1
CAPITOLO 1: INTRODUZIONE
1.1 POLIMERI PER APPLICAZIONI BIOMEDICHE E
FARMACEUTICHE
L’uso di polimeri naturali e sintetici per applicazioni in campo biomedico e farmaceutico sta assumendo proporzioni sempre più ampie. Da una parte, le esigenze della bioingegneria, focalizzate sullo studio dell’evoluzione delle protesi nel tempo, e dall’altra, la formulazione di sistemi in grado di definire i profili cinetici e topologici di un rilascio controllato di farmaci, o di fornire farmaci macromolecolari con un’elevata selettività recettoriale, costituiscono i principi alla base di questo settore della chimica macromolecolare, volto alla produzione di materiali innovativi.
1.1.1 POLIMERI BIODEGRADABILI PER USO BIOMEDICO
I materiali biodegradabili sono stati ampiamente studiati per applicazioni biomediche come ad esempio scaffolds temporanei [1] e matrici per rilascio controllato di agenti terapeutici [2,3]. Recentemente il termine “degradazione” o “biodegradazione” è diventato una parola chiave nello sviluppo di nuove tecnologie, non solo per applicazioni mediche e farmaceutiche, ma anche per l’agricoltura e applicazioni ambientali.
Il concetto di “biodegradabilità di un polimero” è stato definito in vari modi da diversi autori:
a) cambiamenti nelle proprietà superficiali o perdita di resistenza meccanica[4] b) assimilazione da parte di microrganismi [5]
c) degradazione da parte di enzimi[6,7]
d) rottura della catena polimerica con susseguente riduzione del peso molecolare [8,9]
e) estrazione di materiale a basso peso molecolare che porta a difetti di superficie
Tuttavia, il processo di degradazione “in vivo” non può essere comparato con il termine “biodegradazione” per il fatto che quest’ultima implica la partecipazione attiva di entità
2 biologiche come enzimi e microrganismi [10] e risulta difficile identificare il coinvolgimento e il ruolo di specie biologiche in un processo degradativo “in vivo”. Infatti, sia processi idrolitici che enzimatici possono contribuire in diverse proporzioni ad un processo di degradazione: il primo stadio potrebbe, ad esempio, essere idrolitico ma in conseguenza di variazioni strutturali sulla superficie polimerica o dell’aumentata permeabilità, potrebbe in seguito dominare un processo di degradazione enzimatica. Così il termine biodegradazione comprende in genere tutti i tipi di degradazione sia idrolitici che metabolici. Inoltre per processi “in vivo”, la degradazione di materiali polimerici, viene descritta oltre che in termini di biodegradazione, anche in termini di BIORIASSORBIMENTO, BIOASSORBIMENTO, BIOEROSIONE, e BIODETERIORAMENTO. Sebbene non sia stata stabilita una chiara differenza tra questi termini, si possono dare le seguenti definizioni:(libro )
1) BIODEGRADAZIONE: conversione del materiale in prodotti intermedi meno complessi o prodotti finali tramite solubilizzazione, idrolisi, o azione di entità biologiche che possono essere enzimi o altri prodotti dell’organismo.
2) BIORIASSORBIMENTO: degradazione del materiale in composti a basso peso molecolare che possono essere eliminati dal corpo attraverso vie naturali. 3) BIOASSORBIMENTO: scomparsa del materiale dal suo iniziale sito di
applicazione con o senza degradazione delle macromolecole disperse.
4) BIOEROSIONE: conversione di un polimero insolubile in acqua, ad un polimero solubile in acqua o a piccole molecole. L’erosione può avvenire solo sulla superficie del materiale (surface erosion), o può coinvolgere l’intera massa (bulk erosion).
5) BIODETERIORAMENTO: cambiamento indesiderato delle proprietà meccaniche, fisiche, o chimiche del materiale.
L’utilizzo di polimeri sintetici biodegradabili nel campo dei biomateriali, presenta forti attrattive dato che, variando la struttura o lo schema di sintesi, è possibile ottenere materiali con specifiche caratteristiche fisiche e chimiche. Diversi tipi di polimeri biodegradabili sono stati sviluppati per una vasta gamma di applicazioni [12]. Quelli maggiormente studiati per applicazioni biomediche e farmaceutiche sono i poli(α-esteri) come gli omo e copolimeri dell’acido glicolico e dell’acido lattico e i poli(idrossialcanoati). È stato dimostrato che questi polimeri sono non tossici e
3 biocompatibili così come i loro prodotti di degradazione, in molti casi presenti naturalmente nel corpo[13].
1.2 POLIURETANI BIODEGRADABILI PER USO BIOMEDICO
Nella vasta gamma di polimeri biodegradabili sviluppati, relativamente pochi sono i polimeri elastomerici [14]. Elastomeri degradabili potrebbero trovare applicazione ad esempio nel settore del soft tissue engineering (pelle artificiale e sistema vascolare) [15]. L’utilizzo di elastomeri è comunque conveniente in tutti quei casi in cui il materiale polimerico viene sottoposto ad un qualche stress meccanico.I poliuretani segmentati, costituiscono un ottimo esempio di polimeri con una grande versatilità chimica. Questi materiali sono stati utilizzati, grazie alla loro buona biocompatibilità, come biomateriali soprattutto in applicazioni a lungo termine [16]. Minor interesse è stato invece rivolto allo sviluppo di poliuretani biodegradabili dato che i diisocianati comunemente utilizzati come loro precursori, forniscono prodotti tossici o cancerogeni per degradazione [17]. Quest’ostacolo è stato tuttavia superato, grazie all’ottenimento di diisocianati basati su L-lisina o su altri aminoacidi. Polimeri degradabili tramite enzimi, sono stati sintetizzati, introducendo unità strutturali disponibili in natura come α-amminoacidi e peptidi. In questo modo è possibile ottenere prodotti di degradazione non tossici e facilmente metabolizzabili in vivo. Huang e collaboratori [6] hanno descritto la sintesi di amminoacidi-α-ω-alchilendiesteri per ottenere poliesteruree che vengono attaccate in vitro dall’enzima α-chimotripsina. In seguito sono stati utilizzati diesteri di α-amminoacidi per ottenere poliesterammidi [18], poliesteruree e poliuretani non segmentati [19,20], tutti soggetti ad attacco enzimatico in vitro.
Scarja e Woodhouse [21,22] hanno recentemente riportato la sintesi di un diestere a base di fenilalanina usato come estensore di catena nella preparazione di poliuretani segmentati dei quali è stata studiata in particolare la degradabilità in funzione della natura e del peso molecolare dei segmenti soft impiegati.
Ciardelli e collaboratori [23], hanno sintetizzato una serie di poliuretani e poliuretani-uree segmentati, utilizzando come diisocianato un composto alifatico ottenuto a partire dal cloridrato di L-lisina; come macrodiolo, una serie di copolimeri a blocchi poli(caprolattone)-poli(etilenglicol)-poli(caprolattone) con idonee
4 caratteristiche di biocompatibilità o poli(caprolattone)diolo; come estensore di catena, una diammina di sintesi a base di fenilalanina o cicloesandimetanolo. Lo studio sulla degradabilità è stato condotto in soluzione tampone e in una soluzione dell’enzima α-chimotripsina.
1.3 MICROSFERE PER RILASCIO CONTROLLATO DI
FARMACO
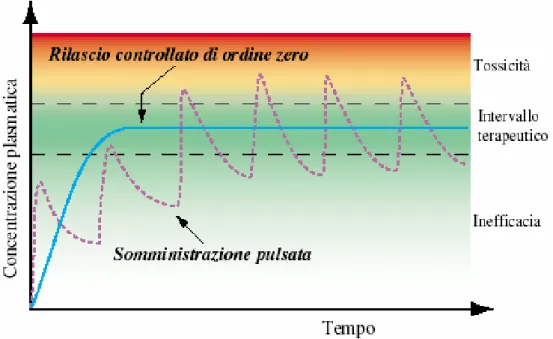
I convenzionali metodi di somministrazione di farmaci, ad esempio per via orale, non consentono di ottenere nel tempo, un profilo di concentrazione plasmatica costante dell’agente terapeutico. Una somministrazione ripetuta presenta spesso, un profilo cinetico pulsato con delle punte di massimo che possono superare l’intervallo terapeutico, raggiungendo livelli di tossicità in conseguenza di un accumulo di farmaco nell’organismo, e delle punte di minimo al di sotto dell’intervallo terapeutico, in concentrazione tale che il farmaco risulti inefficace. In un sistema a rilascio controllato, il farmaco viene invece rilasciato in maniera progressiva con una velocità pressoché costante garantendo così un prolungato effetto terapeutico (Figura 1)
FIGURA 1. Ipotetica concentrazione plasmatica di farmaco nel tempo con forme di dosaggio convenzionali e con un sistema a rilascio controllato.
5 L’idea del rilascio controllato di farmaci da matrici polimeriche risale agli anni ’60 con l’impiego di polietilene [24]. La mancanza di degradabilità in tale sistema, implica la rimozione chirurgica dell’impianto alla fine del processo di cessione. Negli anni ’70 è stato proposto l’impiego di matrici polimeriche biodegradabili per evitare la rimozione del sistema[25].
Come definizione generale, il termine microsfera identifica qualsiasi sistema di natura particellare contenente all’interno un materiale solido, liquido o gassoso. Per scopi biomedici le microsfere sono costituite essenzialmente da una matrice polimerica sintetica, o naturale al cui interno è presente un principio farmacologicamente attivo.
Si possono distinguere:
a) microsfera omogenea nella quale il farmaco si trova disciolto nella matrice polimerica
b) microsfera monolitica in cui il farmaco è finemente disperso nella matrice polimerica
c) microsfera a riserva in cui il farmaco costituisce un nucleo interno immerso nella matrice polimerica
d) microsfera a doppia parete che presenta un rivestimento polimerico esterno con caratteristiche diverse da quelle della matrice interna
La velocità di rilascio del farmaco, dipende dal suo grado di diffusione attraverso le maglie del reticolo polimerico. Nel caso in cui le microsfere siano composte da materiale bioerodibile, la velocità di rilascio dipende principalmente da fenomeni di degradazione in seguito al contatto con i fluidi biologici. Tuttavia, i profili cinetici di rilascio di microsfere bioerodibili, vengono spesso significativamente influenzati anche da fenomeni diffusivi.
1.3.1 PREPARAZIONE DI MICROSFERE
Diverse tecniche sono disponibili per la preparazione delle microsfere e la scelta del metodo più idoneo dipende sia dalla natura dei materiali utilizzati, sia dal tipo di applicazione a cui esse vanno destinate. La preparazione può essere effettuata a partire dai monomeri, o dal polimero già formato.
6 In tabella 1 sono riportati alcuni metodi di preparazione di microsfere a partire dai monomeri.
TABELLA 1 Tecniche per la preparazione di microsfere a partire da monomeri METODI DI PREPARAZIONE DIMENSIONI (µm) Polimerizzazione in emulsione 0.01-1
Polimerizzazione in dispersione 0.5-10 Polimerizzazione in sospensione 50-500 Polimerizzazione per sedimentazione >1000
La polimerizzazione in emulsione è generalmente usata per ottenere sfere di dimensioni nanometriche (nanosfere). La tecnica si basa sulla dispersione del monomero in acqua insieme ad un iniziatore solubile in ambiente acquoso. Viene anche impiegato un tensioattivo per ottenere la formazione di micelle uniformi. La polimerizzazione ha luogo nelle micelle e non nelle gocce di monomero disperso dato che l’iniziatore è in esso insolubile. Le gocce polimeriche risultanti possono essere così uniformi su scala nanometrica, da poter dare fenomeni di diffrazione della luce visibile [26].
Con la polimerizzazione in dispersione è possibile ottenere sfere di dimensioni 0.5-10 µm. Tutti i reagenti, comprendenti monomero, iniziatore e stabilizzante, vengono disciolti in un mezzo organico. L’iniziatore deve essere solubile nel monomero e il polimero risultante deve essere insolubile nel solvente organico utilizzato. La polimerizzazione ha luogo nelle gocce di monomero essendo l’iniziatore solubile in esse. Le particelle di polimero precipitano essendo insolubili nel mezzo organico, e lo stabilizzante ne previene la flocculazione [27].
La polimerizzazione in sospensione viene utilizzata per ottenere particelle di dimensioni comprese tra 50 e 500 µm. Il monomero viene disperso in mezzo acquoso con uno stabilizzante. L’iniziatore utilizzato è solubile nella fase monomerica in cui ha luogo la polimerizzazione. La dimensione e la quantità delle particelle è determinata dalla dimensione e dalla quantità delle gocce di monomero disperso oltre che dall’efficienza dell’agitazione meccanica [28]
7 La maggior parte delle microsfere utilizzate per il rilascio controllato di farmaci, viene comunque preparata a partire da polimeri già formati. Alcune tecniche di preparazione sono:
• Reticolazione in sospensione • Coacervazione
• Spray drying
• Precipitazione del polimero
• Evaporazione-estrazione del solvente
Tra quelle citate, le tecniche più comunemente utilizzate sono reticolazione in sospensione, spray drying ed evaporazione-estrazione del solvente.
La reticolazione in sospensione, è comunemente utilizzata per preparare microsfere a base di proteine o poli(saccaridi). La tecnica comprende due stadi fondamentali. Il primo consiste nella formazione di una dispersione stabile, ottenuta aggiungendo una soluzione polimerica contenente il farmaco disciolto o disperso, ad un liquido immiscibile che funge da mezzo sospendente. Per ottenere una microsospensione, le due fasi vengono sottoposte ad agitazione meccanica mediante un omogenizzatore, oppure vengono sottoposte all’azione di ultrasuoni. Il secondo stadio prevede la conversione diretta delle microgocce di soluzione polimerica in microparticelle di polimero. Un eventuale fenomeno di coalescenza viene inibito grazie all’utilizzo di uno stabilizzante che forma un film sulla superficie delle gocce polimeriche impedendone l’aggregazione. Per sospensioni olio in acqua può essere utilizzato, come agente stabilizzante, il poli(N-vinil-2-pirrolidinone) mentre per emulsioni acqua in olio, acetato di cellulosa. L’indurimento delle gocce di soluzione polimerica e la seguente formazione di particelle di polimero, avvengono in seguito ad un processo di reticolazione delle catene macromolecolari che può aver luogo tramite gruppi funzionali presenti nelle macromolecole oppure per aggiunta di composti polifunzionali alla sospensione. Le particelle possono infine essere isolate, in dipendenza dalla loro dimensione, per filtrazione, decantazione o centrifugazione [29].
La tecnica dello spray drying consente di ottenere microsfere generalmente porose di dimensioni comprese tra 5 e 600 µm. Il polimero viene sciolto in un solvente volatile e il farmaco viene disperso o disciolto in tale soluzione che viene successivamente
8 nebulizzata. Lo spray così ottenuto, viene miscelato, in una camera di miscelazione, con dell’aria calda che fornisce il calore necessario per l’evaporazione e l’essiccamento delle gocce. Le particelle asciutte vengono infine separate dal flusso d’aria. L’inconveniente di tale tecnica, spesso, è l’ottenimento di particelle che danno luogo ad aggregati e presentano inoltre una morfologia non ottimale. La qualità delle particelle può essere comunque migliorata mediante l’aggiunta di plastificanti, come ad esempio acido citrico, che permettono di ottenere microparticelle con una morfologia migliore [30].
La tecnica di evaporazione-estrazione del solvente, viene utilizzata soprattutto per la preparazione di microsfere a base di polimeri sintetici. Il polimero e il farmaco vengono disciolti in un solvente organico. Tale fase viene dispersa in acqua e sottoposta ad agitazione meccanica, ottenendo così un’emulsione bifasica olio in acqua. Spesso viene utilizzata una doppia emulsione, specialmente quando il farmaco risulta essere completamente insolubile nel solvente organico. In questo caso, il farmaco viene disciolto in acqua e successivamente la fase acquosa viene dispersa in un solvente organico contenente il polimero solubilizzato. Viene così formata la prima emulsione acqua in olio. La dispersione di questa prima emulsione in acqua, forma l’emulsione finale olio in acqua. In entrambi i casi (emulsione singola o doppia emulsione) la fase acquosa disperdente può contenere un agente stabilizzante, come ad esempio polivinilalcol, che previene l’agglomerazione delle particelle. L’indurimento delle microsfere avvengono per evaporazione del solvente a pressione ridotta, mediante riscaldamento, o semplicemente, se il solvente è sufficientemente volatile, per agitazione meccanica che va comunque mantenuta durante tutto il processo di evaporazione per evitare l’agglomerazione delle particelle. La velocità di evaporazione del solvente deve essere lenta per garantire l’omogeneità superficiale delle microsfere. Alla fine del processo le microparticelle possono essere isolate dall’ambiente di reazione, mediante filtrazione o centrifugazione in dipendenza dalle dimensioni [31,32].
9
1.3.2 FATTORI DI CONTROLLO PER MICROSFERE A RILASCIO DI
FARMACO
Il controllo e l’aumento della quantità di farmaco incapsulato nelle microsfere, è un importante fattore da prendere in considerazione per evitare quanto più possibile, perdita di farmaco durante la preparazione e per aumentare così la durata del trattamento terapeutico. La quantità di farmaco incapsulata nelle microsfere può essere descritta da due quantità: la prima, che è quella più comunemente usata, viene detta Efficacia d’Incapsulamento (EE) ed è definita come [33]:
EE = ∆D/DT dove:
DT = quantità totale di farmaco utilizzato.
∆D = differenza tra DT e la quantità di farmaco non incapsulata.
La seconda quantità viene detta Capacità di Carico (LC) ed è definita come [33]:
LC = ∆D/SW dove:
SW = peso delle sfere.
∆D = quantità di farmaco effettivamente incapsulato
Due parametri che influenzano sensibilmente l’incapsulamento del farmaco sono la temperatura di formazione delle sfere e la natura del polimero. Yang e collaboratori [34] hanno trovato che le più alte EE si hanno alla più alta e alla più bassa temperatura di preparazione testata. Questo comportamento suggerisce che meccanismi differenti governano l’incapsulamento del farmaco a temperature diverse. A basse temperature, aumenta l’immiscibilità tra le gocce di soluzione polimerica e la fase acquosa, con il risultato di una rapida formazione dello strato esterno delle sfere che possono così intrappolare velocemente il farmaco prima del processo di evaporazione del solvente [35]. A temperature più alte, l’aumento del grado di evaporazione del solvente porta ad un rapido indurimento dello strato esterno delle sfere che possono così incapsulare il farmaco all’inizio del processo di evaporazione. Considerando la relazione tra EE e
10 polimero, Ghaderi e collaboratori [36] hanno trovato un aumento dell’incapsulamento del farmaco al crescere della concentrazione del polimero nella fase organica, mentre Le Corre e collaboratori [37] hanno ottenuto differenti EE per poli(acido lattico) e poli(acido lattico-co-acido glicolico) preparando le rispettive microsfere nelle stesse condizioni operative.
Un altro fattore da tenere in considerazione nella preparazione di microsfere, è la loro dimensione che può essere influenzata da:
• Concentrazione del polimero nell’emulsione finale • Temperatura
• Viscosità
• Velocità di agitazione dell’emulsione finale • Quantità di emulsionante utilizzato
Per quanto riguarda la concentrazione del polimero, vari lavori riportano che la dimensione delle microsfere cresce al crescere della concentrazione polimerica nell’emulsione finale [38,39].
La dimensione delle particelle è anche dipendente dalla temperatura di lavoro: a basse e alte temperature, vengono prodotte microsfere di dimensioni maggiori, mentre a temperature intermedie, si ottengono particelle più piccole. Due differenti meccanismi possono essere distinti: a basse temperature, la maggior viscosità della soluzione, porta alla formazione di microsfere di dimensioni maggiori, a causa della difficoltà di movimento delle macromolecole che non riescono così a formare aggregati più piccoli [40]. A temperature elevate, le maggiori dimensioni delle microsfere, sono dovute all’aumento del grado di evaporazione del solvente. Infatti, la pressione esercitata dal solvente in via di evaporazione, all’interno della goccia polimerica in via di indurimento, tende a spingere il materiale macromolecolare verso l’esterno, portando così alla formazione di microsfere con un diametro maggiore [41].
La dimensione delle microsfere è inoltre influenzata dalla velocità di agitazione dell’emulsione finale: aumentando tale velocità è possibile ottenere particelle più piccole a causa della formazione di un’emulsione più fine. Cambiamenti nelle dimensioni particellari possono anche essere notati variando la quantità di emulsionante
11 utilizzato [42] anche se in questo caso, è difficile fare delle considerazioni di carattere generale.
Un ulteriore fattore da tenere in considerazione nella preparazione delle microsfere è la loro porosità superficiale che può incidere in modo determinante sulla cinetica di rilascio del farmaco. Per metodi di preparazione implicanti una doppia emulsione, la porosità microparticellare può essere controllata dalla prima emulsione acqua in olio: la porosità delle sfere aumenta all’aumentare della quantità di acqua utilizzata [43]. Tuncay e collaboratori [44] hanno utilizzato metanolo al posto dell’acqua nella prima emulsione, ottenendo una significativa riduzione della porosità superficiale delle microsfere. Anche il grado di rimozione del solvente, influenzato da parametri come temperatura e pressione, può incidere sulla porosità: operando a temperature relativamente elevate, in modo da favorire il rapido indurimento della superficie delle microsfere, si ottiene una limitata porosità superficiale; a temperature più alte, la rapida evaporazione del solvente organico porta ad una maggiore porosità delle particelle che in questo caso possono anche presentare un nucleo cavo all’interno [44,45,46].
Un altro parametro rilevante relativo ai sistemi microparticellari, è la loro disperdibilità. Quasi tutti i materiali particolati o microscopici, a contatto con un liquido, acquisiscono una carica elettrica sulla loro superficie. Un importante e funzionale indicatore di tale carica, è rappresentato dal potenziale Zeta che può essere utilizzato per controllare la stabilità di una sospensione colloidale o di un’emulsione [47]. Il potenziale Zeta è definito come la differenza di potenziale elettrico tra lo strato di ioni primariamente adsorbito sulla superficie delle particelle e il mezzo liquido disperdente in cui sono immerse [47,48]. Esso può essere quantificato, seguendo la migrazione delle particelle sottoposte all’azione di un campo elettrico. Può essere asserito che maggiore, in valore assoluto, è il valore del potenziale Zeta, e più stabile è la sospensione, a causa del fatto che le particelle ugualmente cariche superficialmente, tendono a respingersi per interazione elettrostatica inibendo la naturale tendenza all’aggregazione. Al contrario, un basso potenziale Zeta, in valore assoluto, indica instabilità colloidale che può portare all’aggregazione delle particelle. Il potenziale Zeta può essere influenzato da parametri come concentrazione salina, pH, concentrazione e natura del tensioattivo [47].
12
1.3.3 FATTORI CHE INFLUENZANO IL RILASCIO DI FARMACO
Il rilascio controllato di farmaco da parte di matrici polimeriche in forma di microsfere è influenzato da vari fattori che ruotano attorno alla struttura della matrice in cui il farmaco è contenuto e alle proprietà chimiche del polimero e del farmaco.
Differenti profili di rilascio possono comunque essere discussi in termini di proprietà fisiche e chimiche delle microsfere.
Tali proprietà comprendono:
• Peso molecolare del polimero • Cristallinità
• Caratteristiche del farmaco incapsulato • Natura del materiale macromolecolare • Porosità
• Dimensione particellare
L’influenza del peso molecolare del polimero sulla cinetica di rilascio si nota soprattutto per le micrisfere costituite da materiale degradabile. Makino e collaboratori [48] hanno trovato differenti cinetiche di rilascio di farmaco per microsfere preparate da poli(acido lattico-co-acido glicolico) di diverso peso molecolare. Polimeri a basso peso molecolare, mostrano un profilo di rilascio relativamente costante. Al crescere del peso molecolare, la linearità del rilascio viene meno, e si presenta con un andamento pulsato. Tali cinetiche suggeriscono che per microsfere derivanti da polimeri a basso peso molecolare, dopo uno stadio iniziale in cui si osserva un rilascio molto rapido (effetto
burst) dovuto al farmaco presente sulla superficie della microsfera, la degradazione è il
meccanismo predominante [49]. Per microsfere preparate invece da polimeri ad alto peso molecolare, i primi stadi di rilascio piuttosto lenti sono dominati da un meccanismo di diffusione, mentre in uno stadio successivo, la maggior parte del farmaco viene rilasciata a causa della degradazione [49]. Possono quindi essere preparate microsfere a partire da una miscela di polimeri ad alto e basso peso molecolare, per modulare convenientemente la cinetica di rilascio [50].
La cristallinità delle microsfere viene studiata sia rispetto al farmaco incapsulato, sia rispetto alla matrice polimerica costituente le particelle. Spesso l’eventuale cristallinità
13 del farmaco, osservabile analizzando una miscela fisica di farmaco e matrice polimerica, non viene più rilevata una volta che l’agente farmacologico viene incapsulato dal polimero. In questo caso il farmaco è disciolto nella matrice polimerica e si presenta in forma amorfa [51,52]. In altri casi è invece possibile individuare un certo grado di cristallinità del farmaco anche quando quest’ultimo è incapsulato nella particella, situazione indicante che l’agente farmacologico si trova finemente disperso nella matrice. I profili cinetici di rilascio differiscono sensibilmente nei due casi: sperimentalmente si ottiene in vitro un rilascio più conveniente quando il farmaco si presenta in forma amorfa nella matrice macromolecolare [53].
Per quanto riguarda invece la cristallinità del polimero, Edlund e Albertsson [54] hanno suggerito che la degradazione avviene prima nella regione amorfa delle microsfere seguita da una degradazione più lenta delle regioni cristalline. Si deduce quindi che il profilo cinetico di rilascio può essere modulato manipolando la cristallinita delle microsfere che può essere anche influenzata dal processo di preparazione come ad esempio, nel caso della tecnica di evaporazione-estrazione del solvente, dalla velocità di evaporazione del solvente stesso [55].
Il farmaco incapsulato nella matrice polimerica può influenzare la cinetica di rilascio sia per le sue caratteristiche chimiche e strutturali, sia per il modo in cui esso è distribuito nella matrice. In alcuni casi l’agente farmacologico impiegato può indurre una scissione delle catene polimeriche attraverso una “degradazione nucleofilica”. Ciò si osserva tipicamente quando il farmaco contiene gruppi amminici i quali si comportano da nucleofili specialmente se non sono stericamente impediti [56]. La distribuzione del farmaco all’interno della microsfera influenza sensibilmente la cinetica di rilascio: il farmaco viene infatti rilasciato inizialmente dalla superficie della sfera e in un secondo tempo dal suo strato interno. Il profilo cinetico risulta quindi variabile, in dipendenza della quantità di farmaco presente nella superficie della particella e al suo interno, oppure, per sistemi controllati principalmente dalla diffusione, il profilo cinetico varia in base alla distanza diffusionale che il farmaco deva percorrere per poter essere rilasciato [57,58].
Per microsfere preparate da un polimero o da un copolimero, la cinetica di rilascio differisce sensibilmente da quella di microsfere preparate da una miscela di polimeri. In particolare è possibile modulare la cinetica di rilascio utilizzando miscele di polimeri idrofili e idrofobi: all’aumentare della quantità di polimero idrofilo, aumenta la velocità di degradazione della microsfera e si ottiene così un rilascio di farmaco più rapido.
14 Inoltre i polimeri idrofili vanno incontro essenzialmente ad una degradazione superficiale, mentre quelli idrofobi subiscono una degradazione interna. Ciò si traduce, per particelle preparate da suddetti polimeri, in un profilo di degradazione e conseguentemente di rilascio, a due stadi: nel primo si nota un rilascio rapido dovuto alla veloce degradazione del polimero idrofilo; nel secondo, il farmaco viene rilasciato più lentamente dal polimero idrofobo [59]. Un esempio di differente cinetica di rilascio tra copolimero e miscela fisica di polimeri, è stato riportato da Edlund e Albertsson [54], mettendo a confronto microsfere preparate a partire da una miscela di poli(L-acido lattico) e poli(1,5-diossepan-2-one) con microsfere preparate dal corrispondente copolimero. È stato trovato che le particelle di copolimero risultavano porose e di dimensioni maggiori rispetto a quelle preparate dalla miscela dei due omopolimeri. Durante la degradazione, la perdita di peso molecolare risultava più lenta per le particelle derivanti dalla miscela a causa della loro maggiore densità. Inoltre, i diversi profili di rilascio sono stati attribuiti anche alle differenze morfologiche e di cristallinità tra i due tipi di sfere [54].
Porosità superficiale e dimensioni delle microsfere, sono altri due fattori che influenzano sensibilmente la cinetica di rilascio, ed entrambi possono essere modulati variando i parametri operativi nella fase di preparazione. Una maggiore porosità superficiale porta a rilasci più rapidi favorendo i processi diffusivi, talvolta in modo eccessivo, tale da ottenere un effetto “burst iniziale” troppo accentuato e perdendo così le caratteristiche di un sistema a rilascio controllato [60,61]. Per quanto riguarda le dimensioni particellari, è stato riscontrato che il grado di rilascio di farmaco diminuisce all’aumentare della dimensione delle sfere [62]. Per ottimizzare la cinetica di rilascio è a volte conveniente utilizzare sistemi costituiti da miscele di microparticelle con un’opportuna distribuzione dimensionale [63].
15
1.4 RILASCIO MIRATO
Il rilascio di un farmaco verso specifici organi, tessuti o cellule, rappresenta la sfida principale nello sviluppo di sistemi a rilascio di farmaci. Mediante la maggior parte dei metodi di somministrazione attualmente in uso l’agente farmacologico risulta distribuito quasi uniformemente in tutto il corpo. In questo modo il farmaco per raggiungere il sito di azione deve attraversare molte barriere biologiche, come altri organi, cellule e compartimenti intracellulari, dove può essere inattivato. Inoltre il raggiungimento della concentrazione terapeutica nell’organo o cellula bersaglio comporta la somministrazione di farmaco in quantità elevata, per compensare le perdite nei normali tessuti, con conseguente possibile manifestazione di effetti tossici a livello sistemico. Il rilascio mirato (drug targeting) può rappresentare la soluzione a questi problemi. Per
drug targeting si intende la capacità di concentrare il farmaco in organi o tessuti in
maniera selettiva e quantitativa, indipendentemente dal sito e metodo di somministrazione. I vantaggi derivanti da questo tipo di approccio sono evidenti: i protocolli di somministrazione possono essere semplificati, la quantità di farmaco necessaria per ottenere effetti terapeutici può essere ridotta, così come il costo della terapia e la concentrazione di farmaco nel sito può essere aumentata senza provocare effetti indesiderati nei tessuti sani.
Attualmente il concetto di rilascio mirato è legato all’azione coordinata di tre componenti: il farmaco, una molecola segnale capace del riconoscimento selettivo e un
carrier usato per aumentare il numero di molecole di farmaco per la singola molecola
segnale. I carriers farmaceutici comprendono: polimeri solubili, microparticelle, microcapsule, cellule, lipoproteine, liposomi e micelle.
I principali schemi di rilascio mirato attualmente studiati e applicati comprendono: applicazione diretta del farmaco, targeting di tipo passivo, targeting fisico, targeting magnetico, oppure targeting di tipo attivo mediante l’utilizzo di molecole con elevata affinità verso la zona affetta (molecole segnale).
16
1.4.1 APPLICAZIONE DIRETTA DEL FARMACO
In alcuni casi, un rilascio mirato può essere ottenuto semplicemente tramite somministrazione del farmaco direttamente nella regione affetta (organo o tessuto). Esempi riusciti relativi a questo tipo di approccio comprendono la somministrazione intra-articolare di farmaci ormonali nella cura di artriti [64] o l’infusione intracoronarica di enzimi trombolitici nella cura di infarto del miocardio indotto da trombi [65]. Tuttavia le possibilità di applicazione di questo metodo diretto sono piuttosto limitate.
1.4.2 TARGETING PASSIVO
Per targeting passivo si intende l’accumulo di farmaco o sistema a rilascio di farmaco causato da particolari condizioni anatomiche. Ad esempio è stato osservato un aumento della permeabilità dell’endotelio vascolare o effetto EPR (Enhanced Permeability and
Retention) in tumori solidi e danneggiamento delle pareti vasali in seguito a
angioplastica. In queste regioni con aumentata permeabilità vascolare anche particelle come micelle e liposomi di diametro 10-500 nm possono accumularsi negli spazi interstiziali. Utilizzando particelle caricate con farmaci opportuni si può avere rilascio del farmaco nella zona d’interesse terapeutico per degradazione del carrier. Essendo il limite dimensionale per la penetrazione interstiziale diverso da caso a caso, le dimensioni delle particelle caricate con il farmaco costituiscono il parametro selettivo alla base del targeting passivo. Un requisito fondamentale per questo tipo di rilascio è che le particelle rimangano in circolo per il tempo necessario a garantirne l’accumulo nell’organo bersaglio. Liposomi caricati con farmaci antitumorali e ricoperti di polietilenglicol per eludere il RES (sistema reticolo endoteliale) e quindi capaci di rimanere in circolo per tempi più lunghi, hanno mostrato ottimi risultati nella terapia dei tumori [66].
17
1.4.3 TARGETING FISICO
L’idea alla base del targeting fisico è che le regioni affette da patologie possono differire dai tessuti sani in alcune caratteristiche chimico-fisiche come temperatura e/o pH. E’ noto ad esempio che regioni con infiammazioni e presenza di neoplasie mostrano spesso acidosi e ipertermia. Ciò rende possibile l’uso di carrier in grado di rilasciare il farmaco a valori di pH inferiori o temperature più elevate rispetto a quelle dei normali tessuti. Esempi di microsfere e liposomi a rilascio di farmaci, sensibili alla temperatura e al pH sono noti in letteratura [67].
1.4.4 TARGETING MAGNETICO
Un interessante esempio di rilascio mirato dovuto a forze esterne di natura fisica è il trasporto di farmaco per orientamento magnetico. A questo scopo il farmaco è disperso in un carrier particellare con proprietà ferromagnetiche, dovute alla presenza nella matrice polimerica di derivati a base di alluminio, nickel, cobalto, rame, argento, manganese o platino. Dopo somministrazione intravenosa le particelle vengono selettivamente dirette verso il sito specifico mediante l’azione di un campo magnetico extracorporeo applicato in corrispondenza dell’organo bersaglio. La capacità di concentrare le particelle magnetiche in una specifica regione dipende dal flusso sanguigno nella stessa e dall’intensità del campo magnetico esterno [68].
1.4.5 USO DI MOLECOLE SEGNALE
Gli approcci al rilascio mirato appena descritti non sono generalizzabili. La somministrazione diretta di un farmaco in un organo o tessuto malato può essere tecnicamente difficile o il sito della malattia può essere delocalizzato. Spesso la regione malata non differisce molto dai normali tessuti in termini di permeabilità vascolare, temperatura o valore di pH locale. Per quanto riguarda il rilascio mirato mediante orientamento magnetico ci sono dei limiti legati alla portata di flusso sanguigno nel sito-bersaglio. Il modo più generale per dotare di affinità verso il bersaglio un farmaco non specifico, è il legame del farmaco stesso con un’unità (molecola segnale) capace di riconoscimento specifico e legame con il sito da raggiungere. Le sostanze utilizzate
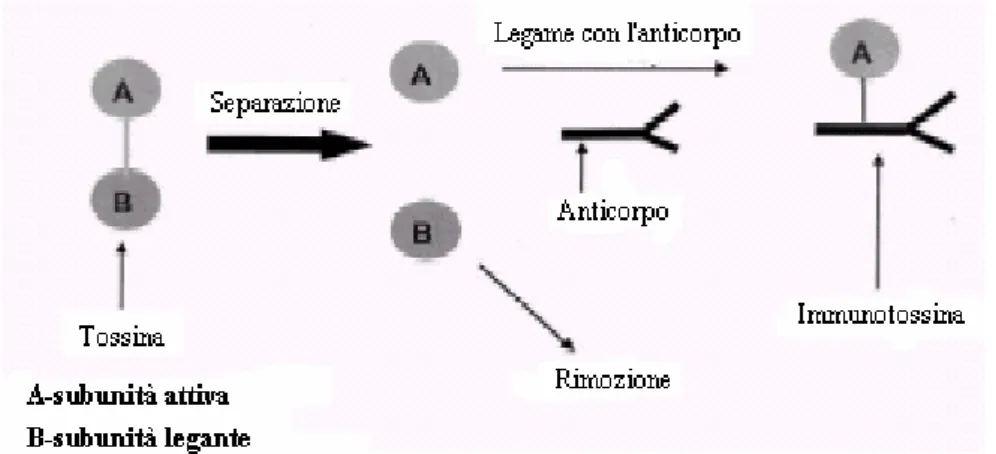
18 come molecole segnale sono: anticorpi e loro frammenti, proteine, lipoproteine, ormoni, poli(saccaridi) e alcuni ligandi a basso peso molecolare. Un metodo diretto per preparare un sistema mirato consiste in un legame del farmaco con la molecola segnale. L’uso di immunotossine [69] rappresenta un esempio di questo approccio. Come mostrato in Figura 2, una tossina naturale può essere divisa in due unità, di cui una attiva e l’altra responsabile del legame con la cellula. La prima viene separata e coniugata con un anticorpo monoclonale. Come risultato, l’unità tossica può essere rilasciata solo in cellule che esprimono uno specifico antigene (di solito cellule cancerose), mentre cellule prive dell’antigene non vengono riconosciute dall’immunotossina e non vengono danneggiate.
Figura 2. Schema di preparazione dell’immunotossina
In alternativa possono essere utilizzati carriers solubili e insolubili, caricati con molte unità attive e coniugati con la molecola segnale, secondo lo schema riportato in Figura 3. Il processo chimico di coniugazione deve essere tale da mantenere la struttura originale del farmaco (affinché non venga persa l’attività terapeutica) e l’attività biologica della molecola segnale, responsabile della specifica interazione ligando-recettore in vivo.
19 Figura 3. Schema di preparazione di un sistema mirato utilizzando un
carrier polimerico
Ad oggi sono stati realizzati sistemi a rilascio mirato verso vari compartimenti del corpo e per diverse patologie, tra cui: componenti del sistema cardiovascolare (sangue, pareti vascolari, cuore), sistema reticolo endoteliale (fegato, milza), sistema linfatico (linfonodi e vasi linfatici), tumori, infiammazioni, infezioni. Diversi tipi di polimeri biocompatibili possono essere utilizzati come carriers solubili, mentre la famiglia dei
carriers insolubili, comprende microparticelle, nanoparticelle, liposomi e micelle.
In particolare per quanto riguarda micro e nanosfere, è possibile fornire loro un orientamento di tipo attivo modificando la loro superficie per ridurre la possibilità che esse vengano riconosciute dall’organismo come corpi estranei, ed evitando conseguentemente la captazione da parte del RES. Possono inoltre essere presenti delle funzionalità che consentono interazioni specifiche con organi o cellule bersaglio. La modifica superficiale viene effettuata mediante un processo di adsorbimento, o tramite legami covalenti sulla superficie delle particelle (Figura 4).
20 FIGURA 4. Rappresentazione schematica di una microsfera superficialmente
funzionalizzata.
Ad esempio l’impiego di tensioattivi come poli(sorbati) e alcanoli polietossilati consentono di eludere il RES permettendo l’aumento della localizzazione delle particelle in organi quali cuore, tratto gastrointestinale e cervello .
1.4.6 MOLECOLE BERSAGLIO E MOLECOLE SEGNALE
Attualmente tutti i tentativi di rilascio mirato attivo prevedono l’impiego di varie e differenti molecole segnale che possono indirizzare il sistema contenente il farmaco verso siti specifici del corpo.
• Molecole bersaglio
Le strategie di rilascio mirato verso cellule specifiche possono essere progettate utilizzando la presenza di un’ampia varietà di molecole bersaglio; sostanzialmente si può agire a tre livelli principali:
1) Recettori.
La presenza di recettori sulla membrana consente di potenziare il rilascio mirato non solo indirizzando i sistemi drug-carrier verso singole cellule bersaglio, ma anche
21 facilitandone il trasferimento all’interno delle stesse, mediante endocitosi mediata da recettore. Alcuni dei recettori più utilizzati sono:
a) Recettori per l’acido folico
Una delle strategie più di successo nella terapia tumorale è l’utilizzo dell’acido folico per sistemi a rilascio di farmaci specifici per i tumori. L’acido folico è una vitamina a basso peso molecolare che si lega specificatamente ai recettori del folato (FR) che si trovano sulla superficie delle cellule tumorali. Gli FR sono sovra-espressi sulla superficie di cellule cancerose nel caso di neoplasie epiteliali come quelle ovariche, colorettali e del seno mentre sono espressi a livelli bassi nei tessuti normali. L’acido folico è un composto stabile, poco costoso, non immunogenico ed ha un’alta affinità con il recettore della superficie cellulare.
b) Recettori LDL;
I recettori per le lipoproteine a bassa densità (LDL) sono una famiglia di 9 recettori che trasportano le LDL, ricche in colesterolo all’interno delle cellule mediante il processo di endocitosi mediata da recettore. Un notevole numero di farmaci e di sistemi basati su lipidi sono stati proposti per indurre il rilascio mirato di agenti antitumorali verso cellule cancerose sfruttando l’interazione con i recettori LDL: alcuni esempi sono i liposomi anionici [70] e i liposomi arricchiti con apolipoproteine (ApoE) [71].
Un problema che si riscontra con questo tipo di approccio tuttavia è la non specificità dei recettori LDL che sono normalmente presenti nella maggior parte degli organi.
c) Recettori di peptidi;
Un’alternativa interessante per il rilascio mirato come terapia antimitotica prevede l’impiego di recettori per i peptidi che sono espressi in quantità maggiore in certi tipi cellulari. Alcuni esempi di recettori per peptidi sono gli analoghi della somatostatina, peptidi intestinali vasoattivi, colecistochinine.
2) Componenti lipidici di membrana
Una classe emergente di molecole bersaglio utili per trasferire il farmaco direttamente alla cellula tumorale è rappresentata dai componenti lipidici di membrana. E’ stato osservato che, l’interazione di alcuni analoghi fosfolipidici sintetici con le membrane cellulari modifica una serie di parametri quali la composizione dei lipidi, la permeabilità e la fluidità di membrana e, come conseguenza, influenza i meccanismi di traduzione del segnale ed induce l’apoptosi. Recentemente è stata scoperta una classe di recettori e di mediatori essenziale per il processo di migrazione e maturazione delle cellule
22 muscolari lisce vascolari (VSMC) dell’intima che coinvolge il lipide sfingosina-1-fosfato (S1P) ed il suo recettore Edg-1 [72].
3) Antigene/proteine di superficie
L’espressione di diversi tipi di proteine sulla superficie cellulare rappresenta la chiave di lettura delle cellule che può essere decifrata mediante l’impiego di anticorpi monoclonali diretti contro queste stesse proteine. Al momento si stanno facendo grossi passi in avanti, per identificare e validare le molecole antigeniche che sono espresse specificatamente nelle cellule tumorali. Un antigene tumorale specifico ideale sarebbe quello che viene espresso solo ed esclusivamente nella cellula tumorale e distribuito omogeneamente in tutte le cellule che formano la massa del tumore. Alcuni esempi noti sono l’antigene specifico della prostata ed il fattore di crescita erbB-2 espresso specificatamente nell’adenocarcinoma umano del seno.
• Ligandi
Il targeting attivo dei farmaci può essere realizzato mediante l’impiego di agenti attivi o ligandi che interagiscono in modo esclusivo con gli specifici recettori/molecole bersaglio identificati su particolari tipi cellulari. Tra i ligandi efficientemente impiegati per realizzare il drug targeting attivo si annoverano molecole semplici come zuccheri (legame specifico per il recettore dell’asialoglicoproteina), acido folico, lectine, albumina (specifica per le cellule del fegato) e peptidi.
Gli anticorpi monoclonali diretti contro gli antigeni associati ai tumori sono stati utilizzati come agenti per mirare alle cellule mitotiche allo scopo di rilasciare enzimi, farmaci a basso peso molecolare, e proteine aventi azione tossica verso queste cellule. Tuttavia gli anticorpi monoclonali sono molecole a peso molecolare relativamente elevato che diffondono lentamente limitando così la loro azione mirata nel trattamento della maggior parte dei tumori solidi. I peptidi a lunghezza ridotta che riconoscono selettivamente le cellule tumorali possono rappresentare la soluzione a questi problemi. La capacità dei corti peptidi sintetici di penetrare efficientemente nel tessuto, di dare origine ad un legame selettivo e di essere internalizzati nelle cellule cancerose sono tutti elementi a favore dell’impiego di questi composti per il rilascio di agenti terapeutici quali oligonucleotidi, tossine e molecole radioattive. Inoltre, i peptidi sono pressoché invisibili al sistema immunitario e generalmente non provocano effetti collaterali nocivi.
23 Tradizionalmente l’identificazione dei ligandi peptidici specifici procedeva attraverso l’analisi strutturale sulla base delle loro caratteristiche funzionali; ciò comportava la sintesi di un notevole numero di peptidi e la loro caratterizzazione funzionale mediante test in vitro ed in vivo.
Tuttavia lo sviluppo della tecnologia della libreria peptidica combinatoriale ha reso possibile identificare specifici ligandi, senza la necessaria conoscenza dei relativi recettori, mimando le strategie di selezione cellulare [73]. Queste librerie peptidiche possono virtualmente contenere tutte le possibile sequenze di corti peptidi che sono in grado di imitare le strutture conformazionali di epitopi continui e discontinui. In tabella 2 sono riassunte alcune sequenze peptidiche specifiche per molecole-bersaglio che sono state identificate mediante libreria di visualizzazione fagica (phage display library).
TABELLA 2 Esempi di ligandi peptidici selezionati mediante phage display library MOLECOLA-BERSAGLIO SEQUENZA PEPTIDICA Peptide atriale natriuretico recettore-A MCHFGGRMDRISCYR Concanavalina A MYWYPY
Steptavidina AECHPQGPPCIEGRK Recettore della trombonina MSRPACPNDKYE Recettore umano della trombopoietina IEGPTLRQWLAARA
24
1.5
SISTEMI NANOPARTICELLARI
Per nanosfere si intendono quei sistemi di natura particellare con dimensione inferiore ad 1 µm. Le nanoparticelle per usi biomedici contengono un agente terapeutico incapsulato nella matrice polimerica oppure adsorbito o coniugato superficialmente. Esse costituiscono un versatile sistema a rilascio controllato di farmaco, e a causa delle loro ridotte dimensioni, possono potenzialmente superare le diverse barriere fisiologiche per guidare l’agente terapeutico verso cellule specifiche o compartimenti intracellulari, con un approccio mirato che può essere sia di tipo attivo che passivo. Come per altri sistemi particolati, in seguito a somministrazione intravenosa, le nanoparticelle vengono rimosse dal flusso sanguigno a causa della captazione da parte del RES. Ciò costituisce un vantaggio se si desidera un rilascio mirato verso fegato e milza che sono i maggiori organi del sistema reticolo endoteliale. Tuttavia il riconoscimento delle nanosfere da parte del RES dipende da caratteristiche superficiali quali carica e carattere idrofilo, e in particolare dalle dimensioni. Particelle con superficie idrofila aventi dimensioni inferiori a 100 nm sono in grado di eludere il RES per un discreto tempo. In questo modo possono rimanere a lungo nel circolo sanguigno permettendo, se funzionalizzate con opportuni ligandi, un rilascio mirato verso altri siti specifici.
Vengono di seguito riportati alcuni esempi di polimeri utilizzati per la realizzazione di nanosfere per il rilascio mirato di farmaco.
• Copolimeri a blocchi a base di poliacrilati
Nanoparticelle con un rivestimento superficiale di PEG sono state preparate mediante precipitazione del copolimero di sintesi poli(amminopoli(etilenglicol) cianoacrilato-co-esadecil cianoacrilato) (poli(H2NPEGGCA-co-HDCA). In seguito il gruppo amminico terminale del blocco idrofilo PEG delle nanoparticelle preformate è stato fatto reagire con l’acido folico sotto forma di estere della N-idrossisuccinimmide [74]. Li e collaboratori [75], hanno utilizzato lo stesso copolimero per la preparazione di nanoparticelle contenenti DNA, alle quali è stata selettivamente coniugata la transferrina per reazione con il gruppo amminico terminale delle catene di PEG, ottenendo così un potenziale sistema a rilascio mirato per terapia genetica. Un elegante
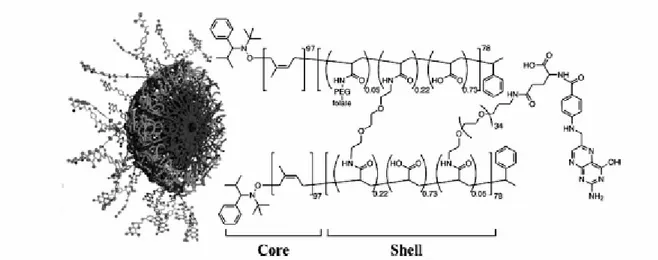
25 strategia è stata seguita da Pan e collaboratori [76]per la preparazione di nanoparticelle reticolate con una morfologia anfifilica di tipo core-shell che le rende innovative come sistema drug carrier biocompatibile e a lunga permanenza nel circolo sanguigno (Figura 5).
Figura 5. Nanoparticelle a base di poli(acido acrilico-b-isoprene)
• Poli(etilenglicol-co-caprolattone) (PEG-PCL)
Un sistema unico per lo studio delle interazioni cellule-materiale è stato realizzato mediante l’impiego di ligandi a base di biotina legati alla superficie di nanoparticelle ingegnerizzate. Le nanoparticelle sono state preparate per nanoprecipitazione di miscele di copolimeri a due blocchi PEG-PCL coniugati e non coniugati con biotina. L’uso potenziale di questi materiali in nanoscala come sistemi a rilascio controllato per la somministrazione orale o intravasculare è ancora oggetto di studio[77].
• Poli(etilenglicol-co-acido lattico) (PEG-PLA)
Olivier e collaboratori, hanno descritto la preparazione di immuno-nanoparticelle a base di metossi-PEG-PLA e maleimmide-PEG-PLA funzionalizzate con anticorpi monoclonali (mAb) specifici per il recettore murino della transferrina [72]. Gli mAb vanno incontro a endocitosi mediata da recettore a livello della barriera ematoencefalica
26 attraverso il sistema di trasporto endogeno della transferrina consentendo il rilascio di farmaco mirato al cervello.
1.6
MALATTIE CARDIOVASCOLARI
Le malattie cardiovascolari sono la principale causa di malattia e mortalità nel mondo industrializzato. La causa scatenante delle malattie cardiovascolari, come l’infarto del miocardio e l’angina instabile, è rappresentata dall’aterosclerosi, un’alterazione cronica che si manifesta con la formazione di placche o ateromi nella parete delle arterie e che progredisce in maniera silente per molti anni.
La stenosi è una riduzione del lume dell’arteria che si assume dovuta alla presenza di una placca ateromatosa complicata o non complicata. In caso di stenosi si ha un ridotto apporto di sangue ai tessuti con conseguente ischemia o addirittura necrosi tissutale. Quest’ultima genera zone infartuate nell’organo interessato inducendo una parziale o totale perdita della sua funzionalità.
Il trattamento delle problematiche cardiovascolari spesso necessita di interventi invasivi molto delicati e difficili da realizzare. Per questo motivo fin dagli anni ’80 sono state studiate molte tecniche di intervento con uno sviluppo particolarmente significativo delle tecniche di angioplastica e degli stents coronarici; più recentemente la ricerca e la sperimentazione clinica hanno posto la loro attenzione sullo studio degli stent a rilascio di farmaco (DES). La ricerca scientifica e clinica relativa ai DES ha preso origine dal fatto che gli stent metallici, pur garantendo buoni risultati clinici, hanno fra gli svantaggi, quello di favorire la formazione di trombi e indurre il processo di restenosi, peggiorando la prognosi a lungo termine. La restenosi è un fenomeno patologico connesso ad un processo di eccessiva cicatrizzazione dovuta alla proliferazione delle cellule muscolari lisce (VSMC) della parete del vaso che crea una ri-occlusione parziale o totale dell’arteria. Tale fenomeno è molto complesso e può essere causato, oltre che dalla predisposizione genetica del paziente alla formazione di placche aterosclerotiche, anche dal verificarsi di lesioni dello strato endoteliale della parete del vaso sanguigno dovuta all’inserimento del dispositivo endovascolare (stent a mezzo catetere). Le manifestazioni endoteliali di aterosclerosi comprendono vari fenomeni tra cui
27 l’aggregazione piastrinica, il rilascio di fattori di crescita, la migrazione di cellule infiammatorie e l’instaurarsi di eventi trombotici.
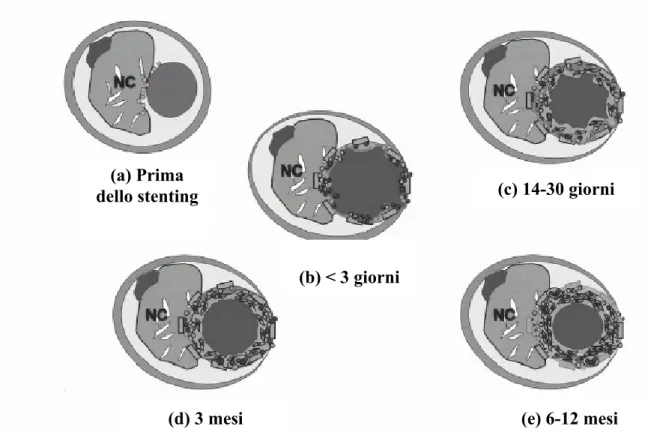
Questi fenomeni possono favorire, a loro volta, la migrazione e la proliferazione delle cellule muscolari lisce (VSCM), che vanno incontro ad un cambiamento del loro fenotipo da contrattile a proliferativo o di “riparazione”, portando alla formazione di una capsula fibrosa a livello della superficie endoteliale in grado di provocare la restenosi. In figura 6 è possibile osservare una schematizzazione dell’azione meccanica dello stent, successiva al suo posizionamento in situ a mezzo catetere, e le fasi che portano all’iperplasia neointimale delle arterie aterosclerotiche [78].
Figura 6. Fasi successive all’impianto dello stent con verificarsi della iperplasia neointimale.
Con la sigla NC si indica la regione non cellulare della placca. Nel primo stadio (a) è illustrata l’arteria prima dell’inserimento dello stent. Successivamente (b) nei primi tre giorni dall’impianto si ha un accumulo di fibrina (proteina insolubile che forma il coagulo) e neutrofili. A 14-30 giorni (c) si ha l’insorgere di un’infiammazione cronica
(a) Prima dello stenting
(b) < 3 giorni
(c) 14-30 giorni
28 (macrofagi, linfociti) e persiste la presenza di fibrina. Durante questa fase, le cellule muscolari lisce iniziano ad occupare le maglie dello stent. A tre mesi (d) è presente sia l’infiammazione cronica che la fibrina. Infine dai 6 ai 12 mesi (e) scompare l’infiammazione cronica e si arresta la fase proliferativa. L’intima dell’arteria appare ricca di cellule muscolari lisce e collagene responsabile della cicatrizzazione dei lembi del tessuto leso.
Essendo la restenosi una patologia vera e propria, essa è curata tramite l’utilizzo di farmaci. Tra i farmaci utilizzati per trattare la restenosi vanno ricordati gli agenti che inibiscono la proliferazione delle cellule e la loro migrazione, ma anche agenti che inibiscono le infiammazioni, che si sospetta siano fra le cause dell’inizio del processo proliferativo delle cellule.
La restenosi può essere trattata mediante le tradizionali terapie farmaceutiche effettuate per via sistemica, tuttavia si ritiene che la somministrazione di un farmaco appropriato, in modo topico, direttamente dallo stent impiantato possa dare risultati ancora migliori. Nel caso di terapia localizzata, l’idea fondamentale è quella di utilizzare “in situ” un agente immunosoppressore e/o antiinfiammatorio con funzione antirestenotica, realizzando quindi un controllo farmacologico localizzato nei confronti della proliferazione neointimale. Per la somministrazione e il rilascio controllato di tale farmaco è possibile utilizzare un rivestimento polimerico (coating) adatto a ricoprire la rete metallica degli stent e in grado di favorire una cinetica di rilascio del farmaco che ne esalti l’efficacia terapeutica.
Sono attualmente allo studio (ed in fase di sperimentazione clinica) stent di tipo DES contenenti diversi farmaci ad azione antirestenotica (paclitaxel, tacrolimus, etc.) con risultati particolarmente soddisfacenti per alcuni di essi.
1.6.1 RUOLO DELLE VSCM NEL PROCESSO DELLA RESTENOSI
Studi farmacologici su modelli animali hanno consentito di individuare, almeno in parte, il comportamento specifico delle VSMC durante il processo di restenosi, e di identificare il ruolo chiave delle molecole interessate a tale processo. Per maggiore chiarezza si distinguono 4 fasi principali [79]:
29
1° fase:
Il danno provocato dall’introduzione del catetere provoca una completa distruzione dell’endotelio e una morte estesa delle VSMC della media. In risposta al danno si assiste, dopo circa 24 ore, alla proliferazione delle VSMC della media indotta dal fattore bFGF rilasciato dalle VSMC lese. Alcuni studi suggeriscono che tra i fattori che controllano la proliferazione delle VSMC della media vi sono il PDGF, un debole mitogeno il cui effetto è più significativo sulla migrazione che non sulla replicazione, gli antagonisti α-adrenergici e l’angiotensina II (AngII).
2° fase:
Le VSMC iniziano a migrare nell’intima, attraversando la lamina elastica interna, già quattro giorni dopo il danno. Sebbene non sia noto il contributo relativo delle molecole coinvolte e l’eventuale presenza di altre, si riconosce a questo livello l’attività dei fattori di crescita PDGF, TGF-β e bFGF.
3° fase:
Si assiste alla replicazione delle VSMC nell’intima; anche se non sono ben note quali siano le molecole mitogene interessate. Indicazioni in questo senso sono suggerite dalla sovra-espressione nell’intima delle molecole PDGF-A PDGF-B anche se non sono chiari i loro meccanismi di azione a questo livello; altre molecole sovra-espresse che controllano la proliferazione nella media sono il recettore di AngII (AT-1), il TGF-β e l’ IGF-1.
4° fase:
Se la neointima è sottoposta in questa fase all’infusione di diversi tipi di mitogeni, si verifica una intensa proliferazione di VSMC che coinvolge sicuramente i fattori TGF-β, bFGF e AngII con funzione di agonisti.
La rigenerazione dell’endotelio può inibire la proliferazione della cellule della neointima sottostante secondo meccanismi attualmente ancora poco chiari. Alcune prove sperimentali, relative all’effetto inibitorio dell’ossido nitroso (NO) prodotto dall’endotelio sulla proliferazione di VSMC in coltura, sembrano suggerire una funzione importante dell’NO nel processo rigenerativo dell’endotelio.
30
1.6.2 RILASCIO MIRATO NEL TRATTAMENTO DELLA
RESTENOSI
La realizzazione di un sistema a rilascio mirato nel trattamento antirestenoico rappresenta un approccio innovativo per orientare in maniera selettiva e quantitativa il farmaco non solo verso il sito vascolare lesionato ma anche verso specifiche popolazioni cellulari coinvolte nel processo di restenosi (cellule bersaglio). La letteratura in merito è tuttavia molto limitata.
L. A. Guzman e collaboratori [80] hanno proposto l’uso di nanoparticelle come carriers per il rilascio locale di Dexamethasone. Gli autori riportano che nanoparticelle infuse mediante catetere possono penetrare nella parete vasale attraverso lo strato endoteliale danneggiato e dare un rilascio prolungato e in situ del farmaco. La somministrazione locale di particelle caricate con Dexamethasone ha come effetto una significativa riduzione dello strato neointimale.
T. Uwatoku e collaboratori [81] hanno riportato un sistema di targeting passivo basato su nanoparticelle core-shell contenenti Dexorubicina in grado di concentrarsi in regioni vascolari con elevata permeabilità (effetto EPR). L’effetto EPR, individuato inizialmente nei tessuti tumorali, si manifesta anche come conseguenza dell’infiammazione locale dovuta a danneggiamento dello strato endoteliale. Le dimensioni e la carica superficiale delle particelle sono i parametri fondamentali per l’accumulo selettivo nei siti vascolari lesionati.
A. C. Thomas e J. H. Campbell [82] hanno realizzato un sistema a rilascio mirato di tre farmaci per la prevenzione della restenosi: Eparina, Eparina frazionata e Rapamycin. Tale sistema ha come bersaglio la fibrina reticolata (presente nell’arteria lesionata), utilizza come molecola segnale un anticorpo della fibrina (H93.7C.1D2/48) e non fa uso di carrier. I farmaci sono stati legati covalentemente all’anticorpo sfruttando la funzionalità amminica o ossidrilica presenti sullo stesso. Sulla base di studi in vivo su conigli con arteria carotidea de-endotelizzata, si sono ottenuti, in alcuni casi, risultati positivi in termini di re-endotelizzazione, riduzione della proliferazione neointimale e del rimodellamento negativo.
La progettazione di un sistema mirato ai recettori integrinici delle piastrine attivate nel processo della restenosi è stata proposta da Lestini e collaboratori [83]. Lo studio si basa
31 sull’affinità di peptidi contenenti la sequenza RGD (Arg-Gly-Asp) verso l’integrina piastrinica GP IIb-IIIa. Liposomi contenenti la sequenza GSSSGRGDSPA legata a una catena di poli(etilenglicol) come spaziatore, sono stati ottenuti per deposizione del film lipidico. Preliminari studi in vitro, per dimostrare che liposomi modificati con RGD sono competitivi con il fibrinogeno per il legame con l’integrina GP IIb-IIIa, sembrano indicare assenza di aggregazione piastrinica a causa dell’affinità del peptide con l’integrina.
32
1.7 SCOPO DELLA TESI
Oggetto di questo lavoro di tesi è la preparazione di micro e nanosfere per rilascio controllato e mirato di farmaco per applicazioni in campo cardiovascolare. Come materiali polimerici verranno utilizzati un copolimero PCL-POE-PCL e una serie di poliuretani segmentati sintetizzati e caratterizzati in un precedente lavoro di tesi.
Come agente farmacologico verrà impiegato il Palitaxel (PXT), un farmaco antiproliferativo, fortemente idrofobo, ampiamente utilizzato nei trattamenti anti-tumorali e recentemente applicato come agente antirestenoico.
Le microsfere saranno preparate con la tecnica di evaporazione-estrazione del solvente ad emulsione singola olio in acqua. Per ogni tipo di polimero i parametri operativi, nella fase di formulazione delle particelle, verranno mantenuti costanti allo scopo di valutare le caratteristiche di quest’ultime in relazione alla struttura delle macromolecole. Le microsfere saranno caratterizzate rispetto alla morfologia, disperdibilità in ambiente acquoso ed efficacia di incapsulamento del farmaco. Verrà inoltre effettuata l’analisi termica per valutare lo stato fisico del farmaco incapsulato. Un confronto tra i termogrammi di microsfere caricate con PXT e microsfere senza farmaco incapsulato (microsfere placebo), aventi la stessa storia termica, consentirà di evidenziare l’influenza esercitata dal farmaco sul potenziale riarrangiamento delle catene polimeriche per formare strutture ordinate.
Il rilascio del PXT dalle matrici polimeriche verrà monitorato per un periodo di 30-40 giorni e il profilo cinetico di rilascio sarà valutato mediante analisi cromatografia. Essendo identiche le condizioni sperimentali nella fase di preparazione delle particelle, le differenze tra le cinetiche di rilascio verranno discusse in relazione alla struttura delle matrici polimeriche, alla maggiore o minore idrofilia e alle differenti proprietà esibite dalle microsfere.
A partire dagli stessi materiali polimerici, verranno preparate nanosfere caricate con PXT con la stessa tecnica utilizzata per la formulazione delle microsfere, variando alcuni parametri operativi. Sulle nanosfere verranno effettuate le stesse caratterizzazioni eseguite per le microsfere.
Nonostante la complessità del meccanismo della restenosi, fenomeno che si verifica in un’alta percentuale di casi in seguito al danno vasale procurato dall’inserimento di un dispositivo endovascolare (stent a mezzo catetere), un ruolo chiave nella formazione
33 della neointima è stato attribuito alla migrazione delle cellule muscolari lisce dalla tunica media alla tunica intima del vaso sanguigno dove, una volta mutato il loro fenotipo da contrattile a proliferativo, iniziano a proliferare in modo incontrollato portando alla riocclusione del vaso [84]. Un sistema per il rilascio mirato di PXT verso tali cellule muscolari lisce prevede, come molecola segnale, l’utilizzo di un ligando che riesca a distinguere non solo le cellule muscolari lisce (VSMC) dalle cellule endoteliali che devono poter proliferare liberamente per completare l’azione riparatoria del vaso [84], ma anche il fenotipo contrattile delle VSMC da quello proliferativo. Grazie alla libreria combinatoriale è stato possibile identificare un ligando peptidico rispondente a tali requisiti [73].
Come approccio verso un sistema a rilascio mirato di questo tipo, verrà effettuata la sintesi di un poliuretano funzionalizzato con un peptide modello utilizzando:
• Come diisocianato un composto alifatico ottenuto a partire dal cloridrato di L-lisina (LDI) già disponibile da un precedente lavoro di tesi
• Come macrodiolo il poli(ε-caprolattone) diolo commerciale
• Come estensore di catena una miscela di L-lisina etil estere e un estensore opportunamente sintetizzato contenente la sequenza peptidica GRGDG
Il polimero verrà caratterizzato mediante analisi termica, NMR, UV, FTIR, SEC.
Tale polimero sarà successivamente convertito in forma microparticellare e verrà caratterizzato con gli stessi metodi utilizzati per le microsfere non funzionalizzate valutando infine l’influenza del ligando sulla cinetica di rilascio del PXT.