Introduzione
1.1
Introduzione
La sostenibilità, l’industria eco-compatibile, l’eco-efficienza e la green chemistry cercano, alla luce delle nuove esigenze, di indicare nuove strade riguardo all’utilizzo di materiali, prodotti e processi che favoriscano lo sviluppo e contemporaneamente salvaguardino le nuove generazioni dalla possibilità di trovarsi in un pianeta esausto. In particolare, la crescente consapevolezza dell’uomo degli effetti che provoca sull’ambiente e delle implicazioni sociali che ciò può avere, unita alla percezione della rapida diminuzione dei combustibili fossili disponibili e al fatto che nuove disposizioni mondiali sono divenute esecutive in materia ambientale, hanno stimolato la ricerca verso nuovi prodotti e nuovi processi, che siano più compatibili con l’ambiente stesso [1]. Si stima che, al giorno d’oggi, il consumo mondiale di petrolio è 100’000 volte più veloce della velocità con la quale la natura potrebbe rimpiazzarlo [2]. Plastiche e composti biodegradabili, generati da materie prime rinnovabili, derivanti dalle biomasse, sono considerati materiali promettenti, che potrebbero rimpiazzare i polimeri sintetici e ridurre la dipendenza dalle risorse combustibili fossili.
Il polimero naturale più abbondante nel nostro ambiente è la cellulosa. La sua naturale produzione annua è stimata in 90 x 109 tonnellate e rappresenta, dunque, la più ovvia risorsa rinnovabile da utilizzare per produrre nuovi bio-materiali [3]. La sua struttura estremamente ordinata è responsabile dell’interesse che esercita per le sue proprietà meccaniche, ma è la causa della elevata difficoltà nel trovare solventi adatti per la sua dissoluzione [4].
I primi tentativi di sciogliere la cellulosa risalgono ai primi anni ’20 [5]. Da allora sono stati scoperti diversi solventi, acquosi e non, che possono sciogliere parzialmente la cellulosa; tutti questi metodi hanno tuttavia la doppia pecca di utilizzare sostanze caratterizzate da una elevata tossicità ambientale, oltre ad una insufficiente capacità solvatante [6]. In generale, il tradizionale processo di dissoluzione della cellulosa richiede condizioni relativamente drastiche e l’uso di
2 solventi costosi e non comuni che, solitamente, non possono essere recuperati al termine del processo [6-10].
Nel 2002, tuttavia Swatloski e coll. riportarono per la prima volta la possibilità di utilizzare un liquido ionico per la dissoluzione e la rigenerazione della cellulosa, evidenziando la possibilità di utilizzare lo stesso sistema per la modificazione chimica del polisaccaride [7]. Il termine liquidi ionici definisce un‘ampia classe di composti ionici che rimangono liquidi a temperature relativamente basse (<100 °C): la natura ionica conferisce loro diverse proprietà chimico-fisiche che li rendono allettanti come solventi eco-compatibili; tra queste, la stabilità termica e chimica, la non-fiammabilità oltre ad una tensione di vapore estremamente bassa [11]. Sebbene siano stati scoperti già nel 1914 da Walden, ci si è resi conto del loro enorme potenziale solamente negli ultimi decenni [12,13].
1.2
Cellulosa
La cellulosa è il polimero organico più comune ed è, a ragione, considerata come una fonte pressoché inesauribile di materiale grezzo in grado di supplire alla crescente domanda di prodotti bio-compatibili ed eco-friendly. Il legno contiene fino al 47% di cellulosa, che costituisce la componente principale della parete cellulare delle piante [14]. Benché la polpa del legno rimanga la materia prima principale dalla quale si ricava la cellulosa, essa può essere estratta anche da alghe, batteri e cereali [15].
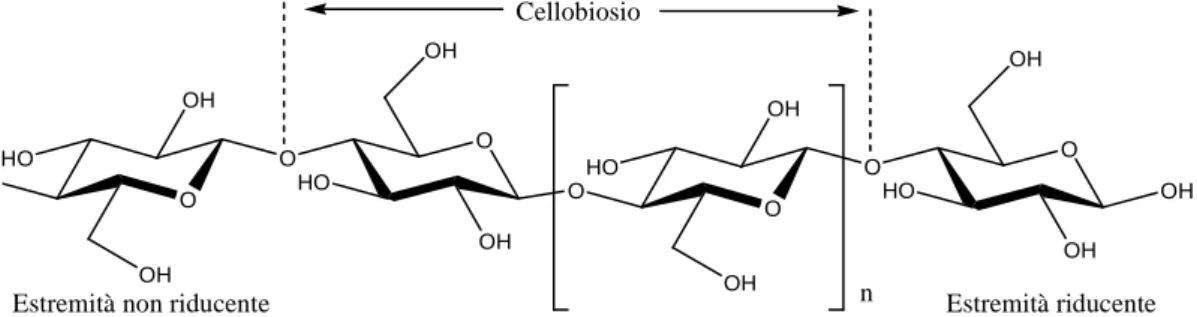
La cellulosa è un polimero lineare di cellobiosio, che consiste di due unità di glucosio legate tramite un legame β-(1→4) glicosidico (C-O-C). La sua struttura è mostrata in Figura 1. Le dimensioni della catena dipendono dal grado di polimerizzazione (DP), che è il numero
Figura 1. Struttura della cellulosa
O HO HO OH OH O O O O HO OH OH O O OH HO OH OH OH HO OH n Cellobiosio
3 delle unità di glucosio che si ripetono. Il DP può variare considerevolmente, da circa 20 per la cellulosa sintetizzata in laboratorio, a più di 10000 unità per la cellulosa batterica [15,16]. Ogni unità di glucosio è ruotata di 180° rispetto alle due alle quali è legata, cosicché la struttura ripete le unità di cellobiosio. La cellulosa ottenuta dal legno contiene circa 104 unità e raggiunge la lunghezza di circa 5µm nelle regioni altamente cristalline [14]. Ogni catena ha una estremità emiacetalica riducente alla posizione C1, mentre l’altra ha una funzione ossidrilica OH alla
posizione C4, che costituisce l’estremità non riducente.
La cellulosa naturale è un polimero lineare semicristallino, composto da regioni cristalline altamente strutturate (micro fibrille), e parti amorfe. Altri polisaccaridi che si ritrovano nelle piante, come le emicellulose, hanno strutture ramificate [17]. Il legame β-(1→4), molto stabile, è ulteriormente rinforzato da legami ad idrogeno intracatena fra l’ossidrile in C3 e l’ossigeno
facente parte dell’anello adiacente, così come fra il gruppo idrossilico in C2 e l’ossigeno del gruppo
idrossi-metilico in C6 (Figura 2) [18]. Nello stato naturale, sono connesse fra loro da 40 a 70 catene
O HO OH OH O O O O HO OH OH O O HO OH OH OH HO OH O HO OH OH O O O O HO OH OH O O HO OH OH OH HO OH O O O O n n
Figura 2. Legami ad idrogeno intra- ed intermolecolari nella cellulosa
di cellulosa, da legami ad idrogeno fra il gruppo idrossi-metilico C6 e l’ossidrile in C3 della catena
adiacente, per formare micro fibrille [19]. Queste micro fibrille sono estremamente rigide, avendo un modulo elastico di circa 20 GPa parallelamente e 15 GPa perpendicolarmente alla direzione della fibra [20]. L’elevata stabilità chimica e meccanica della cellulosa e la sua natura idrofobica sono dovute alla forza dei suoi legami glicosidici e all’enorme numero di legami ad idrogeno intra- ed intermolecolari: essi, difatti, non solo fungono da interconnessioni fra le unità di cellobiosio, ma sono anche responsabili della natura idrofilica di questo bio-polimero [16].
4
1.2.1
Altri polimeri del legno
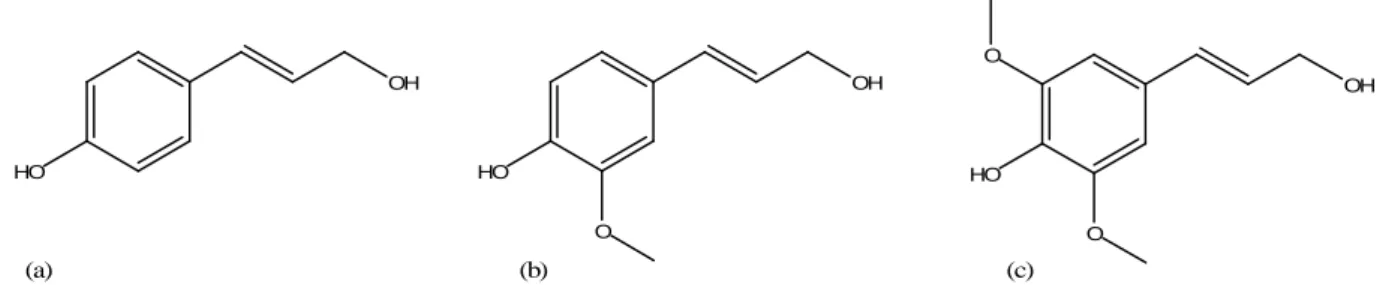
Un materiale ligno-cellulosico è composto anche da lignina (dal 5 al 30%, dipende dal tipo di legno) e da emicellulosa (fino al 35%), oltre che da pectine e glicoproteine che si inseriscono fra le micro fibrille di cellulosa [21]. La lignina è un polimero complesso, composto principalmente da unità fenilpropanoidi, composte essenzialmente dagli alcoli coniferilico, sinapilico e para-cumarilico (Figura 3) [22]. La struttura della lignina, al contrario di quella della
HO OH HO OH O HO OH O O (a) (b) (c)
Figura 3. Fenilpropanoidi: (a) alcol p-cumarilico, (b) alcol coniferilico, (c) alcol sinapilico
cellulosa, esibisce un comportamento da polimero amorfo; inoltre, la struttura della lignina varia considerevolmente al variare della pianta dalla quale si estrae. La lignina annua prodotta dal processo di pulping chimico del legno equivale a 26 milioni di tonnellate, rendendola un ottimo feedstock naturale dal quale partire per successive elaborazioni [23].
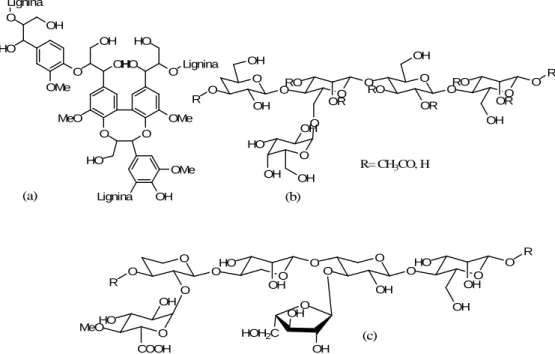
Il terzo grande costituente del legno è la emicellulosa, composta da polisaccaridi di natura xilanica, mannanica, beta-glucanica e xilo-glucanica [24]. Nel legno dolce (derivante dalle conifere), le emicellulose principali sono gli acetil-galatto-glucomannani e gli arabino-4-O-metilglucurono-xilani [25], mentre le più importanti del legno duro (derivante dalle Angiosperme, non Monocotiledoni), sono gli xilani, in particolare il O-acetil-4-O-metilglucurono-d-xilano (Figura 4) [26]. Dalle acque reflue derivanti dal processo di pulping termomeccanico, si possono recuperare circa 5kg di glucomannano per tonnellata di pasta di legno, mediante ultrafiltrazione [27]. Comunque sia, le emicellulose non vengono isolate per un uso industriale [28]. Ci sono varie possibili aree di utilizzo per le emicellulose: esse sono state proposte come dolcificanti, addensanti alimentari ed emulsionanti (fibre), e come building-blocks di partenza per la sintesi farmaceutica [29]. Ad oggi, in larga parte le emicellulose vengono degradate chimicamente, mediante il processo Kraft, per rimanere poi pressoché inutilizzate.
5 HO OH O Lignina OMe O OH OH MeO O HO HO OLignina OMe O HO OMe OH Lignina (a) O O O O O O O OH OH OH OR RO OR RO O OH RO OR O R O O OH HO OH OH R (b) R= CH3CO, H O O O O O O O OH OH HO OH HO O O O OH O R O OH HO COOH R MeO O OH OH HOH2C (c)
Figura 4. Polimeri del legno: (a) lignina, (b) glucomannano, (c) xilano
1.3
Liquidi Ionici
1.3.1
Basi
I liquidi ionici sono sali aventi un basso punto di fusione, generalmente più basso di quello dell’acqua, e sono normalmente costituiti da un catione organico e da un anione poliatomico, organico o inorganico. I cationi di grosse dimensioni determinano una riduzione della simmetria della molecola, fattore che riduce l’impaccamento del composto la cui temperatura di fusione risulta bassa, in genere inferiore alla temperatura ambiente.
Il primo liquido ionico sintetizzato è stato l’etilammonio nitrato [EtNH3][NO3]
nel 1914, con punto di fusione di 12 °C [30]. Nella seconda metà del XX secolo, tuttavia, l’attenzione si è rivolta essenzialmente verso la sintesi ed applicazione di liquidi ionici con cationi eterociclici, in particolare cloruri di piridinio e imidazolio addizionati di AlCl3;
[CnPy][AlCl4] o [CnPy][Al2Cl7] [31]. I sali di piridinio venivano utilizzati nei processi di
elettrodeposizione dell’alluminio e come elettroliti in batterie. I cloroalluminati, sebbene siano dei buoni solventi e dei buoni catalizzatori (possono essere degli efficienti acidi di Lewis), hanno lo svantaggio di essere altamente sensibili all’umidità [32]: l’AlCl3 in presenza di umidità si
ionizza per formare HCl. Il loro utilizzo implica, quindi, l’uso di condizioni rigorosamente anidre ed atmosfere inerti, fattori che hanno favorito lo sviluppo di altre classi di liquidi ionici.
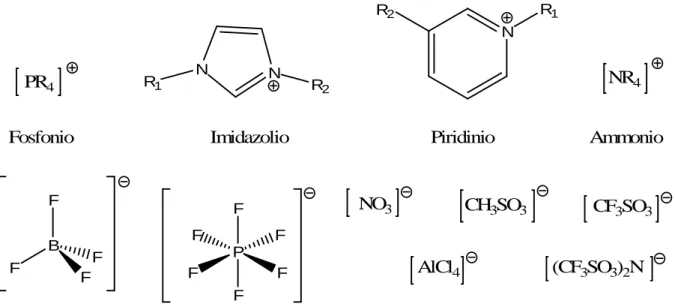
6 Negli anni ’90 sono stati sintetizzati i primi liquidi ionici stabili all’aria e all’acqua [33]; tra questi, i più comuni cationi organici sono dialchilimidazolio (R1R2[M]+), alchilpiridinio
([RPy]+), tetraalchilammonio ([NR4]+), o tetraalchilfosfonio ([PR4]+), mentre i più comuni anioni
utilizzati sono esafluorofosfato (PF6-), tetrafluoroborato (BF4-), nitrato (NO3-), metansolfonato
(mesilato, [CH3SO3-]), trifluorometansolfonato (triflato, [CF3SO3-]), e
bis-(trifluorometansolfonil)-amide ([Tf2N]-), oltre a cloruri, bromuri e ioduri basso fondenti (Figura
5) [34]. Il rapido aumento di interesse è in parte attribuibile all’ampia applicabilità di questi sali, che va dalle reazione catalizzate, all’uso come elettroliti liquidi in processi elettrochimici, a solventi nelle separazione, o nella chimica dei polimeri. Ma la ricerca di nuovi liquidi ionici si è spinta anche oltre, con lo sviluppo di liquidi ionici funzionalizzati, task-specific ionic liquids e liquidi ionici chirali [35].
PR4 R1 N N R2
N R1
NR4
Fosfonio Imidazolio Piridinio
R2 Ammonio F B F F F P F F F F F F NO3 AlCl4 CH3SO3 CF3SO3 (CF3SO3)2N
Figura 5. Cationi e anioni comunemente usati
L’idrofilia dei liquidi ionici è determinata primariamente dall’anione: anioni quali mesilato, tosilato, trifluoroacetato, acetato e dicianammide, danno generalmente liquidi ionici miscibili con l’acqua. Inoltre, tra questi, diversi anioni, con i corrispondenti liquidi ionici, mostrano proprietà di basi di Lewis (dicianammide e acetato) ed hanno dimostrato di poter esercitare effetti interessanti nella catalisi basica.
7
1.3.2
Sintesi e purificazione
La sintesi dei liquidi ionici a base imidazolica può essenzialmente essere divisa in due passaggi: il primo è la formazione del catione desiderato, la seconda la reazione di scambio dell’anione (metatesi), per produrre il prodotto finale, secondo quanto riportato nello Schema 1.
N N N N R X R = Alchile X = Cl, Br, I RX + MY - MX + HY - HX Acido-base Metatesi N N R Y
Schema 1. Tipiche vie sintetiche per la preparazione di liquidi ionici
I più comuni liquidi ionici sono preparati quaternizzando un eterociclo contenente un atomo di azoto con un alogenuro alchilico (reazione di Mentschutkin) [36]. Lo scambio di anione può essere effettuato sia mediante trattamento diretto dell’alogenuro con un forte acido di Lewis per formare anioni complessi, sia mediante addizione di un sale metallico, ottenendo la precipitazione dello ione non desiderato (reazione di Finkelstein) [37]. Altre sintesi sviluppate negli ultimi anni, prevedono l’utilizzo di metodi solvent- e halide-free, l’impiego di microonde e ultrasuoni, o l’uso di reagenti industriali più economici come prodotti di partenza come, per esempio, il detergente sodio ottilsolfato [38].
A seconda del metodo di sintesi scelto, le impurezze che si possono ritrovare nel liquido ionico finale possono essere ammine terziarie, alchil-alogenuri, alchil- solfati (o i prodotti derivanti da reazioni secondarie), e, dopo la metatesi, solfati o alogenuri residui, che possono
8 costituire una notevole fonte di disturbo in alcune reazioni metallo-catalizzate. I metodi di purificazione includono l’estrazione del liquido ionico con solventi polari (ad esempio, etilacetato), estrazione delle soluzioni acquose del IL con solventi organici immiscibili (come il diclorometano), colonne cromatografiche flash di una soluzione di liquido ionico in un solvente organico, o il trattamento del liquido ionico con carbone attivato [37].
1.3.3
Proprietà ed applicazioni
I liquidi ionici offrono un’ampia varietà di proprietà chimico-fisiche, che li rendono interessanti per una vasta gamma di applicazioni. Alcune di queste proprietà sono comuni a tutti gli IL, determinandone caratteristiche comuni. I liquidi ionici con un atomo di azoto quaternarizzato, per esempio, sono non infiammabili e, sebbene fosse stato pensato avessero una bassa tensione di vapore, è stato dimostrato da Earle e coll. che possono essere distillati a basse pressioni ed alte temperature [39]. Comunque sia, queste proprietà sono ampiamente desiderabili in quanto, ad esempio, la pressoché nulla tensione di vapore implica una limitata volatilità del solvente, determinando la diminuzione dell’uso di apparecchiature per la protezione delle vie aeree e di cappe di aspirazione. Questa proprietà inoltre favorisce il loro riciclaggio ed è una delle ragioni principali per cui vengono chiamati green solvents. Studi recenti hanno dimostrato che gli IL hanno un ampio range di tossicità, per cui il termine green solvents dovrebbe essere riconsiderato [40,41].
La diversità dei liquidi ionici li rende utilizzabili in una miriade di applicazioni che includono termometri ottici, biocatalisi e processi di separazione, impiego nella chimica dei polimeri, uso come catalizzatori, elettroliti, biosensori, apparecchi analitici, lubrificanti, come rimpiazzo dei comuni solventi e come telescopi lunari [32,35,42,43,44].
Proprietà come la temperatura di fusione, l’indice di rifrazione, il carattere acido-base, l’idrofilia, la polarità, la densità e la viscosità possono essere adattate di buon grado. Comunque, la presenza di impurezze, derivanti dai reagenti di sintesi, come gli alogenuri, le basi organiche e l’acqua assorbita, possono decisamente alterare le loro proprietà [45,46].
Temperatura di fusione
La temperatura di fusione e la viscosità dei liquidi ionici dipendono in maniera significativa dal tipo di anione, mentre il catione ha una minore influenza [47]. La maggior parte dei liquidi ionici mostra una transizione vetrosa, che avviene quando un liquido sotto raffreddato forma un solido amorfo. I liquidi ionici solidi alla stato cristallino possono contenere alcune regioni
9 amorfe, mostrando quindi sia una temperatura di transizione vetrosa (Tg), sia una temperatura di
fusione (Tfus). Le proprietà termiche sono governate dalle forze di Van der Waals e dalle interazioni
elettrostatiche, che sono determinate essenzialmente dalla dimensione, dalla simmetria, da eventuali legami ad idrogenoe dalla delocalizzazione della carica [48,49].
Simmetria. I cationi scarsamente simmetrici, come 1-butil-3-metilimidazolio
[BMIM]+ possiedono solo una simmetria di tipo C1 e fondono a temperature più basse rispetto ai loro analoghi con maggiore simmetria, come gli 1-butilpirimidinio [BPy]+, sali che possiedono anche una simmetria C2v [13].
Lunghezza della catena alchilica. La lunghezza della catena alchilica influenza
la Tfus. I cationi a formula generale [CnMIM]+ possiedono un core imidazolico sul quale è
concentrata la carica e una coda alchilica laterale idrofobica. Il crescente numero di carboni nella catena laterale (n < 7), diminuisce la temperatura di fusione, a causa della ridotta simmetria complessiva del catione (regioni symmetry-breaking). Comunque la catena laterale con più di sette atomi di carbonio porta ad un incremento della temperatura di fusione, in quanto le forze di Van der Waals fra le catene alchiliche iniziano ad avere un peso maggiore sull’effetto esercitato dalla simmetria.
La Figura 6 illustra la convergenza dei dati sperimentali e uno studio computazionale che ha utilizzato un approccio QSPR (quantitative structure-property relationship) [50].
Figura 6. Temperature di fusione teoriche e sperimentali per una serie di [CnMIM]PF6
Legame ad idrogeno. La presenza o l’assenza di forti legami ad idrogeno
influenza, a sua volta, la temperatura di fusione. L’esistenza di legami ad idrogeno nei liquidi ionici a catione imidazolo venne segnalata nel 1986 [51]. Generalmente, le interazioni C-H…X (X = Cl,
10 Br) sono relativamente deboli, ma diventano veramente forti negli alogenuri a base imidazolica che possiedono, addirittura, un parziale carattere covalente [52]. Questi forti legami ad idrogeno coinvolgono tutti e tre i protoni presenti sull’anello, con conseguente formazione di un reticolo tridimensionale, con interazioni di tipo π-aromatico fra i nuclei etero aromatici (Figura 7) [53]. Anche l’anione [BF4]- può formare legami ad idrogeno negli IL a nucleo imidazolico, sebbene essi
N N H H H H H H H H H H H Cl N N H H H H H H H H H H H Cl N N H H H H H H H H H H H Cl Cl Cl
Figura 7. Rappresentazione schematica dei legami ad idrogeno nel [EMIM]Cl
siano notevolmente più deboli di quelli che si ritrovano nei corrispondenti alogenuri, a causa della più bassa temperatura di fusione dei tetrafluoroborati [52]. In genere gli anioni esafluorofosfati non partecipano a legami ad idrogeno [54].
Viscosità. La viscosità dei liquidi ionici risentono molto di eventuali
contaminanti. La presenza dell’acqua diminuisce la viscosità, mentre i cloruri, residui da eventuali metatesi, hanno l’effetto opposto [45,55]. La presenza di cosolventi sembra decrescere l’aggregazione degli ioni nel liquido, portando ad una diminuzione della viscosità, mentre risultati sperimentali indicano che la natura del cosolvente gioca un ruolo più importante che non la concentrazione stessa [56].
Una delle maggiori barriere alla commercializzazione dei liquidi ionici su larga scala è dovuta alla loro alta viscosità. La viscosità degli IL è più alta dell’acqua e simile a quella degli oli lubrificanti e diminuisce, in genere, all’aumentare della temperatura [57]. La dipendenza della viscosità dalla temperatura su cationi non simmetrici e non portanti alcuni gruppi funzionali
11 sulla catena alchilica, può essere descritta dall’equazione di Arrhenius. Per liquidi ionici aventi un catione simmetrico e una bassa massa molecolare, è più adeguata l’equazione di Vogel-Tammann-Fulcher (VTF). Per altri liquidi ionici, nessun modello può descrivere, in maniera accurata, la dipendenza della viscosità dalla temperatura [57].
Abbott ha predetto il comportamento della viscosità per i liquidi ionici applicando la hole-theory. Per viscosità basse, gli ioni devono essere relativamente piccoli e il liquido deve contenere ampi spazi vuoti. È stato suggerito che liquidi ionici con un’elevata tensione superficiale possiedano queste proprietà; ciò include sali di imidazolio che presentino catene da C4 a C6 [58].
Liquidi ionici con anione nitrato ([NO3]-), che hanno una bassa tensione superficiale, sono un
esempio di IL con bassa viscosità ed elevata conducibilità elettrica [59]. La stessa correlazione fra tensione superficiale e viscosità è stata osservata per gli anioni ([Tf2N]-) e ([PF6-]) [60].
Inoltre, ramificazioni della catena alchilica nei sali con catione 1-allil-3-metilimidazolio ([AMIM]), riduce la viscosità in quanto si riducono le interazioni intermolecolari di tipo dipolo-dipolo. La stessa ragione è valevole per la maggior parte degli anioni perfluorurati [61]. Altri fattori da considerare sono la possibilità di formare legami ad idrogeno e la simmetria [60,62].
Per concludere, la viscosità dei liquidi ionici, aventi i più comuni cationi, decresce nell’ordine [Cl]- > [PF6]- > [BF4]- > [Tf2N]-.
Densità. La densità è una delle proprietà dei liquidi ionici più frequentemente
riportate, probabilmente perché pressoché ogni nuova applicazione sviluppata necessita della conoscenza della densità. In generale, i liquidi ionici sono più densi dell’acqua. La massa molare dell’anione influenza in maniera significativa la densità complessiva dell’intero liquido ionico [61]. I sali con anione bis(metansulfonil)amide [Ms2N]-, per esempio, hanno densità più basse dei
corrispondenti che presentano l’anione [Tf2N]-: posto che i volumi molari di entrambi i composti
sono molto simili, ciò è dovuto alla maggiore massa dell’anione perfluorurato [62]. In più, l’impaccamento è migliore quando lo ione positivo e lo ione negativo hanno dimensioni simili [61].
Tensione superficiale. Su questa proprietà sono stati riportati pochi dati [59,60].
In generale, le tensioni superficiali liquido/aria dei liquidi ionici sono in qualche modo più alte dei comuni solventi organici, acqua esclusa, comprendendo una vasta gamma di valori [61]. I valori della tensione interfacciale variano a seconda della temperatura e, sia l’entropia in eccesso sulla superficie, sia l’energia, sono influenzate dalla lunghezza della catena alchilica, diminuendo al crescere della lunghezza della stessa. Per uno stesso catione, il composto con l’anione più grande
12 ha, in linea di massima, la più alta tensione superficiale [63]. Comunque, i sali di alchilimidazolio [PF6]- hanno valori maggiori delle corrispondenti bis(trifluorometansulfonil)amidi [Tf2N]- [61].
Stabilità termica. La maggior parte dei liquidi ionici mostra una stabilità al
calore relativamente elevata, con reazioni di decomposizione che (Tonset), comprese fra 300°C e
400°C. La decomposizione pare non dipendere dalla natura del catione, ma diminuisce all’aumentare della idrofilia dell’anione [61]. Nella maggior parte dei casi, è stato suggerito che la dipendenza della stabilità dall’anione segua la scala [PF6]- > [BF4]- ≈ [Tf2N]- > [X]- [64]. Altri
fattori influenzano la stabilità: anioni fluorurati, come [PF6]- e [BF4]- per esempio, sono instabili
all’acqua perché decompongono, producendo HF [37,40,65]. Con analisi termo gravimetriche (TGA), si è scoperto che alcuni metalli catalizzano le reazioni di decomposizione: per questo è consigliabile utilizzare recipienti di platino, piuttosto che di alluminio [64,66].
La stabilità dei liquidi ionici è stata riportata recentemente, in quanto è stato scoperto che la degradazione avviene anche a temperature minori, se la temperatura viene mantenuta ad un livello sufficientemente elevato per tempi sensibilmente più lunghi (≈ 10h). Sulla base di queste osservazioni, è suggerita l’introduzione di un nuovo parametro, che descriva la temperatura massima alla quale è possibile lavorare [66,67].
Di solito l’anione decompone via de alchilazione, mentre il catione tende a subire migrazioni di alchili, con successive eliminazioni [66]. In generale, i sali di imidazolo tendono ad essere più stabili dei corrispettivi sali di tetraalchilammonio [64].
Polarità. Per i solventi molecolari, il termine polarità è spesso collegato alla loro
capacità di sciogliere altri composti. La definizione precisa di polarità di un solvente è, in realtà, molto più complesso, in quanto racchiude molte proprietà molecolari responsabili delle interazioni fra le molecole di solvente e le molecole di soluto (forze coulombiane, induttive, di dispersione, capacità di formare legami ad idrogeno, capacità di donare o accettare doppietti elettronici). Quindi, la caratterizzazione quantitativa della polarità di un solvente rimane un problema non completamente risolto per i solventi molecolari [61].
Il metodo più comune per descrivere la polarità di un liquido ionico è la determinazione dei parametri solvatocromici di Kamlet-Taft. Spettri di fluorescenza, indici di rifrazione, o reazioni organiche sono altri metodi con i quali è stata determinata la polarità [68,69]. I parametri che mostrano la linearità fra solvatazione ed energia (LSER), sono empirici e servono a descrivere sia la acidità/basicità dei legami ad idrogeno, sia la loro polarità/polarizzabilità. Come regola empirica, si assume che i liquidi ionici siano solventi polari simili ad alcoli a catena corta o
13 media [68,69]. Eventuali molecole d’acqua presenti come impurezze non influenzano più di tanto la polarità. La basicità dei legami ad idrogeno è regolata in maniera maggiore dall’anione e la polarità del liquido ionico, in qualche modo, dalla polarità dell’ambiente [69,70].
Permittività elettrica. La permittività dielettrica relativa, o costante dielettrica εs,
non può essere utilizzata per la determinazione quantitativa della polarità di un solvente, in quanto questa proprietà non garantisce un’adeguata correlazione con la maggior parte dei solventi molecolari. I metodi più comuni per il calcolo della permettività a non sono applicabili a causa della altra conducibilità elettrica dei liquidi ionici, mentre la misura della permettività ad alte frequenze (f > 5 GHz) e la successiva estrapolazione alla frequenza zero, fornisce una stima delle permittività relative [71-74]. Huang e Weingaertner hanno concluso che le permittività relativi dei sali a base di 1,3-dialchilimidazolio a temperatura ambiente, variano molto leggermente (εs = 10-15 F/m), se
comparate a quelle dei solventi molecolari. Anche se le permittività dei liquidi ionici variano da 11 a 57, esse sono significativamente più basse di quelle dei comuni solventi [71,72]. L’influenza dell’anione sul valore di εs appare essere minore, sia perché la capacità di formare legami ad
idrogeno influenza la permittività e dunque la polarità, pressoché nel solito modo dei liquidi molecolari [72].
Miscibilità con l’acqua. La natura idrofilica o idrofobica di un liquido ionico
determina le sue proprietà di solvatazione che, a loro volta, determinano il recupero del prodotto mediante estrazione con solvente e la contaminazione ambientale attraverso le acque di rifiuto. Una recente pubblicazione mostra un modo per separare liquidi ionici miscibili con l’acqua dalla fase acquosa [75]. La miscibilità dei liquidi ionici con l’acqua dipende principalmente dalla natura idrofilica dell’anione dalla natura idrofobica del catione, determinata dalla lunghezza della catena alchilica [61]. Normalmente la solubilità in acqua diminuisce al diminuire della temperatura. Tutti i liquidi ionici sono igroscopici in qualche modo ed assorbono acqua dall’atmosfera.
Studi IR indicano che l’acqua disciolta all’interno di un liquido ionico è maggiormente presente nello stato free, legato a due anioni mediante legame ad idrogeno. Cammarata e coll. hanno proposto che il grado di idrofilia di un IL possa essere utilizzata come indicatore per la forza di questi legami ad idrogeno [76]. Studi di risonanza magnetica nucleare (NMR) suggeriscono che le molecole d’acqua preferiscano interagire con H-2, H-4 e H-5 del nucleo imidazolico e che, come risultato, il reticolo tridimensionale del liquido ionico risulti indebolito [77].
14
Tossicità. I liquidi ionici hanno rapidamente destato l’attenzione della comunità
scientifica come green solvents. Inizialmente gli studi ecologici ed ambientali furono pochi e solo negli ultimi anni sono stati studiati la citotossicità, la tossicità verso i microorganismi e l’impatto ambientale dei più comuni liquidi ionici [41,78-80]. L’effetto del [BMIM]Cl, il liquido ionico più ampiamente utilizzato, sull’alga marina Vibrio fischeri, sui batteri luminescenti, su linee cellulari leucemiche, o enzimi come l’acetilcolinesterasi (che gioca un ruolo essenziale nel sistema nervoso di tutti gli organismi più evoluti), hanno dimostrato che il liquido ionico non è estremamente tossico (EC50 ≈ 13µM) [78].
Comunque sia, la tossicità dei liquidi ionici aumenta in maniera considerabile all’aumentare della lunghezza della catena alchilica, a causa dell’aumento di lipofilia derivante. Il catione, in particolar modo, determina l’effetto tossico dei liquidi ionici, sebbene pare possa giocare un piccolo ruolo anche l’anione [79]. Più in genere, liquidi ionici a base imidazolica sono più tossici dei più ingombrati stericamente liquidi ionici a base fosfonio ed è preferibile, laddove possibile, inserire un sostituente metilico come catena laterale [80,81].
1.4
Dissoluzione e rigenerazione della cellulosa
1.4.1
Introduzione
Una dissoluzione della cellulosa, efficiente economicamente e ecocompatibile, è fondamentale per sviluppare l’utilizzo del biopolimero ed indirizzarlo verso nuovi materiali, essenzialmente copolimeri cellulosa-plastiche biodegradabili. Idealmente, tutti i legami ad idrogeno presenti all’interno delle micro fibrille di cellulosa dovrebbero essere rotti durante il processo di dissoluzione. Il polisaccaride disciolto dovrebbe poi potersi mescolare con altri biopolimeri (lignina, emicellulose, chitosano, cheratina, fibre, ecc.), come rafforzativi, prima del recupero del solvente; stadio che determinerà la formazione di legami ad idrogeno intra- ed inter-molecolari con sé stesso e gli altri componenti. Il modeling molecolare di interfacce fra la cellulosa e le molecole circostanti, indica che le interazioni interfacciali superficiali sono dominate dalle interazioni di Coulomb, in particolar maniera dai legami ad idrogeno [20].
In realtà, è difficile sciogliere la cellulosa perché la degradazione del legame glicosidico delle singole catene di cellulosa avviene in condizioni drastiche e talvolta il polimero rigenerato ha un grado di polimerizzazione (DP), più basso rispetto a quello del materiale di partenza [82].
15
1.4.2
Dissoluzione della cellulosa con metodi convenzionali
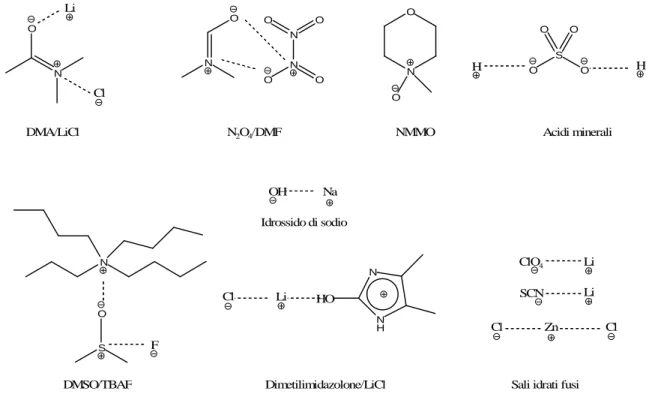
Una dissoluzione efficiente della cellulosa è un obiettivo di vecchia data nella ricerca e nello sviluppo della cellulosa ed ha ancora una notevole importanza. La cellulosa naturale contiene regioni altamente cristalline e il reticolo di legami ad idrogeno, molto ramificato, la rende insolubile in acqua e in molti solventi molecolari. La Figura 8 fa una panoramica sui metodi di
N O Li Cl DMA/LiCl N O N N O O O O N2O4/DMF N O O NMMO O S O O O H H Acidi minerali N S O F DMSO/TBAF Cl Li HO N H N Dimetilimidazolone/LiCl OH Na Idrossido di sodio ClO4 Li SCN Cl Zn Li Cl
Sali idrati fusi
Figura 8. Solventi usati tradizionalmente per disciogliere la cellulosa
dissoluzione della cellulosa più convenzionali [5,48,83-87]. A seconda dell’interazione del solvente con il polisaccaride, essi vengono chiamati derivatizzanti o non derivatizzanti [88]. I solventi derivatizzanti interagiscono chimicamente con i gruppi idrossilici della cellulosa e formano intermedi, mentre i solventi non derivatizzanti non lo fanno. Esempi commerciali di solventi derivatizzanti per la cellulosa sono idrossido di sodio/disolfuro di carbonio (Viscosa), oppure miscele di idrossido di sodio/urea (CarbaCell) [15,88]. La N-metilmorfolina-N-ossido monoidrato (NMMO), viene usata come un solvente non derivatizzante per la dissoluzione diretta della cellulosa nella produzione industriale di fibre (processo Lyocell) [89]. Mediante l’utilizzo di N,N-dimetilacetammide/LiCl e dimetilsolfossido (DMSO)/tetrabutilammonio fluoruro triidrato (TBAF), entrambi agenti non derivatizzanti, è possibile usare una varietà di prodotti e sintesi più ampia [82,90]. Recentemente, sali idrati fusi, come LiX.nH2O (X = I-, NO3-, CH3CO2-, ClO4-), sono stati
16 della cellulosa [85]. Studi sulle modificazioni chimiche avvenute sulla cellulosa disciolta mostrano che è necessario un largo eccesso di reagente: questo è dovuto all’elevato contenuto di acqua dei sali idrati, aumentando la percentuale di prodotti derivanti da reazioni secondarie [82].
La cosa più ovvia che si evince dall’osservazione della Figura 8, è che l’elevata polarità, se non il loro carattere ionico, sono favorevoli per la distruzione della rete di legami ad idrogeno della cellulosa. Inoltre, lo ione cloruro sembra essere uno dei migliori anioni per dissolvere la cellulosa; e, benché siano stati proposti vari meccanismi di interazione fra cloruro e polisaccaride, non ci sono dati specifici che supportino questa tesi [91].
1.4.3
Dissoluzione con liquidi ionici
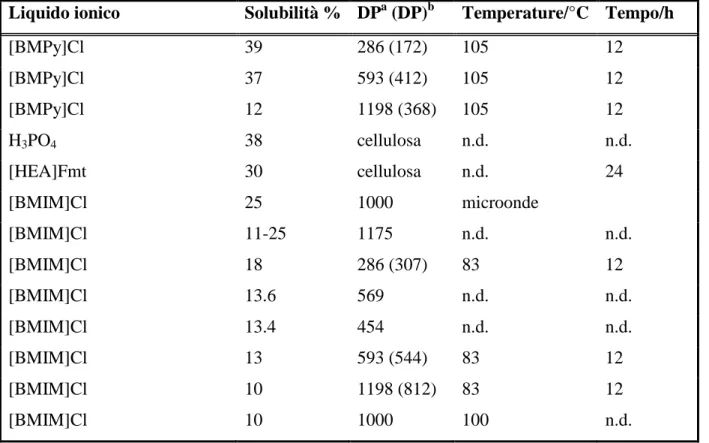
I liquidi ionici sembrano essere molto polari, a causa del loro carattere ionico: ciò risulta in una migliore capacità di dissoluzione del biopolimero. La Tabella 1 dà una prospettiva sulle capacità solventi dei liquidi ionici riportate in letteratura.
Sono molti i fattori che influenzano la dissoluzione della cellulosa e la Tabella 1. prende in considerazione solamente la capacità di dissoluzione di questi solventi e non la cinetica del processo. Liquidi ionici con basse viscosità favoriscono il processo di dissoluzione
Tabella 1. Capacità di dissoluzione della cellulosa da parte di liquidi ionici e loro solventi, per diversi gradi di polimerizzazione (DP)
Liquido ionico Solubilità % DPa (DP)b Temperature/°C Tempo/h
[BMPy]Cl 39 286 (172) 105 12 [BMPy]Cl 37 593 (412) 105 12 [BMPy]Cl 12 1198 (368) 105 12 H3PO4 38 cellulosa n.d. n.d. [HEA]Fmt 30 cellulosa n.d. 24 [BMIM]Cl 25 1000 microonde [BMIM]Cl 11-25 1175 n.d. n.d. [BMIM]Cl 18 286 (307) 83 12 [BMIM]Cl 13.6 569 n.d. n.d. [BMIM]Cl 13.4 454 n.d. n.d. [BMIM]Cl 13 593 (544) 83 12 [BMIM]Cl 10 1198 (812) 83 12 [BMIM]Cl 10 1000 100 n.d.
17
Liquido ionico Solubilità % DPa (DP)b Temperature/°C Tempo/h
[BMIM]Cl 5 1000 80 sonicazione [BMIM]Cl 3 1000 70 n.d. [BMIM]Cl 3 1000 70 n.d. [BMIM]Fmt <20 250 85 n.d. [BMIM]Fmt 8 225 110 n.d. [AMIM]Fmt 20 250 80 n.d. NMMO 17 454 (444) n.d. n.d. NMMO 14.4 569 (537) n.d. n.d. [BMIM]Ac 13.2 569 n.d. n.d. [EMIM]Ac 15 225 110 n.d. [EMIM]Ac 13.5 569 n.d. n.d. [EMIM]Cl 15.8 569 n.d. n.d. [EMIM]Cl 12 268 (329) 90 12 [EMIM]Cl ≈10 cellulosa 100 2 [EMIM]Cl 6 593 (580) 90 12 [EMIM]Cl 4 1198 (1129) 90 12 [AMIM]Cl 14.5 220 80 <0.5 [AMIM]Cl 14.5 650 80 <0.5 [AMIM]Cl 12.5 1085 n.d. n.d. [AMIM]Cl 11 250 100 n.d. [AMIM]Cl 10 250 100 n.d. [AMIM]Cl 10 1175 n.d. n.d. [AMIM]Cl 8 1600 80 <0.5 [AMIM]Cl >3 1176 n.d. n.d. [AMIM]Fmt 21.5 250 85 n.d. [BDMIM]Cl 12.8 569 n.d. n.d. [BDMIM]Cl 9 286 (377) 90 12 [BDMIM]Cl 6 593 (580) 90 12 [BDMIM]Cl 4 1198 (1129) 90 12 [BDMIM]Cl ≈3 1176 n.d. n.d. [ADMIM]Br 12 286 (320) 80 12 [ADMIM]Br 4 593 (599) 80 12 [ADMIM]Br 4 1198 (1203) 80 12
18
Liquido ionico Solubilità % DPa (DP)b Temperature/°C Tempo/h
[EMIM]MP 10 250 40-65 n.d. [HEMIM]Cl 6.8 cellulosa 70 12 [Bu4P]Fmt >5 225 110 n.d. [(C6)3C14P]Dca <0.5 225 110 n.d. [BzDTA]Cl 5 286 (327) 62 12 [BzDTA]Cl 2 593 (527) 62 12 [BzDTA]Cl 1 1198 (966) 62 12 [Bu4N]Fmt 1.5 225 110 n.d. [Amm110]Cl 0.5 225 110 n.d. [Amm110]Fmt 0.5 225 110 n.d. [Amm110]Ac 0.5 225 110 n.d. [Amm110]Dca <0.5 225 110 n.d. [M(OEt)2EtIM]Ac 12 225 110 n.d. [M(OEt)3EtIM]Ac 12 225 110 n.d. [M(OEt)4EtIM]Ac 10 225 110 n.d. [M(OEt)2Et3N]Ac 10 225 110 n.d. [M(OEt)3Et3N]Ac 10 225 110 n.d. [H(OEt)2MIM]Ac 5 225 110 n.d. [M(OEt)7EtIM]Ac 3 225 110 n.d. [H(OEt)3MIM]Ac 2 225 110 n.d. [H(OEt)2MIM]Cl 2 225 110 n.d. [M(OEt)3-MeOEtOMe-IM]Ac 0.5 225 110 n.d. [M(OPr)3EtIM]Ac 0.5 225 110 n.d. [M(OEt)3BuIM]Ac <0.5 225 110 n.d. [MM(EtOH)NH]Ac <0.5 225 110 n.d. [(MeOEt)2NH2]Ac <0.5 225 110 n.d. [MM(MeOEt)NH]Ac <0.5 225 110 n.d. [M(MeOEt)2NH]Ac <0.5 225 110 n.d. [BMIM]Br 5-7 1000 microonde [BMIM]SCN 5-7 1000 microonde [BMIM]BF4 1000 microonde [BMIM]PF6 1000 microonde [C6MIM]BF4 5 1000 microonde
19
Liquido ionico Solubilità % DPa (DP)b Temperature/°C Tempo/h
[C8MIM]BF4 poco solubile 1000 microonde
[C2MIM]Cl ≈10 cellulosa 100 2 [C3MIM]Cl ≈0.5 cellulosa 100 2 [C4MIM]Cl ≈20 cellulosa 100 2 [C5MIM]Cl ≈1.5 cellulosa 100 2 [C6MIM]Cl ≈6.5 cellulosa 100 2 [C7MIM]Cl ≈5 cellulosa 100 2 [C8MIM]Cl ≈4 cellulosa 100 2 [C8MIM]Dca <1 225 110 n.d. [C9MIM]Cl ≈2.5 cellulosa 100 2 [C10MIM]Cl ≈0.5 cellulosa 100 2 [ABIM]Cl >3 1176 n.d. n.d. [DAIM]Cl >3 1176 n.d. n.d. [APIM]Cl <3 1176 n.d. n.d. [BMIM]Tf2N <0.5 225 110 n.d. [BMIM]Sac 1176 n.d. n.d. [BMIM]Ts 1176 n.d. n.d. [BMIM]Ts 1176 n.d. n.d. [BMIM]HSO4 1176 n.d. n.d. [BMIM]Dca 1176 n.d. n.d. [BMIM]Dca 1 225 110 n.d. [BDMIM]SCN 1176 n.d. n.d. [ABIM]Dca 1176 n.d. n.d. [AOMIM]Dca 1176 n.d. n.d. [AOMIM]Cl 1176 n.d. n.d. Ch/urea 1176 n.d. n.d.
a Prima della rigenerazione; b Dopo la rigenerazione; n.d. Dato non disponibile; Cationi: [ABIM] =
1-allil-3-butilimidazolio, [ADMIM] = 1-allil-2,3-dimetilimidazolio, [AMIM] = 1-allil-3-metilimidazolio, [Amm110]Cl =
AMMOENG ™, [AOMIM] = 1-allilossi-3-metilimidazolio, [APIM] = 1-allil-3-propargilimidazolio, [BMIM] = 1-butil-3-metilimidazolio, [BDMIM] = 1-butil-2,3-dimetilimidazolio, [BzDTA] = benziltrimetil(tetradecil)ammonio, [BMPy] = 3-metil-N-butilpiridinio, [EMIM] = 1-etil-3-metilimidazolio, [HEA] = 2-idrossietilammonio, [HEMIM] = 1-(2-idrossimetil)-3-metilimidazolio; Anioni: Ac = acetato, BF4 = tetrafluoroborato, Cl = cloruro, Dca = dicianammide, Fmt
= formiato, MP = metilfosfonato, PF6 = esafluorofosfato, Sac = saccarinato, SCN = tiocianato, Tf2N =
bis-(trifluorometansolfonil)amide, Ts = tosilato; Altri solventi: Ch/urea = colina:urea (1:2), NMMO = N-metilmorfolina-N-ossido
20 (principalmente a causa della mobilità degli ioni), e tempi di dissoluzione più lunghi (> 12 h), non sempre portano a migliori risultati, specialmente ad alte temperature [92-94]. Il rischio di parziali degradazioni ad elevate temperature conta poco, considerando che, molto spesso, la migliore temperatura alla quale effettuare la dissoluzione è 10°C sopra al punto di fusione del liquido ionico [85,95].
Risultati impressionanti si ottengono quando si effettua un riscaldamento mediante microonde e non fornendo calore. Ad esempio la solubilità di una cellulosa con un DP di 1000 può essere incrementata del 150% [7]. Il riscaldamento con microonde è caratterizzato da un processo di riscaldamento interno, dovuto ad un assorbimento di calore diretto da parte delle molecole polari e che differisce significativamente dai metodi convenzionali di riscaldamento, basati sul trasferimento di calore. Questo riscaldamento interno può essere il responsabile della più efficiente rottura del reticolo di legami ad idrogeno presente fra le micro fibrille, anche se si deve prestare attenzione perché il riscaldamento è estremamente veloce e può portare alla pirolisi del polimero altrettanto rapidamente [86]. La dissoluzione mediante sonicazione, invece, sembra avere solo un piccolo effetto positivo sulla dissoluzione [7]. Altri fattori che influenzano la dissoluzione, sono il grado di polimerizzazione della cellulosa, così come la struttura del liquido ionico [93].
I cationi migliori per il processo di dissoluzione della cellulosa sono basati su
cores di natura metilimidazolica e metilpiridinica, con catene allilica, etilica o butilica: la catena
allilica determina una diminuzione della capacità di impaccamento e di ordinamento del sale, per cui sali con questa catena hanno punti di fusione decisamente minori (17°C per [AMIM]Cl, contro i 65°C di [BMIM]Cl) e, soprattutto, una viscosità molto minore (685 MPa.s del [AMIM]Cl, contro gli 11000 MPa.s del [BMIM]Cl a 30°C) [97,98]. Perfino il numero di atomi di carbonio della catena laterale, nella serie da C2 a C20, dà ottimi risultati, rispetto a numeri decisamente maggiori [96]. Il
potere di dissoluzione massimo si raggiunge con una catena laterale butilica (C4); inoltre, sembra
che una funzione idrossilica sulla stessa catena aumenti la solubilità: ciò può essere dovuto alla aumentata polarità dell’anello etero aromatico [86]. I doppi legami sulla catena laterale diminuiscono la viscosità del liquido ionico e lo stesso effetto è osservato se uno degli atomi di carbonio della catena laterale viene rimpiazzato da un atomo di ossigeno, benché questi IL tendono, in genere, a non dissolvere la cellulosa [38a,48].
Il miglior anione in assoluto per la dissoluzione è il cloruro ([Cl]-), sebbene l’acetato ([CH3CO2]-), che è in grado di sciogliere fino al 4% in peso di cellulosa ad 80°C, sia
abbastanza efficiente. Studi sono stati condotti per sviluppare liquidi ionici che avessero una minore viscosità ed il formiato ([HCOO]-) [99], è stato dimostrato essere non solo considerevolmente meno viscoso dei composti con anione cloruro (≈66 cP a 25°C per [AMIM]Fmt), ma addirittura avere una
21 maggiore capacità solvente, a causa della sua maggiore capacità di accettare legami ad idrogeno [13]. Sono stati preparati anche liquidi ionici di natura imidazolica aventi come anioni dimetilfosfati ([Me2PO4]-), metilfosfonati ([MePO2(OMe)]-) e metil metilfosfonati ([MePO3)]-): in particolare
[EMIM][MePO3)]- ha permesso di preparare soluzioni di cellulosa al 10%, riscaldando a 45°C per
30 minuti o, addirittura, soluzioni al 2-4% senza alcun riscaldamento preventivo; nel caso di questo anione, i problemi maggiori derivano dalla tossicità [100].
Studi di rilassamento condotti su 13C e 35/37Cl, indicano che vi è una relazione stechiometrica fra gli anioni cloruro e i gruppi idrossilici della cellulosa [101].
Figura 9. A. Profili di dissoluzione di cellulosa microcristallina in vari percentuali in peso in [EMIM]Ac a 80°C; B. Paragone fra le abilità di sciogliere la cellulosa di vari liquidi ionici ad 80°C; C. Microscopia di EMIM[Ac] con il 4% in peso di cellulosa
Il miglior liquido ionico per sciogliere questo polimero, è stato dimostrato essere [BMIM]Cl ] (Figura 9), capace di raggiungere il 25% in peso di cellulosa. La sostituzione del Cl -con anioni quali Br- e SCN- porta ad una diminuzione della solubilità, mentre anioni come BF4- e
22 PF6-, non coordinanti, non sono adatti a scioglierla; inoltre la sostituzione della catena laterale
butilica con una più ingombrante, porta ad un’ulteriore diminuzione di solubilità. Il problema maggiore dell’anione cloruro e di tutti gli alogenuri in generale, è essenzialmente la loro elevata viscosità e il loro elevato punto di fusione, che ne rende, di fatto, impensabile l’utilizzo per applicazioni di tipo industriale con la cellulosa (che aumenterebbe addirittura la viscosità della miscela): temperature di oltre 80°C, potrebbero portare alla formazione di organo-alogenuri per decomposizione termica parziale del liquido ionico [102]; questi potrebbero essere potenzialmente pericolosi per l’ambiente e gli animali, dopo l’inevitabile contaminazione che seguirebbe il processo industriale [103]. Un altro punto a loro sfavore è il potenziale corrosivo degli alogenuri, non trascurabile. Per questo [EMIM]Ac si ritiene essere un miglior solvente: ha un punto di fusione decisamente molto basso (< -20°C), una bassa viscosità (≈ 145 cP a 25°C), oltre ad un potere corrosivo estremamente minore [104-106]; in più si è dimostrato essere sia meno tossico del corrispettivo alogenuro, sia biodegradabile [107].
Si ritiene che sia il catione che l’anione siano coinvolti nel meccanismo di dissoluzione della cellulosa. Lo Schema 2 mostra il probabile meccanismo della dissoluzione del
OH OH OH Cellulosa Cellulosa [BMIM]Cl O O Cellulosa Cellulosa [BMIM] H Cl H Cl [BMIM]
Schema 2. Meccanismo proposto per la dissoluzione della cellulosa in [BMIM]Cl
polimero nei liquidi ionici: l’atomo di ossigeno e quello di idrogeno della cellulosa formano un complesso elettron-accettore-donatore (EDA, electron donor-electron acceptor), con le specie cariche che costituiscono il liquido ionico; è stato suggerito che ciò accada principalmente fra gruppi idrossilici in C6 e in C3 delle catene di cellulosa vicine [108]. Questa interazione ha come
effetto la separazione dei gruppi idrossilici appartenenti a catene differenti, portando alla dissoluzione della cellulosa nel liquido ionico [9,86].
Heinze e coll. hanno investigato riguardo l’interazione che possono avere i cationi degli IL, ma usando modelli più semplici (cellooligomeri, con un DP compreso fra 6 e 10). Sulla base di studi NMR, è stata suggerita la possibile formazione di un legame covalente di [EMIM]Ac fra il carbonio C1 della cellulosa e il C2 del nucleo imidazolico (Figura 9), il carbonio più acido:
23 O O O O OH N N OH OH HO OH OH HO OH OH HO OH H CH3CO2
-Figura 10. Struttura proposta per un legame covalente fra [EMIM]Ac ed un cellooligomero (DP da 6 a 10)
glucosio, dopo la dissoluzione dello stesso in [EMIM]Ac. Ebner e coll. hanno verificanto questa ipotesi, mediante l’utilizzo di un labeling con 13C e con esperimenti di fluorescence labeling, che indicano la formazione di un legame covalente fra il carbonio C-2 dei liquidi ionici con catione 1-alchil-3metilimidazolio e l’estremità riducente della cellulosa [109]. Sorprendentemente, questo non avviene quando l’oligomero viene disciolto in [EMIM]Cl: dunque, la formazione di questo intermedio è fortemente catalizzata da basi [37]. Una spiegazione a questa dicotomia, può essere trovata se si pensa ad un liquido ionico, [EMIM]Ac, nel quale erano presente impurezze di natura basica (imidazolo), e all’altro, nel quale probabilmente non c’erano, rendendo di fatto il IL incapace di catalizzare la reazione. Un’altra spiegazione di questo fenomeno, può essere il reticolo di legami ad idrogeno del [EMIM]Cl (Figura 7) [37]. Secondo Leipner e coll., che hanno sviluppato la dissoluzione della cellulosa con sali idrati fusi, è necessaria una sfera di coordinazione non saturata del catione, affinché questo possa interagire con la cellulosa [87]. Ma anche se si assume che il catione [EMIM] abbia questa sfera di coordinazione, ciò non spiega il simile potere solvente di questi due liquidi ionici nei confronti del polimero. È stato ipotizzato che la cellulosa disciolta possa esistere con diversi DP che dipendono, fra gli altri, dalla natura del solvente [15,108]. Ciò può spiegare la differente reattività dei liquidi ionici nei confronti di questo biopolimero.
Anche la contaminazione del solvente può influenzare la capacità solvente del liquido ionico. È stato dimostrato che lo 0.01% in peso di acqua nel [BMIM]Cl è sufficiente ad ostacolare la dissoluzione della cellulosa [7]. Pressioni elevate, da 0.2 a 0.9 MPa possono agevolare il processo di dissoluzione, mentre l’addizione di cosolventi può addirittura diminuire la velocità (disolfuro di carbonio), o non avere alcun effetto sulla solubilità, riducendo però la viscosità della miscela (DMSO-d6, CH3Cl, DMF) [92,110-113].
Comunque sia, la fonte principale di cellulosa rimane il legno e il processo Kraft, inventato 120 anni fa, rappresenta ancora il metodo industriale principale per l’estrazione della cellulosa dal legno [114]. I liquidi ionici sono in grado di dissolvere anche la lignina, oltre alla cellulosa [114,115]. Sun e coll. hanno disciolto polvere di legno in [EMIM]Ac o [BMIM]Ac e
24 hanno parzialmente separato la lignina scegliendo una miscela ricostituente di acetone ed acqua (1:1): la lignina pura, priva di contaminazioni saccaridiche, può essere facilmente disciolta in questa miscela, mentre la cellulosa e i materiali ricchi di cellulosa vengono disciolti in liquidi ionici e poi recuperati a parte. Può essere anche possibile incrementare il processo di delignificazione usando liquidi ionici a base colinica: non solo sono più biocompatibili rispetto ai liquidi ionici a base imidazolica, ma ci si aspetta che siano solventi scarsi nei confronti della cellulosa; essendo però ottimi solventi per la suberina e la lignina, possono essere utilizzati nelle separazioni [116].
Mikkola e coll. hanno dimostrato l’elevata efficienza di metodi di riscaldamento non convenzionali, a fronte del comune riscaldamento termico, per la dissoluzione della cellulosa nei due liquidi ionici [AMIM]Cl e [BMIM]Cl: ad esempio, per ottenere una soluzione al 5% di cellulosa in [AMIM]Cl serviva 1 h o più, mentre sono necessari solo due minuti mediante irradiazione con ultrasuoni e si arriva ad un 27% in peso utilizzando ultrasuoni ad impulsi (1 minuto di ultrasuoni seguito da un minuto di pausa) [117]. Swatloski e coll. hanno invece dimostrato che si possono ottenere soluzioni fino al 25% se, accanto ad un normale riscaldamento, si affiancano impulsi a microonde [118].
1.4.4
Rigenerazione della cellulosa
Al giorno d’oggi, le fibre di cellulosa rigenerate derivano dal processo industriale della viscosa, conosciuto da ormai un centinaio di anni: le fibre di cellulosa ottenute con questo processo (Rayon), hanno eccellenti proprietà per una vasta gamma di prodotti, dal Modal (fibra prodotta a partire dalla polpa del legno delle Fagacee, che si sfibra, si restringe e scolorisce in maniera molto ridotta rispetto al cotone), alle fibre tecniche, come quelle impiegate in pneumatici ad alte prestazioni. Tecnologicamente, la via della viscosa è estremamente complessa, nonché problematica dal punto di vista ambientale, per via dell’uso di CS2, dei metalli pesanti durante la
fase di precipitazione e della produzione di sottoprodotti inquinanti [119,120].
Recentemente, fibre precipitate da soluzioni di NMMO monoidrata, stanno competendo con le fibre ottenute mediante il processo viscosa. In particolare, le fibre di Lyocell (o Tencel) esibiscono eccellenti proprietà, come un’ottima resistenza agli sforzi, sia nello stato umido che secco, un buon modulo elastico (capacità di subire deformazioni di tipo elastico), una buona capacità di assorbimento, oltre a lucentezza e morbidezza; inoltre, la particolarità di questo processo, è la completa assenza di reazioni chimiche, con la totale assenza di sottoprodotti. Questo metodo, però, non è ancora riuscito a soppiantare quello della viscosa per vari motivi: la cellulosa necessita di essere attivata, sono necessarie alte temperature, si formano sottoprodotti di
25 decomposizione del solvente se non si aggiungono antiossidanti (sottoprodotti che andranno poi riconvertiti in NMMO), e i costi sono ancora molto elevati [121].
Il concetto sul quale si basa la rigenerazione della cellulosa è sempre il solito, indipendentemente dal tipo di solvente utilizzato. La soluzione contenente il biopolimero viene precipitata mediante l’addizione di un eccesso di un solvente, come acqua, acetone, diclorometano, acetonitrile, o miscele degli stessi [7,11,48,92,93,122,123]. Spesso si preferisce utilizzare acqua deionizzata quando si precipita da sistemi solventi con NMMO, in quanto le fibre coagulate presentano un più alto grado di cristallinità e la matrice rigenerata ha una maggiore forza [124,125].
Il risultato opposto si ottiene quando la cellulosa viene recuperata dai liquidi ionici, con un grado di polimerizzazione finale più basso del 50-75% rispetto a quello del biopolimero disciolto [126]. Sebbene gli studi 1H NMR a bassa risoluzione sulle fibre di cellulosa rigenerata in uno stato idratato mostrino un rapido scambio protonico fra gli idrossili delle catene esterne della cellulosa con l’acqua circostante, questa interazione non è sufficiente per ristabilire completamente il rigido reticolo di legami ad idrogeno della cellulosa microcristallina [127]. Studi recenti indicano che l’idrogel finale è libero da residui di liquido ionico, che potevano contribuire all’abbassamento del grado di cristallinità [128]: sebbene la spettroscopia fotoelettronica a raggi X (XPS), sia di solito usata per analizzare la superficie dei materiali, è stata utilizzata per determinare eventuali cambiamenti nel reticolo di legami ad idrogeno della cellulosa, dopo trattamento con liquidi ionici [129].
La forma del materiale rigenerato varia a seconda del metodo di rigenerazione utilizzato e delle condizioni. Questi film, da 0.1 a 0.2 mm, si ottengono versando la cellulosa su lastre di vetro prima di effettuare una estrazione solido/liquido con acqua [130]; altri gruppi di lavoro hanno spruzzato acqua sulla superficie del gel, in modo da fissare forma desiderata, prima di lasciare la fibra immersa in acqua per alcuni giorni [131]. Duchemin e coll. hanno caratterizzato tutti i composti a base di cellulosa ed hanno investigato sull’effetto che hanno i vari parametri alla quale la si processa, come la velocità di dissoluzione, la velocità di precipitazione, la concentrazione iniziale di solvente nella cellulosa [131]. Questi parametri influenzano le proprietà meccaniche di tutti i materiali composti di cellulosa, attraverso la loro influenza sulla cristallinità, sulla formazione di spazi vuoti e sulla microstruttura laminata: i gel più duri si ottengono seguendo una precipitazione lenta e graduale. Comunque, se si inverte l’ordine con il quale si invertono le soluzioni e se si aggiunge il solvente per la coagulazione alla cellulosa, la cellulosa può essere recuperata in forma flocculata, facilmente separabile per filtrazione [126].
Kosan e coll. hanno plasmato la cellulosa mediante un processo di dry-wet
26 che la cellulosa esiste in vari stati di soluzione, a seconda del tipo di solvente usato e ciò influenza le proprietà delle fibre di polimero rigenerate.
Infine, aerogel nano fibrillari di cellulosa possono essere preparati se il gel rigenerato viene lavato con diossido di carbonio liquido, prima di essiccare in condizioni supercritiche: il diossido di carbonio rilasciato lascia un polimero poroso, la cui struttura corrisponde ad un aerogel (sostanza allo stato solido, simile a gel, in cui il componente liquido è sostituito da gas; il risultato è una schiuma solida) [94]. Metodi simili sono stati usati per ottenere idro- e metano gel [132].
1.4.5
Riciclo e rigenerazione del liquido ionico
L’elevato costo dei liquidi ionici rende necessario il loro riciclo e la loro rigenerazione, in particolar modo se si considerano gli elevati volumi necessari per il trattamento delle biomasse. Al giorno d’oggi, gli IL vengono fabbricati industrialmente sulla scala dei kg e poi venduti a prezzi elevati; la BASF, il maggior produttore al mondo di liquidi ionici, prevede un assestamento dei prezzi, con l’incremento della domanda, intorno ai 40$/kg per [EMIM]Ac, ad esempio. Lee e coll. hanno dimostrato un deterioramento del liquido ionico [EMIM]Ac dopo il quarto riciclo [133]: questo processo prevede l’utilizzo di vari step, quali la precipitazione del materiale cellulosico con anti-solventi, come acqua deionizzata o alcoli, filtrazioni, assorbimento di eventuali impurezze mediante trattamento con carbone attivo, lavaggi con solventi organici e una purificazione finale con allumina neutra attivata [134]. Questo elevato numero di passaggi per ottenere il recupero del liquido ionico, è un problema che deve essere ancora risolto ed ottimizzato.
Comunque sia, alcune opzioni esistono e possono essere sviluppate ed ottimizzate. L’uso di solventi in fase supercritica (SC), può essere interessante. Le proprietà dei solventi esistenti in questa fase possono, infatti, essere facilmente modificabili tramite piccole variazioni di pressione e temperatura: una volta ottenuta l’estrazione del liquido ionico, uno switch verso condizioni al di sotto dello stato supercritico permetterebbero un facile allontanamento del solvente e un facile recupero del prodotto desiderato, il liquido ionico, nel nostro caso. Un’altra opzione è l’utilizzo di resine a scambio ionico, in maniera tale da poter ottenere il nostro IL in forma salina, con solo parziale precipitazione del polimero [135]; se, inoltre, il liquido ionico ottenuto fosse di natura idrofobica ([BMIM]PF6), ci si potrebbe immaginare l’uso di un trasportatore (shuttle), a
temperatura controllata, attraverso una fase micellare fra la fase lipofila, costituita dal nostro liquido ionico, e una fase idrofila, formata da blocchi di glicole polietilenico, come descritto da Lodge e coll. [136]
27
1.5
Funzionalizzazione della cellulosa
Le modificazioni chimiche delle fibre naturali mirano ad un’alterazione delle loro proprietà chimico-fisiche, per soddisfare le esigenze del produttore. Gli esteri della cellulosa con acidi organici e inorganici, così come gli eteri, sono stati i composti pionieri della chimica della cellulosa e rimangono le tecniche di derivazione della cellulosa più importanti. Vengono usate come rivestimenti, leganti, stucchi, lubrificanti per trapani, esplosivi, film ottici, o mezzi per ottenere separazioni, oltre che in applicazioni mediche (dialisi, bioingegneria dei tessuti e delle ossa, medicazione di ferite, rimpiazzo delle articolazioni), nell’industria alimentare, o per l’adsorbimento di metalli pesanti [15,137-139]. Questi derivati possono essere acetati, carbossimetilati, benzoilati, carbammati, metacrilati, carbonati, solfati, solfonati, ftalati, furoati, esteri maleici, o resine esterificate [6,10,15,44,82,113,130,137,139-147].
I liquidi ionici sono stati usati come solventi per molte modificazioni della cellulosa. Derivati del biopolimero acetilati, benzoilati e carbammati, sono stati preparati con successo in [BMPy]Cl, [BMIM]Cl, [BDMIM]Cl, [AMIM]Cl, [ADMIM]Br, [BnMIM]Cl, [EMIM]Cl e [EMIM]Ac [1,10,88,95,130,141,148]. Il grado di sostituzione (DS), può essere influenzato dalla scelta dell’agente acilante, dal tempo di reazione e dai rapporti stechiometrici fra i reagenti, ma dipende anche dallo stato di dissoluzione della cellulosa. Nel caso delle acilazioni, gli acil cloruri sono in genere più efficaci rispetto alle corrispondenti anidridi per le modificazioni [86,95,146]. La presenza di basi, come 1-metilimidazolo, o la piridina, possono aumentare l’efficienza della modificazione [88,112]. Inoltre, la differente reattività dei tre gruppi ossidrilici del monomero glucosio (C6 > C3 > C2), permette l’introduzione regio selettiva di gruppi protettivi,
come gli eteri, per esempio [6,149]. Granström e coll. hanno selettivamente protetto la cellulosa con 4-metossitritile in [AMIM]Cl, ottenendo una diminuzione della reattività del polimero modificato [150]. Queste funzionalizzazioni regio selettive sono un fattore chiave per la formazione di soprastrutture colloidali [15].
28 OH Cellulosa TsCl O Ts O HN O N3 NaN3 H 2NR R N R N N Ammina Solfonato Azide Triazolo O Emiacetale HO R O HN R Carbanilato O R OCN R O SiR3 Sililetere OH SiR3 O R' Etere R' O R'' O HN R' Ammina R' Cl O R' O O R'' O
Schema 3. Modi di funzionalizzazione della cellulosa
Lo Schema 3 è una panoramica semplificata di alcuni modi con cui si funzionalizza attualmente la cellulosa. Il gruppo idrossilico offre un ampio spettro di possibili reazioni con altri gruppi funzionali. Se si escludono eteri ed esteri, si possono trovare dati in letteratura che riguardano solfati, solfonati, deossiderivati, sililati, ammine, carbonilati, tritilati, emiacetali e azidi [6,15,112,147,149,151,152]. Koehler e coll. hanno sintetizzato trimetilsililcellulosa in [BMIM]Cl, [EMIM]Cl e [EMIM]Ac [112]. Lo ftalato di cellulosa è stato preparato in [AMIM]Cl e i florurati sono stati ottenuti mediante modificazione omogenea in [BMIM]Cl [145,146]; i gruppi idrossilici reagiscono con isocianati per ottenere carbonilati, gli ossirani per dare emiacetali e gli alogenuri solfonilici vengono convertiti in solfonati, come mesilati e tosilati, per esempio [15,153]. Questi solfonati sono facilmente rimossi e sono intermedi favorevoli per formare derivati della 6-deossicellulosa, come amidi o azidi [15,151]: le azidi della cellulosa possono essere dei valevoli precursori per derivati triazolici. In più, i solfati di cellulosa, solubili in acqua, sono stati preparati utilizzando diversi reagenti solfonilanti, in [BMIM]Cl, [AMIM]Cl e [AMIM]Ac [113].
Un approccio interessante è l’introduzione di molecole portanti gruppi funzionali ad entrambe le estremità della catena. Hattori e coll. hanno immomibilizzato carbammati sulla cellulosa attraverso un coupling via silano e li hanno copolimerizzati con acido metacrilico o
29 etilenglicol-dimetilacrilato, per ottenere polimeri ramificati, sotto UV [140]; altri hanno sviluppato polimeri elaborati, mediante l’utilizzo di alogenuri terziari, che polimerizzano facilmente con doppi legami reattivi, seguendo una via radicalica [142,154]. Un altro gruppo di ricerca ha fatto reagire i gruppi ossidrilici della cellulosa con 2-bromoisobutirril bromuro e ha fatto copolimerizzare il prodotto ottenuto con metacrilato o stirene via atom-transfer-radical-polymerization (ATRP), ottenendo polimeri living, ancora attivi perfino dopo un anno [155]. Meng e coll. hanno attivato la superficie della cellulosa con allilbromuro o divinilsolfone, prima di effettuare una polimerizzazione di tipo ATRP con polimetilacrilato (PMMA), o polistirene (PS) [156,157]. Oltre ai metodi già citati, l’elaborazione può essere fatta anche per condensazione diretta, ad esempio con silanoli, per ottenere un aumento di compatibilità della superficie con molecole idrofobiche. Sono stati introdotti anche prodotti bifunzionalizzati, che vengono utilizzati in matrici inerti [158].
Alcune modificazioni degne di nota sul biopolimero, includono l’utilizzo di dendroni (dendrimeri, una classe di polimeri macromolecolari monodispersi, nella forma ridotta), acidi boronici e il batterio Acetobacter xylinum: sono stati introdotti macromolecole dendritiche, fino alla quarta ramificazione, sulla superficie della cellulosa [130,153,159-161]: in genere, questi materiali vengono utilizzati per separazioni migliori per la gas cromatografia capillare ad alta risoluzione e nelle microestrazioni capillari [153]. Liebert, per esempio, ha sfruttato la capacità di acidi borici e boronici di reagire con i polioli nei liquidi ionici, mostrando che possono esserci interazioni al C2 e al C3 dei derivati del glucosio: la formazione di un anello a sette membri è stata
confermata, portando alla conclusione che una struttura simile può formarsi nelle macromolecole aventi glucosio come monomero, ovvero cellulosa ed amido [162]. Infine, Pommet e coll. hanno modificato la cellulosa naturale usando batteri, mediante la deposizione di cellulosa batterica nano particolata intorno alle fibre, aumentando la loro adesione a polimeri rinnovabili, come la cellulosa acetato-butirrato (CAB), o l’acido polilattico (PLA) [163].
Nella maggior parte dei casi, il liquido ionico può essere recuperato con successo al termine dell’operazione [88,95], sebbene si debba prestare attenzione a quegli IL che non sono inerti alle condizioni di polimerizzazione, per non ottenere prodotti indesiderati [109,146].
1.6
Materiali a base di legno
Un obiettivo a lungo termine è lo sviluppo di materiali a base di legno, con proprietà simili, o migliori, rispetto agli attuali materiali derivanti dal petrolio. Le fibre naturali offrono vantaggi rispetto a quelle artificiali, come una bassa densità, riciclabilità e biodegradabilità
30 [144]. Lu e coll. hanno proposto di raggruppare i metodi per formare questi materiali: combinatoriale, miscelazione, ammollo, spray [164]. La maggior parte dei materiali contenenti cellulosa sono stati preparati sia mediante miscelazione con materiali compatibili con l’acqua, sia mediante estrusione da una matrice plastica immiscibile con l’acqua [142]. Indipendentemente dal metodo di processazione, è importante distinguere fra due tipi generali di materiali a base di legno: miscele di legno con altri componenti e materiali formati interamente di legno.
1.6.1 Miscele di polimeri
La maggior parte dei polimeri naturali sono idrofili, in quanto contengono sia gruppi idrossilici, che gruppi polari. La segatura di legno e le plastiche riciclate, come il polietilene ad alta densità (HDPE), sono stati utilizzati per formare miscele di polimeri. La materia prima deriva da materiale di rifiuto economico: Adhikary e coll., per esempio, hanno combinato la farina di legno secca, con un granulato di HDPE mediante estrusione ed hanno pressurizzato, da 1 a 5 MPa, per formare un materiale ibrido. L’uso di un agente di coupling, come il polipropilene maleato, all’aumentare del contenuto di polimero, non solo aumenta la stabilità, ma anche la resistenza meccanica del materiale [165].
Un modo differente per fare miscele di biopolimeri, è possibile usando cellulosa disciolta. Il concetto è di formare un reticolo articolato fra le catene di cellulosa e le fibre del polimero sintetico durante il processo di rigenerazione, come illustrato nello Schema 4. La cellulosa
O O HO OH OH * N N O O Br AIBN 80°C, 5 h Materiale composto Cellulosa N N Cl = N N O O Br * * n n = Cellulosa
Schema 4. Miscela di due liquidi ionici usati per formare un copolimero cellulosa/IL
viene disciolta in una miscela di due liquidi ionici, in cui il [BMIM]Cl ha funzione di solvente, mentre il 1-(4-acriloilssi-butil)-3-metilimidazolio bromuro, agisce da monomero per la
![Figura 6. Temperature di fusione teoriche e sperimentali per una serie di [CnMIM]PF6](https://thumb-eu.123doks.com/thumbv2/123dokorg/7546128.108770/9.892.325.539.791.985/figura-temperature-fusione-teoriche-sperimentali-serie-cnmim-pf.webp)
![Figura 7. Rappresentazione schematica dei legami ad idrogeno nel [EMIM]Cl](https://thumb-eu.123doks.com/thumbv2/123dokorg/7546128.108770/10.892.232.658.291.645/figura-rappresentazione-schematica-legami-idrogeno-emim-cl.webp)
![Figura 9. A. Profili di dissoluzione di cellulosa microcristallina in vari percentuali in peso in [EMIM]Ac a 80°C;](https://thumb-eu.123doks.com/thumbv2/123dokorg/7546128.108770/21.892.175.710.398.992/figura-profili-dissoluzione-cellulosa-microcristallina-vari-percentuali-emim.webp)
![Figura 10. Struttura proposta per un legame covalente fra [EMIM]Ac ed un cellooligomero (DP da 6 a 10)](https://thumb-eu.123doks.com/thumbv2/123dokorg/7546128.108770/23.892.109.784.103.275/figura-struttura-proposta-legame-covalente-fra-emim-cellooligomero.webp)
