Capitolo 6
Materiali e metodi
6.1
Materiali
6.1.1 Fango
I fanghi utilizzati nell’impianto di gassificazione provengono dal depuratore situato in località Casolino (Arezzo) e gestito dal Consorzio Nuove Acque. Tale impianto di depurazione è adibito al trattamento di reflui civili ed ha una capacità di circa 90000 abitanti equivalenti.
Il residuo fangoso è prelevato dal Sedimentatore primario e subisce i seguenti trattamenti:
• ispessimento;
• digestione anaerobica; • centrifugazione.
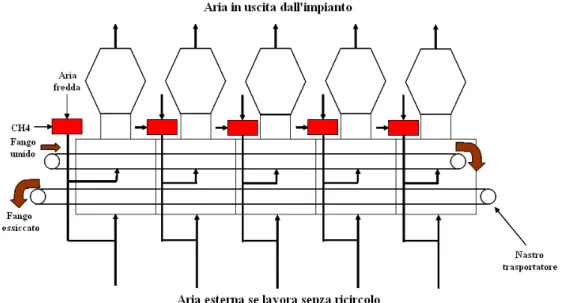
Dopo il trattamento di centrifugazione il fango è inviato ad un impianto di essiccamento ad aria calda, in grado di trattare1 t/h di fango contenente l’80% di umidità. Il fango, distribuito sopra un nastro trasportatore, viene in contatto con aria calda attraversando cinque camere poste in serie. L’aria alimentata, ad una temperatura di circa 50°C, è ottenuta miscelando aria esterna con i fumi ottenuti dalla combustione del metano attraverso cinque bruciatori, uno per ogni sezione di essiccamento. Ciascuna camera è dotata di un ventilatore, la cui portata è pari a 50000 Nm3/h. Nei mesi in cui la temperatura esterna risulta troppo nell’impianto è previsto un riciclo di aria calda e in tal caso il reintegro di aria è di 10000 Nm3/h. Il consumo specifico di metano per tonnellata di fango trattata in ingresso è di 65 m3/h.
Si riporta di seguito lo schema esemplificativo dell’impianto di essiccamento che opera senza ricircolo (Figura 6.1).
Figura 6.1: Schema dell’ impianto di essiccamento
L’impianto è in grado di produrre circa 2000 t/anno di fango essiccato con un tenore di umidità del 20%, sotto forma di pellet aventi dimensioni medie di 1 cm (Figura 6.2).
Figura 6.2: Immagine dei fanghi utilizzati nella sperimentazione
Il campione di fango è stato prima essiccato, per eliminare l’umidità ambientale, e poi macinato, per ottenere una polvere da cui prelevare porzioni su cui eseguire le successive analisi.
6.1.2 Pellet di legno
Il pellet di legno utilizzato nelle prove di avviamento dell’impianto e nelle successive prove sperimentali, in miscela con i fanghi, è del tipo comunemente utilizzato nelle stufe domestiche (Figura 6.3).
Anche il campione di pellet è stato essiccato e macinato prima di essere sottoposto ad analisi.
Figura 6.3: Immagine del pellet di legno utilizzato nella sperimentazione
6.2
Metodi d’analisi
Per la campagna sperimentale, i campioni del fango e del pellet di legno sono analizzati impiegando le seguenti tecniche:
• analisi immediata; • analisi elementare;
• analisi termogravimetrica (TGA);
• analisi spettrometrica con plasma induttivo (ICP).
L’analisi immediata ha consentito la caratterizzazione dei campioni in termini di umidità, sostanze volatili, carbonio fisso, ceneri, mentre l’analisi elementare ha fornito i valori di Carbonio, Idrogeno, Azoto e Ossigeno.
Le analisi termiche sono, invece, quel gruppo di tecniche analitiche aventi in comune il principio operativo di registrare una o più proprietà chimico-fisiche in funzione della temperatura, quando il campione viene riscaldato o raffreddato secondo un determinato programma, o del tempo se il campione viene mantenuto a temperatura costante.
Le tecniche più comunemente impiegate devono soddisfare tre principi basilari:
a) la proprietà fisica di interesse e la temperatura del campione devono essere misurate in maniera continua;
c) la temperatura del campione deve aumentare o diminuire con velocità nota e uniforme entro certi intervalli di temperatura.
L’interpretazione di una curva termoanalitica consiste nel mettere in relazione un certo andamento della particolare proprietà chimico fisica di interesse con possibili trasformazioni che hanno luogo nel campione, quali reazioni chimiche o processi fisici come passaggi di stato.
Sono stati sottoposti a TGA non solo le biomasse processate, ma anche il tar raccolto dalle acque di lavaggio della sezione di depurazione del syngas.
L’analisi ICP ha permesso invece di rilevare il contenuto di metalli come Cd, Cr, Hg, Pb, Ni e Zn nei campioni di pellet di legno, di fango e di ceneri prodotte dalla loro gassificazione.
Per quanto riguarda invece l’analisi dei gas sono state impiegate le seguenti tecniche: • Gas-Cromatografia;
• Spettrometria di massa.
La Gas-Cromatografia ha consentito sia la separazione dei vari componenti gassosi presenti nel gas sia l’analisi quali-quantitativa dei principali componenti quali ad esempio: CO, H2, CO2, CH4, C2H2, C2H4 e C2H6.
La Spettrometria di massa ha consentito invece di effettuare una analisi più dettagliata
dei composti ottenuti dalla gassificazione; in particolare è stata utilizzata per individuare la presenza di ulteriori composti oltre ai principali.
6.3
Analisi immediata
L’analisi immediata (proximate analysis), comprende un gruppo di prove di laboratorio che classifica il combustibile in termini di ceneri, umidità, sostanze volatili e (per differenza) carbonio fisso.
Le ceneri derivano direttamente dai costituenti inorganici presenti nella biomassa di
partenza. L’origine del materiale inorganico può essere duplice: inerente, cioè costituzionale, o di natura accidentale [11].
Per calcolarne il contenuto, è stato effettuata l’analisi secondo la Norma UNI 9903, in base alla quale una quantità nota di fango, opportunamente omogeneizzato è stata messa in una capsula di porcellana. La capsula contenente il campione è stata posta nella
muffola fredda, e si è riscaldato con un gradiente tale da evitare perdite di materiale dovute alla rapida espulsione di sostanze volatili.
La combustione è stata terminata a circa 600 °C. La capsula è stata raffreddata in essiccatore e pesata non appena fredda.
La percentuale di ceneri del campione è stata calcolata secondo la formula seguente: 100 × − = C B A Pc (1) dove:
Pc è il contenuto percentuale di ceneri;
A è la massa della capsula più la massa delle ceneri, in grammi; B è la massa della capsula vuota, in grammi;
C è la massa del campione analizzato, in grammi.
Per umidità si intende la quantità di acqua (libera e legata) presente nel materiale (ad una data temperatura).
Può essere suddivisa in umidità inerente, propria della struttura della biomassa, e umidità superficiale, fortemente influenzata dalla pezzatura e dalla condizioni ambientali. L’umidità della biomassa è una proprietà dinamica: tende a portarsi in equilibrio con l’ambiente e varia in maniera consistente in funzione del tipo di biomassa, delle condizioni ambientali, delle modalità e dei tempi di trasporto e stoccaggio. Il contenuto di umidità, generalmente espresso come percentuale in massa, è una parametro importante sia sotto il profilo tecnico-operativo sia in relazione ad alcune caratteristiche qualitative del materiale [11].
In laboratorio il contenuto di umidità viene determinato calcolando la perdita in massa di un campione sottoposto ad essiccamento, in condizioni normalizzate, alla temperatura di 105°C.
Il contenuto di volatili di un combustibile rappresenta la porzione di materiale rilasciata sottoforma di gas.
A tal fine una quantità nota di campione è stata sottoposta a riscaldamento fino a 950°C, e mantenuta a tale temperatura per un periodo di tempo complessivo pari a 7 min, come previsto dalla Norma UNI 9903.
Le sostanze volatili sono calcolate secondo la seguente formula: 100 × − = A B A SV (2)
dove:
SV è la quantità di sostanze volatili nel campione; A è la massa in grammi del campione analizzato;
B è la massa in grammi del campione dopo il riscaldamento.
Si è tenuto conto del fatto che fino a 100°C le sostanze rilasciate sono quasi esclusivamente acqua, per cui si è preso come base di riferimento, per stimare il valore di A, la massa in grammi del campione dopo riscaldamento a 100°C.
Il contenuto di carbonio fisso è la massa rimanente dopo il rilascio di volatili, escludendo le ceneri e il contenuto di umidità; esso è stato valutato secondo la
relazione:
CF = 100- Pc- SV (3) dove:
CF è il contenuto di carbonio fisso; Pc è il contenuto di ceneri;
SV è la percentuale di sostanza volatile.
6.4
Analisi elementare
Il contenuto percentuale di Carbonio, Idrogeno, Azoto e Ossigeno rappresenta la cosiddetta analisi elementare.
Le informazioni che è possibile trarre dall’analisi elementare si basano sostanzialmente sul rapporto percentuale tra i principali elementi e contribuiscono a definire la formula minima della biomassa.
L’analisi elementare viene inoltre utilizzata come supporto per valutare la qualità energetica della biomassa. Come andamento generale, ad alti contenuti di Carbonio e Idrogeno corrispondono elevati poteri calorifici; elevate concentrazioni di ossigeno e azoto hanno un effetto opposto. Azoto, Zolfo e Cloro infine, sono importanti soprattutto ai fini della valutazione delle emissioni inquinanti, delle prestazioni in impianto,con particolare riferimento a fenomeni corrosivi, e delle problematiche connesse. Il Cloro in particolare, oltre ad essere un precursore per la formazione di diossina durante la combustione, svolge un ruolo primario anche nella formazione di ceneri particolarmente aggressive.
Il campione di cui deve essere determinata la composizione percentuale di C, H, e N, viene pesato in contenitori di stagno. Il contenitore viene inserito in un campionatore, mediante il quale viene introdotto in un reattore di combustione mantenuto alla temperatura di 1050 °C.
I gas prodotti dalla combustione, trasportati da un flusso costante di elio, sono cataliticamente trasformati allo stato ossidato: CO2, H2O, N2, NXOY.
Un secondo reattore, mantenuto alla temperatura di 650 °C e riempito con rame, riduce gli ossidi di azoto ad azoto elementare. Una colonna cromatografia separa quindi N2,
CO2 e H2O.
Ogni componente, unitamente al gas di trasporto, fluisce in un rivelatore a conducibilità termica che genera un segnale elettrico proporzionale alla sua concentrazione. Tale segnale, opportunamente amplificato ed elaborato, viene visualizzato come picco cromatografico da un registratore potenziometrico. Dall’integrazione dei picchi è possibile risalire al contenuto di C, H, e N nel campione, attraverso un confronto con i valori forniti da un campione di riferimento analizzato nelle medesime condizioni.
Dalla misura eseguita sui campioni di riferimento si ricava il fattore K che viene calcolato per ciascun elemento da determinare secondo la seguente equazione:
1 % − ⋅ ⋅ = t Ws I K (4) dove : t
% = % teorica del campione di riferimento Ws = Peso del campione di riferimento in mg
I = Integrale del segnale relativo al campione di riferimento
La percentuale di ciascun elemento nel campione si ottiene dalla seguente equazione: I W K⋅ ⋅ = −1 % (5) dove :
K = Fattore medio riferito ciascun elemento I = Integrale del campione
W = Peso del campione espresso in mg
6.5
Analisi termogravimetrica (TGA)
Un parametro fisico particolarmente adatto allo studio dei cambiamenti chimici in un materiale è la variazione di peso che si verifica quando un materiale interagisce con l’atmosfera circostante evolvendo o assorbendo gas. Nell’analisi termogravimetrica (TGA) viene infatti misurata la variazione di peso che si registra in conseguenza del riscaldamento o raffreddamento di un certo campione secondo un determinato programma di temperatura.
Il campione del materiale in esame viene posto in un ambiente a temperatura controllata su un apposito sistema di pesatura in cui le variazioni di temperatura e di peso sono registrate in continuo.
L’apparecchiatura necessaria deve quindi essere dotata di una bilancia di precisione e di un forno riscaldato elettricamente la cui temperatura possa essere controllata con sufficiente precisione.
Negli strumenti moderni il forno è alimentato elettricamente attraverso un riscaldamento resistivo, le resistenze sono posizionate attorno ad un supporto tubolare, isolato elettricamente, che è un buon conduttore termico all’interno del quale viene posto il campione.
L’esterno della fornace deve essere isolato, inoltre di norma sono presenti specifici sistemi di raffreddamento ad aria (ventole) e/o termostatazione a circolazione di un opportuno fluido refrigerato (termostatato).
L’atmosfera intorno al campione può essere condizionata mediante l’immissione controllata di un gas con l’ausilio di opportune valvole e di un sistema di vuoto, in tal modo è possibile condurre prove sotto vuoto, in atmosfera statica o sotto flusso di gas che, a seconda delle esigenze, può essere inerte, riducente o ossidante e per certe particolari applicazioni anche corrosivo.
Il principale svantaggio che si ha utilizzando un’atmosfera statica deriva dalla possibile condensazione dei prodotti di reazione sulle parti più fredde dello strumento, ciò può causare corrosione nei meccanismi della bilancia o errori di pesatura se essi si depositano sugli elementi della bilancia. Inoltre in questo caso si possono avere reazioni secondarie tra i prodotti e il campione residuo.
La granulometria del campione influenza la perdita di peso dal momento che ha effetto
Figura 6.4: Dipendenza della curva TG dalla granulometria del campione
I campioni possono essere in forma di solido polverizzato (compresso sotto forma di pellet o sparso finemente sopra la superficie del crogiolo), di film sottile o di liquido. Generalmente sono preferiti i liquidi e le polveri fini, queste ultime non dovranno essere però eccessivamente fini poiché altrimenti potrebbero essere asportate dal flusso di gas.
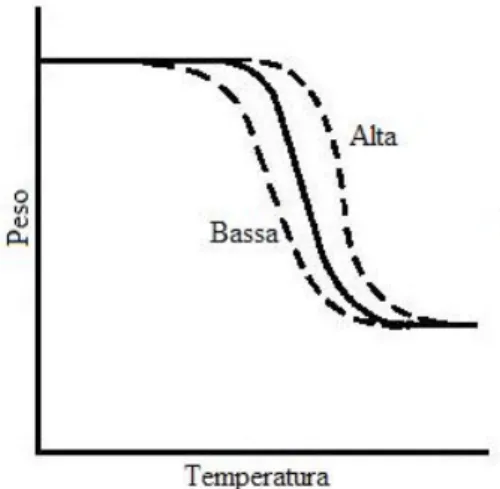
Il peso del campione, come la sua forma, può influenzare la curva TG incidendo sulla diffusione, e sul trasferimento di calore (Figura 6.5).
Figura 6.5: Dipendenza della curva TG dal peso del campione
Quando si opera con campioni di peso intorno a pochi milligrammi la spinta idrostatica del gas sul campione o la presenza di correnti convettive possono manifestarsi come disturbi nella curva termogravimetrica o apparenti variazioni di peso.
In particolare scaldando un portacrogiolo vuoto la spinta idrostatica causa un apparente aumento di peso pari al peso del gas spostato dal portacrogiolo.
Poiché la temperatura cambia, anche la densità del gas cambia e ciò provoca una variazione della spinta idrostatica che si traduce in una apparente variazione di peso anche se di norma trascurabile. Per questo motivo per analisi accurate è necessario
effettuare delle prove in bianco nelle stesse condizioni di analisi e sottrarre dalla curva dell’analisi quella della prova in bianco.
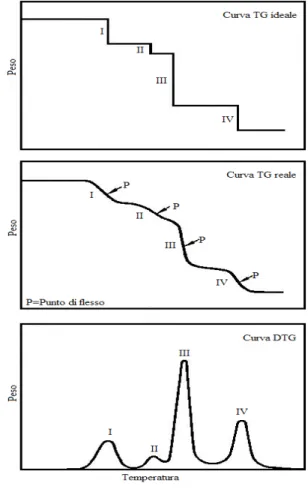
Una curva termogravimetrica di norma mostra, in un grafico in cui si riporta la perdita di peso in funzione della temperatura, una serie di scalini più o meno accentuati che possono essere separati da tratti ad andamento costante del peso; alcuni processi però possono avvenire in un intervallo di temperatura più ampio tale da sovrapporsi ad altri fenomeni così da rendere non risolti i vari stadi di perdita di peso.
In questi casi risulta utile ricorrere alla termogravimetria derivativa (DTG) grazie alla quale è possibile individuare una serie di picchi che corrispondono ai vari stadi della perdita di peso e i cui massimi corrispondono ai punti di flesso della curva TG (Figura 6.6).
Inoltre l’area dei picchi è proporzionale alla perdita di peso di ogni singolo stadio.
Figura 6.6: Curva TG ideale, curva TG reale, curva DTG
Un riscaldamento veloce aumenta la temperatura alla quale ha luogo una reazione e alla quale la velocità di perdita di peso raggiunge il massimo (Figura 6.7), inoltre aumenta
l’intervallo nel quale è osservata la perdita di peso; quest’ultimo effetto si verifica anche nel caso di bassa velocità di riscaldamento, in tal caso però diminuisce la temperatura alla quale appare la reazione.
Figura 6.7: Dipendenza della curva TG dalla velocità di riscaldamento
Tra i possibili impieghi della termogravimetria appare interessante lo studio del comportamento termico dei materiali in relazione alla loro degradazione.
La degradazione termica provoca infatti lo sviluppo di sostanze volatili che si allontanano dal campione in fase gassosa, pertanto la termogravimetria si presta particolarmente bene per seguire l’evolversi delle materie volatili mentre un dato campione viene sottoposto a riscaldamento.
Ciò permette di valutare la temperatura al di sopra della quale un dato materiale perde le sue originali caratteristiche e quindi permette anche di definire il campo di impiego entro certi intervalli di temperatura.
Le analisi termogravimetriche sono state effettuate utilizzando uno strumento Netzsch 409C. Per fango e pellet di legno è stato utilizzato un campione di circa 20 mg posto in un crogiolo di Platino di circa 1 cm di diametro, sottoposto al seguente programma di riscaldamento in atmosfera di azoto, per evitare l’ossidazione:
• Preriscaldamento a 40°C;
• Da 40°C riscaldamento fino a 950°C, con una velocità di riscaldamento di 20°C/min;
• A 950°C mantenimento della temperatura per 60 minuti.
Lo strumento consente di registrare la variazione del peso del campione in funzione della temperatura, in tal modo si ricavano le curve termogravimetriche (TG); per
derivazione via software di queste ultime si ottengono le curve DTG dette profili di perdita di peso.
Per il tar è stato utilizzato lo stesso assetto descritto in precedenza (20 mg di campione in crogiolo di Platino) ed è stato sottoposto al seguente programma di riscaldamento in atmosfera di azoto, suggerito dalla letteratura [37]:
• periodo isotermo di 15 min a 40°C;
• riscaldamento da 40°C fino a 247°C, con una velocità di riscaldamento di 4°C/min;
• periodo isotermo di 10 min a 247°C.
6.6
Spettrometria ad emissione atomica con plasma
induttivo (ICP-AES)
Per ICP (Inductively Coupled Plasma) si intende un plasma di argon ad una temperatura di 8000 K, nel quale viene introdotto il campione per nebulizzazione a pressione ambiente.
Il plasma si forma per interazione del gas con un campo di radiofrequenze.
L’accoppiamento tra il plasma e il generatore di radiofrequenza è ottenuto da una spira avvolta attorno alla torcia di quarzo che contiene il flusso d'argon.
La torcia consiste di tre tubi di quarzo concentrici che contengono tre flussi separati d’argon. Il campione viene introdotto come aerosol fine o gas trasportato attraverso il tubo più interno. Il flusso di gas più esterno serve invece come gas di raffreddamento per proteggere il quarzo dalla fusione, e anche come gas per sostenere il plasma. Il flusso di gas ausiliare, che passa nel tubo di mezzo, serve a mantenere il plasma caldo lontano dall'estremità del capillare centrale di iniezione. Sia il gas di raffreddamento sia quello ausiliario vengono introdotti da tubi laterali per creare un flusso vorticoso. Le spire che avvolgono la torcia sono fatte da 2-4 giri di tubi di rame raffreddati con acqua, posizionati pochi millimetri dietro la bocca della torcia. Un generatore di radiofrequenza fornisce corrente alle spire, con una frequenza di 27.12 o 40.68 MHz, in modo da indurre un campo magnetico con asse coincidente a quello della torcia.
L’energia elettrica fornita alle spire viene convertita in energia cinetica degli elettroni del plasma. A pressione atmosferica il cammino medio di un elettrone non è superiore
ad 1 µm: infatti gli elettroni collidono con gli atomi d’argon dando luogo ad una scarica luminosa. La maggior parte dell’energia viene immagazzinata nella parte più esterna del plasma (detta zona di induzione), mentre l’apporto del campione nebulizzato viene diretto verso la zona centrale più fredda. Nella zona di induzione si raggiungono 10000 K, mentre il canale centrale, riscaldato principalmente per radiazione e conduzione, ha temperature tra 7000 e 9000 K in corrispondenza della bocca della torcia. Il fatto che la zona che risente dell’energia elettrica sia diversa da quella in cui viene introdotto il campione rende possibile mantenere costanti le caratteristiche generali del plasma anche quando si abbiano grandi variazioni nella composizione del campione.
L’aerosol del campione, dopo aver lasciato il capillare di iniezione, viene rapidamente essiccato dando luogo a microparticelle solide. A temperature crescenti queste particelle vengono vaporizzate, atomizzate e ionizzate durante il tragitto di pochi millisecondi nel plasma.
Il grado di ionizzazione dipende sia dalla condizioni del plasma sia dai potenziali di ionizzazione di ciascun elemento. Le condizioni del plasma saranno perciò ottimizzate in modo diverso in funzione della tecnica di rivelazione adottata.
Il campione può essere prelevato da una bottiglia attraverso una pompa peristaltica e viene introdotto nel canale centrale della torcia ICP come gas o aerosol fine di particelle liquide o solide, ottenuto utilizzando un nebulizzatore pneumatico, nel quale un flusso di gas ad alta velocità trasforma la soluzione del campione in goccioline finemente disperse. Il comparto composto da pompa peristaltica, nebulizzatore e camera spray può risentire del calore riflesso dal cono di campionamento e quindi deve essere ben isolato dal comparto della torcia ICP.
Poiché nel plasma tutti gli atomi sono eccitati simultaneamente, la luce emessa dal plasma è una combinazione di tutte le lunghezze d’onda dei singoli atomi e ioni presenti nel campione e nel gas del plasma. La raccolta radiale del segnale ottico può essere effettuata ad altezze variabili del plasma, in modo da ottimizzare l’analisi per ciascun elemento. Questa emissione a banda larga deve essere raccolta e separata nelle singole lunghezze d'onda, che vengono inviate su un rivelatore (fotomoltiplicatore o a stato solido) che produce un segnale elettrico di intensità proporzionale all’intensità delle righe di emissione. Le intensità di emissione vengono rilevate, simultaneamente o in sequenza, e la concentrazione di analita presente nel campione viene determinata per confronto con una soluzione di riferimento a concentrazione nota.
La tecnica ICP-AES è soggetta sia ad interferenze spettrali sia a interferenze non-spettrali o fisiche. Queste ultime sono associate al trasporto fisico del campione nello strumento, cioè le differenze di viscosità e tensione superficiale tra il campione e gli standard acquosi di calibrazione. Invece una interferenza spettrale è dovuta alla presenza di luce alla lunghezza d’onda dell’analita che non è dovuta all’analita o al semplice rumore di fondo. Essa può avere diverse cause, ad esempio la deriva del fondo e l'interferenza diretta di un'altra specie elementare o ionica.
Il modello usato per le analisi svolte è un ICP-AES Parkin-Elmer 400.
La preparazione eseguita sui campioni di ceneri da sottoporre all’analisi è la seguente: • viene preparato 1 g di campione macinato ed essiccato;
• si prepara una soluzione acquosa di HCl/HNO3 3:1 in volume;
• si miscela il tutto e si riscalda a 160°C per 2 ore;
• si filtra il campione e si diluisce con acqua fino ad un volume di 100 ml; • si sottopone il campione all’analisi ICP.
La procedura usata per pellet di legno e fanghi è la stessa riportata sopra: cambia tuttavia la quantità di campione, pari questa volta a 2 g.
6.7
Test di lisciviazione delle ceneri
In accordo alla normativa UNI 12457-2 sono stati effettuati dei test di lisciviazione sul residuo solido raccolto nell’impianto alla fine delle prove di gassificazione. La cessione di composti solubili al contatto con l’acqua è considerato infatti uno dei principali meccanismi di rilascio in grado di produrre un potenziale rischio per l’ambiente durante il riutilizzo o lo smaltimento dei rifiuti. Lo scopo di tali prove è quindi quello di identificare le proprietà di lisciviazione delle ceneri ottenute da gassificazione di fanghi da depurazione, in modo da stabilirne la pericolosità in ottica del successivo smaltimento.
La procedura utilizzata per il test è la seguente:
• viene preparato un campione di 90 g di residuo solido macinato ed essiccato: il materiale deve avere una granulometria per il 95% inferiore ai 4 mm. Tuttavia il materiale deve essere più omogeneo possibile, cercando di evitare la formazione di particelle eccessivamente fini;
• Il campione solido viene collocato all’interno di una bottiglia di vetro o di polietilene (HDPE)/polipropilene (PP) del volume nominale di 1 L;
• si aggiunge l’agente lisciviante (acqua distillata, demineralizzata o de-ionizzata) in quantità tale che il rapporto tra liquido e solido sia pari a 10 (per 90 g di campione, 900 ml di acqua);
• si colloca la bottiglia tappata in un agitatore a rotazione (o a rovesciamento) che imponga alla bottiglia una velocità rotazione di circa 10 giri/min;
• dopo 24 ore si filtra l’eluato su una membrana filtrante da 0,45 µm, utilizzando un dispositivo di filtrazione sotto vuoto o a pressione;
• si suddivide l’eluato in un numero di sottocampioni, a diluizione desiderata, su cui saranno eseguite le analisi chimiche d’interesse.
Nel caso del presente lavoro di ricerca si è ritenuto opportuno effettuare un indagine per valutare il contenuto di metalli pesanti, cloruri, solfati e nitrati ceduti dal residuo all’agente lisciviante, in modo da poter stabilire il grado di pericolosità delle ceneri come rifiuto da smaltire in discarica dedicata.
6.8
Gas-cromatografia
6.8.1 Generalità
La cromatografia é una tecnica di separazione di vari componenti di una miscela, essa consiste nello sfruttare in modo particolarmente efficiente la diversa attitudine che ogni molecola o ione possiede nel distribuirsi tra due differenti fasi (una stazionaria e una mobile).
Nella tecnica cromatografica una fase viene immobilizzata (fase stazionaria) e l’altra viene fatta scorrere sopra di essa (fase mobile, o 'eluente'); é così possibile condurre l’estrazione in modo continuo. Una specie chimica depositata sulla fase stazionaria e immessa nella corrente di fase mobile si distribuirà infatti dinamicamente tra le due fasi, in misura proporzionale alla diversa affinità che possiede per esse.
La fase stazionaria può essere costituita da un solido o un liquido opportunamente supportato.
La fase mobile è costituita da un fluido (che si muove sopra la fase stazionaria) che può essere sia un liquido che un gas.
Supponendo di avere una colonna riempita uniformemente di un materiale solido in granuli di dimensioni omogenee (la cosiddetta fase stazionaria, o fase fissa), il principio su cui si basa la cromatografia è descritto di seguito.
All'inizio della colonna si deposita la miscela contenente le sostanze da separare.
Si fa scorrere poi un solvente (la fase mobile, detta eluente): la fase mobile trascinerà in modo diverso le diverse sostanze lungo la colonna, a seconda della loro affinità verso le due fasi.
Alla fine della colonna viene posto un rivelatore in grado di evidenziare le varie sostanze che fuoriescono in tempi diversi emettendo un segnale con una intensità proporzionale alla loro concentrazione, il segnale viene registrato da uno strumento che darà così luogo al cromatogramma, che riporta le concentrazioni di sostanza in uscita in funzione del tempo o del volume di eluente (Figura 6.8).
Figura 6.8: Schema di funzionamento del principio alla base della cromatografia
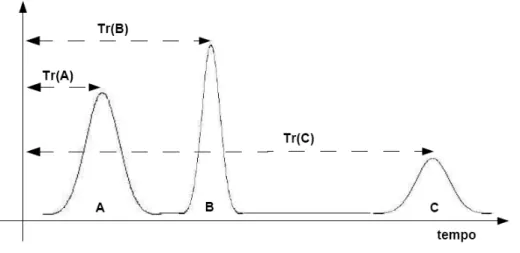
Ogni sostanza iniettata in colonna (in determinate condizioni di flusso di carrier, di fase stazionaria e di temperatura) è caratterizzata dal tempo di ritenzione Tr (Figura 6.9), che corrisponde al tempo che intercorre fra l'introduzione del campione e la sua uscita dalla colonna (50% della quantità totale).
Il tempo di ritenzione viene di solito riferito ad una sostanza non trattenuta dalla colonna, quale ad esempio l'aria; le sostanze più trattenute dalla fase stazionaria della colonna avranno un tempo di ritenzione alto e viceversa.
Figura 6.9: Cromatogramma che illustra vari tempi di ritenzione
Fissate tutte le condizioni operative (temperatura, lunghezza colonna, diametro, fase stazionaria, velocità…) un certo componente esce ad un certo tempo di ritenzione che è riproducibile; questo vuol dire che fissate le condizioni operative se tale componente è presente in concentrazioni diverse e in miscele diverse esce sempre a tale Tr.
Se si considera un sistema formato da due fasi in cui viene introdotta una sostanza: la sostanza si distribuirà fra le due fasi a seconda delle sue particolari proprietà chimico-fisiche.
Indicando con Cm e Cs le sue concentrazioni nella fase mobile e nella fase stazionaria rispettivamente, e supponendo che le condizioni sperimentali siano tali da conseguire il raggiungimento di equilibri successivi del tipo:
Cs Cm←→K
K prende il nome di coefficiente di distribuzione e corrisponde alla costante di equilibrio:
K = Cs / Cm
E’ dal valore di K che dipende il tempo di ritenzione, infatti il tempo che una sostanza trascorre nella colonna dipende dal valore di Cs rispetto a Cm; un’elevata concentrazione nella fase stazionaria, rispetto a quella nella fase mobile, indica una maggiore affinità per la prima.
In altre parole, l’eluente (fase mobile) incontrerà una certa difficoltà nel trascinare con sé alcune sostanze, mentre altre, relativamente più affini ad esso e meno verso la fase stazionaria, verranno più facilmente dislocate dalle posizioni che occupano e trasportate
così verso la fine della colonna, separandosi sempre di più dalle sostanze maggiormente trattenute.
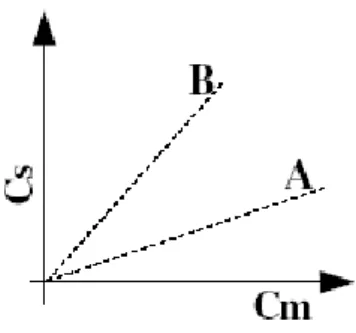
L’equazione K = Cs / Cm è l’equazione di una retta, la cui pendenza è data da tg α = K, tale retta è detta isoterma di distribuzione (Figura 6.10).
Nel caso in cui si abbiano due sostanze A e B per le quali Ka < Kb si ha che la sostanza B è più affine per la fase fissa di quanto lo sia la sostanza A. Se queste due sostanze A e B percorrono insieme la colonna, accade che A uscirà per prima e quanto maggiore è la differenza di K tanto migliore sarà la separazione tra le sostanze.
Figura 6.10 : Rappresentazione della isoterma di distribuzione per due composti distinti
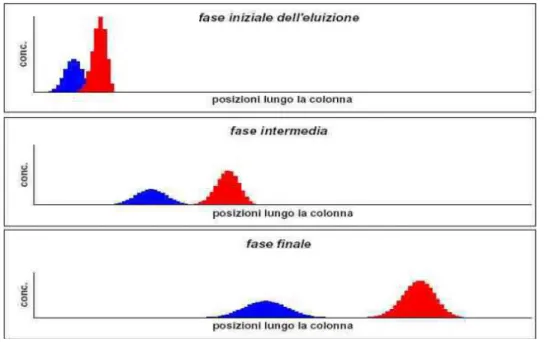
Nella realtà le isoterme non hanno l’andamento teorico visto (Figura 6.11), ad un certo punto la fase fissa non trattiene la stessa quantità di sostanza rispetto alla fase mobile e si avvicina alla saturazione, cioè la capacità solvente della fase fissa può dirsi esaurita; per questa ragione le singole zone del cromatogramma non hanno contorno preciso e simmetrico.
Figura 6.11: Rappresentazione della isoterma di distribuzione reale
Nella tecnica gas-cromatografica la fase mobile é un gas che fluisce attraverso una colonna in cui si trova la fase stazionaria, la quale può essere un solido granulare poroso oppure un liquido. Secondo lo stato fisico della fase stazionaria, la gas-cromatografia si
può suddividere in cromatografia gas solido (GSC) e in cromatografia gas liquido (GLC).
L’unica limitazione della gas-cromatografia é la necessità di rendere volatili i campioni da analizzare, per cui in alcuni casi essa é soppiantata dall’ HPLC.
I meccanismi di separazione relativi alla GC sono sostanzialmente due:
I principali meccanismi chimico-fisici della separazione cromatografica per i gas si basano su:
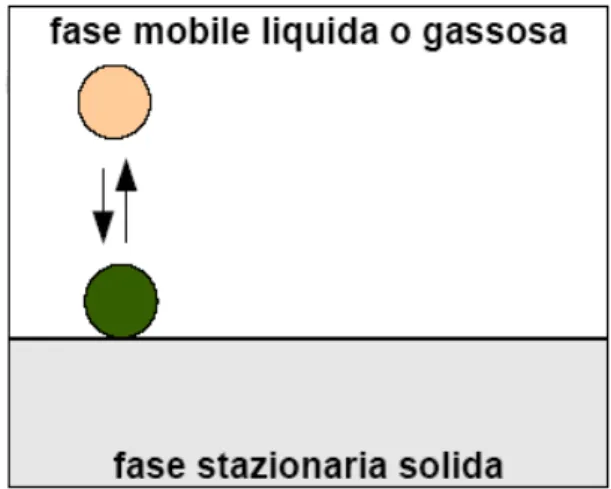
• Adsorbimento: la fase stazionaria é un solido sulla cui superficie si trovano dei siti attivi in grado di stabilire legami secondari (dipolo-dipolo, ponte di idrogeno, Van der Waals) con le diverse molecole della miscela da separare (Figura 6.12). Se la fase mobile é un liquido si parla di cromatografia liquido-solido (LSC), se invece é un gas, di cromatografia gas-liquido-solido (GSC).In genere, le molecole che più facilmente vengono fissate sono quelle che presentano gruppi polari, anche se la natura dell’adsorbente influisce sul fenomeno. L’aumento di temperatura agisce negativamente sull’adsorbimento in quanto provoca una maggior agitazione termica.
Figura 6.12 : Rappresentazione del meccanismo di adsorbimento
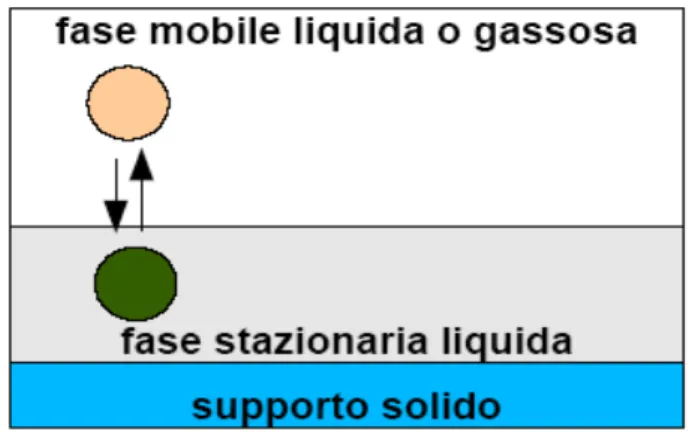
• Ripartizione: la fase stazionaria é un liquido, immiscibile con la fase mobile. Si ha una ripartizione dei soluti tra le due fasi, in questo meccanismo ci rientra l’assorbimento (Figura 6.13). Se la fase mobile é un gas si parla di cromatografia gas-liquido (GLC), se invece é un liquido, di cromatografia liquido-liquido (LLC).
Figura 6.13 : Rappresentazione del meccanismo di ripartizione
6.8.2 Strumentazione
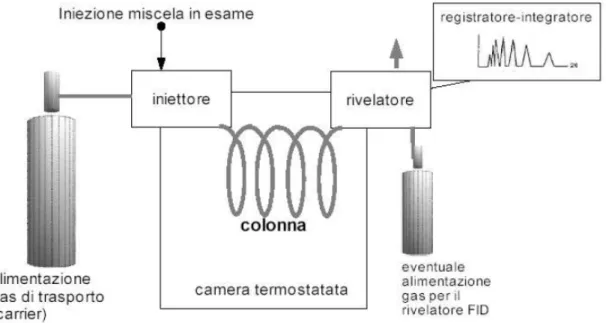
Lo schema essenziale di un tipico gas-cromatografo è riportato in Figura 6.14. • Sistema di alimentazione gas di trasporto (carrier)
Lo scopo principale é quello di trascinare i componenti della miscela in analisi lungo la colonna cromatografica. I gas più comunemente usati come carrier sono: azoto, elio, argon, talvolta può essere utilizzato anche l’idrogeno. Per una questione di reperibilità e di costi, generalmente si usa l'azoto. Prima comunque che venga introdotto nella colonna é necessario che vengano eliminate le tracce di ossigeno e di anidride carbonica che, se non eliminate, potrebbero interagire con le sostanze delle miscele che si vogliono separare. Inoltre é anche necessario anidrificare il carrier, in quanto la presenza di acqua determinerebbe, nel caso di una gascromatografia di adsorbimento, una disattivazione dei siti attivi della fase stazionaria.
Figura 6.14 : Schema di un tipico gas-cromatografo
• Sistema di alimentazione dei gas per il rivelatore FID
Qualora si utilizzi un rivelatore a ionizzazione di fiamma (FID) è necessario alimentare un combustibile ed un comburente (ad esempio idrogeno ed aria). • Iniettore o camera di iniezione
Il suo compito é assicurare l’istantanea vaporizzazione del campione liquido. Poiché con l’uso di colonne capillari la quantità di campione liquido da iniettare é dell’ordine dei nanolitri, e misurare queste quantità con siringhe é praticamente impossibile, sono state messe a punto particolari tecniche di iniezione.
Spesso si utilizzano quindi opportune tecniche (split, ...) che consentono di far entrare effettivamente in colonna solo una parte del liquido iniettato. La camera di iniezione è corredata da un sistema di resistenze variabili attraverso le quali è possibile fissare la temperatura ritenuta più adatta per la vaporizzazione della miscela. L’introduzione del campione viene effettuata con una iniezione su un apposito disco di gomma al silicone, posto tra una ghiera metallica e il dispositivi di attacco alla colonna.
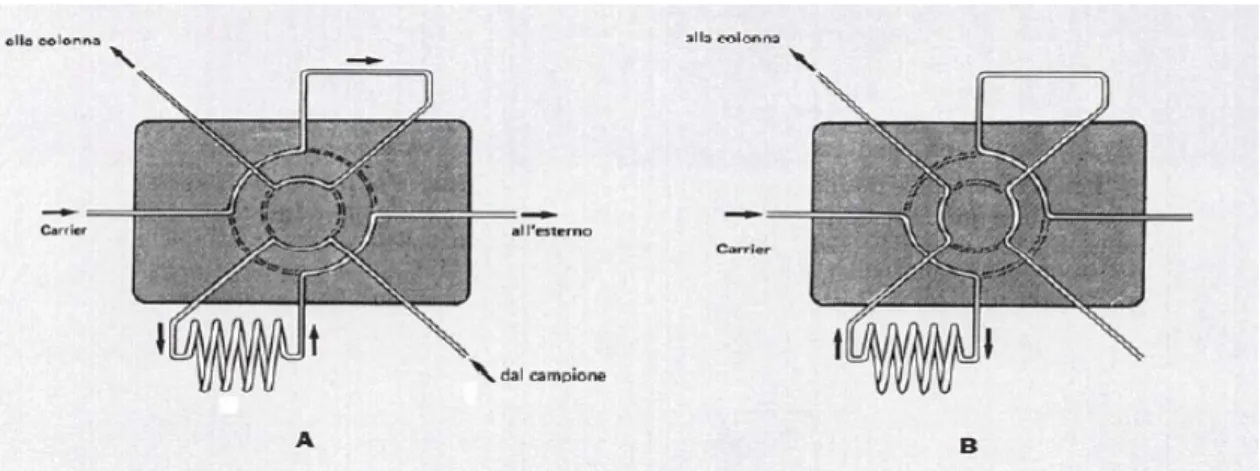
Per quanto riguarda l’ introduzione di gas si utilizza un sistema di valvole come riportato in Figura 6.15.
Osservando la figura si nota che nella configurazione A in colonna fluisce solo il gas di trasporto e il campione passa attraverso il serpentino riempiendolo. Quando la parte centrale ruota si ha la configurazione B dove il campione è escluso e entra
e esce senza interferire con niente; il gas di trasporto invece va sempre in colonna,ma prima passa dentro il serpentino trascinando con se il campione che si è qui accumulato.
Figura 6.15: Rappresentazione del sistema di valvole per l’introduzione del gas
• Colonna
La colonna può essere di due tipi:
- colonna impaccata (diametro interno 2-4 mm, lunghezza 1-4 m): usata nella gas-cromatografia classica, comporta una separazione in colonna di acciaio o di vetro(due metri circa) riempita di materiale inerte (supporto per la fase stazionaria) sul quale è distribuita una pellicola sottile di liquido (fase stazionaria) continuamente attraversata da un gas (fase mobile) detto gas di trasporto. Il processo di separazione è limitato dalla lentezza di eluizione della molecole del campione lungo la colonna.
- colonna capillare (diametro interno 0,1-0,8 mm, lunghezza 10-100 m), ormai di uso comune, sono realizzate in silice fusa, rappresenta un’importante innovazione per la sua rapidità di eluizione e per una migliore risoluzione (il numero di picchi risolti, in metà tempo, è superiore di oltre quattro volte quello della colonna impaccata). Essa è molto più lunga dell’impaccata di diametro molto minore e quindi contiene una quantità molto minore di fase stazionaria, per cui la quantità di campione da iniettare è molto più piccola e viene fluita prima.
Le colonne sono alloggiate in una camera termostatica, in genere a circolazione di aria calda, con questo sistema viene assicurata una buona stabilità di temperatura. Un dispositivo permette all’operatore di fissare la temperatura, la quale può essere
mantenuta costante per tutta la durata dell’analisi (isoterma) oppure fatta variare (programmata).
• Rivelatore
I dispositivi in grado di rivelare la presenza di una sostanza estranea nel gas di trasporto, a valle della colonna, possono dividersi in universali e selettivi. I primi consentono di individuare tutti i componenti di una miscela, i secondi rivelano solo particolari categorie di composti.
Tra i rivelatori più usati, si segnalano:
- Rivelatore a ionizzazione di fiamma (FID): si tratta di un rivelatore universale ma distruttivo in quanto i campioni vengono bruciati per ottenerne la trasformazione in ioni allo stato gassoso (Figura 6.16). Il carrier viene convogliato verso un ugello a cui giungono anche idrogeno ed aria, necessari per alimentare una piccola fiammella. Una resistenza posta accanto all’ugello provoca l’accensione della fiammella. Quest’ultima si trova circondata da un collettore cilindrico caricato positivamente; il secondo elettrodo del circuito, quello caricato negativamente, é costituito dall’ugello stesso. La microfiamma provoca una debolissima corrente ionica tra gli elettrodi, che vengono mantenuti sotto una differenza di potenziale di circa 300V. Questa corrente, elaborata, amplificata e misurata, viene inviata ad un opportuno registratore e costituisce il rumore di fondo. Quando un componente della miscela raggiunge la fiamma, viene subito ionizzato con conseguente aumento dell’intensità di corrente e quindi rivelato con un segnale più intenso. Come già detto questo rivelatore é di tipo universale, sono poche infatti le sostanze che hanno potenziali di ionizzazione così alti da non poter essere ionizzate nelle normali condizioni di lavoro (tra queste abbiamo acqua, solfuro di carbonio, anidride carbonica, ossido di carbonio, ossidi di azoto, ammoniaca, acido solfidrico, biossido di zolfo, acido formico, gas nobili, azoto e ossigeno). La sensibilità di questo rivelatore é molto elevata, infatti si può arrivare ai nanogrammi.
Figura 6.16: Schema di un rilevatore FID
- Rivelatore a cattura di elettroni (ECD): si tratta di un rivelatore selettivo e non distruttivo (Figura 6.17). Esso é costituito da una sorgente radioattiva (63Ni) che
emette radiazioni beta (elettroni). Gli elettroni, detti primari, emessi dalla
sorgente, vengono a trovarsi in un campo elettrico di cui la sorgente costituisce
l’anodo, mentre il catodo si trova verso l’uscita. Gli elettroni primari colpiscono il
carrier formando ioni positivi ed elettroni secondari. Il flusso di queste cariche
costituisce la corrente di fondo e dipende dalla differenza di potenziale tra i due
elettrodi. Quando insieme al carrier é presente un’altra sostanza elettroaffine, cioè
in grado di catturare gli elettroni secondari, si verifica una diminuzione di corrente
di fondo. La corrente, elaborata, amplificata e misurata, viene inviata ad un
registratore. I limiti di rilevabilità possono essere molto bassi, ad esempio per i
pesticidi cloro-organici o derivati del fosforo, si può arrivare a rivelare i
picogrammi. Le sostanze maggiormente rivelate sono quelle contenenti alogeni.
- Rivelatore a conducibilità termica (TCD): si tratta di un rivelatore universale e non distruttivo. Si basa su due sensori contenenti un filamento la cui resistenza elettrica varia al variare della temperatura. La temperatura dipende a sua volta dalla conducibilità termica dei gas con cui sono a contatto i filamenti (e che varia con la composizione dei gas stessi). Un sensore è lambito dal carrier puro mentre l'altro è sull'uscita della colonna: un accurato sistema elettrico rileva ed amplifica le differenze dei due segnali. La sensibilità di questo rivelatore non è elevata ed inoltre costringe all'uso di carrier più costosi (ad esempio elio e argon).
Figura 6.17 : Schema di un rilevatore ECD
- Rivelatore a spettrometria di massa: si tratta di un rilevatore che permette l’ identificazione e la caratterizzazione strutturale delle molecole, sarà analizzato in modo dettagliato successivamente.
• Registratore e integratore
Il segnale in uscita dal rivelatore passa ad un registratore che ha il compito di realizzare il tracciato
cromatografico. I moderni strumenti sono corredati anche di un integratore che permette il calcolo automatico delle aree dei picchi, operazione indispensabile per effettuare analisi di tipo quantitativo.
• Cromatogramma
Ogni sostanza in uscita dalla colonna genera un segnale che verrà registrato sotto forma di 'picco'.
Ogni picco è caratterizzato da:
- altezza del picco: è la distanza fra il massimo del picco e la sua base, misurata perpendicolarmente all’asse dei tempi.
- ampiezza del picco: è il segmento delimitato sulla base del picco dai punti di intersezione delle tangenti tracciate nei punti di flesso di ambedue i lati (Figura 6.18).
La successione dei vari picchi, corrispondenti alle varie sostanze in uscita dalla colonna, costituisce il 'cromatogramma'. Il cromatogramma riporta in ordinate la risposta del rivelatore e in ascisse i tempi di uscita delle varie sostanze.
Figura 6.18 : Rappresentazione di un picco
A questo punto, dal grafico (oltre a altezza e ampiezza dei picchi) si determinano due parametri essenziali:
- tempo di ritenzione:
– è il tempo impiegato tra l’iniezione del campione e la registrazione del massi- mo del picco;
– dipende dalla natura della sostanza, dalla colonna e dalle condizioni operative; – è fondamentale per le analisi qualitative.
- area del picco:
– è la superficie delimitata dal contorno del picco e la linea di base;
– dipende dalla quantità di sostanza in uscita e dalle caratteristiche del rivelatore;
– è fondamentale per le analisi quantitative.
Dall’analisi del cromatogramma si vanno a definire parametri fondamentali relativi alla separazione cromatografia. L’esame di questi parametri (selettività, efficienza, risoluzione) é fondamentale per la scelta delle colonne e della temperatura.
Per colonne capillari non é necessario cercare grande selettività in quanto la loro grande efficienza può compensare una minor selettività.
- Selettività: è definita come la capacità di una colonna di fornire picchi distanziati e dipende dalla temperatura e dalla natura della fase stazionaria.
In Figura 6.19 sono riportati due cromatogramma, di una miscela di due composti, ottenuti con due diverse fasi stazionarie: nel secondo caso si ha una maggior
Figura 6.19 : Rappresentazione di due picchi per comprendere il concetto di selettività
- Efficienza : è la capacità del sistema cromatografico di mantenere compatta la
banda di eluizione di una sostanza lungo tutto il percorso della fase mobile. Ciò significa ottenere picchi alti e stretti all’uscita
della colonna. La cosa é di grande importanza, perché qualora due sostanze avessero tempi di ritenzione molto vicini se ne potrebbe ottenere ugualmente la separazione. Quindi, quanto più stretti sono i picchi tanto più efficiente risulta la colonna. In Figura 6.20 si riportano due cromatogrammi di una miscela di due sostanze effettuati con colonne diverse; in ambedue i casi si ha la stessa selettività, ma nel secondo caso si ha una maggior efficienza.
- Risoluzione: questo fattore tiene conto sia della selettività che dell’efficienza, e indica il grado di effettiva separazione ottenuto per due sostanze in un processo cromatografico.
Dal punto di vista numerico si ottiene dalla relazione:
2 2 ) ( ) ( B A r r W W A T B T R + − =
Per avere una buona separazione, dal punto di vista quantitativo, si deve avere risoluzione almeno 0,8 . Per capire il significato di tale relazione si consideri l’esempio riportato in Figura 6.21.
Figura 6.21: Rappresentazione dei parametri alla base della risoluzione
6.8.3 Analisi qualitativa
L’interpretazione dei cromatogrammi rappresenta l’operazione più lunga. E’ necessario innanzitutto avere la più completa serie di informazioni sulla natura e l’origine della miscela da analizzare.
I metodi utilizzabili per l’individuazione delle sostanze sono:
- basarsi su dati di letteratura, quali i tempi di ritenzione; tali valori dipendono da molti fattori quali le caratteristiche dello strumento, le condizioni operative e l’operatore.
- effettuare un confronto dei tempi di ritenzione tra la miscela in esame e sostanze pure o miscele di composizione nota.
- metodo basato sull’arricchimento. Quando si ritiene che un determinato picco corrisponda ad una sostanza nota, si aggiunge alla miscela una certa quantità di sostanza pura. Se compare un altro picco, siamo sicuri che la specie nota non é presente nella miscela, mentre se un picco risulta più alto, potrebbe essere presente e per questo é necessario effettuare altre analisi cambiando condizioni operative. - impiego di strumenti ausiliari. Il gas in uscita da un rivelatore non distruttivo può essere fatto gorgogliare in appositi solventi e la soluzione indagata con altri metodi strumentali.
Inoltre é possibile collegare direttamente il gas-cromatografo ad uno spettrometro di massa , in questo modo si può registrare lo spettro di massa il quale é univoco per una certa specie chimica (e fornisce poi importanti indicazioni strutturali).
6.8.4 Analisi quantitativa
Tale metodo si basa sul confronto delle aree dei picchi.
Si deve però tenere conto di una serie di possibili complicazioni:
- non è detto che tutte le sostanze presenti nel campione si vedano nel cromatogramma;
- i rivelatori possono presentare diverse risposte per diverse sostanze; - non tutti i picchi potrebbero essere ben separati;
- non è facile conoscere accuratamente la quantità di miscuglio effettivamente immesso in colonna.
A causa di ciò esistono varie metodologie di studio quantitativo, quali: • Confronto diretto dell’area dei picchi
Risulta il caso più semplice e si può avere ad esempio usando un FID per la rivelazione di idrocarburi. Se la risposta del rivelatore é uguale per tutti i componenti e se questi sono rappresentati tutti nel cromatogramma da picchi ben distinti e risolti, si ha che il rapporto tra area picco e concentrazione del componente é uguale per tutti i picchi.
In questo caso, la % in massa di ciascun componente si ottiene dividendo l’area del rispettivo picco per la somma delle aree di tutti i picchi, rapportando il valore a 100, cioè la % in massa corrisponde alla % delle aree.
Nel caso in cui le condizioni per utilizzare questo metodo non fossero verificate si procede con gli altri metodi.
• Taratura diretta
Con questo metodo é possibile determinare la concentrazione dei soli componenti che interessano.
La procedura è la seguente:
- si inietta un volume noto del campione e si registra il cromatogramma;
- si inietta lo stesso volume di una miscela a concentrazione nota ('standard') dei componenti da determinare, e si registra il cromatogramma;
- Si procede al calcolo diretto; ad esempio per una sostanza A:
campione A campione A dard s A dard s A C S C S tan : tan = :
E’ importante fare in modo che la concentrazione nello standard e nel campione non siano molto diverse. Il principale inconveniente sta nella misurazione del volume da iniettare, per questo risulta migliore fare diverse iniezioni calcolare la media delle aree. Il notevole vantaggio di questo metodo é che non obbliga a lavorare su tutti i componenti della miscela.
6.9
Spettrometria di massa
6.9.1 Generalità
La spettrometria di massa è una tecnica analitica di informazione strutturale basata sulla ionizzazione di una molecola e sulla successiva frammentazione in ioni di diverso rapporto massa/carica (M/z).
Il principio del metodo si basa sul fatto che una molecola è ionizzata per espulsione di un elettrone; il catione radicalico che si forma (ione molecolare) in parte si frammenta dando molecole e/o radicali neutri (che lo strumento non rileva), in parte generando cationi e/o radicali cationici (ione frammento).
Lo ione molecolare e i vari ioni che si originano per frammentazione vengono discriminati sulla base del loro rapporto massa/carica e rivelati da un detector.
La tecnica di spettrometria di massa è caratterizzata dunque da tre fasi principali: - ionizzazione di una molecola in fase gassosa;
- separazione dei diversi ioni prodotti; - rilevazione delle specie ioniche prodotte.
Il risultato finale visivo è lo spettro di massa, che rappresenta l’abbondanza relativa degli ioni in funzione del loro rapporto M/z. Di seguito (Figura 6.22) si riporta a titolo esemplificativo lo spettro di massa del n-decano.
Figura 6.22 : Spettro di massa del n-decano
La spettrometria di massa consente di misurare le masse molecolari (sia nominali che esatte) e di ottenere dei profili di frammentazione che sono specifici per ciascun composto, di cui costituiscono quindi un’impronta digitale.
Si può così individuare la formula di struttura di composti sconosciuti, anche avendone a disposizione piccole quantità.
L’interpretazione dello spettro di massa consiste dunque nello studio dei segnali dovuti agli ioni generati nella prova con l’obiettivo di:
- individuare le sostanze presenti in una miscela ignota di partenza;
- misurare le masse molecolari e le formule di struttura dei composti più complessi.
6.9.2 Strumentazione
Lo spettrometro di massa può essere schematizzato nel seguente modo (Figura 6.23):
Come si può notare l’apparecchiatura lavora sottovuoto; inoltre il vuoto generato deve essere molto spinto (dell’ordine di 10-6÷10-5 torr) in modo da impedire una perdita di ionizzazione causata da urti con gas atmosferici.
Verranno di seguito descritte ed analizzate le singole parti che costituiscono lo spettrometro di massa.
• Introduzione del campione
L’introduzione del campione nella camera di ionizzazione avviene in ragione di pochi microgrammi. In queste condizioni la maggior parte degli analiti è in fase gassosa; i solidi che non sublimano verranno invece vaporizzati inserendoli accanto al filamento di ionizzazione. Se lo strumento è collegato in uscita ad una colonna cromatografia, come è ormai prassi in molti casi, il campione entra nello strumento al termine dell’eluizione o direttamente in fase gassosa o tramite un dispositivo di vaporizzazione immediatamente a valle della colonna.
Queste tecniche, note come GC-MS e HPLC-MS, sono estremamente utili nall’analisi di miscele di prodotti.
• Camera di ionizzazione
Questa sezione si basa sul fatto che se una molecola è investita in fase vapore da un fascio di elettroni di notevole energia si può avere per urto la sua ionizzazione a ione positivo o negativo.
In genere gli strumenti sono regolati per lavorare unicamente con ioni positivi, i quali possono spontaneamente o per urto decomporsi in una serie di frammenti di massa inferiore e questi a loro volta in altri.
Ogni molecola avrà quindi una frammentazione caratteristica e specifica che dipenderà sia dalla natura delle molecole sia dalle condizione operative di ionizzazione.
Il campione viene ionizzato in un’apposita camera di ionizzazione, in cui il fascio di elettroni viene prodotto da una sorgente ionica che varia a seconda della tecnica utilizzata.
In genere gli elettroni sono emessi da un filamento caldo di tungsteno o renio, e passano attraverso un condotto, che crea il raggio, nella parte centrale della camera che contiene il campione gassoso.
La frazione di elettroni che non urta contro le molecole è separata da un campo magnetico perpendicolare al fascio di ioni (camera di raccolta), le molecole non
ionizzate sono allontanate dalla pompa ad alto vuoto, mentre quelle ionizzate sono accelerate e convogliate verso l’analizzatore.
Il sistema di ionizzazione svolge un ruolo essenziale nella spettrometria di massa, perché da esso dipende anche il numero, la natura e l’abbondanza dei frammenti molecolari che compaiono nello spettro di massa.
In base al tipo di sorgente utilizzata la ionizzazione primaria del campione viene realizzata in vario modo; le tecniche più usate sono:
- impatto ionico (E.I.) - ionizzazione chimica (C.I.)
Tali tecniche di ionizzazione vengono di seguito presentate nel dettaglio. - Impatto elettronico (E.I.)
Questo tipo di ionizzazione è la tecnica più comunemente impiegata (Figura 6.24). Un filamento di tungsteno incandescente emette un fascio di elettroni che, accelerati verso un anodo posto dalla parte opposta al filamento, acquistano un’elevata energia (circa 70 eV). Quando questi elettroni vengono a contatto con la sfera elettronica di una molecola (impatto elettronico), le trasferiscono la loro energia provocando l’espulsione di un elettrone con formazione di un radical-catione (ione molecolare) M++.
Siccome l’energia necessaria per ionizzare una molecola organica è di circa 13-14 eV, i radical-cationi sono prodotti ad un’energia vibrazionale molto alta, che ne può determinare la frammentazione con formazione di un radicale e un catione. Tutti gli ioni positivi (cationi e radical-cationi) sono respinti da una piastra, tenuta ad un potenziale positivo, verso una serie di piastre forate, tenute a potenziale positivo crescente, dette piastre acceleratrici.
Nel loro tragitto gli ioni subiscono un’accelerazione proporzionale al potenziale V delle piastre acceleratici e vengono espulsi attraverso una fenditura in uscita. Si può far percorrere agli ioni la giusta traiettoria per giungere al rivelatore variando l’intensità del campo magnetico B, oppure quella del potenziale delle griglie V (di solito si fa variare B).
- Ionizzazione chimica (C.I.)
La ionizzazione chimica viene utilizzata quando gli ioni molecolari prodotti con il metodo dell’impatto elettronico sono troppo poco stabili e si frammentano completamente.
Tale tecnica infatti si basa sull’interazione del campione vaporizzato con un reagente ionizzato, che di solito è un acido di Bronsted gassoso.
I più usati reagenti di questo tipo sono quelli che derivano dalla ionizzazione ad impatto elettronico del metano
3 5 4 4 CH CH CH CH•+ + → + + · 2 5 2 4 3 CH C H H CH+ + → + + 2 5 3 4 5 2H CH C H 2H C + + → + +
Se la molecola M ha un’affinità per il protone più alta di quella del metano, allora si avrà la formazione dello ione M-H+.
[
]
4 5 M M H CH CH+ + → − + +[
]
2 4 5 2H M M H C H C + + → − + +Gli ioni M-H (detti quasimolecolari) non possiedono un’energia elevata e quindi subiscono una minore frammentazione.
In genere la ionizzazione a impatto elettronico dà dei frammenti più significativi di quanto non faccia la ionizzazione chimica. Infatti in seguito alla C.I. i legami C-C tendono a rompersi solo se il prodotto della scissione è particolarmente stabile. Frequentemente lo scheletro degli atomi di carbonio rimane intatto e la rottura è limitata a legami di tipo C-O, C-S o C-N.
Ne deriva che la C.I. è particolarmente adatta a molecole come idrocarburi, alcoli, esteri, ammine, amminoacidi e piccoli peptidi che in condizioni di E.I. darebbero
In pratica il metodo consiste nell’introdurre, insieme al campione, del metano in forte eccesso. Statisticamente sarà il metano ad essere ionizzato per impatto elettronico, generando •+
4
CH e proseguendo con le reazioni già presentate: il radical-catione di metano, incontrando un’altra molecola di CH4, forma CH3· e
CH5+, che funziona da acido per una molecola organica M generando a sua volta
l’acido coniugato MH+. Tale specie non si forma ad un livello vibrazionale eccitato, e non frammenta.
La particolarità è che nello spettro vedremo lo ione molecolare +1. • Analizzatore
L’analizzatore consente di differenziare gli ioni generati, in base al loro rapporto massa/carica.
Le tipologie maggiormente impiegate sono le seguenti: - analizzatore magnetico
- analizzatore a quadrupolo - analizzatore a tempo di volo
Ciascuno di essi sarà di seguito presentato nel dettaglio. - Analizzatore magnetico
È quello più usato, in quanto consente di ottenere le risoluzioni migliori.
L’analizzatore magnetico è costituito da un tubo lungo circa 1 metro (Figura 6.25), piegato con un raggio di curvatura r’ ed immerso in un campo magnetico H. Gli ioni che escono dalla camera di ionizzazione entrano nel tubo analizzatore e, per effetto del campo magnetico, subiscono una deviazione della loro traiettoria rettilinea (deflessione). La nuova traiettoria curvilinea ha un raggio di curvatura r che è direttamente proporzionale alla quantità di moto dello ione (m·v) e inversamente proporzionale al campo magnetico H.
La relazione tra il raggio di deflessione e la massa dello ione risulta la seguente:
⋅ ⋅ = V H r z m 2 2 2 dove:
- z: carica degli ioni
- V: potenziale di accelerazione - m: massa dello ione
Da qui si può notare che per un certo valore della coppia H e V esiste un solo valore della massa m per cui il raggio di deflessione r coincide con il raggio di curvatura del tubo r’.
Gli ioni che hanno questo valore di massa escono dal tubo, gli altri no.
Operando a potenziale V costante e facendo una scansione di campo H è possibile far uscire dal tubo gli ioni a diversa massa in tempi diversi.
Per misurare ad alta sensibilità, atta a rilevare la presenza di uno ione preciso è anche possibile fissare il valore di V sul valore corrispondente alla massa desiderata (monitoraggio del singolo ione o selected ion monitoring, SIM)
Figura 6.25 : Schema di uno spettrometro con analizzatore magnetico
- Analizzatore a quadrupolo
È costituito da quattro barre cilindriche metalliche, lunghe circa 20 cm, che delimitano lo spazio percorso dagli ioni provenienti dalla camera di ionizzazione e diretti al detector (Figura 6.26).
Le barre sono mantenute ad un potenziale elettromagnetico oscillante, in modo che quando le due barre verticali hanno potenziale positivo quelle orizzontali hanno potenzialo negativo, e viceversa.
Il flusso di ioni attraversa questo spazio a sezione quadrata; il campo oscillante li fa muovere secondo traiettorie sinusoidali consentendo solo a quelli di una data massa di attraversare l’intero quadrupolo e giungere al rilvelatore.
Operando una scansione di frequenza di oscillazione del campo è possibile far uscire ioni a massa molecolare crescente.
Figura 6.26: Rappresentazione di analizzatore a quadrupolo
Gli strumenti con questo tipo di separatore risultano essere più compatti nelle dimensioni e generalmente meno costosi di quelli basati sul campo magnetico statico.
• Rivelatore
Come collettore e rivelatore degli ioni si usa comunemente un moltiplicatore elettronico, costituito da una serie di elettrodi in cascata.
Quando uno ione arriva sul primo elettrodo questo emette un fascio di elettroni che vanno a colpire il secondo elettrodo, il quale a sua volta emette una quantità maggiore di elettroni e così via.
Il risultato è una forte amplificazione del segnale che viene poi digitalizzato ed elaborato infine dal calcolatore dello spettrometro per la presentazione dello spettro di massa.
L’uso di calcolatori permette inoltre di combinare rapidamente la gestione dei parametri dello strumento con la ricerca bibliografica in librerie di spettri in
formato elettronico, in modo da automatizzare l’identificazione dei composti in base al loro spettro e alle condizioni operative in cui è stata condotta l’analisi. È possibile inoltre procedere ad un’integrazione dei singoli picchi in modo da effettuare un’analisi quantitativa delle specie chimiche corrispondenti.
- Risoluzione dello strumento
Il potere risolutivo dello strumento determina la capacità di separare tra di loro ioni di uguale massa nominale ma diversa massa esatta. Nell'esame delle caratteristiche di uno strumento e' necessario stabilire cosa si intenda per "separare". Nella Figura 6.27, gli ioni sono in entrambi i casi separati, ma ovviamente la risoluzione e' maggiore nel caso A. I dati di potere risolutivo sono per convenzione misurati su coppie di segnali separati tra di loro da una valle (h) alta il 10% dell'altezza (H).
Figura 6.27 : Esempio di picchi separati con risoluzione maggiore (A) e minore (B)
Il potere risolutivo (PR) è definito nel modo seguente:
1 2 1 Massa Massa Massa PR − =
La risoluzione di uno strumento può essere regolata agendo su fenditure micrometriche che restringono la dispersione del fascio ionico. Riducendo l'ampiezza delle fenditure aumenta la risoluzione (fino al limite dello strumento), ma diminuisce la sensibilità (meno ioni raggiungono il rivelatore).
• Spettro di massa
Lo spettro di massa si presenta come un insieme di linee verticali (picchi) di intensità diversa, ciascuna corrispondente al valore di massa di uno ione frammento. Il picco a valore di massa più elevato non sempre coincide con quello relativo allo ione molecolare. In genere, la corrente ionica è normalizzata a 100, ossia il picco più alto (picco base) ha valore 100, indipendentemente dal suo valore assoluto. Dallo spettro di massa si può risalire dunque alla struttura di un composto incognito, attribuendo ai singoli ioni una composizione elementare e ricostruendo i meccanismi di frammentazione seguendo schemi tipici per le varie classi di composti. Nell’interpretazione di uno spettro si segue una procedura abbastanza semplice:
- identificazione dello ione molecolare. - identificazione di ioni caratteristici.
- identificazione di processi di frammentazione caratteristici.
- ricostruzione della struttura della molecola sulla base della conoscenza di meccanismi di frammentazione standard
6.10
Accoppiamento di Gas-cromatografia e Spettrometria di
massa
6.10.1 Metodo di analisi scelto per il caso in esame
La strumentazione che si è deciso di utilizzare vede principalmente l’uso di un gas-cromatografo (Figura 6.28).
Esso è stato allestito con tre diversi rivelatori in modo da analizzare il più alto numero possibile di composti presenti nel gas in uscita dall’impianto. I tre rivelatori utilizzati sono:
• TCD; • FID;
• Spettrometro di massa.
Al fine di far arrivare ad ogni rivelatore miscele di gas differenti, la strategia seguita è stata quella di utilizzare due diverse colonne capillari che operano in parallelo.
Le colonne impiegate e il loro rispettivo scopo sono descritti di seguito.
• SUPELCO Carboxen GC PLOT 1010: separa i gas permanenti, con particolare interesse a CO, CO2, H2, O2; la corrente in uscita viene analizzata sia dal TCD
che dal FID.
• HP GS-GasPRO: capace di separare gas leggeri e idrocarburi fino a C20; la
corrente in uscita viene analizzata dallo Spettrometro di massa. I gas di trasporto utilizzati sono:
• Elio; • Argon.
Oltre a questi gas si utilizza anche aria e idrogeno per alimentare la fiamma del FID.
Figura 6.28: Immagine del Gas-Cromatografo e dello Spettrometro di massa utilizzati
6.10.2 Configurazione adottata
Il campionamento avviene mediante l’impiego di due valvole identiche che operano in serie per quanto riguarda il riempimento dei due loop e in parallelo per quanto riguarda invece l’iniezione del campione in colonna.
Questa configurazione risulta necessaria per permettere l’introduzione del campione simultaneamente nelle due colonne per mezzo dei due carrier impiegati.
• Posizione OFF delle valvole
L’ Elio è immesso nel sistema attraverso la sorgente 1 di gas (S1) ed entra nella porta 4 della valvola 1 (V1-4); il collegamento interno corrispondente conduce il
carrier fuori dalla valvola mediante la porta 5; da qui va ad un iniettore split-splitless che introduce il gas nella colonna HP GS-GasPRO, collegata allo spettrometro di massa.
Il percorso del campione è invece il seguente: esso entra nella porta 1 della valvola V1, esce da V1-6, riempie il LOOP-1, rientra in V1-3 ed esce da V1-2. Da qui il campione è inviato alla valvola V2, in cui segue un percorso del tutto analogo a quello precedente: entra nella porta 1 della valvola V2, esce da V2-6, riempie il LOOP-2, rientra in V2-3, esce da V2-2 e infine è espulso dallo scarico.
Il carrier che attraversa la valvola V2 è argon ed è immesso nel sistema per mezzo della sorgente 2 di gas (S2): esso, con un meccanismo identico a prima, entra dalla porta 4, esce dalla porta 5 ed è immesso direttamente nella colonna SUPELCO Carboxen GC PLOT 1010.
Quest’ultima è collegata ad un three-ways splitter che divide la corrente in due e permette di alimentare ciò che esce dalla colonna ai due rivelatori FID e TCD, mentre la terza uscita è mantenuta chiusa. Allo splitter arriva anche una corrente ausiliaria di argon dalla sorgente 3.
Figura 6.29: Posizione OFF delle due valvole V1 e V2
• Posizione ON delle valvole
Quando la parte centrale delle valvole gira in senso orario i collegamenti interni tra le porte passano da 5-4 1-6 3-2 a 5-6 3-4 1-2.
In questa configurazione l’elio entra in V1-4, esce da V1-3 e passa dal LOOP-1 trascinando con sé il campione accumulato, che attraverso le porte 6 e 5 è