7.
PARTE SPERIMENTALE
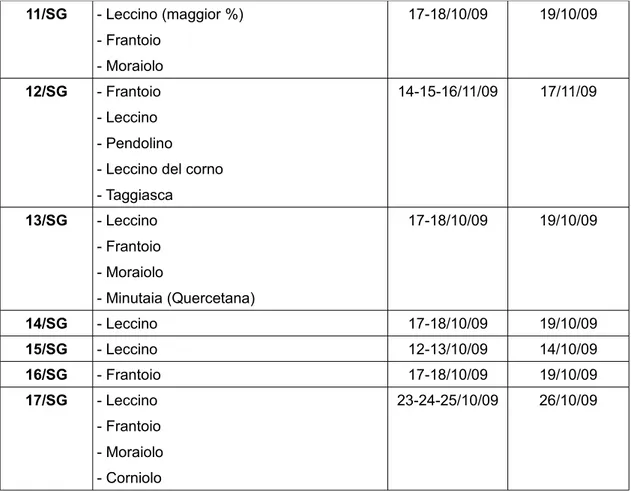
Lo studio è stato condotto su 17 campioni di olio d'oliva, forniti dall'associazione Nostrato, provenienti tutti dalla zona di Montignoso (MS) ed appartenenti a diverse cultivar, come mostra la seguente tabella.
Numero Campione
Cultivar Data raccolta Data frangitura 01/SG - Minutaia (Quercetana) - Frantoio - Leccino - Pendolino 30-31/10/09 02/11/09 02/SG - Minutaia (Quercetana) 20-21/11/09 23/11/09 03/SG - misto 16/10/09 19/10/09 04/SG - Leccino - Frantoio - Moraiolo - Minutaia (Quercetana) 16/10/09 19/10/09 05/SG - Frantoio - Leccino 28-29-30/10/09 31/10/09 06/SG - Leccino - Frantoio - Moraiolo 23-24-25/09/09 26/09/09 07/SG - Minutaia (Quercetana) - Frantoio - Leccino - Pendolino 17-18/10/09 19/10/09 08/SG - Minutaia (Quercetano 100%) 31/10/09 01/11/09 02/11/09 09/SG - Pendolino - Leccino
- 4 cultivars di tipologia sconosciuta chiamati localmente “Grossinaio” - maturazione precoce
04-05/10/09 06/10/09
10/SG - Minutaia (Quercetano 100%) 31/10/09 - 01/11/09
11/SG - Leccino (maggior %) - Frantoio - Moraiolo 17-18/10/09 19/10/09 12/SG - Frantoio - Leccino - Pendolino - Leccino del corno - Taggiasca 14-15-16/11/09 17/11/09 13/SG - Leccino - Frantoio - Moraiolo - Minutaia (Quercetana) 17-18/10/09 19/10/09 14/SG - Leccino 17-18/10/09 19/10/09 15/SG - Leccino 12-13/10/09 14/10/09 16/SG - Frantoio 17-18/10/09 19/10/09 17/SG - Leccino - Frantoio - Moraiolo - Corniolo 23-24-25/10/09 26/10/09
Tabella 11: denominazione dei campioni in studio e relative caratteristiche
Come mostrano i dati in tabella, in tutti gli oli esaminati, non trascorrono più di tre giorni fra la data di raccolta delle olive, che è avvenuta nel periodo compreso fra ottobre e novembre, e la data di frangitura.
7.1. DETERMINAZIONE DEGLI ACIDI GRASSI LIBERI,
METODO A FREDDO (ACIDITA')
Reg CEE 2568/1991 11/07/1991 GU CEE L247 05/09/1991 All II CE 702/2007 21/06/2007 GU CE L161 22/06/2007
Questa analisi esprime la percentuale di acidi grassi liberi presente nel prodotto. Gli acidi liberi si riscontrano soltanto dopo l'estrazione dell'olio dal frutto, poiché all'interno del frutto sono legati e quindi neutri. Essi derivano come prodotto di alcune reazioni innescate da enzimi lipolitici, durante la maturazione del frutto o a causa di una cattiva conservazione del frutto stesso. Proprio per questo motivo l'analisi dell'acidità viene considerata come parametro per stabilire la qualità di un olio di oliva. La presenza elevata di questi acidi grassi liberi (superiori al 3-4%) rende un olio non commestibile poiché avrebbero un'azione irritante sulla mucosa gastroenterica oltre che una sgradevole sensazione in bocca.
Dal punto di vista analitico si tratta di una titolazione acido-base. Sono stati pesati esattamente circa 10 g di olio in beuta a collo largo utilizzando una bilancia tecnica Orma BC 400. L'olio è stato disciolto in una soluzione 1:1 di etere dietilico ed alcool etilico assoluto, a cui sono state aggiunte 2 gocce di soluzione alcolica di fenolftaleina 1%. La determinazione dell'acidità è stata effettuata mediante titolazione colorimetrica con una soluzione di idrossido di sodio 0,1 M utilizzando una buretta di vetro da 10 ml graduata 0,02, fino a comparsa della colorazione rosa persistente. Dalla titolazione è stata ricavata l'acidità espressa in % di acido oleico. La formula da applicare è:
v x M x 28,2 % acido oleico =
P
v = ml di soluzione di NaOH usati M = molarità della soluzione di NaOH P = massa in g dell'olio.
La reazione di neutralizzazione che avviene, riferita all'acido oleico può essere così schematizzata:
CH3(CH2)7CH = CH(CH2)7COOH + NaOH CH3(CH2)7CH = CH(CH2)7COONa + H2O
7.2.
DETERMINAZIONE DEL NUMERO DI PEROSSIDI
Reg CEE 2568/1991 11/07/1991 GU CEE L247 05/09/1991 All III
Questa analisi esprime la quantità di ossigeno già assorbita dall'olio, il quale quindi ha già iniziato una propria attività ossidativa; tale attività, durante l'invecchiamento, porta ad un irrancidimento del prodotto che ne conferisce odori e sapori sgradevoli. Attraverso questa analisi possiamo quindi determinare il grado di ossidazione di un olio di oliva: più alto è il suo valore e più avanzato è lo stadio di irrancidimento del prodotto.
Dal punto di vista analitico si tratta di una titolazione redox. Sono stati pesati esattamente circa 2 g di olio in beuta a collo smerigliato utilizzando una bilancia tecnica Orma BC 400. L'olio è stato disciolto in una soluzione 3:2 di acido acetico glaciale e cloroformio, a cui sono stati aggiunti circa 0,5 ml di soluzione satura di ioduro di potassio. La beuta è stata tappata e mantenuta al buio, lontana da fonti di calore, per 5 minuti, terminati i quali sono stati aggiunti circa 40 ml di acqua demineralizzata ed 1 ml di soluzione acquosa di salda d'amido 10 g/l. E' stata effettuata la titolazione dello iodio liberato con una soluzione di tiosolfato di sodio 0,01 N, utilizzando una buretta da 25 ml, graduata 0,05. Il numero di perossidi (P.V.), espresso in milliequivalenti di ossigeno attivo per kg, è dato dalla formula:
P.V. = (v x N / P) x 1000
v = è il numero di ml della soluzione standardizzata di tiosolfato di sodio usata per la prova, corretto in modo da tener conto della prova in bianco
N = è la normalità esatta della soluzione di tiosolfato di sodio usata P = è il peso in g della sostanza da analizzare.
7.3. ANALISI
SPETTROFOTOMETRICA
UV
(NELL'ULTRAVIOLETTO)
Reg CEE 2568/1991 11/07/1991 GU CEE L247 05/09/1991 All III CEE 183/1993 29/01/1993 GU CEE L22 30/01/1993
L’analisi di un olio mediante spettrofotometria UV-visibile è effettuata correntemente per la valutazione della sua genuinità e per identificare eventuali adulterazioni. Questo tipo di analisi è espresso mediante dei coefficienti K che rappresentano l'assorbimento da parte dell'olio all'esposizione di luce ultravioletta in particolari condizioni. L’assorbimento di luce di una sostanza è regolato dalla legge di Lambert-Beer
A = εbC
Dove A è Assorbanza espressa come Log (Io/I), Io/I è il rapporto tra intensità della luce trasmessa e incidente sul mezzo attraversato; ε, assorbività molare o coefficiente di estinzione molare, è l’assorbanza di una soluzione 1M; b è il cammino ottico in cm e C la concentrazione molare della sostanza che assorbe la luce.
I doppi legami nell’olio vergine non sono mai vicini o coniugati. La presenza di doppi legami coniugati è dovuta all’azione ossidativa di alcuni
reagenti impiegati fraudolentemente, che causa lo slittamento dei doppi legami. Questo slittamento comporta un assorbimento caratteristico all'ultravioletto attorno a 270 nm dove si ha un picco in presenza di olio rettificato (olio di sansa e semi) per la presenza di dieni formatisi per slittamento.
Il coefficiente di estinzione molare alle lunghezze d'onda di 230 nm e 270 nm, indica lo stato ossidativo dell'olio, poiché si possono formare dieni e trieni coniugati durante l'ossidazione del prodotto.
Figura 21: esempio di dieni e trieni coniugati durante l'ossidazione del prodotto
Sono state pesate esattamente 4 gocce di olio, precedentemente filtrato, in matraccio tarato da 10 ml, utilizzando una bilancia analitica Orma BCA 120. L'olio è stato immediatamente disciolto e portato a volume con isoottano per spettrofotometria, per minimizzare il fenomeno ossidativo provocato dalla luce. Un'aliquota di campione così preparato è stata introdotta in apposita celletta di quarzo, con cammino ottico da 1 cm e analizzata mediante spettrofotometro a doppio raggio UV-Vis Perkin-Elmer modello Lambda 25, dopo azzeramento con il bianco, costituito esclusivamente dal solvente.
L'esame spettrofotometrico dell'olio di oliva prevede la determinazione dell'estinzione specifica alle lunghezze d'onda di 232 e 270 nm e la determinazione del ΔK calcolato secondo la seguente formula:
(Km – 4) + (Km + 4)
ΔK = Km -
Dove Km è l'estinzione molare specifica alla lunghezza d'onda m, lunghezza
d'onda di massimo assorbimento intorno a 270 nm.
7.4. ANALISI GASCROMATOGRAFICA DEGLI ESTERI
METILICI DEGLI ACIDI GRASSI
Reg CE 796/2002 06/05/2002 GU CE L128 15/05/2002 All XB + Reg CEE 2568/1991 11/07/1991 GU CEE L248 05/09/1991 All XA Reg CEE 1429/1992 26/05/1992 GU CEE L150 02/06/1992
L'utilità della determinazione degli acidi grassi, dal punto di vista analitico, è dovuta al fatto che da una sola procedura viene ricavata una notevole quantità di parametri, che sono peraltro specifici, cioè corrispondono a singoli componenti chimici presenti in un olio.
Sono state introdotte in una provetta con tappo a vite 5 gocce di campione che è stato poi disciolto in 3 ml circa di n-esano RPE. Alla soluzione sono stati aggiunti 0,2 ml circa di soluzione metanolica di idrossido di potassio 2 N. La provetta è stata tappata e sottoposta ad energica agitazione per almeno 30 secondi. Il campione così preparato è stato lasciato a riposo finché la soluzione superiore non è diventata trasparente. In questo modo gli esteri metilici si formano per transesterificazione con una soluzione metanolica di idrossido di potassio. A stratificazione avvenuta è stata iniettata una quantità della stessa compresa fra 1 e 1,5 μl nel gascromatografo.
L'analisi gascromatografica è stata effettuata con gascromatografo HRGC 5300 Mega Series e colonna capillare Select Fame, lunga 50 m e con diametro di 0,25 mm, rivestita internamente da uno strato di cianopropilsilicone spesso 0,25 μm. Le temperature operative sono:
colonna 190°C iniettore 290°C FID 290°C
Terminata la corsa gascromatografica è stato valutato il grafico mediante il sistema di integrazione Turbochrom 4.1 e, quando necessario, i picchi sono stati integrati manualmente.
Figura 22: cromatogramma relativo all'analisi gascromatografica degli esteri metilici degli acidi grassi
Per il calcolo della composizione in acidi grassi, sono stati presi in considerazione i seguenti acidi grassi:
Miristico (C14:0)
Palmitico (C16:0): somma delle aree dei picchi che corrispondono agli esteri metilici ed etilici.
Palmitoleico (C16:1): somma delle aree dei picchi che corrispondono agli isomeri w9 e w7 dell'estere metilico.
Stearico (C18:0) Elaidinico (C 18:1)
Oleico (C18:1): somma delle aree dei picchi che corrispondono agli isomeri w9 e w7 dell'estere metilico, dell'estere etilico.
Linoleico (C18:2): somma delle aree dei picchi che corrispondono agli esteri metilici ed etilici.
Linoleico (C18:2): isomeri trans dell'estere metilico. Arachico (C20:0).
Linolenico (C18:3): somma delle aree dell'estere metilico. Linolenico (C18:3): isomeri trans dell'estere metilico. Eicosanoico (C20:1).
Beenico (C22:0). Lignocerico (C24:0).
Lo squalene non viene preso in considerazione per il calcolo del totale dell'area, poiché, nonostante il picco relativo allo stesso compaia nel cromatogramma, non si tratta di un acido grasso.
Il calcolo della percentuale di ogni acido grasso è stato effettuato in base alla formula che segue:
% X = (Area X x 100) / ( area totale )
7.5. DETERMINAZIONE DEI POLIFENOLI TOTALI
Metodo interno
E' stato pesato esattamente, alla bilancia analitica Orma BCA 120, direttamente in una colonna SPE Mega BE Varian (non endcapped C18 OH – 1 g – 6 ml), circa 1 g di olio. L'olio è stato lasciato adsorbire completamente dalla fase solida ed è stato poi lavato con 3 aliquote da 5 ml di n-esano, evitando sempre di portare a secco la colonna. L'esano è poi stato allontanato completamente con flusso di azoto dal basso verso l'alto. Infine i polifenoli sono stati estratti con alcool metilico raccogliendo in un matraccio tarato da 10 ml e portando a volume direttamente.
Per la determinazione quantitativa sono stati prelevati 2 ml della soluzione così ottenuta e sono stati trasferiti in matraccio tarato da 10 ml. Contemporaneamente è stato preparato un bianco dei reattivi. Il volume di ogni campione è stato integrato con acqua distillata per raggiungere 5 ml circa. Ad ogni matraccio sono stati aggiunti 0,5 ml di reattivo di Folin-Ciocalteau e dopo 3 minuti, 1 ml di soluzione di carbonato di sodio satura circa al 35%. Ogni palloncino è stato portato a volume e conservato al buio per 60 minuti per permettere lo sviluppo del colore. Terminati i 60 minuti il contenuto di ogni matraccio è stato trasferito in provetta da centrifuga e centrifugato per permettere la precipitazione del sedimento.
E' stata poi effettuata la lettura spettrofotometrica con spettrofotometro a doppio raggio UV-Vis Perkin-Elmer modello Lambda 25, a 725 nm contro il bianco dei reattivi. La lettura viene effettuata rispetto ad una curva di taratura esterna fatta con acido caffeico.
Per il calcolo dei risultati è stato utilizzato il software WinLab, questi vengono espressi come mg/Kg di olio.
7.6. DETERMINAZIONE DELLA COMPOSIZIONE E DEL
CONTENUTO
IN
STEROLI
MEDIANTE
GASCROMATOGRAFIA CON COLONNA CAPILLARE +
DETERMINAZIONE
DELL'ERITRODIOLO
E
DELL'UVAOLO
Reg CEE 2568/1991 11/07/1991 GU CEE L247 05/09/1991 All V Reg CEE 183/1993 29/01/1993 GU CEE L22 31/01/1993
Reg CEE 2568/1991 11/07/1991 GU CEE L247 05/09/1991 All VI
Preparazione dell'insaponificabile
Utilizzando una bilancia analitica Orma BCA 120, sono stati pesati esattamente circa 1,2 g di olio in provetta con tappo a vite e 100 μl di soluzione allo 0,1446 % m/m di α-colestanolo. Sono stati aggiunti circa 4 ml di soluzione etanolica di idrossido di potassio 2 N circa, la provetta è stata tappata e posta in piastra riscaldante per COD a circa 100°C, dove è stata lasciata fino a completa saponificazione. Il contenuto della provetta è stato travasato quantitativamente, aiutandosi con acqua, in una provetta con tappo a vite da 40 ml e diluito con acqua. La soluzione così ottenuta è stata poi suddivisa fra due provette ed ulteriormente diluita con acqua. E' stato aggiunto etere etilico a ciascuna provetta fino a riempimento e ciascuna provetta è stata poi sottoposta, dopo essere stata tappata, ad agitazione per circa 30 secondi e lasciata a riposo per far avvenire la stratificazione delle due fasi: acquosa sottostante ed eterea sovrastante. La fase eterea è stata prelevata, fatta passare attraverso allumina basica per allontanare eventuali tracce di acidi e attraverso sodio solfato anidro per eliminare eventuali ulteriori residui di acqua e filtrata. La fase eterea è stata essiccata sotto azoto. Il procedimento di estrazione è stato eseguito due volte. Il materiale essiccato che si trova nella provetta è stato ripreso con etere e fatto nuovamente passare su sodio solfato anidro per eliminare le ultime tracce di acqua dalla frazione insaponificabile. In seguito è stato di nuovo fatto evaporare il solvente sotto azoto.
Separazione della frazione sterolica
Sono state preparate delle lastre al gel di silice da TLC basificate, mediante immersione in una soluzione etanolica 0,2 N di idrossido di potassio per 10 secondi, evaporazione della soluzione sotto cappa aspiratrice e riscaldamento in stufa a 100°C.
La lastra è stata prelevata dalla stufa ed è stata tracciata una linea retta con il lapis a circa 1 cm dal margine inferiore della stessa. La frazione
insaponificabile è stata ripresa con 2-3 gocce di etere etilico ed è stata seminata con pennello pulito lungo la linea tracciata sulla lastra. La lastra è stata posta nella camera di sviluppo che contiene, come eluente, una miscela 65:35 di n-esano ed etere etilico. L'eluizione è stata fatta procedere fino a che il fronte del solvente non ha raggiunto 1 cm circa dal bordo superiore della placca. La placca è stata quindi rimossa dalla camera di eluizione e lasciata sotto cappa per permettere l'evaporazione del solvente. L'eluizione è stata ripetuta una seconda volta.
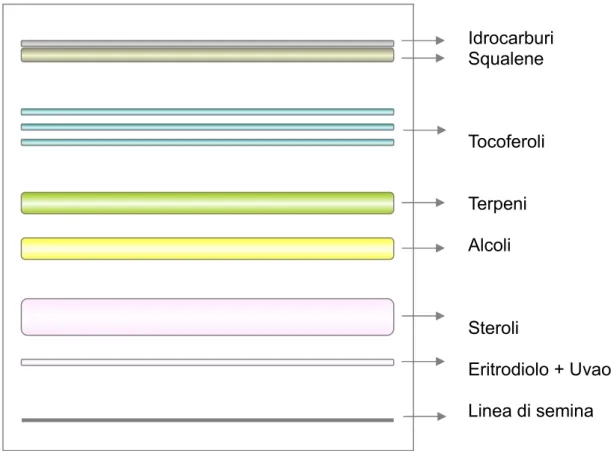
La lastra essiccata è stata spruzzata debolmente ed uniformemente con 2',7'-diclorofluoresceina. E' stata individuata e delimitata con un tratto di lapis la banda relativa agli steroli (Figura 25), individuata precedentemente mediante semina ed eluizione dello standard α-colestanolo, essa è stata poi raschiata con una spatola metallica.
Idrocarburi Squalene Tocoferoli Terpeni Alcoli Steroli Eritrodiolo + Uvaolo Linea di semina
Figura 25: rappresentazione di una TLC su cui viene effettuata la separazione della frazione sterolica dagli altri composti
Il materiale asportato, finemente sminuzzato, è stato trasferito in un bicchiere da 50 ml, all'interno del quale sono stati aggiunti circa 10 ml di etere etilico per permettere l'estrazione della frazione sterolica dal gel di silice. Trascorsi circa 15 minuti il contenuto del bicchiere è stato filtrato e trasferito in una provetta da centrifuga da 10 ml.
Preparazione dei trimetilsilileteri
E' stato allontanato il solvente dalla provetta mediante evaporazione sotto azoto e, in tale provetta, contenente la frazione sterolica essiccata, sono state aggiunte due gocce del reattivo per la sililazione, costituito da una miscela di piridina-esametildisilazano-trimetilclorosilano in rapporto 9:3:1 (Sigma Aldrich). La provetta è stata tappata e agitata cautamente, senza capovolgerla, fino a completa solubilizzazione degli steroli e lasciata a riposo per 13 minuti a temperatura ambiente.
Analisi gascromatografica
Trascorsi i 13 minuti a è stata iniettata una quantità di frazione sterolica silanizzata compresa fra 1 e 1,5 μl nel gascromatografo.
L'analisi gascromatografica è stata effettuata con gascromatografo HRGC Mega 2 Series e colonna capillare Agilent Technologies HP-5MS lunga 30 m, con diametro di 0,25 mm e spessore del rivestimento interno di 0,25 μm.
La temperatura operativa è diversa da zona a zona:
colonna: 260°C ± 5°C
iniettore: 280°CLa registrazione è stata effettuata fino a completa eluizione dei TMSE degli steroli presenti.
Terminata la corsa gascromatografica è stato valutato il grafico mediante il sistema di integrazione Turbochrom 4.1 e, quando necessario, i picchi sono stati integrati manualmente. Il coefficiente di risposta dell'α-colestanolo è stato considerato come unitario.
Il contenuto di ogni singolo sterolo, espresso in mg/ml di sostanza grassa, è stato calcolato secondo la seguente formula:
Ax x ms x 100
sterolo x =
As x m
In cui Ax è l'area del picco dello sterolo x, As è l'area del picco
dell'α-colestanolo, ms è la massa di α-colestanolo aggiunta, espressa in milligrammi e
m la massa di campione prelevato espressa in grammi.
Espressione dei risultati
I contenuti dei singoli steroli sono stati riportati come percentuale, mentre il valore relativo agli steroli totali è stato espresso come mg/1000 g di sostanza grassa. Il contenuto percentuale di ogni singolo sterolo è stato calcolato dal rapporto fra l'area del picco corrispondente e la sommatoria delle aree dei picchi degli steroli.
Ax
% dello sterolo x = x 100 ΣA
7.7. DETERMINAZIONE DELLE SOSTANZE VOLATILI
Metodo interno
Figura 26: cromatogramma relativo all'analisi GC-MS delle sostanze volatili
Il principale processo di alterazione degli oli vegetali è costituito dall’autoossidazione dei grassi insaturi che può modificarne le caratteristiche organolettiche, attraverso la formazione di composti odorosi, e le virtù nutrizionali attraverso reazioni secondarie. In confronto con altri oli vegetali, l’olio di oliva vergine mostra più resistenza all’ossidazione per via del basso tenore di acidi grassi poliinsaturi e della presenza di antiossidanti naturali come polifenoli e tocoferoli. Tuttavia, i processi di irrancidimento sono comunque possibili e tendono quantomeno a diminuire il contenuto di antiossidanti. L’identificazione di un processo di irrancidimento è perciò utile ed è possibile evidenziarlo attraverso il riconoscimento di sostanze marcatori, che nell’olio di
oliva risultano essere in particolare il nonanale, l’ottano, il pentilfurano e il 2-etilfurano, sostanze che possono anche non essere attive dal punto di vista aromatico ma sono comunque indice di un processo di alterazione in corso.
nonanale 2-pentilfurano
Figura 27: sostanze marcatori del processo di irrancidimento dell'olio
Per l'esecuzione dell'analisi è stato utilizzato un dispositivo SPME Supelco rivestito di poli-(dimetilsilossano) necessario per campionare lo spazio di testa di circa 10 ml di olio, contenuto in provetta di vetro. Il dispositivo è stato lasciato per 30 minuti ad equilibrarsi. Terminato il campionamento la fibra è stata trasferita nell'iniettore del sistema GC-MS. L'analisi GC-MS è stata eseguita con un gascromatografo Varian CP-3800 equipaggiato con colonna capillare DB-5 e come rivelatore uno spettrometro di massa a trappola ionica Varian Saturn 2000. Le condizioni analitiche utilizzate sono le seguenti: l'iniettore è stato mantenuto a 250°C, mentre la temperatura del forno è stata fatta variare da 60°C a 240°C mediante una programmata, l'incremento di temperatura è stato di 3°C/min.
L'identificazione dei costituenti è stata eseguita mediante confronto fra i tempi di ritenzione ottenuti nell'analisi dei campioni con quelli contenuti dalla biblioteca del software in dotazione con lo strumento, dalla biblioteca NIST e dalla letteratura.