 0.5 ml di campione sono stati aggiunti a 5 ml di una miscela di esano:isopropanolo 3:2 e, dopo agitazione per circa un minuto, la miscela è stata centrifugata a 2000 giri per 5 minuti a freddo. L’assorbanza del surnatante prelevato è stata determinata allo spettrofotometro alla lunghezza d’onda di 233 nm (contro un bianco costituito da acqua distillata). La differenza di assorbanza a 233 nm rappresentava il contenuto in dieni coniugati, calcolato utilizzando un coefficiente di estinzione molare di 25200 M-1 cm-1 (Srinivasan et al., 2003, modificato).
3.17 Contenuto di perossidi
Il campione (1 g) è stato omogeneizzato mediante Ultra-Turrax con 15 ml di una soluzione cloroformio:metanolo 2:1 e filtrato con appositi filtri. Sette ml di filtrato sono stati miscelati con 2 ml di una soluzione NaCl (0.5%). La miscela è stata centrifugata a 4°C e successivamente sono stati prelevati 1.5 ml dalla frazione inferiore. A questa soluzione sono stati aggiunti 1 ml di miscela cloroformio:metanolo 2:1, 12.5 μl di una soluzione di ammonio tiocianato 30% e 12.5 μl di una soluzione di cloruro di ferro (II). Dopo 20 minuti l’assorbanza è stata determinata a 500 nm. Il PV (Peroxides Value) è stato espresso come mg di cumene idroperossido/kg di campione (Shanta et al, 1994; modificato da Maqsood et al, 2012).
3.18 Valutazione dell’ossidazione degli acidi grassi (test TBARs)
Il grado di ossidazione degli acidi grassi è stato determinato mediante test TBARs. Sono stati pesati 5 g di campione e omogeneizzati con 40 ml di soluzione al 5% di TCA (acido tricloroacetico) per circa 90 secondi. Dopo centrifugazione per 45 minuti a 5000
95 giri, il surnatante è stato prelevato e filtrato. Due ml dell’estratto filtrato sono stati fatti reagire con 2 ml di soluzione 40 mM di TBA (acido tiobarbiturico); la miscela è stata incubata a bagnomaria a 93°C per 45 minuti e, una volta raffreddata, è stata centrifugata per 10 minuti a 4500 giri. L’assorbanza del campione è stata determinata a 532 nm contro un bianco ed espressa come equivalenti di malondialdeide (mg/kg) (Salih et al., 1987).
3.19 Analisi statistica
Le differenze tra il seme e il panello al tempo T0 e le variazioni indotte nel panello dal tempo di conservazione nei due diversi tipi di packaging sono state determinate mediante analisi statistica della varianza (ANOVA) a una via.
Le differenze statistiche tra le medie sono state determinate tramite il test Tukey-Kramer, al livello di significatività dei dati p < 0.05, utilizzando il pacchetto NCSS versione 07.1.21 per Windows.
I dati riportati rappresentano la media ± ES (Errore Standard) di 3 repliche per i carotenoidi e le clorofille e di 2 repliche per tutte le altre analisi effettuate.