Capitolo 2
Sintesi di nuovi liquidi ionici
Durante il corso di questa tesi è stata preparata una serie di liquidi ionici a base pirazolio, una classe di composti fino ad ora poco studiata ma spesso menzionata nella letteratura brevettuale. Data la loro analogia strutturale con i sali di imidazolio, sicuramente i cationi organici più studiati nell’ambito dei liquidi ionici, e che normalmente presentano le migliori proprietà chimico-fisiche (più bassa viscosità, più elevata conducibilità, elevata stabilità chimica, ecc), ritenevamo altamente probabile che i sali di pirazolio, basati anch’essi su un sistema aromatico a cinque termini contenente due atomi di azoto, potessero mantenere alcune delle loro peculiarità. Tuttavia, non si poteva escludere che la presenza dei due atomi di azoto in posizione vicinale, modificando la distribuzione di carica, potesse determinare la variazione anche sostanziale di alcune proprietà del sistema. In particolare, considerando che il protone sul carbonio tra i due atomi di azoto nel sistema imidazolio, C(2)−H, è in genere considerato il sito “acido”, ed a questo protone è attribuita la capacità di formazione di legami ad idrogeno dei sali imidazolio 1,3-dialchil-sostituiti, eliminare questo sito, passando alla struttura pirazolica, poteva rappresentare un metodo efficace per ottenere una nuova classe di liquidi ionici con proprietà solventi diverse (più bassa capacità di funzionare da donatori di legame ad idrogeno) e con proprietà strutturali diverse; la formazione del legame ad idrogeno tra il
35 catione imidazolio ed il controanione è un fattore determinante per la possibilità dei liquidi ionici di dare strutture tridimensionali organizzate.[10]
L’assenza di questo protone poteva inoltre modificare anche la stabilità chimica del liquido ionico risultante, in particolare in condizioni ossidative. Sicuramente, tra le applicazioni più rilevanti dei liquidi ionici è da menzionare l’utilizzo come solventi ed elettroliti in dispositivi elettrochimici, ed in particolare nelle batterie a litio di seconda generazione, dove questi fluidi possono garantire, rispetto ai solventi molecolari, il raggiungimento di due requisiti fondamentali, una praticamente assoluta non-volatilità e una bassa o totalmente assente infiammabilità. Tuttavia, sebbene i sali di imidazolio 1,3-dialchilsostituiti siano i liquidi ionici più spesso applicati, il problema della stabilità catodica rende questi sali non utilizzabili nelle batterie di seconda generazione (batterie a litio) e diversi tentativi sono stati effettuati per verificare la possibilità di incrementarne la stabilità in condizioni riducenti, introducendo ad esempio gruppi elettron-donatori in posizione 2, e catene alchiliche in posizione 3, in maniera tale da favorire la delocalizzazione della carica positiva sul catione imidazolico, o utilizzando additivi in grado di formare film protettivi sulla superficie del litio. In alternativa, è stato proposto l’uso di sali di ammonio (in particolare, sistemi a base pirrolidinio e piperidinio), solfonio, ecc. I sali di pirazolio, mancando del protone in posizione 2, probabilmente se opportunamente sostituiti, potrebbero tuttavia rappresentare un’importante alternativa a quelli di imidazolio per applicazione nelle batterie a litio. A tale proposito è da rilevare che negli ultimi anni, alcuni sali di pirazolio sono stati studiati in ambito elettrochimico, tuttavia in questo settore, più che i semplici liquidi ionici a base pirazolio, hanno attirato attenzione alcuni sali “solidi” a temperatura ambiente o a temperature questa prossime. L’interesse per questa classe specifica di composti può essere ricollegato al fatto che, recentemente, considerando i problemi di sicurezza e la possibilità di ottenere un’alta densità energetica, da più parti è stato proposto l’utilizzo per le batterie a litio di elettroliti allo stato solido in alternativa a quelli allo stato liquido, focalizzando l’attenzione sopratutto su elettroliti di tipo polimerico. Tuttavia, nonostante le buone proprietà meccaniche che molti di questi presentano, al momento non è stato possibile raggiungere, a temperatura ambiente, valori di conducibilità superiori a 10-5 S cm-1 per cui, nelle batterie
36 attualmente in commercio si utilizzano in genere miscele di elettroliti liquidi e polimerici, definiti sistemi elettroliti gel-polimerici.
In alternativa a questi sistemi, è stato proposto recentemente l’uso di composti solidi che si trovano in uno stato metastabile della materia, spesso definiti come plastic crystal.[11] In questi composti, la fase plastica cristallina è generalmente caratterizzata da:
- Un più elevato grado di disordine strutturale, che determina una maggiore diffusività e plasticità;
- Una bassa entropia di fusione;
- Una bassa costante dielettrica, proprietà che rendono tale fase più simile ad uno stato liquido.
I materiali solidi che presentano questo stato sono principalmente costituiti da ammine eterocicliche o alifatiche, e sono in grado di raggiungere conducibilità a temperatura ambiente nello stato plastico superiori a 10-3 S cm-1.
Alcuni sali di pirazolio mostrano questa proprietà, e sebbene il settore sia ancora in una fase di sviluppo primordiale, alcuni esempi sono stati testati per l’applicazione nelle batterie a litio. Un liquido ionico che mostra una fase plastica cristallina offre infatti il vantaggio di mantenere le proprietà di elettrolita anche a temperature inferiori al punto di fusione ed è in grado di sostenere, in queste condizioni, le prestazioni della batteria. L’1,2-dietil-3-metilpirazolio bistriflimide, e le sue miscele con LiTf2N, presentano alte conducibilità, sia allo stato solido che allo stato liquido. Quando applicato nelle batterie a litio questo sale si è dimostrato in grado di sostenere una prestazione, allo stato liquido corrispondente all’83% della capacità teorica, e in fase plastica cristallina al 67%, della capacità teorica. Il confronto con un altro sale di pirazolio, l’1,2-dietil-pirazolio bistriflimide, che non presenta una fase plastica cristallina ha inoltre evidenziato come, sebbene quest’ultimo liquido ionico sia in grado di fornire prestazioni elevate ad alte temperature, queste diminuiscono rapidamente con il diminuire della temperatura, fino a scomparire al di sotto del punto di fusione.
37
2.1
Metodi di sintesi
I sali di pirazolio sintetizzati nell’ambito di questa tesi sono stati preparati utilizzando come composti base due prodotti commerciali, il pirazolo e 3,5-dimetilpirazolo. Poiché è noto che il sistema eterociclico pirazolo è meno reattivo dell’analogo imidazolo le reazioni di N-alchilazione sono state condotte utilizzando ioduri alchilici anziché bromuri o cloruri. In fig. 2.1 è riportato lo schema di reazione.
Figura 2.1.Alchilazione pirazoli.
La metodologia riportata in letteratura prevedeva l’utilizzo di iodometano come agente alchilante, K2CO3 come base e acetone come solvente. Effettuando la reazione in queste condizioni, tuttavia il processo di metilazione richiedeva tempi lunghi e, nel caso dell’alchilazione con ioduri a catena più lunga, 1-iodoetano e 1-iodobutano, le rese rimanevano basse anche dopo giorni di riscaldamento alla temperatura di riflusso.[12] In tab. 2.1 sono riportate le condizioni e le rese delle reazioni di alchilazione.
Tabella 2.1. Alchilazione con K2CO3 (2,2 eq) in acetone.
Alchilante t (gg) T (°C) Rese (%) Pirazolo BuI 1,6 eq 4 Rlfx 50 MeI 1,6 eq 3 ’’ 60 EtI 1,6 eq ’’ ’’ 50 3,5-dimetilpirazolo BuI 1,6 eq 8 ’’ 15 EtI 1,1 eq 5 ’’ 90 MeI 1,1 eq ’’ ’’ 80
38 Per cercare di incrementare la velocità del processo di alchilazione, le stesse reazioni di sostituzione nucleofila sono state testate in acetonitrile, utilizzando come base KOH. In queste condizioni è stato possibile ottenere con elevata resa e in tempi più brevi gli attesi prodotti di N-alchilazione. Inoltre, considerando la maggior reattività del 3,5-dimetilpirazolo, rispetto al non sostituito pirazolo, con questo composto è stato possibile effettuare la reazione di alchilazione anche utilizzando 1-bromoetano e 1-bromobutano.
Una volta definite le condizioni ottimali per ciascun substrato, la reazione è stata ripetuta su scala delle decine di grammi ed i prodotti sono stati isolati per estrazione dalla miscela basica con etere etilico. Eliminati i prodotti volatili (acetonitrile, alogenuri alchilici) per evaporazione a pressione ridotta gli N-alchil-pirazoli isolati sono stati caratterizzati mediante analisi di risonanza magnetica nucleare (NMR) ed utilizzati come tali nella successiva reazione di quaternizzazione. In tab. 2.2 sono riportate le condizioni e le rese delle reazioni di alchilazione ottenute utilizzando KOH come base.
Tabella 2.2. Alchilazione con KOH (1,9 eq) in acetonitrile.
Alchilante (eq) t (gg) T (°C) Rese (%)
Pirazolo BuI (1,02) 2 60 87 ” 3 ’’ 89 EtI (1,02) ” ’’ 60 MeI (1,02) 1 ’’ 67 3,5-dimetilpirazolo ” ” 50 90 EtI (1,02) 2 ’’ 88 EtBr (1,02) 2,5 ’’ 85 BuBr (1,02) ’’ ’’ 99
Sebbene, la metilazione del sistema eterociclico (pirazolo, imidazolo, ecc.) costituisce in genere il primo stadio nel processo di trasformazione di questi composti in liquidi ionici, l’utilizzo di reagenti relativamente tossici, come il metilioduro, viene spesso considerato, a giusta ragione, un fattore che va a detrimento della sostenibilità del prodotto finale, il liquido ionico. Un solvente “green” non solo deve essere eco-compatibile ma la sua sintesi deve essere basata sull’utilizzo di reagenti quanto più possibile non tossici e deve essere effettuata in condizioni di massima sostenibilità. Poiché tra gli agenti metilanti,
39 negli ultimi anni, il dimetilcarbonato (DMC) ha attirato un notevole interesse per la sua non tossicità, abbiamo deciso di testare la possibilità di utilizzare questo composto nel processo iniziale di N-metilazione.[13]
2.2
N-metilazione di Pirazoli con DMC
Gli alogenuri metilici (alogeno: I, Br, Cl) e il dimetilsolfato sono sicuramente gli agenti metilanti più noti e più spesso utilizzati. Purtroppo, questi reagenti altamente efficaci sono anche altamente tossici e spesso corrosivi. Il DMC è attualmente considerato una valida alternativa, eco-sostenibile, agli agenti metilanti sopra menzionati, dal momento che non è tossico e le reazioni di metilazione con esso condotte non producono sali inorganici. Il gruppo uscente, carbonato di metile, si decompone facilmente nella stessa miscela di reazione dando come unici sottoprodotti MeOH e CO2. Il DMC è classificato come un liquido infiammabile, che non ha tuttavia effetti mutageni o irritanti per contatto o inalazione. Pertanto la reazione di metilazione condotta con questo reagente non richiede le attenzioni particolari necessarie per maneggiare lo ioduro di metile o il dimetilsolfato, entrambi tossici e mutageni.
Il processo industriale di sintesi del DMC era inizialmente basato sulla reazione del metanolo con fosgene, mostrata in fig. 2.2, un’altro reagente efficace ma estremamente tossico.
Figura 2.2. Sintesi del DMC con fosgene.
A partire dalla metà degli anni ’80, tuttavia, la ditta Enichem ha messo a punto un nuovo processo di carbonilazione ossidativa (fig. 2.3) per la sintesi industriale del DMC che ha dato un impulso significativo allo sviluppo della chimica di questo composto.
40 Le caratteristiche più importanti di questo processo sono infatti:
- Basso costo delle materie prime, largamente disponibili e di bassa tossicità; - Elevata velocità di produzione;
- Il processo nel suo insieme è definibile come non tossico e i sottoprodotti sono facilmente eliminabili (CO2 e H2O);
- I prodotti che si isolano hanno alta qualità.
Più recentemente, sono stati sviluppati anche nuovi processi, più economici, che non portano alla formazione di sottoprodotti. Tutto questo ha ovviamente contribuito ad incrementare l’uso di tale reagente.
Il DMC possiede due centri attivi, la porzione alchilica e il gruppo carbonilico, la reattività dell’uno o dell’altro può essere modulata variando la temperatura di reazione. Un generico nucleofilo Nu- può quindi interagire con l’uno o l’altro centro, come sopra riportato, dando un prodotto di metilazione o di carbossimetilazione
Me O O Me Nu -O Me O O Me O NuMe + MeO-+ CO2 Nu- NuCOOMe + MeO -Figura 2.4. Meccanismi DMC.
41 La reazione dei sistemi eterocicli, contenenti gruppi NH, in presenza di una base (organica o inorganica) segue la prima via descritta in fig. 2.4 e porta ad un prodotto di metilazione.
In letteratura, le reazioni di metilazione sono in genere condotte a temperature elevate, > 100 °C. Poiché il DMC ha un punto di ebollizione di 90 °C, le reazioni di metilazione del pirazolo e dell’imidazolo sono state condotte per gocciolamento del DMC all’eterociclo riscaldato a 140 °C, in un intervallo temporale di 8 ore e successiva agitazione della miscela per 2 ore. Il sottoprodotto formato durante la reazione, MeOH, veniva distillato e allontanato durante il processo stesso. Questa reazione è riportata dare l’atteso N-metilimidazolo con resa del 98% e l’N-metilpirazolo con resa del 60%. In alternativa, è riportato la reazione in autoclave a 160 °C.
Tentativi di effettuare la reazione di metilazione del pirazolo e del 3,5-dimetilpirazolo in condizioni analoghe a quelle sopra descritte, in assenza di solvente, sfruttando la solubilità del pirazolo nel DMC, portavano alla formazione quasi istantanea di un solido, da cui era possibile isolare l’atteso prodotto con resa basse.
Poiché la formazione del solido rendeva inefficace il processo di agitazione, e questo poteva contribuire alle basse rese di reazione, abbiamo effettuato la stessa reazione utilizzando l’acetonitrile come cosolvente. Inizialmente, sono state effettuate una serie di prove per trovare le condizioni ottimali, variando le quantità relative di DMC, base e solvente. In accordo con i risultati ottenuti, e con quanto detto precedentemente sulla maggior reattività del 3,5-dimetilpirazolo, la reazione del 3,5-dimetilpirazolo con DMC può essere effettuata a temperature inferiori, in tempi più brevi ed in assenza di cosolvente. In fig. 2.5 è riportato lo schema generale della metilazione con DMC.
42
Tabella 2.3. Reazioni di metilazione del pirazolo con DMC.
DMC (eq) MeONa (eq) MeCN (ml) t (h) T (°C) Resa (%)
pirazolo (0,5 g) 8,5 1,5 - 3 rflx 31 ” 2 - ” ” 53 23 ” - ” ” 71 17 1,5 - 6 ” 77 ” ” 3 13 ” 65 8,5 ” 5 6 ” 75 ” ” 3 ” ” 81 6,4 ” 5 ” ” 78 12,8 ” 3 ” ” 69 ” 2 ” ” ” 80 14,2 ” 5 13 ” 83 ” 1,5 ” ” ” 68 6,4 2 ” ” ” 81
Tabella 2.4. Reazioni di metilazione di 3,5-dimetilpirazolo con DMC.
DMC (eq) MeONa (eq) MeCN (ml) t (h) T (°C) Resa (%)
3,5-dimetilpirazolo (0,5 g) 6,4 2 3 3 70 73 4,3 ” 4 ” ” 56 2,1 ” ” ” ” 41 ” ” 3 ” ” 60 6,4 1,5 4 ” ” 47 ” ” 3 ” ” 86 4,3 ” ” ” ” 86 2,1 ” ” ” ” 76 6,4 ” ” ” rflx 87 4,3 ” ” ” ” 76 6,4 1,2 ” ” ” 81 4,3 ” ” ” ” 81 6,4 1,0 ” ” ” 77 ” 1,2 2 ” ” 78 4,3 ” ” ” ” 87 ” ” 3 1 ” 67 6,4 1,0 ” 13 70 68 8,5 1,2 - 3 ” 83 ” 1,5 - ” ” 97
43 Nelle tab. 2.3 e 2.4 sono riportate le varie condizioni utilizzate e le relative rese ottenute per il pirazolo e il 3,5-dimetilpirazolo rispettivamente.
Una volta definite le condizioni ottimali per ciascun substrato, la reazione è stata ripetuta su scala delle decine di grammi ed i prodotti sono stati isolati per estrazione dalla miscela basica con etere etilico. Eliminati i prodotti volatili (acetonitrile, metanolo, DMC) per evaporazione a pressione ridotta gli N-metil-pirazoli isolati sono stati caratterizzati mediante analisi di risonanza magnetica nucleare (NMR) ed utilizzati come tali nella successiva reazione di quaternizzazione.
2.3
Quaternizzazione
Considerando la bassa reattività del sistema pirazolico, le reazioni di quaternizzazione sono state effettuate a temperature comprese tra gli 80-100 °C, in acetonitrile, utilizzando i più reattivi ioduri alchilici. Come atteso, l’ N-metilpirazolo dava conversioni più basse rispetto al corrispondente derivato 3,5-dimetilato, ed anche dopo 5 giorni non è stato possibile raggiungere con questo substrato conversioni superiori al 20%.
La reazione di quaternizzazione su entrambi i substrati è stata effettuata utilizzando sia l’1-iodobutano che l’1-iodobutirronitrile per ottenere pirazoli funzionalizzati in catena laterale (tab. 2.5). La funzionalizzazione della catena alchilica è una procedura ampiamente utilizzata nella chimica dei liquidi ionici in quanto permette di ottenere mezzi di reazioni dotati di proprietà aggiuntive; spesso in grado di funzionare da solventi e catalizzatori o da solventi e leganti o stabilizzatori di catalizzatori. Questa classe di liquidi ionici, generalmente definiti task specific ionic liquids, è stata ampiamente utilizzata nel caso dei composti a base imidazolo e piridinio nella sintesi organica, nei processi di separazione di materiali specifici, nella preparazione di materiali nanostrutturati. I liquidi ionici si possono definire funzionalizzati quando il gruppo funzionale introdotto è legato covalentemente al catione o all’anione. Generalmente è il catione che supporta meglio l’introduzione di gruppo funzionale reattivo, e gli esempi di liquidi ionici in cui la catena funzionalizzata si trova sull’anione sono molto pochi.
44
Tabella 2.5. Reazioni di quaternizzazione.
Alchilante (eq) Tempo (gg) Temperatura (°C) Resa (%) N-MetilPirazolo BuI (1,02) 5 80 20 IPrCN (1,02) 4 100 26 1,3,5-triMetilPirazolo BuI (1,5) 3 80 57 BuI (1,02) 5 ” 26 BuOMs - IPrCN (1,03) 4 90 75
Per quanto concerne la presenza di un gruppo CN in catena laterale, è da sottolineare che liquidi ionici a base imidazolo, piridinio e pirrolidinio, contenenti la funzionalità nitrilica, sono già stati utilizzati con successo come solventi-ligandi nella chimica organometallica. Poiché l’1-iodobutirronitrile non è un composto commerciale, esso è stato sintetizzato a partire dall’1-clorobutirronitrile mediante reazione di sostituzione nucleofila con sodio ioduro.
È infine da rilevare che il tentativo di funzionalizzare l’1,3,5-trimetilpirazolo utilizzando il mesilato di butile non ha portato all’isolamento di alcun prodotto, anche protraendo la reazione per oltre una settimana.
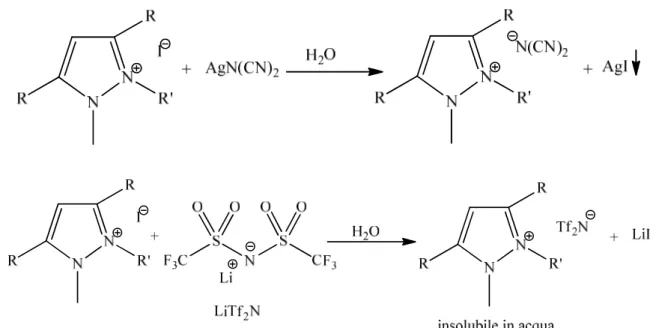
In tutti i casi i prodotti di reazione, i sali ioduro, sono stati isolati dalla soluzione acetonitrilica per evaporazione del solvente, previa addizione di acqua ed estrazione dei composti non reagiti. I sali ioduro, solidi a temperatura ambiente, sono stati quindi disciolti in acqua e sottoposti a reazioni di scambio anionico per ottenere i sali desiderati a base dicianammide e bis-trifluorometansulfonimmide (bistriflimide) (fig. 2.6). Questi anioni in genere permettono di ottenere sali liquidi a temperatura ambiente.
45
Figura 2.6. Reazioni di scambio.
Nella reazione di scambio per introdurre l’anione dicianammide si utilizza il corrispondente sale d’argento, AgN(CN)2, e si sfrutta la bassa solubilità dello ioduro di argento in acqua per isolare, in forma pura, il liquido con anione dicianammide, che normalmente è solubile in acqua.
Nella preparazione dei sali di pirazolio aventi come anione la bistriflimide, Tf2Nି, si sfrutta, per separare il liquido ionico formato, l’idrofobicità di quest’ultimo. La reazione viene condotta in acqua, utilizzando LiTf2N; dopo circa un giorno a temperatura ambiente si osserva la separazione di due fasi, il liquido ionico puro e la fase acquosa contenete LiI.
2.4
Parametri solvatocromici
La polarità è una delle caratteristiche più importanti di un solvente che può fornire informazioni quali- e quantitative sulla capacità di un mezzo di interagire con specie in esso disciolte; substrati, intermedi o stati di transizione. La polarità, essendo una proprietà sommatoria di più effetti, è in genere difficile da esprimere in maniera quantitativa
46 utilizzando un unico parametro. Tuttavia, la costante dielettrica rappresenta sicuramente il parametro chimico-fisico più spesso utilizzato dal chimico per valutare la capacità di un certo solvente di interagire con uno specifico soluto e, basandosi sul principio che simile scioglie il simile, di valutare la capacità di solvatare e quindi “sciogliere” una sostanza chimica specifica. D’altra parte anche i più semplici modelli computazionali, spesso utilizzati per descrivere le interazioni elettrostatiche, che sono alla base del processo di solvatazione, considerano i solventi come un continuum omogeneo non strutturato e utilizzano la costante dielettrica, insieme ad altre costanti fisiche, quali il momento di dipolo permanente e l’indice di rifrazione, per valutare quantitativamente gli effetti solvente.
In realtà, le interazioni soluto/solvente avvengono a livello molecolare, all’interno di un discontinuum strutturato in singole molecole di solvente, ciascuna in grado di interagire con il soluto e con le altre molecole del solvente stesso. L’approccio puramente elettrostatico, quello basato sui sovra menzionati parametri chimico-fisici, non tiene conto delle specifiche interazioni soluto/solvente e, spesso, le correlazioni tra gli effetti chimici determinati dal solvente (es. velocità di reazione, selettività di reazione, ecc) e la polarità del solvente, espressa in termini dei soli parametri chimico-fisici, si sono dimostrate non in grado di descrivere il fenomeno: non poche volte, quindi l’approccio elettrostatico è risultato un fallimento.
Per avere una descrizione più soddisfacente dell’effetto solvente è necessario prendere in considerazione tutte le interazioni specifiche e non-specifiche soluto/solvente, solvente/solvente e, quando il soluto è a concentrazione elevata, anche soluto/soluto. In altri termini è necessario definire la polarità del solvente sulla base della capacità globale di solvatazione, che dipende da tutte le interazioni possibili; interazioni non-specifiche, specifiche, intermolecolari tra ioni o molecole di soluto e molecole di solvente, escludendo tutti quei fenomeni che possano condurre ad alterazioni chimiche irreversibili degli ioni o delle molecole di soluto (protonazione, ossidazione, riduzione o formazione di complessi chimici).
47 La polarità dei solventi non può essere quindi descritta quantitativamente da un singolo parametro chimico-fisico. Tuttavia, la mancanza di un’espressione teorica globale per la previsione degli effetti solvente sulla reattività chimica e l’inadeguatezza della definizione di polarità in termini di caratteristiche fisiche, ha portato nel tempo all’introduzione di molteplici parametri empirici che quantificano la polarità dei solventi, e allo sviluppo di un numero altrettanto elevato di scale di polarità. Tra queste sono da menzionare le scale di polarità basate su misure di assorbimento UV-vis di alcune sostanze chimiche, spesso dei coloranti, normalmente definiti “probes”. A seconda della struttura chimica del “probe” è possibile ottenere una valutazione quantitativa di un certo tipo di interazione e quindi l’uso di più probes può fornire dati importanti sulle diverse interazioni intermolecolari soluto/solvente che definiscono la polarità di un certo mezzo.[14]
Gli spettri di assorbimento UV-vis dei componenti chimici sono infatti influenzati dal mezzo circostante e i solventi possono determinare un cambiamento nella posizione, intensità e nella forma della banda di assorbimento. Questo fenomeno prende il nome di solvatocromismo ed è determinato dalla differente solvatazione tra lo stato fondamentale e il primo stato eccitato della molecola. Se, all’aumentare della polarità del solvente, è stabilizzato meglio lo stato fondamentale, questo determinerà un effetto solvatocromico negativo che si traduce in uno shift ipsocromico (verso il blu) della banda di assorbimento in esame, viceversa l’effetto sarà positivo quando è stabilizzato più efficacemente il primo stato eccitato ottenendo uno shift batocromico (verso il rosso). Normalmente, i composti organici che funzionano da sonde solvatocromiche sono composti relativamente complessi, in grado di dare più tipi di interazioni soluto/solvente (interazioni dipolo, dipolo-carica, legame ad idrogeno, interazioni π−π, ecc), e quindi solo la valutazione combinata degli effetti di uno stesso solvente su più probes rende possibile definire in maniera quali/quantitativa la polarità del solvente stesso tenendo conto di tutte le possibili interazioni.
Parametri di Kamlet-Taft
La definizione della polarità di un solvente secondo Kamlet-Taft si basa su l’uso di più sonde solvatocromiche in maniera tale da poter distinguere e valutare, per uno stesso
48 solvente, i diversi tipi di interazioni che questo può esercitare sul soluto e la loro importanza relativa. In particolare, mediante l’utilizzo di tre sonde solvatocromiche è possibile determinare tre parametri che misurano, rispettivamente, la polarizzabilità del solvente (parametro π*), lacapacità di fungere da donatore di legami ad idrogeno (HBD), parametro α, e la capacità di fungere da accettore di legami ad idrogeno (HBA), parametro β.
Più in particolare, il parametro π* misura gli effetti delle interazioni soluto/solvente definibili come interazioni dipolo/dipolo e dipolo/dipolo indotto. Questo parametro, quindi, misura la capacità di un solvente di stabilizzare una carica netta o dei dipoli indotti in virtù di interazioni dielettriche aspecifiche. Il valore π* può essere considerato pertanto una combinazione della polarizzabilità/dipolarità del solvente. Questa scala prende il suo nome dalla transizione π-π* dei probes inizialmente utilizzati (composti nitroaromatici) per determinare questo tipo di effetto solvente. La scala varia da π* = 0 per il cicloesene a π* = 1,00 per dimetilsolfossido.
Generalmente, il valore π* è calcolato sul massimo di assorbimento della N,N-dietil-4-nitroanilina, utilizzando l’equazione (1):
vmax = v0 + s π* (1)
dove;
vmax = lunghezza d’onda al massimo di assorbanza
v0 = 27,52 x 10
3 cm-1 s = -3,128
Il parametro α si determina sulla base del confronto degli shift indotti dal solvente sulla banda di assorbimento π-π* di due probes simili (omomorfici), di cui uno non può funzionare né da accettore di legami ad idrogeno né da donatore (N,N-dietil-4-nitroanilina) mentre per l’altro funziona da accettore di legame ad idrogeno (dye di Reichardt, definito anche come betaina 30, riportato in fig. 2.7).
49
Figura 2.7. Dye di Reichardt.
L’espressione (2) può essere utilizzata per calcolare α:
α = [ET(30)- 14,6 (π*-0,23σ)-30,31]/16,5 (2)
È da rilevare che ET(30)rappresenta un’altro parametro, ampiamente utilizzato per definire la polarità dei solventi, che si determina sulla base del massimo di assorbimento del dye di Reichardt (betaina 30), così come definito dall’eq (3):
ET(30)(kcal mol−1) =28 591.5/λmax (nm) (3)
dove, λmax è il massimo di assorbimento della banda ad energia inferiore, corrispondente al trasferimento di carica intramolecolare (CT) π−π* della molecola di betaina 30 nella sua forma zwitterionica. A causa della sua struttura, le proprietà solvatocromiche della betaina 30 sono fortemente influenzate da due effetti solvente; la dipolarità/polarizzabilità e dalla capacità di dare legame ad idrogeno, poiché entrambi questi effetti stabilizzano maggiormente lo stato fondamentale dipolare rispetto al meno dipolare stato eccitato di Frank-Condon.
Nella fig. 2.8 sotto riportata, è rappresentato lo stato fondamentale ed eccitato della betaina 30, ed in particolare è evidenziato come la diversa solvatazione dello stato fondamentale ed eccitato passando dai solventi non polari, o poco polari, ai solventi polari determina un aumento dell’energia di transizione che si esplica in uno spostamento della banda di assorbimento a lunghezze d’onda inferiori.
50
Figura 2.8. Stato fondamentale ed eccitato della betaina 30.
Infine il parametro β, che è una misura empirica della capacità del solvente di funzionare da accettore di legami ad idrogeno (HBA), analogamente alla determinazione del valore α, è misurato comparando gli shift indotti sulla banda di assorbimento π-π* di due molecole simili, una che funziona da donatore di legame ad idrogeno (4-nitroanilina) e l’altra alchilata sull’azoto che non funziona da donatore di legami ad idrogeno (N,N-dietil-4-nitroanilina). β ha valore 0 per quei solventi non HBA e non EPD, come gli idrocarburi alifatici, mentre β ≈ 0,1 per gli idrocarburi aromatici.
β= [1,035 v(2)max – v(1)max + 2,64 x 103 cm-1]/2,8
v(1)max = assorbanza massima della 4-nitroanilina
v(2)max = assorbanza massima dell’N,N-dietil-4-nitroanilina
Tabella 2.6. Parametri solvatocromici di alcuni dei più comuni solventi organici.
Solvente π* α β Esano -0,08 Cicloesano Cloroformio 0,58 0,34 Dietiletere 0,27 0,47 Acetone 0,71 0,08 0,48 DMSO 1,00 0,76 MeCN 0,75 0,19 0,31 DMF 0,88 0,69 Etanolo 0,54 0,83 0,77 Acqua 1,09 1,17 0,18
51 I parametri di Kamlet-Taft, noti per la maggior parte dei solventi molecolari (alcuni valori sono riportati in tab. 2.6) sono stati determinati anche per alcuni liquidi ionici (tab. 2.7). Dall’esame dei valori riportati nelle due tabelle, si può notare che i liquidi ionici sono generalmente caratterizzati da valori di π* superiori alla maggior parte dei solventi molecolari, da cui si desume che i solventi ionici hanno una più elevata capacità di indurre un dipolo elettrico nei composti in essi disciolti. Il valore di π* varia poco nell’ambito dei liquidi ionici, tuttavia i valori più bassi sono normalmente caratteristici dei sali aventi come anione la bistriflimide, Tf2Nି; probabilmente, la maggior delocalizzazione della carica negativa, che differenzia questo anione dai comunemente usati anioni tetrafluoroborato o esafluorofosfato, determina una riduzione della capacità dei corrispondenti liquidi ionici di interagire con la betaina 30 mediante forze coulombiane.
Tabella 2.7. Parametri solvatocromici di alcuni ILs.
IL π* α β [bmim][PF6] 1,02 0,65 0,25 [bmim][BF4] 0,98 0,67 0,45 [bm2im][BF4] 1,08 0,40 0,36 [emim][Tf2N] 0,98 0,66 0,24 [hmim][Tf2N] 0,97 0,65 0,26 [C3CNmim][BF4] 1,08 0,64 0,34 [bm2im][Tf2N] 1,00 0,41 0,24
I valori di α variano in un ambito più ampio, e nel caso dei liquidi ionici non portanti gruppi funzionali in grado di fungere da specifici donatori di legame ad idrogeno (OH, NH2), i sali di imidazolio non sostituiti sul carbonio eterociclico C2 presentano valori di α più elevati rispetto ai sali di piridinio e pirrolidinio. Sebbene questo parametro sia determinato essenzialmente dalla natura del catione, l’anione esercita una certa influenza. In particolare, gli anioni più basici, che danno quindi luogo a più forti interazioni anione-catione, riducono la capacità del catione di fungere da donatore di legame ad idrogeno. È da sottolineare che la capacità del catione o dell’anione di un liquido ionico di interagire con una specie in esso disciolta deve essere sempre considerata un processo in competizione con la capacità dello stesso catione o anione di interagire con le specie ioniche di carica opposta ad esso adiacenti.
52 Infine, il parametro β è determinato dalla natura dell’anione e aumenta all’aumentare della basicità di quest’ultimo.
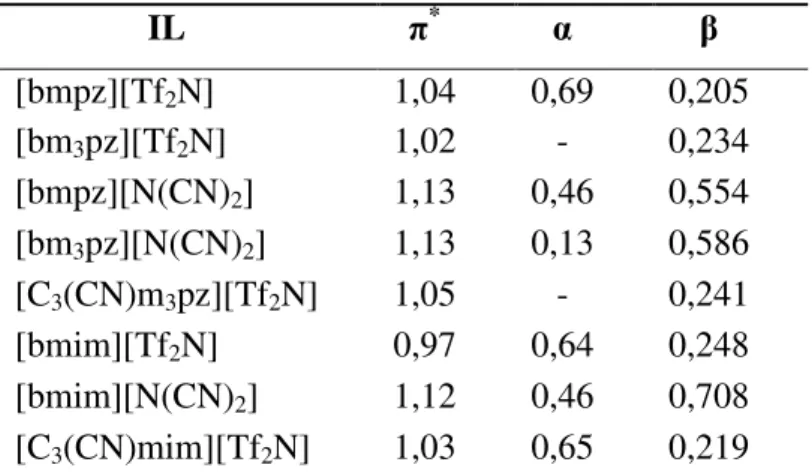
In tab. 2.8 sono riportati i parametri di Kamlet-Taft determinati nell’ambito di questa tesi per i liquidi ionici a base pirazolio. Questi parametri sono stati determinati a 25 °C, utilizzando tre sonde solvatocromiche (dye di Reichardt, N,N-dietil-nitroanilina e 4-nitroanilina) registrando gli spettri di soluzioni di tali composti nei liquidi ionici investigati nell’intervallo di lunghezze d’onda compreso tra 250-600 nm.
Tabella 2.8. Parametri solvatocromici dei sali di pirazolio.
IL π* α β [bmpz][Tf2N] 1,04 0,69 0,205 [bm3pz][Tf2N] 1,02 - 0,234 [bmpz][N(CN)2] 1,13 0,46 0,554 [bm3pz][N(CN)2] 1,13 0,13 0,586 [C3(CN)m3pz][Tf2N] 1,05 - 0,241 [bmim][Tf2N] 0,97 0,64 0,248 [bmim][N(CN)2] 1,12 0,46 0,708 [C3(CN)mim][Tf2N] 1,03 0,65 0,219
Per favorire il confronto, nella stessa tabella sono stati inseriti anche i valori di alcuni liquidi ionici a base imidazolio aventi strutture fortemente correlate ai sali di pirazolio investigati. Purtroppo, per alcuni liquidi ionici a base bistriflimide non è stato possibile determinare il parametro α a causa dell’immediata scomparsa della colorazione dopo aggiunta della betaina 30. La banda di assorbimento a lunghezze d’onda maggiori di questo probe, che determina la colorazione della soluzione, è nota per essere particolarmente sensibile alla presenza di acidi o di cationi metallici (es. Ag+), che possono protonare o interagire fortemente con la porzione fenolica. Tuttavia, i liquidi ionici in questione non dovrebbero presentare valori di acidità tali da determinare la protonazione del probe e sicuramente, dopo i molteplici lavaggi a cui sono stati sottoposti, non contenevano cationi metallici in grado di complessare l’anione fenolato. La causa di questo comportamento è quindi al momento ancora non nota e sotto studio.
53 Sulla base dei dati riportati in tab. 2.8, si può affermare che, analogamente ai liquidi ionici con catione imidazolio, i liquidi ionici pirazolio presentano un’elevata polarizzabilità/dipolarità. E’ inoltre da notare che i liquidi ionici a base 1,2-dialchilpirazolio presentano una significativa capacità di funzionare da donatori di legame ad idrogeno; i valori di α sono paragonabili a quelli degli analoghi sali di imidazolio. Quindi, nei liquidi ionici con catione eterociclico a cinque termini, l’acidità non è una peculiarità del protone compreso tra i due atomi di azoto del sistema imidazolico. Questa ipotesi era stata formulata sulla base dei valori di α misurati per liquidi ionici con catione, 1-metil-3-butilimidazolio, bmim+, 1,2-dimetil-3-butilimidzolio, bm2im+ (tab. 2.7); i secondi mostrano valori di α significativamente più bassi, fatto attribuito alla mancanza del protone in C2. È da notare che l’introduzione di altri due gruppi metilici sull’anello pirazolico nelle posizioni 3 e 5 (adiacenti ai due atomi di azoto) riduce drasticamente la capacità del catione pirazolio di funzionare da donatore di legame ad idrogeno; il valore di α calcolato per [bm3pz][N(CN)2], avente un solo protone aromatico, è notevolmente inferiore a quello del [bmpz][N(CN)2]. Infine, il confronto dei valori di α per i liquidi ionici a base bistriflimide con quelli a base dicianammide conferma il fatto che la capacità del catione di funzionare da donatore di legame ad idrogeno è una proprietà modulata dalla basicità dell’anione; il [bmpz][Tf2N] è caratterizzato da un valore di α notevolmente superiore a quello misurato per [bmpz][N(CN)2].
2.5
Viscosità, Densità e Conducibilità
Per tutti i liquidi ionici a base pirazolio alcune proprietà fondamentali quali la viscosità (
η
) e la conducibilità (κ
) sono state misurate nell’intervallo di temperature compreso tra 20 e 80 °C, dopo accurato essiccamento, evitando quanto più possibile il contatto con l’aria durante il trasferimento e le misure. I valori di viscosità e conducibilità alle diverse temperature sono riportati in tab. 2.9 e 2.12.A parità di anione e sostituenti alchilici i sali di pirazolio sintetizzati sono caratterizzati da valori di viscosità paragonabili agli analoghi sali di imidazolio.
54
Tabella 2.9. Andamento della viscosità (cP) con la temperatura
Liquido Ionico 20 (°C) 30 (°C) 40 (°C) 50 (°C) 60 (°C) 70 (°C) 80 (°C) cP cP cP cP cP cP cP [bmpz][Tf2N] 89,5 51,1 37 25,5 18,5 14,5 11,5 [bm3pz][Tf2N] 101,9 58,9 40,4 27,0 19,3 15,3 11,7 [bmpz][N(CN)2] 37,2 24,4 17,5 12,9 9,9 8,1 6,4 [bm3pz][N(CN)2] 90,0 46,7 33,0 21,8 15,5 11,3 8,8 [C3(CN)mpz][Tf2N] 1488,0 650,9 304,6 171,6 99,2 65,3 47,4 [C3(CN)m3pz][Tf2N] 722,2 303,8 165,8 92,2 57,8 38,4 27,7
Analogamente ai sali di imidazolio, inoltre, nel caso dell’1-metil-3-butilpirazolio la viscosità aumenta passando dal liquido ionico ad anione dicianammide a quello con anione bistriflimide. Nel caso delle dicianammidi, inoltre, la viscosità è sensibile alla presenza o meno di gruppi metilici sull’anello pirazolico; la viscosità del [bm3pz][N(CN)2] è notevolmente superiore a quella del [bmpz][N(CN)2]. D’altra parte, la presenza dei metili nelle posizioni 3 e 5 sul catione pirazolio influenza in maniera meno significativa la viscosità degli analoghi liquidi ionici a base bistriflimide. Infine è da notare che, sebbene la presenza di un gruppo nitrile in catena laterale determina, come nel caso dei liquidi ionici a base imidazolo, un drastico aumento della viscosità, in questo caso l’effetto della metilazione del catione nelle posizioni 3 e 5 sembra giocare un ruolo opposto: la viscosità del [bm3pz][Tf2N] è minore di quella del [bmpz][Tf2N]. In fig. 2.9 e 2.10 sono riportati gli andamenti grafici della viscosità in funzione della temperatura.
Indubbiamente, la viscosità è una proprietà complessa e, allo stato attuale delle conoscenze, sebbene i liquidi ionici a base imidazolo siano stati sicuramente studiati in maniera più ampia ed approfondita dei liquidi ionici a base pirazolio, non esistono neanche per questi delle correlazioni certe tra la struttura e la viscosità.
55
Figura 2.9. Viscosità dei sali di pirazolio con catena alchilica.
Figura 2.10. Viscosità dei sali di pirazolio con catena funzionalizzata.
Tuttavia, nel tentativo di ottenere ulteriori informazioni sulla struttura dei liquidi ionici dai dati di viscosità abbiamo analizzato l’andamento di questa proprietà in funzione della temperatura, utilizzando adeguati modelli matematici. Inizialmente, i dati di viscosità misurati tra 293 e 363 K sono stati interpolati utilizzando la forma logaritmica
56 dell’equazione di Arrhenius, che normalmente è utilizzata per descrivere la dipendenza della viscosità dalla temperatura nel caso di elettroliti liquidi non dissociati:
lnη = lnη∞ + Eη/RT
dove Eη è l’energia di attivazione per un flusso viscoso, lnη∞ è la viscosità a temperatura infinita ed R è la costante dei gas. Tutti i liquidi ionici investigati seguivano “approssimativamente” l’equazione di Arrhenius (in fig. 2.11, è riportato l’andamento dei sei liquidi ionici studiati mentre in fig. 2.12 sono rappresentati i singoli andamenti); le deviazioni maggiori si osservano nel caso dei liquidi ionici funzionalizzati.
2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 1,5 2,0 2,5 3,0 3,5 4,0 4,5 5,0 5,5 6,0 6,5 7,0 7,5 [bm3pz][Tf2N] [bmpz][Tf2N] [bmpz][N(CN)2] [bm3pz][N(CN)2] [C3(CN)mpz][Tf2N] [C3(CN)m3pz][Tf2N] L n η , c P 1000/T (K)
Figura 2.11. Andamento della viscosità in funzione della temperatura secondo l’eq di Arrhenius.
In ogni caso, i valori di Eη, lnη∞ ed il parametro di “fitting” (R) sono riportati in tab. 2.10.
Per questi ILs, i dati di viscosità sono stati analizzati anche sulla base dell’equazione di Vogel-Tamman-Fulcher (VTF) (2):
57 dove η0 (cP), B (K) and T0 (K) sono parametri di “fitting”. I migliori parametri e i coefficienti di correlazione associati sono riportati in tab. 2.10.
2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 2,5 3,0 3,5 4,0 4,5 L n η , c P 1000/T (K) [bmpz][Tf2N] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 2,5 3,0 3,5 4,0 4,5 5,0 L n η , c P 1000/T (K) [bm3pz][Tf2N] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 1,8 2,0 2,2 2,4 2,6 2,8 3,0 3,2 3,4 3,6 3,8 L n η , c P 1000/T (K) [bmpz][N(CN)2] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 2,0 2,5 3,0 3,5 4,0 4,5 L n η , c P 1000/T (K) [bm3pz][N(CN)2] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 3,5 4,0 4,5 5,0 5,5 6,0 6,5 7,0 7,5 L n η , c P 1000/T (K) [C3(CN)mpz][Tf2N] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 3,0 3,5 4,0 4,5 5,0 5,5 6,0 6,5 7,0 L n η , c P 1000/T (K) [C 3(CN)m3pz][Tf2N]
58 290 300 310 320 330 340 350 360 0 20 40 60 80 100 Model: Exp3P1 Equation: y =a*exp(b/(x+c)) Weighting: y No weighting Chi^2/DoF = 2.09349 R^2 = 0.99817 a 0.91753 ±0.62081 b 356.36479 ±126.27141 c -215.1272 ±16.23982 η , c P T (K) [bmpz][Tf2N] 290 300 310 320 330 340 350 360 0 20 40 60 80 100 Model: Exp3P1 Equation: y =a*exp(b/(x+c)) Weighting: y No weighting Chi^2/DoF = 0.96049 R^2 = 0.99938 a 0.40729 ±0.23716 b 523.81819 ±128.36866 c -198.12462 ±13.3425 η , c P T (K) [bm3pz][Tf2N] 290 300 310 320 330 340 350 360 5 10 15 20 25 30 35
40 Model: Exp3P1Equation: y =a*exp(b/(x+c)) Weighting: y No weighting Chi^2/DoF = 0.02439 R^2 = 0.99986 a 0.2988 ±0.06987 b 513.92816 ±58.00114 c -186.44667 ±6.91382 η , c P T (K) [bmpz][N(CN)2] 290 300 310 320 330 340 350 360 0 20 40 60 80 100 Model: Exp3P1 Equation: y =a*exp(b/(x+c)) Weighting: y No weighting Chi^2/DoF = 3.18471 R^2 = 0.99741 a 0.76314 ±0.6286 b 327.2823 ±136.38648 c -224.32613 ±16.91677 η , c P T (K) [bm3pz][N(CN)2] 290 300 310 320 330 340 350 360 0 200 400 600 800 1000 1200 1400 1600 Model: Exp3P1 Equation: y =a*exp(b/(x+c)) Weighting: y No weighting Chi^2/DoF = 54.32187 R^2 = 0.99987 a 0.04465 ±0.04393 b 1189.91579 ±245.68949 c -178.74875 ±12.83206 η , c P T (K) [C3(CN)mpz][Tf2N] 290 300 310 320 330 340 350 360 0 100 200 300 400 500 600 700 800 Chi^2/DoF = 29.25277 R^2 = 0.99969 a 0.45001 ±0.3165 b 569.16873 ±123.79242 c -215.87187 ±9.47455 η , c P T (K) [C3(CN)m3pz][Tf2N]
59
Tabella 2.10. Parametri relative all’analisi della viscosità in funzione della temperatura secondo l’equazione
di Arrhenius e Vogel-Tamman-Fulcher (VTF). Liquidi Ionici Eηηηη kJ mol−1−1−1−1 lnηηηη∞ cP R η η η η0 cP B K T0 K R 2 [bmpz][Tf2N] 28,26 -7,3 0,995 0,91 356 215 0,998 [bm3pz][Tf2N] 29,93 -7,9 0,996 0,40 523 198 0,999 [bmpz][N(CN)2] 24,60 -6,5 0,997 0,29 513 186 0,999 [bm3pz][N(CN)2] 32,34 -8,9 0,995 0,76 327 224 0,997 [C3(CN)mpz][Tf2N] 49,21 -13,0 0,994 0,046 1187 178 0,999 [C3(CN)m3pz][Tf2N] 46,55 -12,7 0,994 0,45 569 215 0,998
Per questi composti, considerando le forti interazioni elettrostatiche tra anione e catione, le energie di attivazione Eη possono essere considerate come le barriere energetiche da superare per permettere agli ioni di muoversi uno rispetto all’altro e quindi possono essere dare informazioni sulle interazioni catione-anione. E’ innanzitutto da notare che il valore di Eη del [bmpz][Tf2N] è paragonabile a quello dell’analogo sale di imidazolo Eη = 25,40. Inoltre, sia i valori di Eη che B (B è in realtà un parametro empirico più simile ad un’energia libera che di attivazione) dimostrano una dipendenza dalla struttura; in particolare, i valori elevati di Eη, associati ai valori elevati e negativi di lnη∞ trovati per i liquidi ionici funzionalizzati con un gruppo nitrile suggeriscono per questi sistemi un più elevato grado di associazione e di ordine. Tabella 2.11. Densità a 25 °C. Liquidi Ionici ρρρρ (g/mL) [bmpz][Tf2N] 1,45 [bm3pz][Tf2N] 1,38 [bmpz][N(CN)2] 1,07 [bm3pz][N(CN)2] 1,05 [C3(CN)mpz][Tf2N] 1,56 [C3(CN)m3pz][Tf2N] 1,45
Come ci si aspettava, i pirazoli aventi la dicianammide come anione hanno valori di densità più bassi, paragonabili a quelli degli analoghi sali di imidazolio. Anche i Tf2Nି presentano valori paragonabili a questi ultimi. L’incremento repentino della viscosità
60 associato alla presenza in catena laterale della funzionalità nitrilica non è accompagnato da un aumento paragonabile dei valori di densità. Inoltre non vi sono differenze sostanziali dovute alla presenza dei due metili sull’anello aromatico.
In tab. 2.12 sono riportati i valori di conducibilità misurati per i sali di pirazolio a diverse temperature, comprese tra 20 e 80 °C.
Sebbene la viscosità da sola non sempre riesce a rendere conto della conducibilità, normalmente esiste una correlazione inversa tra questi due parametri. In accordo con i dati di viscosità, l’1-metil-3-butilpirazolo dicianammide è il liquido ionico caratterizzato dalla più elevata conducibilità, mentre i liquidi ionici funzionalizzati ([C3(CN)mpz][Tf2N] e ([C3(CN)m3pz][Tf2N]) dai più bassi valori di conducibilità. Le differenze tra i valori di conducibilità ad una certa temperatura in funzione della struttura, o per uno stesso composto in funzione della temperatura, non sono tuttavia sempre in accordo con i dati di viscosità.
Come evidenziato da Ohno et. al.1, la conducibilità è determinata non solo dalla forma e raggio degli ioni ma anche dal loro grado di associazione anione-catione. Il moto correlato di coppie ioniche o aggregati di dimensioni maggiori riduce la conducibilità; per questi sistemi, la viscosità non è un buon indicatore delle dinamiche molecolari.
Tabella 2.12. Andamento della conducibilità (mS/cm) con la temperatura.
Liquidi Ionici 20 (°C) mS/cm 30 (°C) mS/cm 40 (°C) mS/cm 50 (°C) mS/cm 60 (°C) mS/cm 70 (°C) mS/cm 80 (°C) mS/cm [bmpz][Tf2N] 2,90 3,70 4,41 5,15 5,91 6,69 7,36 [bm3pz][Tf2N] 2,36 2,94 3,68 4,56 5,22 5,98 6,80 [bmpz][N(CN)2] 7,74 9,29 11,04 12,83 14,55 16,40 18,10 [bm3pz][N(CN)2] 2,60 3,80 5,24 6,61 8,16 9,74 11,48 [C3(CN)mpz][Tf2N] 0,24 0,42 0,64 0,95 1,28 1,75 2,17 [C3(CN)m3pz][Tf2N] 0,36 0,62 0,93 1,31 1,76 2,40 2,93 In fig. 2.14 è riportato il grafico della conducibilità in relazione alla temperatura dei sali di pirazolio.
Figura
L’andamento della conducibilità in funzione della temperatura di natura del liquido.
2,8 2,9 -2 -1 0 1 2 3 L n κ , m S /c m
Figura 2.15. Grafici di Arrhenius per conducibilità dei sei liquidi ionici a base pirazolio
Come atteso, sulla base dell’andamento della viscosità in funzione della temperatura, i sei liquidi ionici a base pirazolio seguivano “approssimativamente” l’equazione di Arrhenius:
Figura 2.14. Conducibilità in funzione della temperatura.
L’andamento della conducibilità in funzione della temperatura di
2,9 3,0 3,1 3,2 3,3 3,4 3,5 [bmpz][Tf [bm3pz][Tf [bmpz][N(CN) [bm3pz][N(CN) [C3(CN)mpz][Tf [C3(CN)m 1000/T (K)
Grafici di Arrhenius per conducibilità dei sei liquidi ionici a base pirazolio
atteso, sulla base dell’andamento della viscosità in funzione della temperatura, i sei liquidi ionici a base pirazolio seguivano “approssimativamente”
lnκ = lnA + (Eκ/RT)
61 L’andamento della conducibilità in funzione della temperatura dipende dalla
[bmpz][Tf2N] pz][Tf2N] [bmpz][N(CN)2] pz][N(CN)2] (CN)mpz][Tf2N] (CN)m3pz][Tf2N]
Grafici di Arrhenius per conducibilità dei sei liquidi ionici a base pirazolio
atteso, sulla base dell’andamento della viscosità in funzione della temperatura, i sei liquidi ionici a base pirazolio seguivano “approssimativamente”
62 (in fig. 2.15, sono riportati i grafici di Arrhenius dei sei liquidi ionici studiati mentre in fig. 2.16 sono rappresentati i singoli andamenti); le deviazioni maggiori anche in questo caso si osservavano per i liquidi ionici funzionalizzati.
2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 1,0 1,2 1,4 1,6 1,8 2,0 L n κ , m S /c m 1000/T (K) [bmpz][Tf2N] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 0,8 1,0 1,2 1,4 1,6 1,8 2,0 L n κ , m S /c m 1000/T (K) [bm3pz][Tf2N] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 2,0 2,2 2,4 2,6 2,8 3,0 L n κ , m S /c m 1000/T (K) [bmpz][N(CN)2] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2 2,4 2,6 L n κ , m S /c m 1000/T (K) [bm3pz][N(CN)2] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 -1,5 -1,0 -0,5 0,0 0,5 1,0 L n κ , m S /c m 1000/T (K) [C3(CN)mpz][Tf2N] 2,8 2,9 3,0 3,1 3,2 3,3 3,4 3,5 -1,0 -0,5 0,0 0,5 1,0 L n κ , m S /c m 1000/T (K) [C3(CN)m3pz][Tf2N]
63 Anche i dati di conducibilità sono stati quindi analizzati sulla base dell’equazione di Vogel-Tamman-Fulcher (VTF):
κ = Α exp[−B/(T−T0)] (2)
dove A (mS/cm), B (K) and T0 (K) sono parametri di “fitting”. I migliori parametri e i coefficienti di correlazione associati sono riportati in tab. 2.13, insieme ai parametri di Arrhenius.
Tabella 2.13. Parametri relative all’analisi della densità in funzione della temperatura secondo l’equazione di Arrhenius e Vogel-Tamman-Fulcher (VTF). Composti Eκκκκ kJ mol-1 lnA cP R A cP B K T0 K R 2 [bmpz][Tf2N] 13.05 6.5 0.995 29.4 209 202 0.998 [bm3pz][Tf2N] 15.05 7.1 0.996 39.5 282 193 0.999 [bmpz][N(CN)2] 12.06 7.0 0.998 96.0 298 174 0.999 [bm3pz][N(CN)2] 20.78 9.6 0.993 79.0 271 213 0.997 [C3(CN)mpz][Tf2N] 30.76 11.4 0.995 43.0 424 211 0.999 [C3(CN)m3pz][Tf2N] 29.51 11.2 0.995 63.0 456 204 0.998
Il fatto che i liquidi ionici funzionalizzati presentino valori di Eκ superiore agli altri liquidi ionici a base pirazolio conferma la presenza in questi sistemi di un più elevato grado di aggregazione.
64 290 300 310 320 330 340 350 360 3 4 5 6 7 8 Chi^2/DoF = 0.00178 R^2 = 0.99954 a 29.46097 ±3.79518 b -209.53544 ±31.10862 c -202.4913 ±8.77324 κ , m S /m c T (K) [bmpz][Tf 2N] 290 300 310 320 330 340 350 360 2 3 4 5 6 7 Model: Exp3P1 Equation: y =a*exp(b/(x+c)) Weighting: y No weighting Chi^2/DoF = 0.00318 R^2 = 0.99919 a 39.54974 ±9.19763 b -282.32074 ±60.8543 c -193.53308 ±13.85283 κ , m S /c m T (K) [bm3pz][Tf2N] 290 300 310 320 330 340 350 360 6 8 10 12 14 16 18 Chi^2/DoF = 0.00652 R^2 = 0.99969 a 96.09329 ±14.36195 b -298.1815 ±44.49237 c -174.98452 ±10.94861 κ , m S /m c T (K) [bmpz][N(CN) 2] 290 300 310 320 330 340 350 360 2 4 6 8 10 12 Chi^2/DoFR^2 = 0.99991= 0.00136 a 79.07682 ±5.99477 b -271.16054 ±16.99288 c -213.52722 ±3.4343 κ , m S /c m T (K) [bm3pz][N(CN)2] 290 300 310 320 330 340 350 360 0,0 0,5 1,0 1,5 2,0 2,5 Chi^2/DoF = 0.00084 R^2 = 0.99889 a 43.21641 ±19.92697 b -424.01886 ±108.70835 c -211.85724 ±14.87495 κ , m S /c m T (K) [C3(CN)mpz][Tf2N] 290 300 310 320 330 340 350 360 0,0 0,5 1,0 1,5 2,0 2,5 3,0 Chi^2/DoF = 0.00206 R^2 = 0.99847 a 63.20908 ±36.28283 b -456.17451 ±143.01275 c -204.85019 ±19.24362 κ , m S /c m T (K) [C3(CN)m3pz][Tf2N]
Figura 2.17. Grafici di VTF per la conducibilità dei singoli ILs a base pirazolio
2.6
Finestra elettrochimica
Il criterio chiave per selezionare un solvente per applicazioni elettrochimiche è la stabilità elettrochimica del solvente stesso che si determina misurando l’intervallo entro cui esso è inerte. L’entità di tale finestra dipende dalla stabilità ossidativa e riduttiva del composto utilizzato come solvente, e nel caso dei liquidi ionici essa è determinata
65 primariamente dalla stabilità del catione alla riduzione e dell’anione all’ossidazione (non rientrano in questa classe i liquidi ionici con anioni clorometallati, per i quali spesso è la riduzione dell’anione il processo catodico limitante).
Il metodo più utilizzato per determinare la finestra elettrochimica è la voltammetria ciclica. In un sistema a tre elettrodi, il potenziale di un elettrodo inerte di lavoro è variato nel tempo verso potenziali più grandi positivi (anodici) e negativi (catodici) fino a che il valore base della corrente non cresce in maniera repentina a causa dell’ossidazione o riduzione del solvente. I potenziali limite ossidativi e riduttivi vengono assegnati quando la corrente di base raggiunge dei valori soglia. Poiché la scelta del valore limite è soggettiva, la definizione della finestra elettrochimica presenta un certo grado di incertezza, generalmente intorno a 0,2 V. E’ inoltre da rilevare che nel caso dei liquidi ionici la presenza di impurezze (spesso acqua e alogenuri) può avere un profondo effetto sui potenziali limite che determinano la finestra elettrochimica. In particolare gli alogenuri, presenti quale conseguenza di una non completa reazione di scambio, sono più facilmente ossidabili degli anioni fluorurati (es. bistriflimide) spesso utilizzati per applicazioni elettrochimiche, e sono in grado di determinare una riduzione del potenziale anodico. Per quanto concerne l’acqua, questa essendo in grado sia di ossidarsi e che ridursi può influenzare in maniera sostanziale la finestra elettrochimica. Per evitare quindi i problemi derivanti dalla presenza di acqua, che può essere presente in quantità non trascurabile anche nei liquidi ionici idrofobici, i campioni una volta posti nella cella elettrochimica, prima di effettuare le relative misure, sono stati sottoposti a un processo di ri-essiccamento, mantenendoli per 24 ore a 60-80 °C sotto vuoto.
Le finestre elettrochimiche dei sali di pirazolio, determinate usando come elettrodo di lavoro un elettrodo al platino, sono riportate in tab. 2.14.
66
Tabella 2.14. Finestre elettrochimiche dei sali di pirazolio.
IL E(catodico)/V E(anodico)/V E(totale)/V
[bmpz][Tf2N] -2,21 +2,53 4,74 [bm3pz][Tf2N] -2,35 +1,95 4,30 [bmpz][N(CN)2] -2,40 +1,60 4,00 [bm3pz][N(CN)2] -2,43 +1,27 3,70 [C3(CN)mpz][Tf2N] -1,22 +2,98 4,20 [C3(CN)m3pz][Tf2N] -1,03 +2,47 3,50 [bmim][Tf2N] -2,50 +2,12 4,62
Poiché la natura dell’elettrodo di lavoro può influenzare i potenziali limite, a scopo di confronto è stata misurata anche la finestra elettrochimica del [bmim][Tf2N], per il quale sono riportati i relativi valori in letteratura. I valori di tab. 2.14 dimostrano che i sali di pirazolio, come la maggior parte dei liquidi ionici, hanno un’ampia finestra elettrochimica. La finestra elettrochimica dei [bmpz][Tf2N] non presenta sostanziali differenze rispetto al corrispondente sale di imidazolo, [bmim][Tf2N]. L’introduzione dei gruppi metilici nell’anello pirazolico determina tuttavia una diminuzione del potenziale anodico con un restringimento della finestra, che risulta più marcato nel [bm3pz][N(CN)2] e nel [C3(CN)m3pz][Tf2N]. Le finestre elettrochimiche meno ampie si hanno per i liquidi ionici con dicianammide. È infine da notare che l’introduzione della funzione nitrilica causa un aumento del potenziale catodico e anodico.








![Sintesi e caratterizzazione biologica di nuovi
3,6-diazabiciclo[3.1.1]eptani quali potenti e selettivi
ligandi per i recettori neuronali nicotinici α4β2 e α7](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)