2. MATERIALI E METODI
2.1. Siti di campionamento e trattamento dei campioni
Gli individui di Paracentrotus lividus analizzati nel presente lavoro provengono da località situate sia all’interno del bacino del Mar Mediterraneo che sulle coste atlantiche di Irlanda (da un impianto di acquacoltura) e Spagna, per un totale di quindici stazioni di campionamento (Fig. 2.1, Tab. 2.1). Per ogni località sono stati analizzati cinque individui, per un totale di settantacinque. Il disegno di campionamento è stato scelto in funzione dell’elevato potenziale di dispersione di questa specie. Le singole località sono state quindi scelte cercando di mantenere una “maglia” larga il più possibile e cercando di avere campioni rappresentanti dei vari distretti biogeografici del Mediterraneo, in modo da evidenziare eventuali relazioni tra la diversità genetica dei campioni, le distanze geografiche e le principali correnti superficiali del Mar Mediterraneo e dell’Oceano Atlantico. Il disegno di campionamento è stato, inoltre, infittito a livello del Mare Adriatico in base ai risultati ottenuti in uno studio pilota comprendente campioni di cinque località (Quercianella, Alghero, Giardini Naxos, Ancona e Galway - Irlanda), che facevano ipotizzare una situazione particolare per questo bacino (Barbieri et al., 2005).
La raccolta degli individui di P. lividus è stata effettuata a partire dalla zona subtidale e talvolta anche nelle pozze di scogliera, scalzando gli animali dal substrato con un coltello o, più semplicemente, con le mani indossando guanti in cuoio o neoprene. La raccolta degli esemplari per il presente lavoro è stata effettuata per mezzo di immersioni in apnea o con autorespiratore ad aria (ARA).
Gli animali raccolti sono stati sezionati trasversalmente, all’incirca alla metà del corpo, per mezzo di forbici oppure di un coltello; le gonadi sono state quindi prelevate in numero variabile per ogni individuo da una a cinque, a seconda delle loro dimensioni, e trasferite, per mezzo di pinzette, in provette sterili da 10 ml contenenti etanolo assoluto. Le pinzette sono state accuratamente ripulite e flambate prima di ogni prelievo al fine di non inquinare i tessuti prelevati. I campioni di tessuto così fissati, una volta giunti in laboratorio, sono stati etichettati e conservati a – 20 °C.
500 Km
N
Gal
Bai
Anc
Que
Mlj
Pit
Ust
Les
Pal
Bri
Sca
Cde
Nax
Alg
Rho
Fig. 2.1. Siti di campionamento.
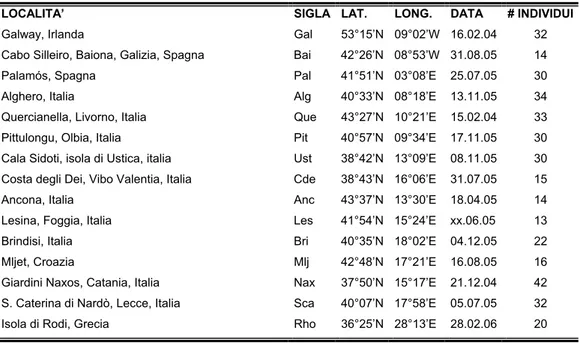
Tab. 2.1. Località di campionamento di P. lividus, sigle di riconoscimento delle località, coordinate geografiche del sito di campionamento, data di campionamento e numero di individui raccolti.
LOCALITA’ SIGLA LAT. LONG. DATA # INDIVIDUI
Galway, Irlanda Gal 53°15’N 09°02’W 16.02.04 32 Cabo Silleiro, Baiona, Galizia, Spagna Bai 42°26’N 08°53’W 31.08.05 14 Palamós, Spagna Pal 41°51’N 03°08’E 25.07.05 30 Alghero, Italia Alg 40°33’N 08°18’E 13.11.05 34
Quercianella, Livorno, Italia Que 43°27’N 10°21’E 15.02.04 33 Pittulongu, Olbia, Italia Pit 40°57’N 09°34’E 17.11.05 30
Cala Sidoti, isola di Ustica, italia Ust 38°42’N 13°09’E 08.11.05 30 Costa degli Dei, Vibo Valentia, Italia Cde 38°43’N 16°06’E 31.07.05 15
Ancona, Italia Anc 43°37’N 13°30’E 18.04.05 14 Lesina, Foggia, Italia Les 41°54’N 15°24’E xx.06.05 13 Brindisi, Italia Bri 40°35’N 18°02’E 04.12.05 22 Mljet, Croazia Mlj 42°48’N 17°21’E 16.08.05 16 Giardini Naxos, Catania, Italia Nax 37°50’N 15°17’E 21.12.04 42 S. Caterina di Nardò, Lecce, Italia Sca 40°07’N 17°58’E 05.07.05 32 Isola di Rodi, Grecia Rho 36°25’N 28°13’E 28.02.06 20
2.2. Estrazione del DNA
L’estrazione del DNA è una tappa fondamentale per la maggior parte delle analisi genetiche e, dato che la maggior parte delle tecniche molecolari è basata sulla reazione a catena della polimerasi, il DNA estratto deve avere caratteristiche qualitative tali da garantire una buona amplificazione. In letteratura esistono diversi protocolli di estrazione del DNA; la scelta del protocollo più adatto dipende dall’organismo in esame e anche dal tipo di tessuto. Le tecniche di estrazione si dividono in tre categorie fondamentali, ognuna delle quali presenta una varietà di protocolli applicativi:
1. estrazione chimica
2. estrazione mediante ultracentrifugazione e purificazione 3. estrazione tramite colonne e kit di separazione.
L’estrazione chimica è la tecnica normalmente più utilizzata, l’ultracentrifugazione è utilizzata per ottenere DNA altamente purificato, mentre i kit vengono prodotti ed ottimizzati per specifici tessuti animali o vegetali, permettendo normalmente di ottenere, in maniera piuttosto veloce, un prodotto puro, ma a basso peso molecolare (Procaccini & Maltagliati, 2003).
La prima fase del lavoro ha riguardato la messa a punto di un protocollo di estrazione specifico che garantisse ripetitività ed una resa discreta, sia in termini di quantità che di qualità del DNA estratto. Questa fase ha presentato più problemi di quanto previsto. Inizialmente diversi kit commerciali e protocolli standard sono stati saggiati su due diversi tipi di tessuto dell’animale, al fine di stabilire quale fosse il più adatto. I tentativi si sono concentrati sulle gonadi, data la relativa abbondanza di tessuto, e sui pedicelli ambulacrali. Scartati subito i pedicelli, dai quali non è stato possibile ottenere DNA amplificabile in quantità soddisfacente, gli sforzi si sono concentrati sulle gonadi che risultavano, inoltre, molto più facili da asportare e più voluminose, più adatte, quindi, nel caso si volessero ottenere maggiori quantità di DNA, in prospettiva di futuri approfondimenti dello studio in oggetto tramite altri marcatori genetici. Fra i diversi protocolli di estrazione utilizzati, quelli risultati particolarmente efficaci sono stati essenzialmente due, di seguito indicati come
“protocollo A” e “protocollo B”. Il primo è specificamente rivolto all’estrazione di DNA mitocondriale (mtDNA), previa purificazione dei mitocondri, mentre il secondo permette di ottenere l’intero DNA genomico (totDNA), comprendente, quindi, anche la frazione nucleare.
2.2.1. Protocollo A
Le preparazioni di mitocondri erano ottenute da omogeneizzati di gonadi di P.
lividus per mezzo di centrifugazioni differenziali (Lansman et al., 1981). Una porzione
di tessuto di circa 300 mg era omogeneizzato in 4 ml di una soluzione omogeneizzante (0.25 M saccarosio, 5 mM HEPES, 1 mM EDTA pH 7.2) e quindi centrifugato a 500 x g per 10 minuti a 4 °C, al fine di eliminare la componente nucleare ed i residui cellulari. Il sopranatante era recuperato e centrifugato a 9.400 x
g per 10 minuti a 4 °C. I mitocondri erano quindi recuperati e sospesi in 400 µl di una soluzione tampone (50 mM glucosio, 10 mM EDTA, 25 mM Tris-HCl pH 8.0). La procedura per isolare mtDNA era eseguita con un’estrazione alcalina, secondo il metodo di Palva & Palva (1985). Per la lisi delle membrane mitocondriali e la denaturazione dell’eventuale DNA nucleare contaminante presente, erano aggiunti 2 volumi (800 µl) di una soluzione alcalina (0.2 M NaOH, 1% SDS) e la sospensione così ottenuta era mescolata. Dopo un’incubazione in ghiaccio per 5 minuti, erano aggiunti 300 µl di 3 M acetato di potassio ed i contenuti della provetta erano mescolati. La provetta era conservata a -80 °C per 5 minuti e quindi centrifugata a 10000 x g per 10 minuti a 4 °C. Dopo aver prelevato il sopranatante, il mtDNA era precipitato aggiungendo un volume di isopropanolo e la provetta era posta a – 80 °C per 5 minuti. Il precipitato era raccolto mediante centrifugazione a 16100 x g per 15 minuti a 4 °C, sottoposto a due “lavaggi” con etanolo 70% e asciugato, mediante l’utilizzo di una pompa a vuoto. Il “pellet” di mtDNA era quindi sospeso in 400 µl di TE (1 mM EDTA, 10 mM Tris-HCl pH 8,0), incubato con 40 µl di DNase-free RNase (1 mg/ml) per un’ora a 37 °C ed estratto con un volume di fenolo e successivamente con un volume di cloroformio. Il mtDNA era, infine, di nuovo precipitato con due volumi di etanolo in presenza di 1/10 di volume di 4 M LiCl e conservando la provetta a –20 °C per almeno 2 ore. Il precipitato era raccolto mediante centrifugazione a
16100 x g per 15 minuti a 4 °C, sottoposto a due “lavaggi” con etanolo 70%, asciugato, mediante l’utilizzo di una camera sotto vuoto, e sospeso in un appropriato volume di TE.
2.2.2. Protocollo B
Il totDNA è stato estratto secondo il protocollo “fenolo/cloroformio” (Sambrook et
al., 1989), opportunamente modificato ed ottimizzato per l’estrazione da gonadi di P. lividus. Una porzione di tessuto di circa 20 mg era posta in una provetta da 1.5 ml e
omogenizzata meccanicamente in 500 µl di H2O. Alla sospensione così ottenuta
erano aggiunti 150 µl di NDS (0.5 M EDTA, 0.1 M Tris-HCl pH 9.5, 1% SDS), precedentemente riscaldato a 55 °C, e 65 µl di Proteinasi K (10 mg/ml). La miscela era quindi incubata a 55 °C per 2 ore o per tutta una notte. Dopo aver aggiunto un volume di PEG 12% /NaCl 1.2 M, la preparazione era incubata in ghiaccio per un’ora. Seguivano una centrifugazione di 15 minuti a 4° C a 16000 x g e due lavaggi con etanolo 70 %. Il “pellet” era asciugato con una pompa a vuoto e successivamente risospeso in 500 µl di H2O sterile. La sospensione era incubata per
un’ora a 37 °C in presenza di 5 µl di RNasi A (1 mg/ml). Il DNA era, quindi, estratto una volta con un volume di fenolo/cloroformio (1:1) e due volte con un volume di cloroformio. La preparazione di DNA così ottenuta era nuovamente precipitata, aggiungendo un volume di PEG 12% /NaCl 1.2 M e incubando in ghiaccio per un’ora. Seguivano una centrifugazione di 15 minuti a 4 °C e due lavaggi con etanolo 70%. Il “pellet” era asciugato all’interno di una camera sotto vuoto e successivamente risospeso in 100 µl di H2O sterile. Il DNA così ottenuto era, quindi, precipitato
aggiungendo 1/10 di volume di acetato di sodio 3M pH 4.8 e 2 volumi di etanolo freddo e conservato a -20 °C per 2 ore o per una nottata. Seguivano una centrifugazione di 15 minuti a 4 °C a 16000 g e due lavaggi con etanolo 70%. Il “pellet” era asciugato con una pompa a vuoto e successivamente risospeso in un appropriato volume di TE. Allo scopo di determinarne la concentrazione, una piccola aliquota dei campioni di DNA (sia di mtDNA che di totDNA) estratti era sottoposta a lettura spettrofotometrica alle lunghezze d’onda di 260 e 280 nm. Per valutarne lo stato di “integrità”, circa 1 µg di DNA era sottoposto a corsa elettroforetica su gel di
agarosio 1.2%, utilizzando una differenza di potenziale costante di 100 V e TAE 1X (1 mM EDTA, 40 mM Tris-acetato), come tampone di corsa; dopo colorazione per circa 10 minuti in TAE contenente bromuro di etidio (10 mg/ml), il gel era visualizzato mediante l’utilizzo di un trans-illuminatore a luce ultravioletta e fotografato con un apparato fotografico digitale collegato a quest’ultimo.
2.3. Sintesi di oligonucleotidi
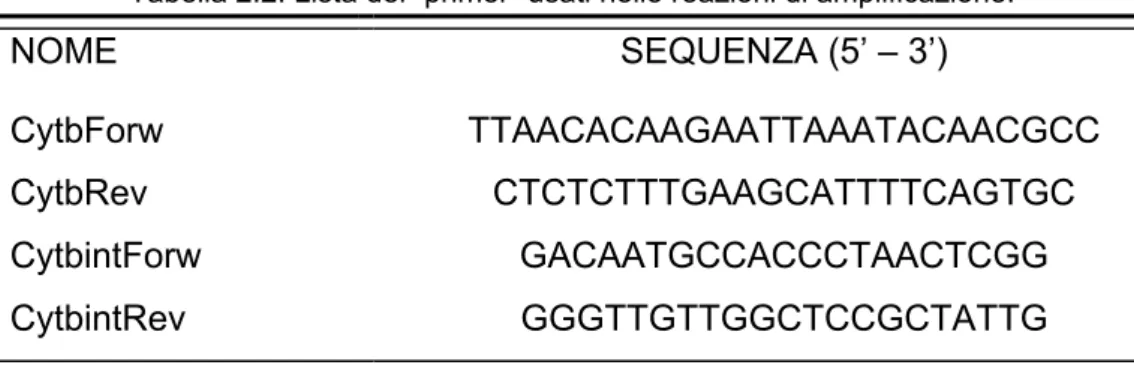
Gli oligonucleotidi usati come “primer”, cioè come sequenze di innesco complementari a regioni fiancheggianti il gene bersaglio (Cytb), nelle reazioni di amplificazione sono stati sintetizzati sulla base della sequenza totale del genoma mitocondriale di P. lividus disponibile nelle banche dati GenBank/EMBL (n° di accesso: J04815) e ivi depositata da Cantatore et al. (1989). Essi erano il primer “forward” CytbForw e quello “reverse” CytbRev (Tab. 2.2). Gli oligonucleotidi usati come primer interni nelle reazioni di sequenziamento diretto dei prodotti amplificati erano anch'essi costruiti sulla base della medesima sequenza disponibile nelle banche dati ed erano il primer “forward” CytbintForw e quello “reverse” CytbintRev (Tab. 2.3.1).
Tabella 2.2. Lista dei “primer” usati nelle reazioni di amplificazione.
NOME SEQUENZA (5’ – 3’)
CytbForw TTAACACAAGAATTAAATACAACGCC
CytbRev CTCTCTTTGAAGCATTTTCAGTGC CytbintForw GACAATGCCACCCTAACTCGG CytbintRev GGGTTGTTGGCTCCGCTATTG
2.4. Reazioni di amplificazione e sequenziamento
Le reazioni di amplificazione sono state condotte con la tecnica della reazione a catena della polimerasi (Polymerase Chain Reaction, o PCR) e sono state eseguite con una fase iniziale di denaturazione di un minuto a 94 °C, 35 cicli comprendenti
una fase di denaturazione di 30 secondi a 94 °C, una fase di “annealing” di 30 secondi a 55 °C e una fase di estensione di 2 minuti a 72 °C, seguiti da una fase terminale di estensione di 7 minuti a 72 °C. La dimensione dei prodotti amplificati è stata stimata mediante comparazione con un opportuno DNA di riferimento, dopo corsa elettroforetica. I prodotti amplificati, prima di essere sottoposti a reazioni di sequenziamento “diretto”, sono stati purificati su colonnine cromatografiche di poliacrilammide, al fine di eliminare eventuali residui di primer e desossinucleotidi, seguendo le indicazioni della ditta fornitrice.
Le reazioni di sequenziamento “diretto” dei prodotti amplificati sono state eseguite utilizzando un kit commerciale, seguendo le istruzioni della ditta fornitrice, ed un sequenziatore automatico multicapillare.
2.5. Trattamento dei dati
2.5.1. Allineamento delle sequenze
Le sequenze ottenute sono state allineate con il programma CLUSTALX (Thompson et al., 1997). Gli allineamenti sono stati visualizzati con il programma BioEdit (Hall, 1999); il medesimo software era usato anche per la conversione delle sequenze da nucleotidiche ad amminoacidiche. Per selezionare l’appropriato modello di sostituzione da usare per stimare le distanze genetiche, è stato eseguito un “hierarchical likelihood ratio test” utilizzando il programma Modeltest 3.06 (Posada & Crandall, 1998).
2.5.2. Stime di diversità genetica all’interno dei campioni
La diversità genetica presente all’interno di ciascuna località è stata stimata calcolando la diversità aplotipica (h) e la diversità nucleotidica (π). Il primo parametro è analogo all’eterozigosità attesa, ottenibile con i dati diploidi (ad esempio, alloenzimi e microsatelliti), ed è definita come la probabilità che due aplotipi scelti a caso siano differenti nel campione (località):
h = —— n - 1 n
(
1 –Σ
pi2)
k
i = 1
dove n è il numero di copie del gene nel campione, k è il numero di aplotipi, e pj è la
frequenza dell’i-esimo aplotipo nel campione. Se c’è completa omogeneità e tutti gli individui hanno lo stesso aplotipo, il valore di h è nullo, mentre se tutti gli individui hanno aplotipi differenti, h assume il suo massimo valore, cioè 1. La diversità nucleotidica (π) è la probabilità che due nucleotidi omologhi scelti a caso siano differenti. È equivalente alla diversità aplotipica a livello nucleotidico:
πn = —————— i j ij i = 1 1<
Σ Σ
p p dk
L
dove dij è una stima del numero di mutazioni che sono avvenute dalla divergenza
degli aplotipi i e j, k è il numero di aplotipi e pi è la frequenza dell’i-esimo aplotipo.
2.5.3. Stime di diversità genetica tra campioni
Una prima analisi dell'eterogeneità genetica tra i campioni provenienti dalle diverse località è stata effettuata mediante un test esatto basato sul metodo “MonteCarlo Markov Chain” (MCMC), che valuta la significatività della differenziazione genetica tra i campioni nel loro complesso. Inoltre l’eterogeneità genetica totale tra i campioni e tra ciascuna coppia di campioni è stata stimata usando l’estimatore dell’indice di fissazione FST di Weir & Hill (2002). È stata quindi
applicata l’analisi della varianza molecolare (AMOVA) (Excoffier et al., 1992) per ripartire la diversità genetica nelle sue diverse componenti. In una prima fase è stata indagata solamente la varianza all’interno e tra i campioni; l’AMOVA è stata poi eseguita anche aggiungendo un ulteriore livello, costituito da gruppi di località appartenenti alla medesima macroregione geografica. Sono stati considerati quattro gruppi corrispondenti all’Atlantico nord-orientale, al Mediterraneo occidentale, al Mediterraneo orientale e all’Adriatico. Per queste analisi è stato usato il programma ARLEQUIN versione 3.01 (Excoffier et al., 2005).
“network” sia con il metodo della massima parsimonia (Templeton et al., 1992, Crandall et al., 1994), sia con l’algoritmo del “median-joining” (Bandelt et al., 1999; 2000). La prima analisi è stata effettuata utilizzando il software TCS versione 1.21 (Clement et al., 2000); mentre per la seconda è stato utilizzato il programma NETWORK versione 4.1.1.2 (disponibile gratuitamente al sito internet
http://www.fluxus-technology.com). Si è preferito usare i network invece degli alberi e
dei dendrogrammi, in quanto i processi microevolutivi intraspecifici e i rapporti filogeografici tra gli individui e le popolazioni non necessariamente seguono le dicotomie caratteristiche degli alberi e dei dendrogrammi. I network quindi forniscono una rappresentazione migliore delle relazioni di tipo “reticolato” che caratterizzano i rapporti filogenetici e filogeografici presenti tra popolazioni naturali conspecifiche (Bandelt et al., 1999; Clement et al., 2000; Posada & Crandall, 2001).
Le distanze genetiche di Tamura & Nei (1993) sono state analizzate mediante la tecnica multivariata del multidimensional scaling (MDS) (Kruskal & Wish, 1978). Si tratta di un metodo di ordinamento spaziale non parametrico che permette di creare una grafico delle osservazioni (singoli aplotipi o centroidi dei campioni) in un numero finito di dimensioni, in maniera tale che la distanza tra ogni coppia di punti sul grafico rispetti il più possibile i rispettivi valori di distanza genetica. Inizialmente, l’algoritmo alla base dell’MDS pone in modo casuale i punti in uno spazio le cui dimensioni vengono da noi definite (di solito due, talvolta tre) e, successivamente, con un procedimento iterativo dal numero di cicli definito, ridefinisce le posizioni dei punti stessi per ottenere la rappresentazione che più rispecchi i valori delle distanze genetiche. L’accordo tra la distanza tra i punti nel grafico e la matrice delle distanze genetiche è espresso dal coefficiente di stress. Lo stress equivale a zero nel caso di perfetta concordanza (Clarke, 1993). Clarke & Warwick (1994), secondo un criterio di natura empirica, suggeriscono che quando lo stress è compreso tra 0.00 e 0.05, la rappresentazione MDS è “completamente attendibile”; quando lo stress è compreso tra 0.05 e 0.15 la rappresentazione MDS è “buona”; quando lo stress è compreso tra 0.15 e 0.20 la rappresentazione MDS è “accettabile”; infine, quando lo stress è maggiore di 0.20 la rappresentazione MDS non è accettabile. Normalmente, per facilità di interpretazione, si usano rappresentazioni bidimensionali, ma quando il coefficiente di stress supera il valore di 0.20 risulta opportuno scegliere una
configurazione grafica con un più alto numero di dimensioni per abbassare lo stress (Clarke & Warwick, 1994).
Stime del flusso genico sono state ottenute grazie al programma DnaSP (Rozas
et al., 2003) calcolando il parametro γST (Nei 1982), analogo all’ FST, utilizzando la
relazione γST = 1 / (1 + 2Nm). Le stime sono state applicate per il totale dei dati, per il
solo Mediterraneo escludendo i campioni atlantici, e considerando separatamente Mediterraneo occidentale, Mediterraneo orientale e Mar Adriatico
L’ipotesi della presenza di “isolamento da distanza”, cioè la possibile relazione tra divergenza genetica e distanza geografica per ciascuna coppia di campioni è stata vagliata con un’analisi di regressione dei valori di FST sui valori di distanza
geografica. Le distanze geografiche tra le località sono state misurate come le distanze nautiche più brevi (in chilometri) tra le località ottenute utilizzando un atlante geografico digitale. Per valutare la significatività della regressione è stato applicato il test non parametrico di Mantel (1967) per il confronto di due matrici di distanza. Questo test permette di determinare l’eventuale presenza di correlazione delle due matrici (nel caso del presente lavoro la matrice dell’indice di fissazione - FST - e
quella di distanza geografica). Il valore del parametro di associazione Z (coefficiente di Mantel) è calcolato a partire dai dati reali e poi comparato alla serie di pseudovalori ottenuti per permutazione di righe (o colonne) di una delle due matrici. L’ipotesi nulla è l’indipendenza tra le due matrici . Se non c’è relazione tra le due matrici, il valore di Z ottenuto sui dati reali non si discosterà significativamente dalla distribuzione degli pseudo-Z ottenuti con le permutazioni. Nel caso contrario, sarà rigettata l’ipotesi nulla e quindi le matrici risulteranno significativamente correlate. I valori di probabilità nel presente lavoro sono stati ottenuti mediante 10000 permutazioni. Il test di Mantel e i grafici di regressione sono stati ottenuti mediante il programma IBD Web Service ver. 2.1, disponibile online (Jensen et al., 2005).
2.5.4. Aspetti di demografia storica
La comparazione di sequenze nucleotidiche all’interno delle specie fornisce un valido approccio per determinare aspetti rilevanti della loro storia evolutiva (Cann, 2001). Variazioni della dimensione di una popolazione lasciano nel DNA tracce
particolari che possono essere individuate nelle sequenze nucleotidiche. Ciò ha consentito lo sviluppo di test statistici per valutare l’espansione demografica di una popolazione. Per valutare la possibilità di eventi recenti di espansione demografica in
P. lividus è stata eseguita un’analisi della distribuzione “mismatch” che si basa sul
modello dell’espansione improvvisa di Rogers & Harpending (1992). Questo modello, applicabile a sequenze di DNA non ricombinante, prevede una popolazione di femmine, con dimensione iniziale N0, in equilibrio tra mutazione e deriva genetica; si
suppone poi che la popolazione cresca (o si restringa) fino alla dimensione finale N1
dopo t generazioni. Solo la dimensione della popolazione femminile è importante ai fini del modello, perché i maschi non trasmettono il genoma mitocondriale. Nello specifico, N0 e N1 non si riferiscono al numero assoluto di femmine nella
popolazione ma al loro “numero efficace”, definito come il reciproco della probabilità che due individui scelti a caso abbiano la stessa madre (Rogers, 1995). Il modello dell’espansione improvvisa mantiene una buona affidabilità, non solo nel caso dell’espansione improvvisa, ma anche in quello di una crescita continua della popolazione (Rogers & Harpending, 1992). Questo modello è in grado di approssimare la storia demografica di una popolazione basandosi sui tre parametri
N0, N1 e t; ognuno dei quali è però dipendente da µ, cioè la sommatoria dei tassi di
mutazione per sito della sequenza in esame. Quindi la distribuzione mismatch può fornirci una stima per i tre parametri solamente nella forma combinata al fattore µ; per cui si ha: θ0 = 2µN0, θ1 = 2µN1, e τ = 2µt (Rogers, 2005). Questi parametri
misurano la taglia della popolazione femminile in unità pari a 1/2µ individui, e il tempo in unità 1/2µ generazioni. DnaSP (Rozas et al., 2003) mostra in forma di tabella e di grafico la distribuzione delle differenze nucleotidiche per ciascuna coppia di sequenze (distribuzione mismatch) e i valori attesi per popolazioni in espansione o regressione demografica. I valori attesi per una popolazione in espansione si dispongono graficamente a formare una curva dalla forma approssimabile ad un’onda (Rogers & Harpending, 1992). La posizione della cresta dell’onda è determinata da τ ed è informativa sul tempo relativo all’espansione in unità 1/2µ: assunto che l’onda procede lungo l’asse delle ascisse con un tasso di 2µ, la cresta sarà a τ = 2µt dopo t generazioni (Fig. 2.2).
FIG. 2.2. Evoluzione della distribuzione delle differenze per coppie di basi. Distribuzioni calcolate per θ0 = 2, θ1 = 200, e
valori variabili di τ, assumendo che θ aumenti dal valore iniziale θ0, al
valore θ1, a τ unità di tempo
mutazionale prima del presente. Da Rogers & Harpending, 1992.
Popolazioni stabili dovrebbero, in teoria, originare una disposizione rettilinea ma generalmente presentano una disposizione disordinata, una spezzata, scarsamente
attinente alla disposizione attesa. Nel caso di popolazioni in espansione, la curva delle
osservazioni segue più o meno fedelmente la distribuzione attesa o comunque presenta onde evidenti (Fig. 2.3) (Rogers & Harpending, 1992).
FIG. 2.3. Esempio di curva relativa ad una popolazione in espansione: la linea continua indica la distribuizione attesa, i cerchietti indicano la distribuzione osservata. Da Rogers & Harpending, 1992.
Per valutare la significatività statistica delle espansioni demografiche, è stato applicato il test R2 (Ramos-Onsins & Rozas, 2002), utilizzando il programma DnaSP
(Rozas et al., 2003). Questo metodo, noto come R2 di Ramos-Onsins & Rozas, si
basa sulla frequenza delle mutazioni segreganti all’interno delle sequenze, tipiche dei fenomeni di espansione demografica. La statistica R2 rileva l’eccesso di mutazione
sui rami esterni di una linea genealogica e quindi quell’eccesso di mutazioni recenti e proprie di una singola sequenza (“mutazioni private”) che deriva dalla crescita di una popolazione. La statistica R2 è definita dalla seguente espressione:
R2 = ——————————— S i = 1
(
Σ
(
U
i – k / 2)
/
n
)
2 1/2 nDove n è la dimensione del campione, S è il numero complessivo di siti segreganti, k è il numero medio differenze nucleotidiche tra due sequenze e Ui è il
numero di mutazioni esclusive dell’i-esima sequenza. Recentemente è stato dimostrato che i due test più potenti per individuare l’espansione demografica delle popolazioni sono il test FS di Fu (1997) e quello R2 di Ramos-Onsins & Rozas (2002).
Il comportamento di quest’ultimo risulta però superiore all’altro nel caso di campioni a bassa numerosità (Ramos-Onsins & Rozas, 2002). L’analisi della distribuzione mismatch e il test R2 sono stati eseguiti sia sul totale dei dati (meno i dati relativi al
campione proveniente dall’impianto di acquacoltura di Galway, Irlanda), sia su sottoinsiemi di dati, di cui alcuni corrispondenti a quelli inseriti nell’AMOVA e gli altri derivanti dall’unione di alcuni di essi.