Chapter 4
Stereoselective Synthesis of N-Cbz-Imino Glycal-Derived Allyl Epoxides and 2,3-Unsaturated-Aza-O-Glycosides
4.1. Introduction
As shown in the previous sections of this thesis, in our laboratory, the diastereoisomeric allyl epoxides 2.1α and 2.1β, derived, from D-allal and D-galactal, respectively, were synthesized (Scheme 4.1) and their reactivity in the presence of different types of nucleophiles, particularly O-nucleophiles, was studied. The reaction of 2.1α and 2.1β with simple alcohols and partially protected monosaccharides, led to the corresponding O-glycosides through a completely regio- and stereoselective 1,4-addition process affording α-O-glycosides from 2.1α and β-O-glycosides from 2.1β, in a new, uncatalyzed, substrate-dependent stereospecific glycosylation process.1
Scheme 4.1. Stereoselective addition of O-Nucleophiles to allyl epoxides 2.1α and 2.1β
These results prompted us to verify if the observed regio- and stereoselectivity could be also maintened in the presence of important structural modifications, as the substitution of the endocyclic oxygen with a N-substituted nitrogen, thus transforming a glycal system into an imino glycal system and transferring our studies from sugars to azasugars.
The aim was to synthetize the racemic diastereoisomeric allyl epoxides 4.1α and 4.1β, the aza analogues of 2.1α and 2.1β, derived, from D,L-imino allal and D,L-imino galactal respectively (Scheme 4.2), and to study the regio- and stereoselective behavior, in nucleophilic addition
O BnO O 2.1! O BnO O 2.1"
ROH = MeOH, EtOH, i-PrOH, t-BuOH, benzyl alcohol, dihydrocholesterol, phenol, diacetone-D-glucose, 1,2;3,4-di-O-isopropylidene-D-galactopyranose
ROH (3 equiv) ROH (3 equiv) benzene benzene ! " O OBn O O R H 1 #+ 1 O OBn O O R H O OR BnO HO !-O-glycoside #+ O OR BnO HO "-O-glycoside " !
reactions with alcohols, in order to check if these new previously not described allyl oxirane systems behave similarly to 2.1α and 2.1β.
As shown in Scheme 4.2, a N-Cbz functionality was chosen as the endocyclic nitrogen-containing group. The choice of an urethane functionality, instead of a corresponding tertiary amine group, as the nitrogen-bearing group, was based on the consideration that an urethane moiety in that position was believed easier to handle under the different reaction conditions and characterized by chemical and electronic properties comparable to those of endocyclic oxygen of the corresponding epoxides 2.1α and 2.1β.
Scheme 4.2. N-Cbz-Imino glycal-derived allyl epoxides 4.1α and 4.1β
Our interest in imino glycals systems derived also from an examination of the relevant literature, which showed that the only studies found on these systems concerned their partial application of Ferrier’s reaction.2 As previously mentioned, the results of these studies have indicated a large or complete facial selectivity with exclusive or almost exclusive formation of the corresponding β-anomers, due to a preferential pseudoaxial attack of the nucleophile, in a sort of product-dependent selectivity (Scheme 4.3 and Chapter 3).
Scheme 4.3. Product-depend selectivity under Ferrier reaction on tri-O-acetyl imino glucal On the basis of this literature result, it appeared interesting to check not only whether the new imino glycal-derived epoxides 4.1α and 4.1β were able to glycosylate alcohols, but also to investigate the facial selectivity of this process and the regio- and stereochemical outcome of the nucleophilic attack. N BnO O 4.1! N BnO O 4.1" C OBn C O O OBn N N C C
Tri-O-acetyl-D-iminoglucal !-anomer (24-72% de)
OAc AcO AcO Nu AcO AcO O OR O OR Nu BF3.Et 2O ! ! R = 9-fluorenylmethyl
Finally the possibility to elaborate the double bond present in the addition products in order to obtain “true” azasugars made, in our opinion, these compounds interesting substrates for a systematic study of their reactivity.
A retrosynthetic analysis had indicated that the trans diol 4.2, derived from D,L-imino glucal, was a valuable synthetic intermediate for the synthesis of epoxides 4.1β, which, in turn, could be useful for the synthesis of trans diol 4.3, derived from D,L-imino gulal, a useful precursor for epoxide 4.1α (Scheme 4.4).
Scheme 4.4. Retrosynthetic analysis for the synthesis of epoxides 4.1α and 4.1β
Trans diol 4.2 could be easily prepared by means of two synthetic procedures developed by
Dransfield,2 and Comins.3
Scheme 4.5. Dransfield’s protocol for the synthesis of the corresponding tri-acetyl-Fmoc-derivative (4.10) of trans diol 4.2 O HO HO OH a) NaH, PMBCl, DMF b) Hg(OAc)2, THF-H2O, NaBH4 O PMBO PMBO OPMB OH c) Ph3P CH2, toluene d) TPAP, NMO, 4A sieves, CH2Cl2 PMBO O OPMB OPMB
e) HONH2.HCl, Py, EtOH, 60°C f) LiAlH4, Et2O, t.a.
g) FmocCl, K2CO3, THF:H2O (3:1) PMBO OPMB OPMB NH Fmoc h) CF3CO2H, CH2Cl2 i) Ac2O, Py, t.a. AcO NH OAc OAc Fmoc AcO NH OAc OAc Fmoc j) O3, -78°C, CH2Cl2, Me2S, t.a. k) (COCl)2, Et3N, DMF, CH2Cl2 N AcO AcO OAc Fmoc 4.4 4.5 4.6 4.7 4.8 4.9 4.10 D-glucal N BnO HO OH Cbz 4.2 N BnO O 4.1! N BnO O 4.1" COBn OC OBn O N BnO 4.2 C OBn O HO OH N BnO 4.3 C OBn O HO OH
Actually, the procedure described by Dransfield led to the corresponding tri-acetyl-Fmoc-derivative (4.10) of the trans diol 4.2 (Scheme 4.5). However this protocol could be reasonably be adapted to the direct synthesis of the trans diol 4.2. Following the Dransfield’s protocol,2 the tri-acetyl-Fmoc-derivative 4.10 was synthesized (11 steps) starting from the commercially available D-glucal, initially O-protected and then hydroxylated on the double bond to give the hemiacetal 4.5. Wittig olefination followed by TPAP oxidation of the resulting secondary alcohol furnished ketone 4.6 which was converted into amine 4.7 by LiAlH4 reduction of the corresponding oxime and further Fmoc protection. Amine 4.7 was obtained as an unseparable 77:23 mixture of isomers in favour of the required (6R)-diasteroisomer. After the PMB ether protecting groups had been switched to acetates, triacetate 4.8 could be separated from its C(6) epimer 4.9. Ozonolytic cleavage of the terminal double bond, followed by dehydration of the resulting hemiacetal using oxalyl chloride, led to the diastereomerically pure triacetate 4.10, having the right relative configuration as in the desired trans diol 4.2 (Scheme 4.5).
An appropriate modification of this protocol, including a) monobenzylation of the primary alcoholic functionality of D-glucal 4.4, and b) use of N-Cbz protection instead of N-Fmoc one, makes the above described procedure useful for the synthesis of the desired N-Cbz protected trans diol 4.2.
The procedure described by Comins3 directly led to the trans diol (2S, 3R, 4R)-4.2, in a completely regio- and enantioselective fashion, starting from 4-methoxy-3-(triisopropylsilyl)pyridine 4.11, as shown in Scheme 4.6.
Scheme 4.6. Enantioselective and stereoselective synthesis of the trans diol 4.2 elaborated by Comins
Since this route appeared easier, quicker and completely stereoselective in every step compared to the one described by Dransfield, we decided to synthetize racemic trans diol 4.2 by a racemic application of enantioselective Comins’s protocol. The choice for a racemic synthesis of these
N OMe TIPS 1) R*OCOCl 2) BnOCH2(2-Th)Cu(CN)Li2 3) H3O+ N O TIPS CO2R* BnO N O H BnO 1) n-BuLi 2) BnOCOCl N O Cbz BnO toluene, reflux Pb(OAc)4 N O Cbz BnO AcO aq. HCl EtOH N Cbz BnO HO O 4.11 (2S)-4.14 (2S, 3R)-4.16 (2S)-4.12 (2S, 3R)-4.15 1) NaOMe 2) H3O+ N Cbz BnO HO OH (2S, 3R, 4R)-4.2 AcOH Me4NBH(OAc)3 CH3COCH3 (2S)-4.13
products derived from the observation that our preliminary studies did not necessitate of chiral, non-racemic compounds which, in addition, turned out particularly expensive to be prepared. 4.2. Synthesis of imino glycal-derived allyl epoxides 4.1α and 4.1β
The synthetic protocol is based on the regioselective addition of a superior order cuprate [(BnOCH2)(2-thienyl)Cu(CN)Li2] 4.19 to the C(2) carbon of the N-Cbz pyridinium chloride 4.18, generated in situ by reaction of 4-methoxypyridine (4.17) with Cbz-Cl. Acid hydrolysis of the intermediate enol ether 4.20 afforded, through the intermediate emiacetal 4.21, the dihydropyridone 4.14 (Scheme 4.7).3
Scheme 4.7. Synthesis of the dihydropiridone 4.14
The superior order cuprate 4.19, necessary for the introduction of the -CH2OBn side chain, was obtained only, in situ, by means of a laborious procedure. Actually, for the preparation of 4.19, the commercially available benzyl chloromethyl ether (4.24), freshly distilled in order to remove the contaminant benzyl chloride, was added to a solution of Bu3SnLi (4.23), on its own obtained by deprotonation of Bu3SnH (4.22) by LDA, at 0°C (Scheme 4.8).4 The obtained (benzyloxymethyl)tributylstannane 4.25 is then purified by chromatography or distillation and subsequently treated, in situ, with BuLi, at -78°C , to give the more reactive organo lithium 4.26. This reagent is allowed to react with the commercially available (Aldrich) Lipshutz so called “cuprate in a bottle” (2-Th)Cu(CN)Li, to give the superior order cuprate 4.19.5
Scheme 4.8. Synthesis of the superior order cuprate 4.19
N OMe CbzCl THF N OMe Cbz Cl -N MeO Cbz BnO H2C2O4 sat.aq -78 °C N O BnO Cbz BnOCH2Cu(CN)]Li2 [2-(Th)] 4.20 4.14 -25 °C + 4.18 65% (4.19) 4.17 N MeO Cbz BnO 4.21 Bu3SnH LDA THF, 0°C Bu3SnLi PhCH2OCH2Cl -78°C t.a. Ph O Sn(Bu3) 4.22 4.23 4.25 4.24 n-BuLi THF, -78°C Ph O Li 4.26 Ph O Cu(CN)Li 4.19 S S Cu(CN)Li -78°C
Once obtained, dihydropyridone 4.14 was regio- and stereoselectively acetoxylated by treatment with Pb(OAc)4 stabilized with AcOH at refluxing in toluene, which determined the exclusive formation of α-acetoxy ketone 4.15 (Scheme 4.9).
Scheme 4.9. Synthesis of the trans diol 4.2.
The stereoselectivity observed in the acetoxylation reactionof enone 4.156 arises by a selective intramolecular acetate delivery to the α-face, from the more favored axial direction, of the intermediate enol-lead triacetate 4.27, that is formed in the acid enviroment (AcOH) (Scheme 4.10). It should be underlined that the reaction intermediate 4.27 is characterized, as all the other imino glycal compounds and synthetic intermediates present in this study, by a preference for that conformer in which the alkylic side chain on C(5) in an axial position. In this way, the system minimizes the negative torsional strain (1,3 allylic strain) present between the side chain and the Cbz group which would be at the maximum level if the side chain is equatorial (See Structures, Conformations and Configurations, Paragraph 4.4 for. As shown by literature data6 and as found by appropriate computational conformational analysis, the benzyloxymethyl side chain is always at the maximum distance possible from the urethane substituent.
Scheme 4.10. α-stereoselective acetoxylation of the dihydropyridone 4.14
Acid hydrolysis of 4.15 afforded the α-hydroxy ketone 4.16, that was diastereoselectively reduced to the trans diol 4.2 by means of Me4NBH(OAc)3 in the presence of AcOH (Scheme 4.9).3,7 The reduction reaction, carried out under Evan’s conditions,7 proceeds through the intermediate formation of an alkoxydiacetoxyborohydride species (4.28) in which the intramolecular hydride delivery to the protonated (from AcOH) carbonyl functionality necessarily occurs from the α-face, that is from the same face where the original hydroxy functionality was directed (Scheme 4.11).
Pb(OAc)4 toluene (reflux) N BnO O O OBn Pb O OAc OAc O Me N O BnO Cbz 4.14 N O BnO Cbz AcO 4.27 4.15 AcOH 5 N O BnO Cbz AcO N O BnO Cbz HO N O BnO Cbz 50 °C 4.14 62% 94% HCl/EtOH Pb(OAc)4 toluene (reflux) 4.15 4.16 AcOH N Cbz OH HO BnO 4.2 AcOH Me4NBH(OAc)3 72% CH3COCH3
Once again, the intermediate 4.28 necessarily adopted a conformation in which the benzyloxymethyl side chain is axial.
Scheme 4.11. Reduction of α-hydroxy ketone 4.16 to trans diol 4.2
After purification by gradient flash chromatography, which turned out to be necessary in order to remove some contaminants which could affect the subsequent reactions, trans diol 4.2, was subjected to selective functionalization of the two hydroxyl groups on C(3) and on C(4) in order to obtain a stable precursors of epoxide 4.1β.
The reaction of the trans diol 4.2 with TBDMS-Cl (1.1 equiv) was not regioselective, as desired, and a 40:23:37 mixture of 3-OTBS- 4.29, 4-OTBS- 4.30, and 3,4-di-OTBS-derivative 4.31 was obtained and subsequently easily separated by flash chromatography (Scheme 4.12).
Scheme 4.12. Reaction of diol 4.2 with TBDMS-Cl
Subsequent treatment of 3-OTBS-derivative 4.29 with MsCl yielded the corresponding mesylate 4.32, subsequently deprotected by TBAF to the trans hydroxy mesylate 4.33 (Scheme 4.13). Base-catalyzed cyclization of 4.33 with t-BuOK afforded, in situ, the desired epoxide 4.1β, which, unexpectedly, turned out to be sufficiently stable to be isolated to the point that, by performing the cyclization reaction in CD3CN, the full characterization of 4.1β by 1H NMR and 13C NMR was possible. AcOH Me4NBH(OAc)3 CH3COCH3 N N Cbz O HO BnO O BnO OH BnO O B OAc OAc H N Cbz OH HO BnO 4.16 4.28 4.2 N BnO Cbz OH HO N BnO Cbz OTBS HO TBDMS-Cl DMF imidazole 4.2 4.29 N BnO Cbz OH TBSO 4.30 N BnO Cbz OTBS TBSO 4.31 40 : 23 : 37 (27%) (14%) (21%)
Scheme 4.13. Synthesis of epoxide 4.1β
The stability of epoxide 4.1β was almost surprising, considering that glycal-derived epoxides 2.1α and 2.1β previously prepared in our laboratory were decidedly unstable and could be obtained only
in situ. Evidently, the reduced electron-donating conjugative effect of the urethane moiety of 4.1β
compared with the same elettronic effect of the vinyl ether oxygen of 4.1β could be responsible of the observed stability of the N-Cbz substituted system by giving less electronic assistance to the oxirane opening process (Scheme 4.14).
Scheme 4.14. Resonance contribution in 4.1β and 2.1β
N BnO Cbz OTBS HO N BnO Cbz OTBS MsO MsCl/Py N BnO Cbz OH MsO 4.32 4.33 82% 75% 4.29 t-BuOK N BnO Cbz 4.1! O N BnO Cbz OH HO 4.2 TBDMS-Cl DMF imidazole TBAF THF 36% (1.5 equiv)TsCl Py t-BuOK MeCN N BnO Cbz OR TsO 4.34, R = H N BnO Cbz OTs HO MeCN 4.34-Ac, R = Ac 4.34-Al Py, Ac2O 90% decomposition product N O O OBn BnO N O OBn BnO N O OBn BnO O 4.1! O O BnO O BnO O 2.1! O
Considering that the silylation of the diol 4.2 was not regioselective, as an alternative, faster synthetic procedure to epoxide 4.1β, we treated the trans diol 4.2 with TsCl (1.5 equiv): the trans hydroxy tosylate 4.34 was regioselectively obtained as the only reaction product (Scheme 4.13). Unfortunately, in this reaction, the monotosylate 4.34 is obtained in an unsatisfactory yield, reasonably due to the concomitant formation of the very reactive and unstable regioisomeric allyl tosylate (4.34-Al) which rapidly decomposes in the reaction medium, determining the loss of the corresponding portion of the starting diol 4.2. In this framework, a further, clear demonstration of the instability of a mesyl group on C(3) carbon of these imino glycal system, was obtained by trying to mesylate (MsCl, 2 equiv, Py) the mono TBS-derivative 4.30 (Scheme 4.13), in which only the allyl hydroxy group is available for substitution. A reaction mixture was obtained containing only the excess of the MsCl without any trace of substitution product.
The trans hydroxy tosylate 4.34 was subsequently cyclyzed under basic conditions to epoxide 4.1β. The exact structure of the trans hydroxy tosylate 4.34 was determined by an 1H NMR study with decoupling and NOE experiments carried out on the corresponding acetyl derivative 4.34-Ac, and on the basis of conformational considerations (Scheme 4.15). Appropriate experiments indicated that there is NOE between the methyl group of the acetate and the –CH2 of the side chain, and between H(5) and the aromatic moiety of the tosyl group, whereas it is absent between the methyl group of the acetate and H(5) proton. Moreover, the unfavorable interaction present between the benziloxycarbonyl chain COOBn) of the urethane moiety and the side chain on the ring (-CH2OBn) present in the all-equatorial conformer 4-34-Ac’ makes the corresponding all-axial conformer 4.34-Ac’’, the only conformer actually present at the equilibrium. This conformational limitation together with the consideration that the acetyl group and the side chain in 4.34-Ac are necessarily in a cis relationship, as a consequence of the consolidated synthetic pathway, make 4.34-Ac, and thus 4.34 as the only structure consistent with the experimental data (1H NMR). The exact regio- and stereochemistry of acetate 4.34-Ac, and thus of 4.34, determines the exact structure of epoxide 4.1β, which is simply obtained by SN2-type cyclization of 4.34 under basic conditions.
Scheme 4.15. Conformational study of the acetate 4.34-Ac
NCbz OBn AcOO H5 N H5 BnO Cbz O 4.34-Ac' 4.34-Ac'' SO2 Me CH3 O SO2 Me
In order to increase the efficiency of the synthetic protocol leading to epoxide 4.1β from trans diol 4.2, other attemps to protect selectively the allylic C(3)-OH group of trans diol 4.2 were done. For example, the regioselective C(3) monosilylation by means of different silylating agent in the presence of different base, as TBDMSOTf, Py CH3CN; TBDMSiCl, NaH, THF; 2,6 lutidine, TBDMOTf, CH2Cl2, or the regioselective C(3) monoacetylation by Py, Ac2O or DMAP, Ac2O, CH2Cl2 protocols, were tried.8 However, in all cases no satisfactory results were obtained.
Following the retrosynthetic analysis initially formulated, for the synthesis of the diastereoisomeric epoxide 4.1α, we initially thought necessary to have the diastereoisomeric trans diol 4.3. On the basis of previous experiences from our laboratory on the corresponding glycal system (Chapter 2), we subjected epoxide 4.1β to the reaction with tetrabutylammonium trimethyl silanolate (Bu3MeN+Me3SiO),1 a hydroxide ion equivalent which, in our opinion, could led to the desired
trans diol 4.3 in a completely, regio- and sterelselective fashion. Unfortunately the reaction of
epoxide 4.1β under these conditions afforded only a complex reaction mixture, probably due to the saponification of the urethane moiety.
Also the use of an alternative, similar reagent, the commercially available tetrabutylammonium acetate (Bu3MeN+AcO), led to a complex reaction mixture, too, without any trace of the desired product or its derivative.
Scheme 4.16. Reaction of epoxides 4.1β with Bu4N+AcO- and Bu4N+Me3SiO
-As a consequence, an alternative synthetic protocol was envisaged for the synthesis of epoxide 4.1α (Scheme 4.17). Mesylate 4.35, obtained by mesylation (MsCl/Py) of α-hydroxy ketone 4.16, was partially isomerized by AcONa in refluxing MeCN for 30 hours to give a 4:6 mixture of epimer 4.36 and starting mesylate 4.35. Attemps to increase the amount of the epimer 4.36 by heating in a microwaves apparatus was not successful to indicate that the 6:4 ratio corresponded to the thermodynamic ratio between the two epimers.
It is worth to note that, definitely, the α-hydroxy ketone 4.16 resulted the common intermediate for the stereodivergent synthesis of both the desired epoxides 4.1α and 4.1β.
After flash chromatography, while the recovered starting mesylate 4.35 was recycled, the pure diastereoisomeric mesylate 4.36 was regio- and stereoselectively reduced by means of the NaBH4/CeCl3 protocol, under Luche’s conditions,9 with the exclusive formation of the trans hydroxy mesylate 4.37 (Scheme 4.17). Cyclization of 17 under basic conditions (t-BuOK) afforded
N BnO Cbz 4.1! O Bu4N+Me3SiO -or Bu4N-AcO -complex reaction mixture N BnO Cbz OH HO 4.3 no trace of
the desired epoxide 4.1α. Contrary of what observed in the case of epoxides 4.1β, the diastereoisomeric epoxide 4.1α turned out to be not sufficiently stable to be isolated and and/or characterized by 1H NMR spectrum: it could only be prepared, in situ, and left it to react immediately with a nucleophile.
Scheme 4.17. Stereoselective synthesis of epoxide 4.1α
As for the reduction of 4.36, the observed complete 1,2-regioselectivity is due to the presence of the Cerium salt, that, by favoring increased the polarization of the carbonyl group, guarantees the exclusive attack of the hydride ion on C(3) carbon. On the other hand, the observed complete stereoselectivity is a consequence of conformational and stereoelectronic factors. Actually in the mesyloxy ketone 4.36, which exists in the only conformer 4.36’ with the -CH2OBn group axial, a pseudoaxial attack of the hydride to give the trans hydroxy mesylate 4.37 is more favored with respect to a corresponding pseudoequatorial attack, in accordance with the common behavior of NaBH4, as a reducing agent (Scheme 4.18).9
Scheme 4.18. Pseudoaxial attack of the hydride ion to the conformer 4.36’
4.3. Addition reactions of epoxides 4.1α and 4.1β with O-nucleophiles
The regio- and stereochemical behavior of the two diastereoisomeric epoxides 4.1α and 4.1βin addition reactions of simple O-nucleophiles, as alcohols, was examined by means of the usual
protocol A and protocol B reaction conditions.
Under protocol A (alcohol as the solvent), the treatment of a MeOH solution of the trans hydroxy mesylate 4.37 with MeONa (1.1 equiv) afforded, through the intermediate formation of epoxide 4.1α, a 75:25 mixture of the anomeric methyl α-4.38α and β-O-glycoside 4.38β (Scheme 4.19).
N Cbz 4.36' BnO O MsO H-BH3 -4.37 Cl3Ce N BnO Cbz MsO O N BnO Cbz MsO O CeCl3 N BnO Cbz MsO OH N BnO Cbz O AcONa MeCN refluxing 4.36 t-BuOK 4.37 4.1! NaBH4 MeCN -40 ºC 88% 34% 97% N BnO Cbz HO O 4.35 4.16 Py, 0°C MsCl 6 : 4
Under the same conditions, epoxide 4.1β (from trans hydroxy tosylate 4.34) afforded an 85:15 mixture of the corresponding methyl β-4.39 and α-O-glycoside 4.39α. The pairs of glycosides 4.38β-4.38α and 4.39α-4.39β were separated by preparative TLC and structurally characterized (1H NMR, FTIR and appropriate NOE experiments, see Paragraph 4.4).
In both cases, the glycosylation reactions resulted completely 1,4-regioselective, with exclusive attack of the nucleophile (MeOH) on the C(1) carbon of the allyl system, while the attack of MeOH on the C(3) was not observed at all.
The reactions were not completely stereoselective: a mixture of α- and β-glycosides were obtained in both cases. However it could be observed that the predominant methyl O-glycoside had the same configuration as the starting epoxide: α-O-glycoside 4.38α from 4.1α and β-O-glycoside 4.39β from 4.1β.
Scheme 4.19. Regio- and stereoselectivity of the addition of MeOH to epoxides 4.1α and 4.1β (protocol A) When the same reactions were carried out under protocol B [MeONa (1.1 equiv), MeOH (3 equiv) in anhydrous MeCN], the methanolysis of epoxides 4.1α and 4.1β became completely regio- and stereoselective, with exclusive formation of the α-O-glycoside 4.38α from 4.1α and the β-O-glycoside 4.39β from 4.1β (Scheme 4.20).
Other O-glycosyl acceptors, such as EtOH, i-PrOH and t-BuOH were utilized in the reactions with epoxides 4.1α and 4.1β. Except for the reaction of 4.1β with t-BuOH, which led to a complex reaction mixture under protocol A, the reactions of epoxides 4.1α and 4.1β were completely regio- and stereoselective with exclusive formation of the corresponding alkyl O-glycoside (4.40-4.42α from 4.1α and 4.43β-4.44β from 4.1β) with configuration the same as the starting epoxide (Scheme 4.21). N BnO Cbz O N BnO Cbz OH MsO N BnO Cbz HO OMe MeONa (1.1 equiv) 4.1! 4.38! (75%) MeOH N BnO Cbz HO OMe 4.38" (25%) + 4.37 N BnO Cbz O N BnO Cbz OH TsO N BnO Cbz HO OMe MeONa (1.1 equiv) 4.1" 4.39" (85%) MeOH N BnO Cbz HO OMe 4.39! (15%) + 4.34
Scheme 4.20. Regio- and stereoselectivity of the addition of MeOH to epoxides 4.1α and 4.1β (protocol B) The complete 1,4-regio- and stereoselectivity observed in the reactions of 4.1α and 4.1β with alcohols was rationalized by hydrogen bonding between the oxirane oxygen and the O-nucleophile. Considering that theoretical calculations indicated that epoxides 4.1α and 4.1β exist as the only corresponding conformer 4.1α’ and 4.1β’ with the benzyloxymethyl group axial (Scheme 4.22, see also Paragraph 4.4), the nucleophile (alcohol) is guided to the α-face in the case of 4.1α, reacting through conformer 4.1α’, or the β-face in the case of 4.1β, reacting through conformer 4.1β’, respectively, suitably arranged for an entropically favored attack on the corresponding C(1) carbon, from the same side as the oxirane ring: an α-directed attack in 4.1α’ (route a, a pseuodoequatorial attack) and a β-directed attack in 4.1β’ (route b, a pseudoaxial attack) (Scheme 4.22).
Scheme 4.21. Regio- and stereoselectivity of the addition of EtOH, i-PrOH and t-BuOH (protocol A) to epoxides 4.1α and 4.1β N BnO Cbz O N BnO Cbz OH MsO N BnO Cbz HO OMe MeONa (1.1 equiv) 4.1! 4.38! MeCN 4.37 N BnO Cbz O N BnO Cbz OH TsO MeONa (1.1 equiv) 4.1" MeCN N BnO Cbz HO OMe 4.39" 4.34 MeOH MeOH N BnO Cbz O N BnO Cbz OH MsO N BnO Cbz HO OR t-BuOK (1 equiv) 4.1! ROH 4.37 N BnO Cbz O N BnO Cbz OH TsO t-BuOK (1 equiv) 4.1" ROH N BnO Cbz HO OR 4.34 4.40, R = Et (>99%) 4.41, R = i-Pr (>99%) 4.42, R = t-Bu (>99%) 4.43, R = Et (>99%) 4.44, R = i-Pr (>99%)
In the framework of the proposed rationalization, only in an excess of the more nucleophilic MeOH (protocol A), can a configurationally opposite attack by the free, non-coordinated nucleophile (route c and d, R = Me) become competitive, as was experimentally observed both with epoxides 4.1α and 4.1β. In all other cases, the reduced nucleophilicity of the alcohol, probably associated with a more marked tendency of the imino glycal system of 4.1α and 4.1β to coordination, is sufficient to afford a completely stereoselective result, even in the presence of a large amount of the nucleophile (protocol A)
Scheme 4.22. Rationalization of the selectivity in the glycosylation of alcohols by allyl epoxides 4.1α and 4.1β In our opinion this result was quite interesting considering that we observed a pseudoaxial attack in the case of epoxide 4.1β and a pseudoequatorial attack in the case of epoxide 4.1 α. This meant that the facial selectivity of this process was indipendent of the nature (pseudoaxial or pseudoequatorial) of the nucleophilic attack, as it is reported in literature for imino glycal systems reacting under Lewis acid mediated Ferrier reaction conditions (Scheme 4.3 and Chapter 3). While in literature is reported a sort of product-dependent selectivity, which led principally to the β-anomer, due to a preferential pseudoaxial attack of the nucleophile, the stereochemical outcome in our process of O-glycosylation depended only on the configuration of the epoxide and the correlated direction of the oxirane oxygen-alcohol coordination, in a sort of a substrate-dependent selectivity to the point that we observed α-anomers for epoxides 4.1α and β-anomers for epoxides 4.1β.
A simple confirmation of how, at least in the reaction of 4.1α with alcohols, the oxirane oxygen-alcohol coordination and related nucleophilic attack goes in the opposite way to the natural tendency of the imino glycal system to undergo preferential pseudoaxial nucleophilic attack at
N O OBn BnO O a H O R N BnO O O R H O OBn 1 b 4.1!' O R H O R H c d 4.38",4.40"-4.42" 4.39!, 4.43!, 4.44! N BnO Cbz HO OR N BnO Cbz HO OR b
R = Me, Et, i-Pr, t-Bu
4.1" 4.1! N BnO Cbz N BnO Cbz a O O 4.1"' "-O-glycosides !-O-glycosides
C(1), is obtained by treating the methyl a-O-glycoside 4.38α (the substrate-dependent selectivity anomer from 4.1α and MeOH) with a 10-5 N H
2SO4-MeOH solution. A clean reaction occurs with complete epimerization of 4.38α to 4.38β, the product-dependent selectivity anomer, by pseodoaxial attack of MeOH on the intermediate protonated species 4.45 (Scheme 4.23). Alternatively the intermediate occurrence of an N-acyliminium carbocation, pseodoaxially attacked by MeOH, cannot be excluded. Analogously, under the same reaction conditions, methyl O-glycoside 4.39α completely epimerizes to 4.39β, whereas 4.38β and 4.39β are stable.
Scheme 4.23. Epimerization of 4.38α to 4.38β and 4.39α to 4.39β
In our opinion, the present results offer the best demonstration of the occurrence and importance of an oxirane oxygen-alcohol coordination in directing the stereoselectivity of the addition reaction of alcohols to the present imino glycal- and related glycal-derived allyl epoxides.
4.4. Structures, conformations and configurations
It has to be underlined that the characterization of the all compounds mentioned by NMR spectroscopy was hampered by the presence of rotamers about the N-Cbz bond. For this reason it was necessary the use of variable temperature NMR spectroscopy (CD3CN as solvent) to complete structural assignments (See Experimental, Paragraph 4.5).
First of all, in order to have information on the conformational equilibrium on epoxides 4.1α and 4.1β, a theoretical conformational study was carried out, for simplicity, on the less computing demanding epoxides 4.1α-Me and 4.1β-Me in which the original side chain (-CH2OBn) present in 4.1α and 4.1β is advantageously substituted with a simple methyl group which has the same steric hindrance but it allows simpler calculations.
N BnO O OBn 4.38! 4.39" (>99%) N BnO Cbz HO OMe OMe MeOH H2SO4 N BnO O OBn O H Me + O H Me -MeOH 4.45 pseudoaxial attack HO HO N BnO Cbz HO OMe 4.39! N BnO Cbz HO OMe 4.39" 10-5 N H 2SO4 MeOH
Similar analysis were, then, conducted on methyl glycosides 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe structurally related to methyl glycosides 4.38α, 4.38β, 4.39α, and 4.39β, respectively (Scheme 4.24), in order to have conformational information not only on imino glycal systems, but also to the obtained 2,3-unsaturated glycosydes.
Scheme 4.24. Compounds used for computazional analysis
Quantum-chemical computations on epoxides 4.1α-Me and 4.1β-Me and methyl glycosides 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe were carried out with the Gaussian 03 set of programs. The geometrical optimizations of stationary points along the potential energy hypersurface (PES) were performed in vacuum at density functional theory level (B3LYP)10 with the 6-31+G(d) basis set.11 Berny analytical gradient optimization routines were used for optimization. All stationary points were characterized by frequency calculations in order to verify that minima have no imaginary frequency. In order to quantify the intramolecular hydrogen bond force, in the case of compounds 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe single point energy and the NBO analysis were performed using the perturbative method MP212 with 6-31+G(d) basis set, on the previously DFT optimized structures. Enthalpy, entropy, ZPE and free energy values were calculated using the standard expressions for an ideal gas in the canonical ensemble at 298.15 K and 1 atm pressure.13 The calculated free energy for all structures, were corrected by adding the ZPE and taking away E-TS rotational and translational terms. These
N BnO HO OMe N BnO HO OMe N BnO HO OMe 4.39! N BnO HO OMe 4.39" O OBn O OBn O OBn O OBn N MeO HO OMe O OMe 4.38"-OMe N MeO HO OMe O OMe N MeO HO OMe O OMe 439!-OMe N MeO HO OMe O OMe 4.38!-OMe H1 H1 H1 H1 H2 H2 H2 H2 1 2 3 4 5 6 4.38" 4.38! 4.39"-OMe N BnO O OBn O N BnO O OBn O N Me O OMe O 4.1!-Me N Me O OMe O 1 2 3 4 5 4.1! 4.1" 4.1"-Me
thermal and entropic corrections were calculated according to Ben-Naim scheme for thermochemical properties in the liquid phase.14
The results obtained revealed that epoxides 4.1α-Me and 4.1β-Me exist only in the ring conformation 4.1aα-Me and 4.1aβ-Me, respectively, characterized by the methyl group axial (Fig. 4.1).
4.1aα-Me-A 4.1aα-Me-B
4.1aβ-Me-A 4.1aβ-Me-B
Fig. 4.1. Rotameric population around the N-CO bond in conformers 4.1aα-Me and 4.1aβ-Me of epoxides
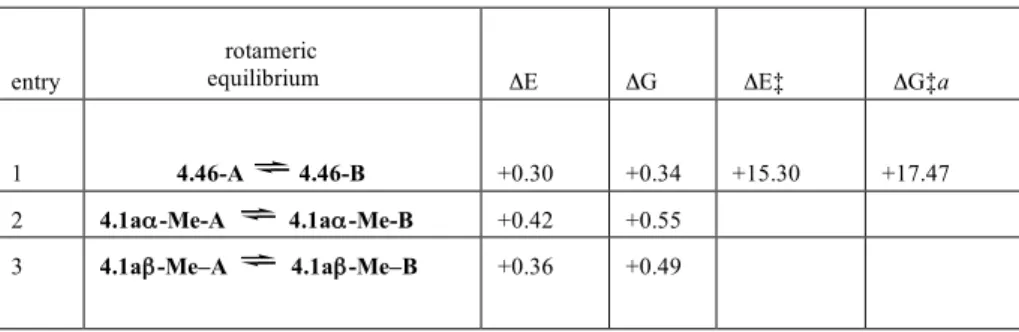
TABLE 4.1. DE, DG, DE‡ and DG‡ (kcal mol-1), for the rotameric equlibrium around the N-CO bond.
B3LYP/6-31+G(d)//B3LYP/6-31+G(d) results
entry
rotameric
equilibrium ΔE ΔG ΔE‡ ΔG‡a
1 4.46-A 4.46-B +0.30 +0.34 +15.30 +17.47
2 4.1aα-Me-A 4.1aα-Me-B +0.42 +0.55
3 4.1aβ-Me–A 4.1aβ-Me–B +0.36 +0.49
a The calculated free energy values in vacuum were corrected by adding the ZPE and taking away E-TS rotational and translational terms.
For each conformer 4.1aα-Me and 4.1aβ-Me, also the rotameric population around the N-CO bond was examined. The results indicated that, among the two optimized structures 4.1aα-Me-A and 4.1aα-Me-B for 4.1aα-Me, and 4.1aβ-Me–A and 4.1aβ-Me-B for 4.1aβ-Me [in rotamer A the C=O bond is proximal to C(5) ring carbon, whereas in rotamer B the C=O bond is proximal to C(1) ring carbon], 4.1aα-Me-A and 4.1aβ-Me–A are the slightly preferred ones (about 70%) (Fig. 4.1). In Table 4.1 are also reported the values for the rotameric equilibrium around the N-CO bond. The rotational barrier around the N-CO bond was calculated on the simplified model 4.46.15 The DG‡ value (about 17 Kcal/mole, Table 4.1, entry 1) is consistent with the signals splitting observed in the 1H NMR spectra, carried out at room temperature, of almost all the N-COOBn functional group-containing compounds described previous in the text.
The results obtained with epoxides 4.1aα-Me and 4.1aβ-Me can reasonably be extended to epoxides 4.1α and 4.1β and allow us to consider that our epoxides are exisiting only in the corresponding conformation with the methyl axial, as previously mentioned in this Chapter. The conformation analysis, which clearly could also be reasonalbly extended to the other imino glycal systems stated in the text, as α-hydroxy ketone 4.16, trans diol 4.2, tosylate 4.24.
The results obtained with the simplified model compounds compounds 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe, structurally related to the addition products, the methyl glycosides 4.38α, 4.38β, 4.39α, and 4.39β indicated that the corresponding conformer with the -CH2OMe
N
O OMe
Me
group axial, 4.38αa-OMe, 4.38βa-OMe, 4.39αa-OMe, and 4.39βa-OMe, respectively, is largely the preferred or almost the only conformer present (Fig. 4.2 and Table 4.2).
The results obtained with the model compounds 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe can reasonably be extended to the addition products, the methyl glycosides 4.38α, 4.38β, 4.39α, and 4.39β and allow us to consider the methyl glycosides 4.38α, 4.38β, 4.39α and 4.39β as existing only in the corresponding with the -CH2OBn substituent group axial
4.38αa-OMe 4.38αb-OMe
4.38βa-OMe 4.38βb-OMe
4.39βa-OMe 4.39βb-OMe
Fig. 4.2. Preferred conformer in model compounds 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe, corresponding to the addition products 4.38α, 4.38β, 4.39α, and 4.39β, with indication of the possible
intramolecular OH….O hydrogen bond.
In Table 4.2 are also reported the values for the conformation equilibrium in the model compounds 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe
entry
conformer
equilibrium DEa DEb DGb,c
conformers ratio (a:b)b 1 4.38αb-OMe 4.38αa-OMe -3.83 -1.81 -1.43 91.8:8.2 2 4.38βb-OMe 4.38αa-OMe -1.87 -1.34 -2.26 97.8:2.2 3 4.39αb-OMe 4.39αa-OMe -2.77 -2.95 -2.97 99.3:0.7 4 4.39βb-OMe 4.39αa-OMe -3.60 -4.09 -5.08 99.98:0.02
a MP2 results. b B3LYP results. c The calculated free energy values in vacuum were corrected by
adding the ZPE and taking away E-TS rotational and translational terms.
The exact configuration of methyl O-glycosides 4.38α, 4.38β, 4.39α, and 4.39β were determined by means of their 1H NMR spectra, appropriate NOE experiments and IR spectra.
First of all the 1,4 addition products were identified considering the diagnostic presence of the two vinyl protons H(2) and H(3) at around δ= 5.95-6.30 ppm, and the absence of the vinylic proton H(1), usually present in the starting glycal system at around δ = 6.75-6.95 ppm. For the determination of the configuration α or β on the C(1), the configuration of the β-product 4.39β was clarified by NOE experiments which had clearly indicated NOE between H(1) and H(5). This
indicates that the two protons are necessarily in a cis relationship, result that is compatible only with a β-anomer structure. As a consequence, the diastereoisomeric anomer 4.39α corresponds to a configuration of α-anomer.
For the determination of the configuration of the methyl-glycosides 4.38α and 4.38β, while NOE experiments did not allow us to determinate the exact configurations, the examination of OH-stretching band of these compounds in dilute CCl4 solution in the 3µ range resulted very useful. The methyl glycosides 4.38α, 4.38β, 4.39α and 4.39β, in fact, indicated that a strong intramolecular hydrogen bond is present in methyl glycosides 4.38α (cis 1,4-OH---O), 4.39α (cis 1,3-OH---O) and 4.39β (cis 1,4-OH---O), whereas no hydrogen bond is present in 4.38β (only free OH is present) (Table 4.3). These experimental results are in accordance with the theoretical conformational study carried out on the model compounds 4.38α-OMe, 4.38β-OMe, 4.39α-OMe, and 4.39β-OMe with examination of the possible intramolecular OH….O hydrogen bond (Figure 4.2) and allowed us to give the right attribution to the O-glycosides compounds.
TABLE 4.3. Spectroscopic data for methyl glycosides 4.38α, 4.38β, 4.39α and 4.39β
IR (CCl4) (OH stretching, cm-1) 1H NMR
(CD3CN) δ
compound cis 1,4-OH---O cis 1,3-OH---O trans 1,3-OH---O free OH H1(J, Hz)
27 3489a 3597b 5.41 (4.4)c
28 3610a 5.50d
29 3443a 5.52 (4.2)c
30 3495a 5.49d
a Strong band. b Weak band. c Doublet. d unresolved singlet
.
4.5. ExperimentalGeneral Procedures. All reactions were performed in flame-dried modified Schlenk (Kjeldahl shape) flasks fitted with a glass stopper or rubber septa under a positive pressure of argon. Air- and moisture-sensitive liquids and solutions were transferred via a syringe. Organic solutions were dried on MgSO4 and concentrated by a rotary evaporator below 40°C at ca. 25 Torr. Flash column chromatography was performed employing 230-400 mesh silica gel (Macherey-Nagel). Analytical TLC was performed on Alugram SIL G/UV254 silica gel sheets (Macherey-Nagel) with detection by 0.5% phosphomolybdic acid solution in 95% EtOH.
Materials. Glacial AcOH, lithium 2-thienylcyanocuprate in diethyl ether, 4-methoxy pyridine, CeCl3.7H2O, MeONa, TBDMS-Cl, 1.0 M TBAF in THF, NaBH4, EtOH (absolute), 1.6 M BuLi in hexane, were purchased from Aldrich and used without purification. Bu3SnH, MsCl, TsCl,
Me4NBH(OAc)3, Pb(OAc)4 stabilized with AcOH (15%), benzyl chloroformate, anhydrous MeCN over molecular sieves, anhydrous pyridine over molecular sieves, anhydrous DMF over molecular sieves, and t-BuOK were purchased from Fluka and used without purification. Pure grade for HPLC acetone, MeOH and i-PrOH were used without any other purification. Toluene, Et2O and THF were distilled from sodium/benzophenone, t-BuOH was distilled from sodium, diisopropyl amine (DIPA) was distilled from calcium hydride and commercial chloromethyl benzyl ether (Fluka) was distilled under vacuum (1.0 mm/Hg)
Instrumentation. Infrared (IR) spectra were obtained using a FTIR spectrophotometer. Data are presented as frequency of absorption (cm-1). Proton and carbon-13 nuclear magnetic resonance (1H NMR and 13C NMR) spectra were recorded at 250 and 62.5 MHz, respectively; chemical shifts are expressed in parts per million (δ scale) downfield from tetramethylsilane and refer to residual protium in the NMR solvent (CHCl3: δ 7.26; CD3CN: δ 1.94). Data are presented as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, m=multiplet and/or multiple resonances), integration, coupling constant in Hertz (Hz). Resonances associated with amide rotamers are indicated with *. Melting points are uncorrected.
(Benzyloxymethyl)tributylstannane (4.19).
Following a previously reported procedure,4 1.6 M n-BuLi in hexane (18 mL, 28.8 mmol) was added dropwise to a stirred solution of diisopropylamine (4.4 mL, 31.6 mmol) in anhydrous THF (58 mL) at 0°C under dry argon. After 30 min, Bu3SnH (7.7 mL, 28.7 mmol) was added and the resulting greenish solution was stirred for 20 min, then cooled to -78°C and treated with freshly distilled chloromethyl benzyl ether (4.0 mL, 28.7 mmol). After 20 min, the reaction was warmed to room temperature and stirred for 2 h. Dilution with hexane and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude reaction product (11.60 g) consisting of 4.19 (1H NMR), which was subjected to flash chromatography. Elution with an 8:2 hexane/CH2Cl2 mixture yielded 4.194a (9.91 g, 84% yield), as an oil: Rf = 0.32 (8:2 hexane/CH2Cl2); FTIR (neat film) ν 1455, 1375, 1085, 1065, 730, 695 cm-1. 1H NMR (CDCl
3) δ 7.20-7.36 (m, 5H), 4.43 (s, 2H), 3.77 (d, 1H, J = 7.4 Hz), 3.73 (d, 1H, J = 7.4 Hz), 1.42-1.74 (m, 6H), 1.20-1.42 (m, 6H), 0.73-1.10 (m, 15H).
(±)-1-(Benzyloxycarbonyl)-2-(benzyloxymethyl)-2,3-dihydro-4-pyridone (14).3
1.6 M n-BuLi in hexane (1.69 mL, 2.70 mmol) was added at -78°C to a stirred solution of (benzyloxymethyl)tributylstannane (4.19) (1.10 g, 2.70 mmol) in anhydrous THF (18 mL). After stirring for 30 min at the same temperature, a 0.25 M solution of lithium 2-thienylcyanocuprate in diethyl ether (10.8 mL, 2.70 mmol) was added dropwise. The solution was stirred for additional 40 min at -78°C (Solution A). In a separate flask, benzyl chloroformate (0.25 mL, 1.80 mmol) in Bu Sn Bu CH2OBn Bu 4.19 N BnO Cbz O 4.14
anhydrous THF (4.4 mL) was added to a solution of 4-methoxy pyridine (0.18 mL, 1.80 mmol) in anhydrous THF (20 mL) at -25°C (Solution B). The reaction mixture was stirred at -25°C for 1 h and then cooled to -78°C. The Solution A was transferred dropwise by a double-tipped needle to the flask containing the Solution B and stirring was continued for an additional 4 h at -78°C. The cooling bath was removed and saturated aqueous oxalic acid (30 mL) was added. The solution was warmed to room temperature and stirring was continued for 12 h. Dilution with Et2O and evaporation of the washed (saturated aqueous NaHCO3, saturated aqueous NaCl) organic solution afforded a crude reaction product (2.20 g) consisting of 4.14 (1H NMR), which was subjected to flash chromatography. Elution with an 8:2 hexane/AcOEt mixture yielded 4.143 (0.41 g, 65% yield), pure as a liquid: Rf = 0.09 (8:2 hexane/AcOEt). FTIR (neat film) ν 1728, 1668, 1606, 1380, 1350, 1280, 1210, 1180, 1050 cm-1.1H NMR (CDCl3) δ 7.78 (d, 1H, J = 8.2 Hz), 7.19-7.42 (m, 10H), 5.29 (d, 1H, J = 8.2 Hz), 5.23 (s, 2H), 4.73-4.91 (m, 1H), 4.51 (d, 1H, J = 12.0 Hz), 4.42 (d, 1H, J = 12.0 Hz), 3.50 (dd, 1H, J = 9.6, 6.1 Hz), 3.40 (dd, 1H, J = 9.6, 6.6 Hz), 2.84 (dd, 1H, J = 16.9, 7.1 Hz), 2.61 (d, 1H, J = 16.9 Hz).
(±)-3-Acetoxy-1-(benzyloxycarbonyl)-2-(benzyloxymethyl)-2,3-dihydro-4-pyridone (4.15).3 Pb(OAc)4 (1.45 g, 3.30 mmol) stabilized with AcOH (15%) was added to a stirred solution of 4.14 (0.71 g, 2.02 mmol) in anhydrous toluene (70 mL), and the reaction mixture was refluxed for 3 h. Additional Pb(OAc)4 (1.45 g, 3.30 mmol) was added and refluxing was resumed for another 18 h. After cooling to room temperature, the solution was filtered through celite with CH2Cl2. Evaporation of the washed (saturated aqueous NaHCO3 with ice, saturated aqueous NaCl) organic solution afforded a crude reaction product (1.60 g) consisting of 4.15 (1H NMR), which was subjected to flash chromatography. Elution with a 7:2:1 hexane/CH2Cl2/AcOEt mixture yielded 4.153 (0.51 g, 62% yield), as a colourness oil: Rf = 0.26 (8:2 hexane/AcOEt); FTIR (neat film) ν 3032, 2867, 1733, 1678, 1605, 1386, 1318, 1266, 1210, 1117, 1030 cm-1. 1H NMR (CDCl 3) δ 7.90 (d, 1H, J = 8.4 Hz), 7.10-7.48 (m, 10H), 5.38 (dd, 1H, J = 8.4, 1.1 Hz), 5.27 (d, 1H, J = 12.2 Hz), 5.21 (d, 1H, J = 12.2 Hz), 5.16-5.31 (m, 1H) 4.72-4.82 (m, 1H), 4.46 (s, 2H), 3.58-3.72 (m, 2H), 2.08 (s, 3H). 13C NMR (CDCl 3) δ 186.7 169.6, 152.7, 143.0, 137.4, 134.8, 129.1, 128.9, 128.6, 128.0, 127.9, 127.6, 105.6, 73.5, 70.1, 69.5, 67.5, 57.6, 21.1. (±)-1-(Benzyloxycarbonyl)-2-(benzyloxymethyl)-3-hydroxy-2,3-dihydro-4-pyridone (4.16).3 10% Aqueous HCl (7 mL) was added to a solution of 4.15 (0.55 g, 1.34 mmol) in absolute EtOH (37 mL), and the reaction mixture was stirred 18 h at 50°C. After cooling to room temperature, dilution with Et2O and evaporation of the washed (saturated aqueous NaHCO3 with ice, saturated aqueous NaCl) organic solution afforded a crude reaction product (0.46 g, 94% yield) consisting of 4.163 N BnO Cbz O AcO 4.15 N BnO Cbz O HO 4.16
practically pure: Rf = 0.09 (7:3 hexane/AcOEt). FTIR (neat film) ν 3373, 1728, 1668, 1601, 1211, 1161, 1028 cm-1. 1H NMR (CDCl 3) δ 7.90 (d, 1H, J = 8.5 Hz), 7.14-7.49 (m, 10H), 5.32 (d, 1H, J = 8.5 Hz), 5.23 (s, 2H), 4.65-4.81 (m, 1H), 4.44 (s, 2H), 4.07-4.15 (m, 1H), 3.56-3.70 (m, 2H). 13C NMR (CDCl3) δ 192.0, 152.7, 142.9, 137.6, 134.9, 129.0, 128.9, 128.6, 128.0, 127.7, 104.2, 73.6, 70.0, 69.5, 67.3, 59.3. (±)-6-O-Benzyl-N-(benzyloxycarbonyl)-imino glucal (4.2).3
Glacial acetic acid (1 mL, 17.28 mmol) was added to a solution of Me4NBH(OAc)3 (2.27 g, 8.63 mmol) in freshly distilled acetone (47 mL), at room temperature. After stirring for 15 min, a solution of 4.16 (0.396 g, 1.08 mmol) in freshly distilled acetone (4 mL) was added dropwise. After stirring for 50 min at room temperature, the mixture was quenched with saturated aqueous ammonium chloride, and one half of the acetone was removed in vacuo. Dilution with Et2O and evaporation of the washed (saturated aqueous Na2CO3, saturated aqueous NaCl) organic solution afforded a crude reaction product (0.403 g) consisting of diol 4.2, which was subjected to flash chromatography. Elution with hexane/AcOEt mixtures from 30:70 to 70:30 yielded 4.23 (0.28 g, 72% yield), pure as a white solid, mp 50-51°C; Rf = 0.10 (1:1 hexane/AcOEt); FTIR (nujol) ν 3412, 1710, 1652, 1397, 1341, 1027 cm-1. 1H NMR (CD 3CN) δ 7.23-7.37 (m, 10H), 6.90 (d, 1H, J = 8.4 Hz), 5.16 (s, 2H), 4.90-5.08 (m, 1H), 4.37-4.55 (m, 3H), 3.92-4.07 (m, 1H), 3.72-3.80 (m, 1H), 3.71 (dd, J = 10.2, 5.9 Hz, 1H), 3.65 (dd, J = 10.2, 6.2 Hz, 1H), 3.40-3.53 (m, 1H), 3.15 (d, 1H, J = 5.2 Hz). 13C NMR (CD3CN) δ 154.2, 139.3, 137.5, 129.4, 129.2, 129.1, 128,8, 125.8, 106.9, 73.5, 70.2, 68.6, 68.3, 65.6, 56.6.
Reaction of trans diol 4.2 with TBDMS-Cl
A solution of diol 4.2 (0.11 g, 0.30 mmol), in anhydrous DMF (1 mL) containing imidazole (0.041 g, 0.60 mmol), was treated at 0°C with TBDMS-Cl (0.050 g, 0.33 mmol). After 18 h stirring at room temperature, dilution with Et2O and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude reaction product (0.160 g) consisting of a 40:23:37 mixture of 3-OTBS derivative 4.29, 4-OTBS derivative 4.30, and 3,4-di-OTBS derivative 4.31 (1H NMR), which was subjected to flash chromatography. Elution with a 3:6:1 CH2Cl2/hexane/AcOEt afforded 3-OTBS derivative 4.29 (0.040 g, 27% yield), 4-OTBS derivative 4.30 (0.020 g, 14% yield) and 3,4-di-OTBS derivative 4.31 (0.038 g, 21% yield). N BnO Cbz HO OH 4.2 N BnO Cbz HO OTBS 4.29 N BnO Cbz TBSO OH 4.30 N BnO Cbz TBSO OTBS 4.31
(±)-6-O-Benzyl-3-O-(t-butyldimethylsilyl)-N-(benzyloxycarbonyl)-imino glucal (4.29), a liquid: Rf = 0.14 (3:6:1 CH2Cl2/hexane/AcOEt); FTIR (neat film) ν 3448, 1710, 1255, 1068 cm-1.1H NMR (CD3CN) δ 7.18-7.46 (m, 10H), 6.86-6.95 (m, 1H), 5.15 (bs, 2H), 4.82-5.08 (m, 1H), 4.37-4.53 (m, 3H), 3.84-4.02 (m, 1H), 3.81-3.84 (m, 1H), 3.56-3.75 (m, 2H), 3.13-3.24 (m, 1H), 0.89 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H). 13C NMR (CD3CN) δ 149.8, 139.0, 134.6, 129.4, 129.2, 129.0, 128.8, 128.4, 128.3, 125.1, 106.7, 73.3, 69.5, 68.3, 66.9, 66.1, 56.1, 26.1, 18.5, -4.4, -4.8. Anal. Calcd for C27H37NO5Si: C 67.05; H 7.71; N 2.90. Found: C 66.71; H, 7.52; N, 2.69.
(±)-6-O-Benzyl-4-O-(t-butyldimethylsilyl)-N-(benzyloxycarbonyl)-imino glucal (4.30), a liquid: Rf = 0.25 (3:6:1 CH2Cl2/hexane/AcOEt); FTIR (neat film) ν 3452, 1712, 1255, 1070 cm-1. 1H NMR (CD3CN) δ 7.27-7.35 (m, 10H), 6.86-6.93 (m, 1H), 5.12-5.27 (m, 2H), 4.93-5.06 (m, 1H), 4.42-4.55 (m, 3H), 4.10-4.17 (m, 1H), 3.62-3.75 (m, 3H), 3.39-3.48 (m, 1H), 0.84 (s, 9H), 0.09 (s, 6H). 13C NMR (CD
3CN) δ 148.6, 145.6, 139.6, 129.5, 129.2, 129.0, 128.7, 128.4, 125.7, 106.9, 73.5, 70.8, 68.3, 65.9, 57.5, 56.6, 25.8, 18.5, -4.6, -4.8. Anal. Calcd for C27H37NO5Si: C 67.05; H 7.71; N 2.90. Found: C 66.84; H, 7.62; N, 2.65.
(±)-6-O-Benzyl-3,4-O-(di-t-butyldimethylsilyl)-N-(benzyloxycarbonyl)-imino glucal (4.31), a liquid: Rf = 0.62 (3:6:1 CH2Cl2/hexane/AcOEt); FTIR (neat film) ν 1714, 1255, 1074 cm-1. 1H NMR (CD3CN) δ 7.24-7.40 (m, 10H), [6.83* (d, J= 8.5 Hz) and 6.90* (d, J= 8.6 Hz), 1H], [5.21* (d, J= 12.5 Hz) and 5.25* (d, J = 11.3 Hz), 1H], [5.11* (d, J= 12.5 Hz) and 5.13*(d, J = 11.3 Hz), 1H], [4.91*(ddd, J= 8.5, 4.8, 1.5 Hz) and 5.00*(ddd, J = 8.4, 4.7, 1.6 Hz), 1H], 4.42-4.58 (m, 2H), 4.43 (dd, 1H, J = 12.5 Hz, 6.0 Hz), [4.09*-4.12*(m) and 4.04*-4.07*(m), 1H], 3.80-3.82 (m, 1H), 3.62-3.67 (m, 2H), 0.87 (s, 9H), [0.83*(s) and 0.84*(s), 9H], [0.09*(s) and 0.10*(s), 6H], [0.08*(s) and 0.06*(s), 6H]. 13C NMR (CD 3CN) δ 154.2, 129.4, 129.3, 129.1, 128.9, 128.8, 128.3, 128.2, 128.1, 125.0*and 125.3*, 106.8*and 107.1*, 73.1*and 73.2*, 70.6*and 70.7*, 68.0*and 68.2*, 67.2*and 67.8*, 57.4*and 56.7*, 26.0, 25.1, 18.5, 18.4, -4.3, -4.5, -4.7, -4.8. Anal. Calcd for C33H51O5NSi2: C 66.29; H 8.60; N 2.34. Found: C 66.01; H, 8.24; N, 2.15.
(±)-6-O-Benzyl-3-O-(t-butyldimethylsilyl)-4-O-mesyl-N-(benzyloxycarbonyl)-imino glucal (4.32).
MsCl (0.035 g, 0.46 mmol) was added at 0° C to a solution of monosilyl derivative 4.29 (0.11 g, 0.23 mmol) in anhydrous pyridine (2 mL). After 4 h stirring at 0° C, dilution with Et2O and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude reaction product (0.065 g) consisting of mesylate 4.32 (1H NMR), which was subjected to flash chromatography. Elution with an 8:2 hexane/AcOEt mixture afforded mesylate 4.32 (0.106 g, 82% yield), pure as a liquid: Rf = 0.40 (8:2 hexane/AcOEt). FTIR (neat film) ν 1682, 1604, 1248, 1056 cm-1. 1H NMR (CDCl3) δ
N BnO Cbz MsO OTBS 4.32
7.10-7.42 (m, 10H), [7.01*(d, J= 9.6 Hz) and 6.89*(d, J= 9.6 Hz), 1H], [5.20* (d, J = 12.0 Hz) and 5.24*(d, J= 11.8 Hz), 1H], [5.14* (d, J= 12.0 Hz) and 5.17*(d, J= 11.8 Hz), 1H], 4.66-5.08 (m, 3H), [4.56*(d, J= 12.3 Hz) and 4.60* (d, J = 12.5 Hz), 1H], [4.36* (d, J= 12.3 Hz) and 4.48* (d, J = 12.5 Hz), 1H], 4.12-4.21 (m, 1H), 3.52-3.81 (m, 2H), [2.91*(s) and 2.98*(s), 3H], 0.85 (s, 9H), [0.08*(s) and 0.10*(s), 3H], 0.06 (s, 3H). Anal. Calcd for C28H39NO7SSi: C 59.87; H 7.00; N 2.49. Found: C 59.98; H, 7.09; N, 2.85.
(±)-6-O-Benzyl-4-O-mesyl-N-(benzyloxycarbonyl)-imino glucal (4.33).
1M TBAF in THF (0.070 mL) was added at 0° C to a solution of 4.32 (0.044 g, 0.070 mmol) in anhydrous THF (2.68 mL). After 1 h stirring at 0° C, dilution with Et2O and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude reaction product (0.040 g) consisting of hydroxy mesylate 4.33, which was subjected to flash chromatography. Elution with an 1:1 hexane/AcOEt mixture afforded hydroxy mesylate 4.33 (0.023 g, 75% yield), pure as a liquid: Rf = 0.22 (1:1 hexane/AcOEt); FTIR (neat film) ν 3455, 1712, 1654, 1396, 1338, 1195, 1091 cm-1.1H NMR (CDCl3) δ 7.12-7.46 (m, 10H), 6.98-7.19 (m, 1H), 4.93-5.25 (m, 3H), 4.36-4.72 (m, 3H), 3.99-4.20 (m, 2H), 3.69-3.94 (m, 2H), 3.02 (s, 3H). Anal. Calcd for C22H25NO7S: C 59.05; H 5.63; N 3.13. Found: C 59.23; H, 5.29; N, 2.98.
(±)-6-O-Benzyl-4-O-tosyl-N-(benzyloxycarbonyl)-imino glucal (4.34).
A solution of 4.2 (0.44 g, 1.22 mmol) in anhydrous pyridine (3 mL) was treated, at 0°C, with TsCl (0.35 g, 1.83 mmol) and the reaction mixture was stirred 2 day at room temperature. Dilution with CH2Cl2 and evaporation of the washed (saturated aqueous NaHCO3, saturated aqueous NaCl) organic solution afforded a complex crude reaction product (0.68 g) consisting of 4.34 and unidentified compounds, which was subjected to flash chromatography. Elution with a 4:4:2 CH2Cl2/hexane/AcOEt afforded hydroxy tosylate 4.34 (0.22 g, 36% yield), pure as a liquid: Rf = 0.42 (1:1 hexane/AcOEt). FTIR (neat film) ν 3421, 1716, 1653, 1456, 1261, 1095, 1030 cm-1.1H NMR (CD 3CN, 50°C) δ 7.80 (d, 2H, J = 8.4 Hz), 7.16-7.46 (m, 12H), 6.90 (d, 1H, J = 8.7 Hz), 5.13 (s, 2H), 4.95-5.09 (m, 1H), 4.86 (s, 1H), 4.25-4.54 (m, 1H), 4.37 (s, 2H), 3.87-4.02 (m, 1H), 3.71 (dd, 1H, J = 10.0, 6.2 Hz), 3.64-3.78 (m, 1H), 3.54 (dd, 1H, J = 10.0 , 6.0 Hz), 2.38 (s, 3H). 13C NMR (CDCl3) δ 152.5, 145.3, 136.6, 135.6, 133.4, 130.2, 127.8, 128.7, 128.6, 128.2, 127.9, 127.8, 103.8*,and 104.2*, 78.7* and 78.5*, 77.8, 73.6*and 73.8*, 68.3*and 68.4*, 62.1, 53.5* and 53.9*, 21.8. Anal. Calcd for C28H29NO7S: C 59.05; H 5.63; N 3.13. Found: C 58.71; H, 5.33; N, 2.98. N BnO Cbz MsO OH 4.33 N BnO Cbz TsO OH 4.34
(±)-3-O-Acetyl-6-O-benzyl-4-O-tosyl-N-(benzyloxycarbonyl)-imino glucal (4.34-Ac).
A solution of 4.34 (0.050 g, 0.095 mmol) in anhydrous pyridine (1 mL) was treated, at 0°C, with distilled Ac2O (0.5 mL) and the reaction mixture was stirred 18 h at room temperature. Dilution with Et2O and evaporation of the washed (saturated aqueous NaHCO3, saturated aqueous NaCl) organic solution afforded a crude reaction product (0.050 g, 90% yield) consisting of acetate 4.34-Ac (1H NMR and NOE), pure as a liquid: Rf = 0.44 (1:1 hexane/AcOEt). FTIR (neat film) ν 3429, 1714, 1651, 1597, 1496, 1338, 1224, 1114, 1018, 968 cm-1.1H NMR (CDCl3) δ 7.72-7.85 (m, 2H), 6.96-7.41 (m, 13H), [5.13*(s) and 5.15*(s), 2H], 5.03-5.08 (m, 1H), 4.78-5.09 (m, 2H), 4.45-4.64 (m, 1H), [4.34*(s) and 4.37*(s), 2H], 3.34-3.60 (m, 2H), [2.31*(s) and 2.36* (s), 3H], 1.88 (s, 3H). 13C NMR (CDCl
3) δ 169, 145.0, 130.0, 128.8, 128.7, 128.6, 128.24, 128.19, 127.9, 127.5, 99.4* and 99.7*, 72.9* and 73.1*, 68.5* and 68.7*, 65.5* and 65.8*, 64.6, 53.6, 52.7, 21.9, 21.0. Anal. Calcd for C30H31NO8S: C 63.70; H 5.52; N 2.48. Found: C 63.53; H, 5.19; N, 2.17. 1H NMR e 13
C NMR confirmation of the formation of epoxide 4.1β.
A solution of hydroxy tosylate 23 (0.015 g, 0.023 mmol) in CD3CN (0.75 mL) was treated, in a NMR tube at room temperature, with t-BuOK (2.8 mg, 0.024 mmol). After 1 h at the same temperature, the registered 1H NMR spectrum of the reaction mixture showed the presence of the pure epoxide 6: Rf = 0.30 (1:1 hexane/AcOEt). 1H NMR (CD 3CN) δ 7.20-7.50 (m, 10H), 6.88 (d, 1H, J = 8.3 Hz), 5.25 (dd, 1H, J = 8.3, 3.3 Hz), 5.14 (s, 2H), 4.55-4.68 (m, 1H), 4.52 (s, 2H), 3.65 (dd, 1H, J = 8.5, 4.9 Hz), 3.42-3.59 (m, 2H), 3.33 (t, 1H, J = 3.9 Hz). 13C NMR (CD 3CN) δ 154, 140, 137.0, 129.58, 129.44, 129.32, 129.11, 128.61, 128.55, 102.76, 73.72, 68.88, 68.22, 51.74, 48.3, 46.2. (±)-1-(Benzyloxycarbonyl)-2-(benzyloxymethyl)-3-mesyl-2,3-dihydro-4-pyridone (4.35). MsCl (0.11 mL, 1.46 mmol) was added at 0°C to a solution of hydroxy ketone 4.16 (0.27 g, 0.73 mmol) in anhydrous pyridine (2 mL). After 3 h stirring at 0°C, diluition with CH2Cl2 and evaporation of the washed (10% aqueous HCl and ice, saturated aqueous NaHCO3 and saturated aqueous NaCl) organic solution afforded a crude reaction product (0.32 g, 97% yield) consisting of mesyloxy ketone 4.16 pure as a liquid: Rf = 0.36 (2:1:7 hexane/i-Pr2O/CH2Cl2); FTIR (neat film) ν 1730, 1662, 1604, 1454 cm-1. 1H NMR (CD 3CN, 50°C) δ 7.98 (d, 1H, J = 8.2 Hz), 7.17-7.48 (m, 10H), 5.35 (d, 1H, J = 8.2 Hz), 5.27 (s, 2H), 4.84-5.00 (m, 2H), 4.49 (d, 1H, J = 11.5 Hz), 4.43 (d, 1H, J = 11.5 Hz), 3.60-3.81 (m, 2H), 3.07 (s, 3H). 13C NMR (CD 3CN) δ 185.2, 153.0, 145.0, 138.6, 136.2, 129.6, N BnO Cbz TsO OAc 4.34-Ac N BnO Cbz O 4.1! N BnO Cbz O MsO 4.35
129.3, 129.2, 128.7, 128.5, 105.0, 76.8, 73.9, 70.2, 67.8, 59.1, 39.1. Anal. Calcd for C22H23NO7S: C 59.32; H 5.20; N 3.14. Found: C 59.58; H, 5.13; N, 2.92.
Epimerization of mesyloxy ketone 4.35
A solution of mesyloxy ketone 4.35 (0.683 g, 1.53 mmol) in anhydrous MeCN (34 mL) was treated at room temperature with AcONa (0.252 g, 3.07 mmol) and the resulting reaction mixture was refluxed for 30h. After cooling, the organic solution was diluted with Et2O, filtered and evaporated to afford a crude reaction product (0.635 g, 93% yield) consisting of an almost 60:40 mixture of mesyloxy ketones 4.35 and 4.36 (1H NMR) which was subjected to flash chromatography. Elution with a 4:4:1 hexane/CH2Cl2/Et2O afforded pure mesyloxy ketone 4.35 (0.290 g, 42% yield) and 4.36 (0.231 g, 34% yield).
(±)-1-(Benzyloxycarbonyl)-2-(benzyloxymethyl)-3-mesyl-2,3-dihydro-4-pyridone (4.36) (0.231 g, 34% yield), pure as a liquid: Rf = 0.38 (2:1:7 hexane/i-Pr2O/CH2Cl2). FTIR (neat film) ν 1730, 1682, 1606, 1261, 1093, 1022 cm-1. 1H NMR (CD3CN) d 7.98 (d, 1H, J = 7.9 Hz), 7.20-7.54 (m, 10H), 5.55 (d, 1H, J = 7.9 Hz), 5.20-5.30 (m, 3H), 4.8-5.00 (m, 1H), 4.48 (d, 1H, J = 12.0 Hz), 4.43 (d, 1H, J = 12.0 Hz 3.9 (dd, 1H, J = 11.0, 5.0 Hz), 3.8 (dd, 1H, J = 11.0, 2.7 Hz), 3.23 (s, 3H). 13C NMR (CD3CN) d 188.0, 153.6, 144.1, 139.0, 129.5, 129.2, 128.4, 128.2, 104.7, 76.4, 73.8, 69.9, 67.5, 57.6, 39.1. Anal. Calcd for C22H23NO7S: C, 59.32; H, 5.20; H, 3.14. Found: C, 59.12; H, 4.83; N, 2.97.
(±)-6-O-Benzyl-4-O-mesyl-N-(benzyloxycarbonyl)-imino gulal (4.37) Solid CeCl3.7H2O (0.406 g, 1.09 mmol) was added to a solution of 4.36 (0.24 g, 0.54 mmol) in anhydrous MeOH (33 mL). After ten minutes, the resulting solution was treated at -40°C with NaBH4 (0.062 g, 1.64 mmol) and the reaction mixture was stirred 30 min at the same temperature. Dilution with Et2O and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude reaction product (0.237 g, 98% yield) consisting of hydroxy mesylate 4.37 (1H NMR) which was subjected to flash chromatography. Elution with 1:1 hexane/AcOEt afforded hydroxy mesylate 4.37 (0.212 g, 88% yield), pure as a liquid: Rf = 0.22 (1:1 hexane/AcOEt); FTIR (neat film) ν 3439, 1714, 1655, 1396, 1338, 1196, 1089 cm-1. 1H NMR (CDCl
3) δ 7.09-7.47 (m, 10H), 6.74-6.99 (m, 1H), 5.00-5.28 (m, 3H), 4.28-4.62 (m, 3H), 4.28-4.61 (m, 3H), 3.51-3.85 (m, 2H), 3.09 (s, 3H). 13C NMR (CDCl3) δ 153.0, 138.0, 135.6, 128.7, 128.5, 128.4, 128.3, 127.7, 127.4, 125.7, 106.0, 80.9, 73.2, 68.5, 66.3, 66.2, 54.4, 38.5. Anal. Calcd for C22H25NO7S: C, 59.05; H 5.63; N 3.13. Found: C, 58.75; H, 5.96; N, 3.48. N BnO Cbz O MsO 4.36 N BnO Cbz MsO OH 4.37
Reactions of epoxides 4.1α and 4.1β with O-Nucleophiles (protocol A) Reaction of epoxide 4.1β with MeOH
Typical Procedure. A solution of hydroxy tosylate 4.34 (0.10 g, 0.20 mmol) in anhydrous MeOH (4 mL) was treated with MeONa (0.012 g, 0.22 mmol) and the reaction mixture was stirred 18 h at room temperature. Dilution with AcOEt and evaporation of the washed (saturated aqueous NaCl) organic solution afforded a crude product (0.060 g, 78% yield) consisting of an 85:15 mixture of methyl β-glycoside 4.38β and methyl α-glycoside 4.38α (1H NMR) which was subjected to preparative TLC with a 4:6 hexane/AcOEt mixture, as the eluant. Extraction of the two most intense bands (the faster moving band contained 4.38β) afforded pure methyl glycosides 4.38β (0.043 g, 56% yield) and 4.38α (0.007 g, 9% yield).
Methyl 6-(O-benzyl)-N-(benzyloxycarbonyl)-2,3-dideoxy-β- D,L-threo-hex-2-enoaza-pyranoside (4.38β), a liquid: Rf = 0.54 (4:6 hexane/AcOEt). FTIR (CCl4) n 3495 cm-1 (cis 1-4-OH---O). 1H NMR (CD3CN, 50°C) δ 7.22-7.43 (m, 10H), 5.64-5.81 (m, 2H), 5.42-5.49 (m, 1H), 5.18 (s, 2H), 4.67-4.81 (m, 1H), 4.48 (s, 2H), 4.40-4.43 (m, 1H), 3.75-3.86 (m, 2H), 3.33 (s, 3H). 13C NMR (CDCl
3) δ 153.8, 137.6, 136.2, 132.3, 128.8, 128.6, 128.5, 128.2, 128.0, 127.8, 124.3, 79.7, 73.5, 69.3, 68.1, 67.4, 56.8, 50.4. Anal. Calcd for C22H25NO5: C, 68.91; H 6.57; N 3.65. Found: C, 69.26; H, 6.24; N, 3.29.
Methyl 6-(O-benzyl)-N-(benzyloxycarbonyl)-2,3-dideoxy-α- D,L-threo-hex-2-enoaza-pyranoside (4.38α), a liquid: Rf = 0.44 (4:6 hexane/AcOEt). FTIR(CCl4) ν 3443 cm-1 (cis 1-3-OH---O). 1H NMR (CD3CN) δ 7.23-7.42 (m, 10H), 6.00 (dd, 1H, J = 10.0, 4.6 Hz), 5.86 (ddd, 1H, J = 10.0, 4.2, 0.9 Hz), 5.52 (d, 1H, J = 4.2 Hz), [5.06*(s) and 5.08*(s), 2H], 4.49 (s, 2H), 3.70-4.13 (m, 4H), 3.25 (s, 3H). 13C NMR (CDCl3) δ 153.8, 142.2, 138.3, 130.0, 128.9, 128.8, 128.7, 128.6, 128.4, 128.0, 127.9, 81.4, 74.0, 73.4, 68.1, 62.4, 53.6, 53.2. Anal. Calcd for C22H25NO5: C, 68.91; H 6.57; N 3.65. Found: C, 68.82; H, 6.34; N, 3.49.
Reaction of epoxide 4.1β with EtOH
Following the typical procedure, the treatment of hydroxy tosylate 4.34 (0.042 g, 0.080 mmol) in anhydrous EtOH (2 mL) with t-BuOK (0.010 g, 0.088 mmol) afforded, after 12 h at room temperature, a crude product (0.028 g,
88% yield) essentially consisting of ethyl
6-(O-benzyl)-N-(benzyloxycarbonyl)-2,3-dideoxy-β-D,L-threo-hex-2-enoazapyranoside (4.43β), (1H NMR), pure as liquid: Rf = 0.21 (7:3 hexane/AcOEt). FTIR (neat film) ν 3450, 1706, 1412, 1330, 1018 cm
-N BnO Cbz HO OEt 4.43! N BnO Cbz HO OMe 4.38! N BnO Cbz HO OMe 4.38!
1. 1H NMR (CD
3CN, 50°C) δ 7.18-7.45 (m, 10H), 5.58-5.82 (m, 2H), 5.55 (s, 1H), 5.17 (s, 2H), 4.65-4.82 (m, 1H), 4.50 (s, 2H), 4.32-4.47 (m, 1H), 3.26-3.87 (m, 4H), 1.06 (t, 3H, J = 7.1 Hz). 13C NMR (CDCl3) δ 154.6, 137.0, 135.8, 131.7, 128.3, 128.2, 128.0, 127.7, 127.6, 127.5, 77.8, 77.0, 73.2, 69.0, 67.6, 66.9, 50.0, 14.9. Anal. Calcd for C23H27NO5: C, 69.50; H 6.85; N 3.52. Found: C, 69.45; H, 6.79; N, 3.22.
Reaction of epoxide 4.1β with i-PrOH
Following the typical procedure, the treatment of hydroxy tosylate 4.34 (0.072 g, 0.14 mmol) in anhydrous i-PrOH (2.4 mL) with t-BuOK (0.017 g, 0.15 mmol) afforded, after 18 h at room temperature, a crude product (0.050 g, 87% yield) consisting of isopropyl glycoside 35 which was subjected to flash chromatography. Elution with a 7:3 hexane/AcOEt mixture afforded isopropyl 6-(O-benzyl)-N-(benzyloxycarbonyl)-2,3-dideoxy-β-D,L-threo-hex-2-eno-azapyranoside (4.44β) (0.030 g, 52% yield), pure as liquid: Rf = 0.25 (7:3 hexane/AcOEt). FTIR (neat film) ν 3458, 1703, 1417, 1332, 1116, 1014 cm-1.1H NMR (CD3CN, 50°C) δ 7.26-7.44 (m, 10H), 5.77-5.79 (m, 1H), 5.64-5.68 (m, 1H), 5.59-5.64 (m, 1H), 5.18 (s, 2H), 4.67-4,78 (m, 1H), 4.51 (s, 2H), 4.35-4.44 (m, 1H), 3.78-4.02 (m, 3H), 3.33 (d, 1H, J = 5.6 Hz), 1.10 (d, 3H, J = 6.2 Hz), 1.02 (d, 3H, J = 6.2 Hz). 13C NMR (CD
3CN) δ 151.3, 140.0, 138.0, 132.1, 129.3, 129.2, 128.7, 128.6, 128.4, 128.3, 76.6, 70.0, 69.3, 68.2, 67.7, 65.9, 53.4, 23.4, 21.6. Anal. Calcd for C24H29NO5: C, 70.05; H 7.10; N 3.40. Found: C, 69.70; H, 6.81; N, 3.90.
Reaction of epoxide 4.1β with t-BuOH
Following the typical procedure, the treatment of hydroxy tosylate 4.34 (0.050 g, 0.095 mmol) in anhydrous t-BuOH (2 mL) with t-BuOK (0.012 g, 0.104 mmol) afforded, after 18 h at room temperature, a crude product (0.032 g) consisting of a complex reaction mixture with no evidences of the presence of addition products (1H NMR).
Reaction of epoxide 4.1α with MeOH
Following the typical procedure, the treatment of hydroxy mesylate 4.37 (0.120 g, 0.27 mmol) in anhydrous MeOH (5 mL) with MeONa (0.016 g, 0.30 mmol) afforded, after 18 h at room temperature, a crude product (0.088 g, 85%) consisting of a 75:25 mixture of methyl α-glycoside 4.39α and methyl β-glycoside 4.39β (1H NMR) which was subjected to preparative TLC with an 1:1 hexane/AcOEt mixture, as the eluant. Extraction of the two most intense bands (the faster moving band contained 4.39α) afforded pure methyl glycosides 4.39α (0.050 g, 48% yield) and 4.39β (0.017 g, 16% yield).
N BnO Cbz HO O-i-Pr 4.44! N BnO Cbz HO OMe 4.39! N BnO Cbz HO OMe 4.39!