85
4. Akt inhibitors
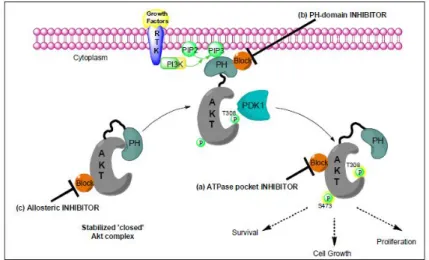
PI3K/Akt/PDK1 pathway plays a critical role in proliferation and survival in tumor cells and is also linked with resistance to radiotherapy, chemotherapy, endocrine therapy and novel anticancer therapies. In this context, Akt might have all the characteristics of a good cancer target. Consequently, an intense search for small molecule inhibitors of Akt as anticancer agents has been in the forefront of the cancer drug discovery. Several sites on the protein provide functionally important regions that are suitable for binding small molecule inhibitors. These include the ATP binding pocket that has been so successfully targeted in other protein kinases, the phosphoinositide binding pocket of the PH domain, a hinge region lying between the PH and protein kinase domains, and the substrate binding groove that lies adjacent to the ATP binding pocket.
Figure 4.0: Akt inhibition.
4.1. ATPase pocket inhibitors
4.1.1 Akt inhibitors derivatives from PKA modulator
Most kinase inhibitors bind in the enzyme active site and display ATP-competitive behavior. Many prototypical heterocyclic frameworks have been identified. Because of the high degree of homology in the ATP binding pocket
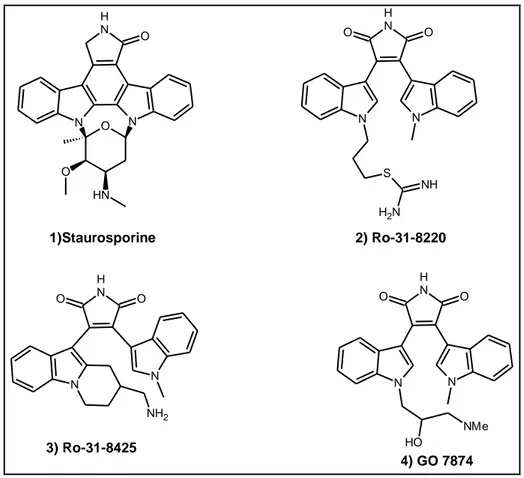
86 between Akt, PKA and PKC, many typical PKA and PKC inhibitors have been identified as inhibitors of Akt. Staurosporine (1) is a non-selective kinase inhibitor1 that is more potent against serine/threonine kinases2. The Akt activity of staurosporine and several analogs have been reported and the results are summarized in Table 4.13. N N N H O O N H O 1)Staurosporine N N N H O O S NH N H2 2) Ro-31-8220 N H O O N N NH2 3) Ro-31-8425 N N N H O O NMe O H 4) GO 7874 Figure 4.1: Structure of Staurosporine and analogs
None of ring-open analogs (2–4) in Table 4.1 are more potent than staurosporine, with compound 2 being the most active (240 nM). Activity of staurosporine against various kinases has been published recently, where it showed an IC50 value of 1.5
nM against Akt1, versus that of 3.6 nM against PKA2.
Table 4.1: Activity of staurosporine and analogs in the Akt1 flashplate assay (ATP: 2 μM).
Compound IC50(nM)
Staurosporine(1) 11
Ro-31-8220(2) 240
Ro-31-8425(3) 1,000
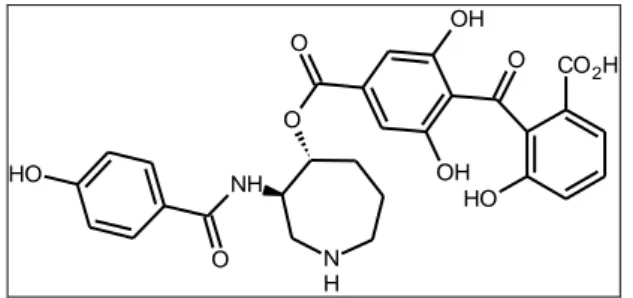
87 A study of analogs of balanol, a non-selective kinase inhibitor, has recently been published. Balanol is potent PKA inhibitor with a Ki value of ∼ 4 nM4
. Its binding to the ATP pocket of PKA has been well characterized. The 4-hydroxybenzamide binds to the hinge via the hydroxyl group and the C-6 hydroxyl group on the benzophenone hydrogen binds to Lys725,6.
N H NH O OH O O H CO2H OH O O H O
Figure 4.2: Structure of Balanol.
Akt inhibitors that were made by replacing the hinge binding phenol of balanol with 4-pyridinyl or other heteroaryl groups were reported by scientists at Hoffmann-La Roche7.
In an effort to improve plasma stability of the balanol, the unstable ester group (5a) was replaced by a number of metabolically less labile linkers8. For example, the half-life in mouse plasma increased from < 1 min to 69 h with the simple substitution of an amide (5b, IC50 = 4 nM) for the ester (5a, IC50 = 5 nM), causing only little change
in Akt activity (Table 4.2). Compounds featuring alternative linkers, such as ether (5c), amines (5d and 5e) and trans-vinyl (5f), also showed improved stability, but with much weaker Akt activity. Regrettably, none of the compounds show selectivity over PKA.
88 N H NH A B O O H F N O OMe Compound A-B IC50(nM) T1/2 (h) Akt1 PKA 5a O O 5 5 <0.02 5b N H O 4 2 69 5c O 355 39 29 5d N H 3000 800 161 5e NH 25.000 45.000 NA 5f 160 360 NA
Table 4.2. Akt and PKA activity and half-lifes in mouse plasma of compounds 5a-f.
4.1.2 Pyridines analogs as Akt inhibitors
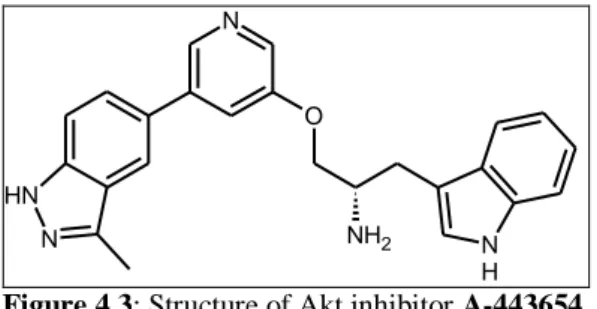
Researchers from Abbott recently reported the identification and development of a novel series of 3,5-disubstiuted pyridines analogs that bind to the ATP-binding site of Akt. This class of compounds is broadly characterized as ATP-competitive, reversible pan-Akt kinase inhibitors that inibit Akt activity in vitro and in vivo. A-443654, an indazole-pyridine based compound, which inhibits Akt1 with a Ki of
160 pM and blocks the growth of pancreatic and prostate tumour xenografts, but only at concentrations that were two-fold lower than the maximally tolerated dose9. Further mechanistic studies suggested that the compound worked by inhibiting the Akt-dependent up-regulation of expression of Aurora A kinase which may have been responsible for the G(2)M cell cycle block observed10.
89 N N N H O NH2 N H
Figure 4.3: Structure of Akt inhibitor A-443654.
Davies and colleagues at Astex Therapeutics have recently successfully obtained crystal structures of A-443654 in complex with Akt2 and PKA-Akt2 chimera. A structural comparison of inhibitor binding to PKA, Akt2 and PKA-Akt chimera revealed that A-443654 adopts a conformation in Akt2 and PKA-Akt chimera that differs from that in PKA. The methylindazole and the pyridine adopt almost identical binding modes in both PKA and Akt. However, the conformation of the indole is very different when bound to Akt, in which the indole ring is directed towards the front of the ATP-binding cliff in a new hydrophobic pocket formed by the side chains of Met282, Phe439 and Val166.
In its new conformation, the indole ring forms non-polar Van der Waals contacts with the side chain of Met282, which might be the driving force for the new conformation because of the absence of such interaction in PKA due to the substitution by Leu173 at the same point. Interestingly, the phenyl ring of Phe163 now occupies the hydrophobic pocket underneath the glycine rich loop, which was filled by the indole ring of A-443654 in the PKA structure.
The indole nitrogen is ∼ 3.5Å to the carboxylate oxygen atoms of Glu236, which explains why N -methylation of the indole is detrimental for Akt activity2.
Figure 4.4. The schematic representations of interactions of compound A-443654 in complex with A.
90 However, further development of this compound is compromised by its poor selectivity for Akt relative to other members of the ACG kinase family11.
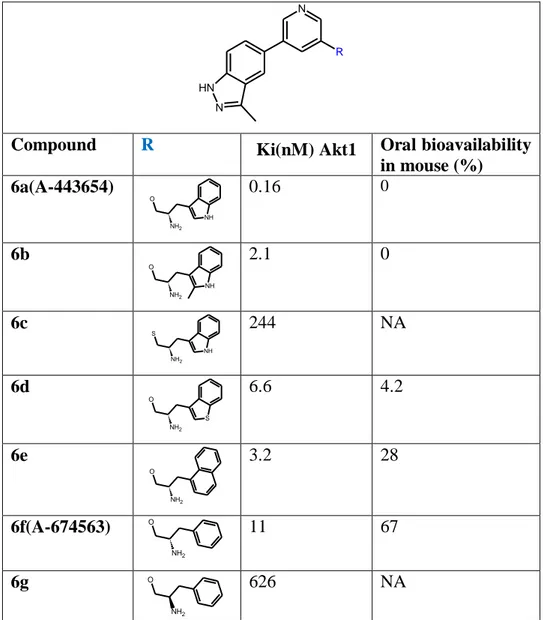
Related Akt inhibitors with a variety of side chains were explored in an effort to improve PK properties (Table 4.3)12. In general, the indole-containing side chain is far superior to other groups. The phenyl analog 6f (A-674563) was identified to have drastically improved PK profile with oral bioavailability of 67% in mouse, but is ∼ 70-fold less active than A-44365412. Its cellular activity against MiaPaCa-2 cells (EC50 = 400 nM) is only fourfold weaker. In a reported PC-3 xenograft model (PTEN
–deficient human prostate carcinoma cell line), A-674563 demonstrated moderate efficacy enhancement when dosed orally in combination with paclitaxel13.
N
N N H
R
Compound R Ki(nM) Akt1 Oral bioavailability in mouse (%) 6a(A-443654) NH O NH2 0.16 0 6b NH O NH2 2.1 0 6c NH S NH2 244 NA 6d S O NH2 6.6 4.2 6e O NH2 3.2 28 6f(A-674563) O NH2 11 67 6g O NH2 626 NA
Table 4.3. Structure–activity relationship of the 3,5-disubstiuted pyridines series with different side
91 More recently, three patent applications were published by scientists from GlaxoSmithKline focusing on the 7-side chain modifications with the rest molecules fixing to the 1H-imidazo[4,5-c]pyridines group14-16. The activity against Akt was provided for several compounds as seen in Table 4.4. The lead compound 7g (GSK690693) was reported to be in a Phase I clinical trial in patients with solid tumors and lymphomas17,18. GSK690693 is a pan-Akt inhibitor with IC50 values of 2
nM, 13 nM and 9 nM against Akt1, Akt2 and Akt3, respectively. It caused dose-dependent reductions in the phosphorylation levels of multiple substrates of Akt in vitro and in vivo, including GSK-3 β, PRAS40 (in vitro) and FOXO3a (in vitro). One of the potential side effects of treatment of GSK690693 included acute increases in blood glucose levels, although it returned to baseline levels as the circulating drug concentration decreased. N N N N O N N H2 R1 R3 R2 4 6 Compound R1 R2 R3 IC50(nM)
Akt1 Akt2 Akt3
7a H NH O 50 270 190 7b H NH O 70 270 510 7c Cl NH O 30 20 10 7d NH2 O 40 20 10 7e Cl NH NH O 8.320 7.940 450 7f Cl OH OH NH O 69 38 32 7g (GSK690693) H2N N H O 2 13 9 7h H2N O N H 2 8 NA 7i H2N O N 2 16 NA
92 A fragment-based drug discovery approach led to the identification of AT7867 (Astex)19 and CCT128930 (Cancer Research UK)20. AT7867 exhibited activity against all three Akt isoforms (IC50 range 17–47 nM) and roughly equally potency
against PKA (20 nM) and p70S6k (80 nM). CCT128930, however, exhibited good potency against Akt isoforms (Akt2 IC50=6 nM) and 20–30-fold less potency
towards PKA and p70S6k. This improved selectivity was proposed to be due to the exploitation of a single amino acid difference between Akt and PKA20. AT7867 and CCT128930 inhibited the growth of human glioblastoma and breast cancer xenografts, respectively. NH Cl N N H N N N H N NH2 Cl AT7867 CCT128930
Figure 4.5: Structure of Akt inhibitors:AT7867 and CCT128930.
4.1.3 Other ATP-pocket binders of Akt
Recently, scientists at Amphora Discovery (now licensed to Genentech) have reported a series of N-(thiazol-2-yl)thiophene-2-carboxamides (8–10) as Akt inhibitors21. Although no specific Akt activity was disclosed, many compounds appeared to be synthesized in several libraries rather than through structure-based design. The 4-aryl group on the thiazole is speculated to be the hinge-binder. The substituted N-aminopropyl groups seemed to be important as it appears in many of the examples disclosed. They have also disclosed another two patents covering the substituted thiophenes22 and 1,2,4-triazoles23 as kinase inhibitors. Again, no activity was disclosed. Based on our knowledge on the ATP-binding site of Akt, these compounds are probably not potent Akt inhibitors, although some of which were said to be active against Akt.
93 S O N N R R S N Ar
Compd Ar Compd Ar Compd Ar
8 O 9 N
MeO
10
N O
Table 4.5.Structure of N-(thiazol-2-yl)thiophene-2-carboxamides
Novartis have discovered a series of imidazo[4,5- c]quinolines as Akt inhibitors with claimed IC50 values of 0.01 – 100 μM24. Of 139 examples prepared, compounds 11–
14 are the only four inhibitors with a provided range for Akt activity (IC50 values <
500 nM). Several 8-aryl groups were used repeatedly, including 2-thienyl, 3-thienyl, 5-indolyl, 2-benzofuranyl, 2-benzothiophenyl and 5-benzo[1,3]dioxolyl groups. Additionally, many compounds with substitutions such as fluoro, chloro, or methyl at C-2 or C-3 on the 1-phenyl group were prepared. Although no selectivity profile was disclosed, based on the activity of closely related analogs against ALK and p70 S6K, compounds 11–14 are also expected to be active against ALK and p70 S6K .
N N N (CH2)n NH2 X Ar 11 Ar = 5-indolyl X= F n= 1 12 Ar = 2-thienyl X= F n= 1 13 Ar = 3-thienyl X= Cl n= 1 14 Ar = 3-thienyl X= F n= 2
Table 4.6.Structure of imidazo[4,5- c]quinolines
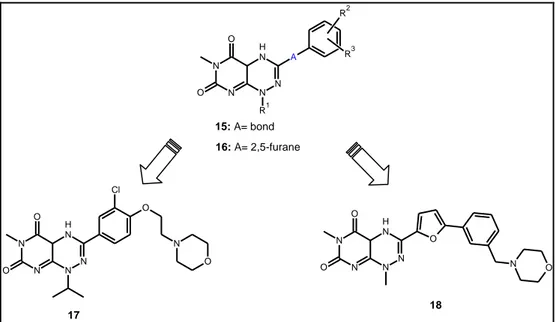
Scientists at Janssen Pharmaceutica have reported a series of 3-aryl and 1-alkyl analogs of toxoflavin as potent Akt and CDC4 inhibitors (15, 16)25,26. The analogs of 3-phenyl (15)25 and 3-(5-phenyl-furan-1-yl) toxoflavins (16)26, as well as their
94 enzymatic (Akt3, Cdk4 and CDC25B) and cellular activities, including cytotoxicity against A2780 human ovarian carcinoma cells and inhibition of Akt3-mediated phosphorylation of MAPK in MDA-MB231 human breast adenocarcinoma cells, were included in the patent.In general, the phenyl series (15) are about five-fold less active than the furanyl series (16). The two representative compounds, 17 and 18, have IC50 values of 110 and 32 nM against Akt3, respectively. Compound 17 showed
similar activity against Cdk4 (IC 50 = 160 nM), but no Cdk4 activity was given for compound 18. N N N H N N A O O R1 R2 R3 15: A= bond 16: A= 2,5-furane N N N H N N O O O N O Cl N N N H N N O O O N O 17 18
Table 4.7.Structure of analogs of toxoflavin
4.2. Phosphoinositide binding pocket of the PH domain
The PH domain of Akt binds PI(3,4,5)P3, the product of PI3 kinase, as well as PI(3,4)P2 which is an immediate metabolite of PI(3,4,5)P3. Both lipids are produced in the plasma membrane in response to PI3K activation, thus inhibition of the PH domain-phosphoinositide interaction would prevent membrane recruitment and thus activation of Akt by PDK1 and PDK2. Based on this hypothesis, 1D-3-deoxyphosphatidylinositol (DPI, 20), which lacks the D-3-hydroxyl group as seen in 1D-phosphatidylinositol (19), was synthesized27,28. Despite showing moderate antiproliferative activity against HT-29 human column carcinoma cells with an IC5095 concentrations up to 250μM29. Like DPI, the 3-deoxy-3-chloro-PtdIns (21) also failed to inhibit PI3K. In contrast to DPI, the 3-deoxy-3-fluoro PtdIns (22) inhibited PI3K with an IC50 value of 30μM, suggesting that the electronic nature and steric
volume of the C-3 group play an important role for recognition by PI3K.
O H O H X OH OH O P O OH O O O O C15H31 C15H31 O 19 X=OH, PtdIns 20 X=H , DPI 21 X=Cl 22 X=F
Figure 4.6: Structure of inhibitors of PIP3 binding
In an effort to increase the stability to phospholipase, a series of 1- O -octadecyl-3-deoxy- or 3-hydroxymethylphosphatidylinositol ether lipids and related carbonate analogs (23a–i) were synthesized. Their structures and activities in inhibiting Akt activation in PDGF-stimulated NIH3T3 cells, in vitro PI3K kinase activity and cell growth of several human cancer cell lines are summarized in Table 4.827,28. Despite the greater variability in activity against Akt activation and PI3K, the growth inhibition of various cancer cell lines was relatively flat, with IC50 values in the
micromolar range for most compounds. The best compound in the series is compound 23a (DPIEL), which is in the late stage preclinical development under a new name PX-316 by Prolx Pharmaceuticals30,31. PX-316 appeared to be the most potent and a specific inhibitor of Akt activation. Its IC50 value against Akt activation
in PDGF-stimulated NIH3T3 cells was 1.5μM, which is ∼ 10-fold lower than its PI3K activity32. It is also active against Akt activation in PDGF-stimulated MCF-7, DU-145 and LNCaP cell lines.
96
A O
R
C18H37
Compound A R In vitro IC50 Antiproliferative IC50
Akt PI3K NIH3T3 HT-29 MCF-7
23a(PX-316 /DPIEL) O H O H OH OH O P O OH O OMe 1.5 14.8 4.5 2.1 7.2 23b HO O H OH OH O P O OH O O H OMe 7.8 31.0 2.0 4.5 5.0 23c HO O H OH OH O P O OH O O H OMe 9.1 18.5 ND 7.5 2.0 23d (DDPIEL) O H OH OH O P O OH O OMe 11.0 0.8 2.0 ND ND 23e HO O H OH OH O P O OH O N N 37.5 14.5 6.5 15.0 6.0 23f HO O H OH OH O P O OH O O H N N 20.5 7.9 8.2 6.5 2.0 23g HO O H OH OH O P OH O OMe ND 5.3 ND 45 ND 23h HHOO OH OH O O O OMe 12.5 15.5 ND 12.5 12.0 23i HO O H OH OH O O O O H OMe 5.0 83 2.0 10.0 1.2
Table 4.8: Structure–activity relationship of phosphatidylinositol ether lipids (μM).
Based on the fact that the metabolism of inositol phospholipids occurs with the formation of 1,2-cyclic phosphate by PI-specific phospholipase C, several compounds (24–26), which were alkylated and dehydroxylated at C-2, were designed and synthesized to block formation of the cyclic phosphate33. The biological activity of these four compounds in H157 and H1703 cells that have high level of endogenous Akt activity was studied34. Compounds 24, 25 and 26 inhibited the phosphorylation of both Thr308 and Ser473 of Akt in H1703 with IC50 values of 4.1,
97 O H O H OH R O P O OH O O C18H37 OMe 24 R=OH, PX-316(DPIEL) 25 R=OMe, PIA5 26 R=H, PIA6
Figure 4.7: Structure of PIAs inhibitors
Perifosine (27, KRX-0401, D-21266, NSC639966), an orally bioavailable alkylphospholipid anticancer agent, is presently in Phase II clinical trials for multiple cancers, including non-small-cell lung cancer (NSCLC), breast cancer, multiple myeloma and sarcoma. It was licensed to Keryx Biopharmaceuticals by Aeterna Zentaris35. Perifosine was shown to perturb several signal pathways, including Akt, which seems to be an important cellular target of perifosine36,37. Like PX-316, perifosine inhibits activation of Akt at concentrations without directly inhibiting PI3K, PDK1 and Akt activity.
N+ O P O O -O
C18H37
Figure 4.8: Structure of Perifosine.
A recent study indicated that perifosine might inhibit binding of Akt PH domain to PtdIns(3,4)P238. In Phase I/II trials, perifosine has shown single agent partial responses or long-term disease stabilizations in solid tumors, including renal, hepatocellular, sarcoma and breast cancer39-42, but it lacked objected responses as a single agent in several recent Phase II trials, including malignant melanoma43, prostate cancer44, head and neck cancer45 and pancreatic adenocarcinoma46. Several Phase II trials to assess the efficacy of daily oral perifosine combined with radiotherapy or with other anticancer agents, such as gemcitabine, paclitaxel, docetaxel, imatinib mesylate, dexamethasone and trastuzumab are ongoing.
98
4.3. Allosteric Akt inhibitors
Merck & Co, Inc have developed a novel class of allosteric inhibitors of Akt. The initial lead compound, 2,3 diphenylquinoxalines, was identified by high through put screening (HTS) and further derivatised to compounds that specifically inhibit Akt1 (Akti-1), Akt2 (Akti-2) or both (Akti-1/2), with minimal activity against Akt347. These compounds do not bind to the ATP binding site or the PH domain, but inhibit Akt activity in a manner that requires the PH domain itself 48,49. As the compounds bind to a site distant from the ATP binding pocket that is likely to be unique to Akt, they exhibit minimal activity towards other protein kinases except CAMK1 through a mechanism that remains to be identified50,51. Akti-1/2 enhances apoptosis induced by doxorubicin and camptothecin,and inhibits IGF-1-stimulated Akt phosphorylation in mice52. Interestingly, the inhibitor preferentially induces caspase-3 activation in tumour cells, with little effect on normal human umbilical vein (HUVEC), normal prostate epithelial (NHPE) or normal human mammary epithelial cells53. This suggests the presence of a reasonable therapeutic window for Akt inhibition in the treatment of cancer.
N N N N NH O Lead compound N N NC N N N N N N H O N N H O N H N 28 3.1 (Akt1) 34 (Akt2) > 50 (Akt3) 29 0.37 (Akt1) 12 (Akt2) > 50 (Akt3) 30 0.76 (Akt1) 3.1 (Akt2) > 50 (Akt3) 31 0.27 (Akt1) 0.16 (Akt2) > 50 (Akt3) 32 0.76 (Akt1) 24 (Akt2) > 50 (Akt3) 33 21 (Akt1) 0.32 (Akt2) 21 (Akt3) N N N N N N R 34 (R=H) 0.06 (Akt1) 0.06 (Akt2) 1.3 (Akt3) 35 (R=Me) 1.1 (Akt1) 1.9(Akt2) > 50 (Akt3) N N N N N N R N N N N H 36 (R=H) 0.02 (Akt1) 0.14 (Akt2) 1.6 (Akt3) 37 (R=Me) 0.06 (Akt1) 0.32(Akt2) > 50 (Akt3) N N N N H 38 0.09 (Akt1) 0.31(Akt2) 2.4 (Akt3) 39 0.06 (Akt1) 0.21(Akt2) 2.2 (Akt3)
99 Further pre-clinical development of this class of compounds was precluded by poor solubility and pharmacokinetics48, however Merck subsequently reported MK2206, a chemical derivative which possesses low nanomolar potency against the three Akt isoforms and inhibits the growth of several tumour xenografts either alone or in combination with other standard chemotherapies54. MK-2206 has recently entered a Phase I clinical trial in patients with solid tumours where it is reasonably well tolerated (the primary drug related toxicity being a skin rash and transient hyperglycaemia). N N N H2 N N H O Figure 4.9: Structure of MK-2206
The compound promoted a sustained fall in Akt and PRAS40 phosphorylation in tumours, blood cells and hair follicles, and evidence for tumour shrinkage was obtained in patients with pancreatic, melanoma and neuroendocrine tumours following MK-2206 administration55-57.
Further developments of this promising first-in-class study are eagerly awaited. Allosteric Akt inhibitors have progressed relatively rapidly into clinical development at least in part because they show good selectivity for Akt. However, our previous finding that Akti-1/2 is equipotent at potentiating platelet aggregation in a manner independent of Akt inhibition may impact of the therapeutic use of these inhibitors by increasing the risk of thrombosis58.
100
4.4. Akt antisense oligonucleotides
Alternative inhibitors of Akt under investigation include antisense oligonuleotides and pseudosubstrtate inhibitors. Before the relatively selective small molecule Akt inhibitors became available, these inhibitors were used as tools to validate Akt as an anticancer target.
RX-0201 is a 20-mer oligonucleotide with a sequence complementary to that of Akt-1 mRNA that inhibits the expression of Akt-Akt-1 in tumor cell lines and bears significant in vitro and in vivo anticancer activity with good safety. A phase I trial with RX-0201 that aimed to determine the maximum tolerated dose and pharmacokinetic and safety profile of RX-0201 was reported at the last ASCO meeting and phase II clinical trials are being planned59.
4.5. Pseudosubstrate and Substrate-mimetic inhibitors
An inhibitor that competes with substrate binding might also be expected to have improved selectivity for Akt over other AGC kinases on the basis that Akt phosphorylates a distinct set of downstream substrates that mediate its biological effects. One such compound, PTR6164, is a peptide that was chemically modified to improve its stability and cell permeability. PTR6164 is relatively stable in plasma, is well tolerated and inhibits the growth and metastatic spread of prostate tumour xenografts in mice60.The compound inhibits Akt with low micromolar potency and is 10-fold selective for Akt over other serine/threonine kinases including PKA and PKC. Despite promising progress, peptide mimics are difficult to progress clinically and issues of therapeutic window, bioavailability, potency and cost could provide significant barriers to implementation.
101
4.6. Inhibitors of Akt with unknown mechanism of
actions
A number of compounds have been reported to inhibit Akt kinase activity through mechanisms that are unclear or without detailed description on how the assays were run. A series of copper complexes of 3-substituted 4-chomones as represented by compound 40 (FPA-124) is the first one in the group61. The compounds were designed based on the structure of genistein, an isoflavonoid found in soy products that has been shown to inhibit Akt activation62, by replacing the 3-(4-hydroxyphenyl) group with the copper complexes of the semicarbazones or thiosemicarbazones63. The best compound in the series, compound 40, inhibited Akt1 kinase activity in vitro with an IC50 value of 100 nM.
However, further studies are needed to determine if the activity is specific for Akt as well as ATP competitive.
O Cu N S N H NH2 Cl Cl O 40 FPA-124
Figure 4.11: Structure of copper complexes
Compounds 41 (API-59CJ-OMe) and 42 (CMEP) are derivatives of the ellipticine alkaloid. They are known to have potent anticancer activity in vitro and in vivo64-66. API-59CJ-OMe was found to significantly inhibit Akt1 kinase activity at 12 μM while showing no effect on Erk-1 kinase activity67. In contrast to API-59CJ-OMe, CMEP seemed to significantly decrease phosphorylation level of Akt in human breast and prostate cancer cell lines at micromolar concentrations68-69. On the other hand, inhibition of Akt kinase activity by CMEP was confirmed to be ATP competitive with an IC50 value of 40, 42 and 30 μM against Akt1, Akt2 and Akt3,
102 respectively, which were much higher than the single-digit micromolar IC50 values
against Akt activation in cells68-69.
N+ N H R CH3 CH3 AcO -41 (API-59CJ-OMe) R=OMe 42 (CMEP) R=Cl
103
Reference chapter 4
1. Rüegg UT, Burgess GM: Trends Pharmacol. Sci. 1989, 10, 218 -220. 2. Li Q, Li T, Zhu GD et al. Bioorg. Med. Chem. Lett. 2006, 16, 1679 -1685. 3. Turek TC, Small EC, Bryant RW et al. Anal. Biochem. 2001, 299, 45-53. 4. Gustafsson AB, Brunton. Mol. Pharmacol. 1999, 56, 377 -382.
5. Narayana N, Diller TC, Koide K et al. Biochemistry. 1999, 38, 2367 -2376.
6. Akamine P, Madhusudan, Brunton LL et al. Biochemistry. 2004, 43, 85 -96.
7. Hoffmann-La Roche: WO03076429, 2003.
8. Breitenlechner CB, Wegge T, Berillon L et al. J. Med. Chem. 2004, 47, 1375 -1390.
9. Y. Luo, A.R. Shoemaker, X. Liu, K.W. Woods, S.A. Thomas, R. de Jong, E.K. Han, T. Li, V.S. Stoll, J.A. Powlas, A. Oleksijew, M.J. Mitten, Y. Shi, R. Guan, T.P. McGonigal, V. Klinghofer, E.F. Johnson, J.D. Leverson, J.J. Bouska, M. Mamo, R.A. Smith, E.E. Gramling-Evans, B.A. Zinker, A.K. Mika, P.T. Nguyen, T. Oltersdorf, S.H. Rosenberg, Q. Li, V.L. Giranda, Mol. Cancer Ther. 2005, 4(6), 977– 986.
10. X. Liu, Y. Shi, K.W. Woods, P. Hessler, P. Kroeger, J. Wilsbacher, J. Wang, J.Y. Wang, C. Li, Q. Li, S.H. Rosenberg, V.L. Giranda, Y. Luo, Neoplasia. 2008, 10(8), 828–837.
11. J. Bain, L. Plater, M. Elliott, N. Shpiro, C.J. Hastie, H. McLauchlan, I. Klevernic, J.S. Arthur, D.R. Alessi, P. Cohen, Biochem. J. 2007, 408(3), 297–315.
12. Thomas SA, Li T, Woods KW et al. Bioorg. Med. Chem. Lett. 2006, 16, 3740 -3744.
13. Luo Y, Shoemaker AR, Liu X et al. Mol. Cancer Ther. 2005, 4, 977 -986.
14. Glaxosmithkline: WO07058850, 2007. 15. Glaxosmithkline: WO07058852, 2007. 16. Glaxosmithkline: WO07058879, 2007.
17. Kumar R, Rhodes N, Knick VB et al. Annual Meeting of American Association for Cancer Research . Los Angeles, USA, 2007 :(Abstract 279).
104 18. Rhodes N, Knick VB, McConnell R et al. Annual Meeting of American Association for Cancer Research . Los Angeles, USA, 2007 : (Abstract 277).
19. K.M. Grimshaw, L.J. Hunter, T.A. Yap, S.P. Heaton, M.I. Walton, S.J. Woodhead, L. Fazal, M. Reule, T.G. Davies, L.C. Seavers, V. Lock, J.F. Lyons, N.T. Thompson, P. Workman, M.D. Garrett, Mol. Cancer Ther. 2010, 9 (5), 1100–1110. 20. T.A. Yap, M.I. Walton, L.J. Hunter, M. Valenti, A. De Haven Brandon, P.D. Eve, R. Ruddle, S.P. Heaton, A.T. Henley, L. Pickard, G. Vijayaraghavan, J.J. Caldwell, N.T. Thompson, W. Aherne, F.I. Raynaud, S.A. Eccles, P. Workman, I. Collins, M.D. Garrett, Mol. Cancer Ther. 2010, 10 (2), 360–371.
21. Amphora Discovery/Genentech: WO06020767, 2006. 22. Amphora Discovery/Genentech: WO05033102, 2005. 23. Amphora Discovery/Genentech: WO05097758, 2005. 24. Novartis: WO05054237, 2005.
25. Janssen Pharmaceutica: WO04007498, 2004. 26. Janssen Pharmaceutica: WO04007499, 2004.
27. Georgtown University and University of Arizona: US6245754, 2001. 28. Georgtown University and University of Arizona: US7153843, 2006. 29. Kozikowski AP, Kiddle JJ, Frew T. J. Med. Chem.1995, 38, 1053 -1056. 30. Meuillet EJ, Ihle N, Baker AF et al. Oncol. Res. 2004, 14, 513 -527.
31. Williams R, Baker AF, Ihle NT et al. Cancer Chemother. Pharmacol. 2006, 58, 444 -450.
32. Hu Y, Qiao L, Wang S et al. J. Med. Chem. 2000, 43, 3045 -3051.
33. Kozikowski AP, Sun H, Brognard J. et al. J. Am. Chem. Soc. 2003, 125, 1144-1145.
34. Castillo SS, Brognard J, Petukhov PA et al. Cancer Res. 2004, 64, 2782 -2792. 35. Aeterna Zantaris: US6172050, 2001.
36. Kondapaka SB, Singh SS, Dasmahapatra GP et al. Mol. Cancer Ther. 2003, 2, 1093 -1103.
105 38. Pordadosu E, Lemmon MA, Keleti D.Annual Meeting of American Association for Cancer Research. Los Angeles, USA, 2007:(Abstract 1645).
39. Crul M, Rosing H, De Klerk GJ et al. Eur. J. Cancer. 2002, 38, 1615 -1621. 40. Van Ummersen L, Binger K, Volkman J et al. Clin. Cancer Res. 2004, 10, 7450 -7456.
41. Knowling M, Blackstein M, Tozer R et al. Invest. New Drugs. 2006, 24, 435 -439.
42. Bailey HH, Mahoney MR, Ettinger DS et al. Cancer. 2006, 107, 2462 -2467. 43. Ernst DS, Eisenhauer E, Wainmann N et al. Invest. New Drugs. 2005, 23, 569 -575.
44. Posadas EM, Gulley J, Arlen PM et al. Cancer Biol. Ther. 2005, 4, 1133 -1137. 45. Argiris A, Cohen E, Karrison T et al. Cancer Biol. Ther. 2006, 5, 766 -770. 46. Marsh RDE W, Rocha Lima CM, Levy DE et al. Am. J. Clin. Oncol. 2007. 30, 26 -31.
47. C.W. Lindsley, Z. Zhao, W.H. Leister, R.G. Robinson, S.F. Barnett, D. Defeo-Jones, R.E. Defeo-Jones, G.D. Hartman, J.R. Huff, H.E. Huber, M.E. Duggan, Bioorg. Med. Chem. Lett. 2005, 15 (3), 761–764.
48. S.F. Barnett, M.T. Bilodeau, C.W. Lindsley, Curr Top Med Chem. 2005, 5 (2), 109–125.
49. J.C. Hartnett, S.F. Barnett, M.T. Bilodeau, D. Defeo-Jones, G.D. Hartman, H.E. Huber, R.E. Jones, A.M. Kral, R.G. Robinson, Z. Wu, Bioorg. Med. Chem. Lett. 2008, 18 (6), 2194–2197.
50. J. Bain, L. Plater, M. Elliott, N. Shpiro, C.J. Hastie, H. McLauchlan, I. Klevernic, J.S. Arthur, D.R. Alessi, P. Cohen, Biochem. J. 2007, 408 (3), 297–315.
51. D. Gilot, F. Giudicelli, D. Lagadic-Gossmann, O. Fardel, Chem. Biol. Interact. 2010, 188 (3), 546–552.
52. C. Cherrin, K. Haskell, B. Howell, R. Jones, K. Leander, R. Robinson, A. Watkins, M. Bilodeau, J. Hoffman, P. Sanderson, G. Hartman, E. Mahan, T. Prueksaritanont, G. Jiang, Q.B. She, N. Rosen, L. Sepp-Lorenzino, D. Defeo-Jones, H.E. Huber, Cancer Biol. Ther. 2010, 9 (7), 493–503.
53. D. DeFeo-Jones, S.F. Barnett, S. Fu, P.J. Hancock, K.M. Haskell, K.R. Leander, E. McAvoy, R.G. Robinson, M.E. Duggan, C.W. Lindsley, Z. Zhao, H.E. Huber, R.E. Jones, Mol. Cancer Ther. 2005, 4 (2), 271–279.
106 54. H. Hirai, H. Sootome, Y. Nakatsuru, K. Miyama, S. Taguchi, K. Tsujioka, Y. Ueno, H. Hatch, P.K. Majumder, B.S. Pan, H. Kotani. Mol. Cancer Ther. 2010, 9 (7), 1956–1967.
55. T.A. Yap, A. Patnaik, I. Fearen, D. Olmos, K. Papadopoulos, N. Tunariu, D. Sullivan, L. Yan, J.S. De Bono, A.W. Tolcher, ASCO Annual Meeting, 2010: (Abstract: 62)
56. T.A. Yap, A. Patnaik, I. Fearen, D. Olmos, K. Papadopoulos, N. Tunariu, D. Sullivan, L. Yan, J.S. De Bono, A.W. Tolcher, J. Clin. Oncol. 2010: (Abstract: 3009) 57. A.W. Tolcher, T.A. Yap, I. Fearen, A. Taylor, C. Carpenter, A.T. Brunetto, M. Beeram, K. Papadopoulos, L. Yan, J. de Bono, J. Clin. Oncol. 2009: (Abstract: 3503) 58. R.W. Hunter, M.T. Harper, I. Hers, J. Thromb. Haemost. 2008, 6 (11), 1923– 1932.
59. Marshall J, Posey J, Hwang S, et al. ASCO Annual Meeting. 2007: (Abstract No. 3564)
60. P. Litman, O. Ohne, S. Ben-Yaakov, L. Shemesh-Darvish, T. Yechezkel, Y. Salitra, S. Rubnov, I. Cohen, H. Senderowitz, D. Kidron, O. Livnah, A. Levitzki, N. Livnah, Biochemistry. 2007, 46 (16), 4716–4724.
61. Wayne State University and University of Pune: WO07035927, 2007. 62. Li Y, Sarkar FH. Clin. Cancer Res. 2002, 8, 2369 -2377.
63. Barve V, Ahmed F, Adsule S et al. :J. Med. Chem. 2006, 49, 3800 -3808. 64. Juray JJ, Haugwitz RD, Varma RK et al. J. Med. Chem.1994, 37, 2190 -2197. 65. Acton EM, Narayanan VL, Risbood PA et al. J. Med. Chem. 1994, 37, 2185 -2189.
66. Arguello F, Alexander MA, Greene JF Jr et al. J. Cancer Res. Clin. Oncol. 1998, 124, 19 -26.
67. Jin X, Gossett DR, Wang S et al. Br. J. Cancer. 2004, 91, 1808 -1812. 68. Zhang M, Fang X, Liu H et al. Cancer Lett . 2007, 252, 244 -258. 69. Zhang M, Fang X, Liu H et al. Biochem. Pharmacol. 2007, 73, 15 -24.

![Table 4.4. Structure–activity relationship of the 1H-imidazo[4,5-c]pyridines.](https://thumb-eu.123doks.com/thumbv2/123dokorg/7569310.111423/7.892.173.789.497.1124/table-structure-activity-relationship-of-the-imidazo-pyridines.webp)

![Table 4.6. Structure of imidazo[4,5- c]quinolines](https://thumb-eu.123doks.com/thumbv2/123dokorg/7569310.111423/9.892.333.623.725.998/table-structure-of-imidazo-c-quinolines.webp)