UNIVERSITA’ DI PISA
DIPARTIMENTO DI MEDICINA CLINICA E SPERIMENTALE
Scuola di Specializzazione in Pediatria
TESI DI SPECIALIZZAZIONE
CORRELAZIONE GENOTIPO - FENOTIPO
NELLA DISCINESIA CILIARE PRIMARIA: ANALISI DI UNA
CASISTICA PEDIATRICA CON MUTAZIONI NEL GENE DNAH11
Candidato: Relatori:
Dott.ssa MARIA DI CICCO
Ch.mo Prof. GIUSEPPE SAGGESE
Dott. MASSIMO PIFFERI
Alla mia famiglia
“Dite: è faticoso frequentare i bambini. Avete ragione. Poi aggiungete: perché bisogna mettersi al loro livello, abbassarsi, inclinarsi, curvarsi, farsi piccoli. Ora avete torto.
Non è questo che più stanca. E' piuttosto il fatto di essere obbligati a innalzarsi fino all'altezza dei loro sentimenti. Tirarsi, allungarsi, alzarsi sulla punta dei piedi.
Per non ferirli.”
Janusz Korczak
I n d i c e
Capitolo I - LA DISCINESIA CILIARE PRIMARIA
1.1 - Discinesia Ciliare Primaria: definizione, cenni storici ed epidemiologia ... 2
1.2 - Funzione ed ultrastruttura delle ciglia dell’apparato respiratorio ... 5
1.3 - Caratteristiche cliniche della Discinesia Ciliare Primaria ... 11
1.4 - Discinesia Ciliare Primaria: metodi diagnostici ... 15
1.4.a - L’Ossido Nitrico nasale: il test di screening della DCP... 15
1.4.b - L’analisi dell’attività ciliare in vitro ... 17
1.4.c - L’esame morfometrico ultrastrutturale delle ciglia dell’apparato respiratorio ... 18
1.4.d - Le colture cellulari e l’immunofluorescenza ... 20
1.5 - Approccio terapeutico e follow-up del paziente con DCP ... 22
Capitolo II - LA GENETICA DELLA DISCINESIA CILIARE PRIMARIA 2.1 - Genetica della Discinesia Ciliare Primaria ... 28
2.2 - PCD-causing genes ... 30
2.2.a - Geni necessari per la corretta struttura e funzione dei bracci esterni di dineina ... 31
2.2.b - Geni necessari per la corretta struttura e funzione dei bracci esterni ed interni di dineina ... 33
2.2.c - Geni necessari per la corretta organizzazione dell’assonema e per la struttura e funzione dei bracci interni di dineina ... 35
2.2.d - Geni necessari per la corretta struttura e funzione dell’apparato centrale e dei radial spokes ... 36
2.2.e - Geni associati a sindromi ... 36
2.3 - Correlazione Genotipo-Fenotipo nella DCP ... 38
2.3 - Il gene DNAH11... 41
Capitolo III - IL NOSTRO STUDIO 3.1 - Scopo dello studio ... 46
3.2 - Materiali e metodi ... 48
3.3 - Risultati ... 54
3.3.a - Popolazione studiata ... 54
3.3.b - Diagnosi di DCP ... 56
3.3.c - Caratteristiche cliniche della popolazione studiata ... 58
3.3.d - Pazienti con mutazioni nel gene DNAH11 ... 62
3.5 - Discussione ... 71
3.5 - Conclusioni ... 78
Capitolo IV - BIBLIOGRAFIA Bibliografia ... 79
Abbreviazioni in ordine alfabetico ... 93
C a p i t o l o I
LA DISCINESIA CILIARE PRIMARIA
1.1 - DISCINESIA CILIARE PRIMARIA: DEFINIZIONE, CENNI STORICI ED EPIDEMIOLOGIA
Con il termine Discinesia Ciliare Primaria (DCP) (OMIM: 244400) si definisce un gruppo di condizioni patologiche congenite, eterogenee dal punto di vista clinico e genetico, alla cui base sono alterazioni della funzione delle ciglia della mucosa respiratoria che determinano, a seguito del conseguente deficit del trasporto muco-ciliare, la comparsa di diversi quadri di patologia a carico delle vie aeree e del parenchima polmonare [1 - 3].
Lo studio delle patologie correlate alle disfunzioni ciliari ebbe inizio nei primi decenni del secolo scorso con la descrizione, da parte di Siewert, di alcuni soggetti in cui erano contemporaneamente presenti un quadro di broncopneumopatia cronica ostruttiva con bronchiectasie e il Situs Viscerum Inversus (SVI) [4], mentre nel 1933 Kartagener esaminò 11 soggetti affetti da sinusite cronica, bronchiectasie e SVI, suggerendo la possibile esistenza di un’eziologia comune per queste tre condizioni: da allora questa triade è nota come Sindrome di Kartagener [5]. Dovettero passare quarant’anni prima che Afzelius descrivesse due fratelli non gemelli con astenozoospermia in cui l’esame ultrastrutturale dei flagelli spermatici dimostrava l’assenza dei bracci di dineina, la cui esistenza e funzione erano state scoperte poco più di dieci anni prima [6, 7]. Nello stesso anno un esperimento sulla valutazione del trasporto muco-ciliare mediante aerosol radiomarcato dimostrò che in due soggetti affetti da Sindrome di Kartagener il trasporto muco-ciliare era praticamente assente [8]. Infine, nel 1976 Afzelius condusse uno studio su 4 uomini (3 con SVI) con astenozoospermia, elevata morbilità infettiva a carico delle vie aeree e trasporto muco-ciliare assente, dimostrando che anche nelle ciglia dell’epitelio
respiratorio di questi soggetti i bracci di dineina erano assenti. Poiché l’alterazione prevalente
sia nei flagelli degli spermatozoi sia nelle ciglia dell’apparato respiratorio era rappresentata dall’assenza dei bracci esterni ed interni di dineina, Afzelius ipotizzò che tale deficit potesse essere alla base di un disturbo generalizzato dell’attività ciliare [9] che Eliasson descrisse nel
1977 in una nuova entità che denominò sindrome delle ciglia immobili: tale condizione venne definita come sindrome congenita con eredità autosomica recessiva caratterizzata da bronchite cronica sin dalla prima infanzia, rinosinusite cronica, sterilità, frequente associazione con bronchiectasie e SVI [10].
Successive analisi funzionali al microscopio ottico a contrasto di fase dimostrarono che in questi soggetti le ciglia non sono necessariamente immobili, bensì possono presentare gradi qualitativamente e quantitativamente variabili di alterata motilità rimanendo invariato l’effetto complessivo negativo sulla capacità di clearance muco-ciliare della mucosa respiratoria. Nel 1980 fu quindi proposto di sostituire la denominazione sindrome delle ciglia immobili con Discinesia Ciliare Primaria per sottolineare come la condizione fosse dovuta alla discinesia e non necessariamente all’immobilità ciliare [11]. Nella denominazione “DCP” (nel mondo anglosassone Primary Ciliary Dyskinesia, PCD) sono attualmente comprese tutte le possibili anomalie ciliari congenite in grado di alterare il trasporto muco-ciliare, per cui la sindrome delle ciglia immobili e la sindrome di Kartagener vengono oggi ritenute due dei numerosi fenotipi della DCP [3].
Allo stesso modo, oggi sappiamo che le alterazioni ultrastrutturali sottostanti all’alterazione della funzione ciliare non si limitano alla dineina, bensì possono interessare ogni singolo componente del ciglio e, secondo recenti stime, nel 10-25% dei casi l’ultrastruttura ciliare può risultare addirittura normale [12, 13]. Negli ultimi dieci anni, inoltre, si è assistito ad un enorme sviluppo delle conoscenze sulla genetica di questa condizione, portando ad identificare, al giugno del 2014, ben 28 geni patogenetici, sebbene si stimi che i geni coinvolti siano ancor più numerosi (dai 250 agli 800 geni) e che le mutazioni descritte fino ad oggi coprano solamente il 50 - 60% dei casi [14]. Non solo: di pari passo alla genetica è cresciuto anche l’interesse per lo studio delle ciglia presenti negli apparati extrarespiratori, a cui è seguita la descrizione di numerose patologie e sindromi che vengono oggi considerate membri della famiglia delle cosiddette “ciliopatie”, di cui ovviamente fa parte anche la DCP [15].
Nonostante ciò, la DCP resta una condizione relativamente “nuova”, della cui prevalenza, incidenza, morbilità e mortalità ad oggi non sono ancora disponibili dati certi. Le stime della prevalenza variano da 1:2.265 (in una popolazione asiatica residente in Inghilterra, con alto grado di consanguineità) a 1:40.000 (in una casistica selezionata per la contemporanea presenza
di SVI e bronchiectasie), con un consenso prevalente su 1:10.000 senza differenza di sesso [16, 17]: la DCP è quindi considerata una “malattia rara”, dal momento che l’Unione Europea ha definito come “rara” qualsiasi patologia che abbia una prevalenza inferiore allo 0,05% della popolazione, ossia inferiore a 1:2.000.
Sulla base dei dati disponibili si può ritenere che in Italia ogni anno nascano circa 70 soggetti affetti da DCP e che complessivamente i malati nel nostro Paese siano circa 4.000, di cui la maggior parte non è stata ancora diagnosticata.
1.2 - FUNZIONE ED ULTRASTRUTTURA DELLE CIGLIA DELL’APPARATO RESPIRATORIO
La porzione più superficiale della tonaca mucosa che riveste le vie aeree è rappresentata, per quasi tutta la sua estensione, da un epitelio ciliato: esso è presente nelle cavità nasali, nei seni paranasali, nelle tube uditive, nell’orecchio medio e nelle vie aeree dalla trachea fino ai bronchioli terminali, con la funzione di assicurare la cosiddetta clearance muco-ciliare. Le ciglia dell’epitelio respiratorio sono, infatti, dotate di movimento, attraverso il quale provvedono al drenaggio del muco prodotto dalle cellule mucipare della mucosa e dalle ghiandole della sottomucosa (in cui sono intrappolati batteri, virus e particelle estranee inalate con ogni atto del respiro) verso l’orofaringe, dove viene successivamente eliminato tramite la deglutizione o l’espettorazione, rappresentando, in tal modo, uno dei meccanismi di difesa dell’apparato respiratorio [18].
Condizioni essenziali per un’efficace clearance muco-ciliare sono la composizione ottimale del muco (glicoproteine, proteoglicani, lipidi ed elettroliti) e una valida attività ciliare: in condizioni normali il muco ha uno spessore di 0,5 - 2 µm e si organizza sulla superficie della mucosa in 2 strati a diversa viscosità di cui quello profondo, periciliare, è costituito per lo più da acqua (fase acquosa) e da soluti, la cui concentrazione è determinata dal trasporto attivo di sodio e cloro (alterato nella Fibrosi Cistica - FC), mentre quello più superficiale costituisce la
fase viscosa del muco, che è in grado di intrappolare gli agenti estranei inalati [18, 19]. Il muco
è prodotto a partire dalle vie aeree cartilaginee fino ai bronchioli terminali e, nell’epitelio respiratorio, per ogni elemento ghiandolare muco-secernente sono presenti 5 cellule ciliate: ciò significa che l’80% delle cellule presenti in tale epitelio sono cellule ciliate, ognuna delle quali presenta 250 - 300 specializzazioni della superficie apicale dette ciglia, con diametro di circa 0,3 µm e lunghezza decrescente da 6 µm a livello della trachea a 3,6 µm a livello della settima generazione bronchiale, a valle della quale si riduce ulteriormente [18].
L’attività delle ciglia respiratorie, visualizzabile mediante l’impiego di videoregistratori di immagini digitali ad elevata risoluzione e ad alta velocità, è caratterizzata da un battito assimilabile ad un colpo di frusta che avviene sempre su un piano perpendicolare all’asse del ciglio e in due fasi distinte: un colpo attivo veloce (1/4 del ciclo), in cui il ciglio è esteso ed in
grado di esercitare il massimo della spinta (colpo efficace o “effective stroke”), seguito da un colpo di recupero lento (3/4 del ciclo), in cui il ciglio si incurva e si irrigidisce dalla base verso l’apice per tornare alla posizione di partenza (fase di recupero o “recovery phase”) [20]. Nella recovery phase il ciglio ritorna nella sua posizione iniziale attraversando la fase acquosa per poi cominciare un nuovo ciclo: il nuovo colpo veloce permetterà l’interazione dell’apice del ciglio con la fase viscosa del muco, con suo conseguente spostamento in direzione dell’orofaringe. Ogni ciclo dura circa 0,1 - 0,2 secondi, pari a circa 8 - 12 flessioni del ciglio al secondo (8 - 12 Hertz) [21]. Le ciglia, inoltre, sono disposte sulla superficie cellulare in file parallele: in ogni fila il battito è coordinato, sincrono ed unidirezionale, mentre le ciglia della fila adiacente battono nella stessa direzione (allineamento ciliare) e nella medesima fase (coordinazione
ciliare), ma con un piccolo ritardo che genera onde unidirezionali che fanno assumere al
movimento complessivo delle ciglia la tipica forma di onda metacronale il cui effetto globale è assimilabile a quello di un “campo di grano battuto dal vento” [22]. Le onde metacronali viaggiano in direzione opposta all’effettivo battito ciliare ed al flusso di muco: in tal modo le secrezioni possono essere spostate contro gravità.
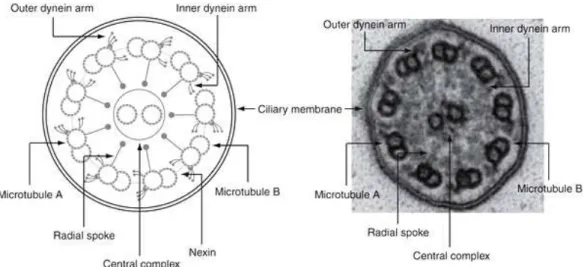
Il movimento delle ciglia è reso possibile da un’ultrastruttura microscopica molto complessa: al microscopio elettronico a trasmissione (TEM), infatti, in sezione trasversale un ciglio appare costituito da una struttura centrale costituita detta assonema, che è delimitata da un’estroflessione della membrana cellulare detta membrana ciliare. I principali costituenti dell’assonema sono i microtubuli, strutture che, oltre a costituire le fondamenta dell’assonema, svolgono anche un importante ruolo in quanto componenti del citoscheletro e del fuso mitotico di tutte le cellule eucarioti. I microtubuli sono strutture tubuliformi il cui costituente principale è la tubulina, eterodimero di 110 KD formato da due monomeri detti alfa (α) e beta (β): i dimeri di tubulina (α + β) allineati in successione danno vita a protofilamenti che si affiancano, disponendosi intorno ad una cavità centrale. L’assonema ciliare è costituito da 9 coppie periferiche di microtubuli e da una coppia centrale (tale organizzazione è detta 9+2): nella coppia centrale i microtubuli sono completi, ovvero costituiti da 13 protofilamenti, mentre la coppia periferica è costituita da due strutture denominate tubulo A e tubulo B, di cui il tubulo A è un microtubulo completo costituito da 13 protofilamenti e il tubulo B è un microtubulo incompleto costituito da 11 protofilamenti. Dai due microtubuli della coppia centrale emergono
sottili bracci proteici incurvati intorno ai due microtubuli quasi a formare un anello, che viene detto guaina centrale (central sheath). Da ogni tubulo A sporge, con una periodicità di 29 nm, un ponte radiale (radial spoke) che termina con un’estremità globulare nei pressi della guaina centrale, connettendo quindi le coppie periferiche a quella centrale. Ciascun tubulo A presenta inoltre due strutture costituite da un’ATPasi chiamata dineina, dette, in base alla loro posizione, braccio interno (IDA) e braccio esterno di dineina (ODA), che si estendono verso il tubulo B della coppia adiacente. Ogni coppia periferica è collegata a quella adiacente da sottili ponti che originano in prossimità dell’emergenza del braccio interno di dineina e che sono costituiti da una proteina elastica detta nexina, che forma legami paragonabili ai cerchi di un barile intorno all’intero assonema [23] (Fig. 1). Le strutture che si trovano nel punto di giunzione tra i radial spokes, il braccio interno di dineina e i legami di nexina vengono oggi considerate parte del cosiddetto Complesso Regolatore Nexina-Dineina (Nexin-Dynein Regulatory Complex - N-DRC), di cui la struttura più importante è il legame di nexina: tale complesso è ritenuto fondamentale per l’ancoraggio del braccio interno ed è necessario per ottenere un movimento ciliare corretto [24, 25].
Figura 1 - Ultrastruttura dell’assonema ciliare (da: Horani A et al. Pediatr Res. 2014; 75: 158-64)
L’assemblaggio dei microtubuli avviene in centri di organizzazione che forniscono una base da cui i microtubuli possono accrescersi: tale funzione è svolta dai corpuscoli basali (basal
bodies), che hanno ultrastruttura identica a quella dei centrioli e che agiscono da stampo per
disporre i microtubuli a coppie con il pattern 9+2. Il corpuscolo basale è una struttura cilindrica posta nella porzione apicale della cellula ciliata e costituita da un anello di 9 triplette di microtubuli, ognuna delle quali è composta da un microtubulo completo (tubulo A) e due incompleti (tubuli B e C). I microtubuli delle coppie periferiche dell’assonema si continuano nei tubuli A e B del corpuscolo basale, mentre quelli della coppia centrale si arrestano 1 µm sopra la superficie cellulare (l’assetto 9+2 dei microtubuli dell’assonema si modifica anche nella porzione apicale del ciglio, dove le coppie periferiche perdono progressivamente il tubulo B e dove, talora, non si riconoscono anche altre strutture quali la coppia centrale ed i bracci di dineina) [26].
Durante la ciliogenesi le ciglia si allungano dal corpuscolo basale mediante l’apposizione di nuove subunità a livello della loro estremità distale. A garantire il corretto assemblaggio e la manutenzione del ciglio è il trasporto intraflagellare, meccanismo complesso che, mediante proteine motrici quali la chinesina, permette il trasporto delle varie subunità da e verso i microtubuli. Tuttavia, non è ancora ben noto il meccanismo per mezzo del quale le proteine vengono trasportate dal citoplasma fino al corpuscolo basale [27].
Come detto, l’assonema ciliare è la struttura che rende possibile il movimento ciliare, il cui motore è la dineina: questa ATPasi ha dimensioni enormi (il suo peso molecolare supera il milione di Daltons) e viene oggi denominata dineina ciliare per distinguerla da un’analoga ATPasi cellulare, detta dineina citoplasmatica, che è coinvolta, invece, nel trasporto degli organelli.
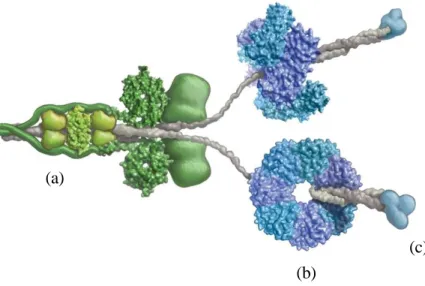
La dineina è costituita da: un’estremità rappresentata da catene leggere e da catene intermedie che ancorano la proteina al microtubulo A e in grado di trasmettere il movimento (a), una regione motrice costituita da una o più catene pesanti che terminano con una testa globulare a sua volta rappresentata da un anello di sei domini AAA (ATPases associated with cellular
activities), di cui 4 in grado di legare e idrolisare l’ATP (b), e un gambo che emerge dal
Figura 2 - Struttura della dineina (a: struttura per l’ancoraggio della dineina al tubulo A, necessaria per
la trasmissione del movimento; b: dominio motore della dineina; c: gambo per il legame al tubulo B)
Nell’assonema i bracci interni ed esterni di dineina differiscono dal punto di vista strutturale e funzionale: nei bracci esterni, che regolano la velocità dello scivolamento dei microtubuli (e quindi la frequenza del battito ciliare), le catene pesanti formano 3 teste globulari e parte dei relativi 3 peduncoli tramite i quali le teste sono collegate alla base comune saldamente ancorata alla superficie esterna del tubulo A. Le teste globulari sono proiettate verso il tubulo B della coppia adiacente ad intervalli regolari di 24 nm (in senso antiorario se osservati dalla base del ciglio). I bracci interni, che invece contribuiscono alla generazione della forza e, insieme ai ponti radiali e alla coppia centrale organizzano l’ampiezza del piegamento e, quindi, determinano la morfologia del battito ciliare, presentano una struttura simile a quella dei bracci esterni, ma sono costituiti da polipeptidi diversi (contengono almeno 7 isoforme di dineina) e possono presentare una o due sole teste globulari [23].
Il movimento ciliare è reso possibile dal reciproco scorrimento delle coppie di microtubuli le une sulle altre, con i bracci di dineina a generare l’energia necessaria a tale scopo. In fase di riposo, in assenza di ATP, il braccio di dineina del tubulo A di una coppia è adeso al tubulo B di quella adiacente; in seguito al legame dell’ATP ai siti di legame sulle teste globulari il braccio si accorcia e si verifica quindi l’idrolisi dell'ATP con conseguente modificazione conformazionale
(a)
(c) (b)
della dineina ed estensione del braccio verso la base del ciglio. A questo punto il braccio esteso aderisce al tubulo B nella nuova posizione mentre si liberano ADP e P e, accorciandosi e ruotando verso l'alto, causa uno scivolamento reciproco delle due coppie periferiche di circa 15 nm [28]. Con l’idrolisi dell’ATP ogni braccio di dineina genera una forza che agisce in modo da spostare verso l’apice del ciglio la coppia di microtubuli adiacenti con cui prende contatto: in presenza di punti fermi di ancoraggio, rappresentati dai legami di nexina e dai ponti radiali, si genera una resistenza elastica che fa sì che il movimento di scorrimento si trasformi in movimento di flessione [29].
Tutte le coppie periferiche sembrano essere funzionalmente equivalenti rispetto allo scivolamento: di conseguenza, se coppie diametralmente opposte fossero attivate insieme, la loro azione si annullerebbe. Inoltre, se tutti i bracci si attivassero nello stesso momento, l’assonema si avvolgerebbe a formare un’elica stretta. Di conseguenza è logico ritenere che solo le coppie di un lato dell’assonema si attivino in un dato istante per produrre la flessione del ciglio e che lo scorrimento dei microtubuli su un lato si alterni con quello sul lato opposto in modo tale che una parte del ciglio si pieghi prima in una direzione e poi in quella opposta. Ciò suggerisce che la direzione del piegamento sia regolata da un meccanismo che attiva di volta in volta lo scivolamento delle diverse coppie in differenti posizioni lungo la circonferenza assonemale, producendo il tipico movimento a frusta del ciglio, che è l’unico efficace. Il meccanismo di propagazione della flessione non è comunque ancora ben compreso mentre è noto che la velocità di scivolamento dei microtubuli, che è legata alla frequenza del battito ciliare, è direttamente proporzionale alla concentrazione di ATP e di dineina, mentre lo sono in maniera minima la flessione e la coordinazione generale del battito. Studi eseguiti sui protozoi hanno dimostrato che la concentrazione di calcio nell'assonema gioca un ruolo fondamentale nel determinare la direzione del battito delle ciglia, probabilmente tramite un meccanismo di modulazione dello scivolamento dei microtubuli che potrebbe essere mediato da un sensore calmodulinico [28 - 30]. Un ruolo nella regolazione della morfologia del battito ciliare è svolto anche dalla guaina centrale, insieme ai legami di nexina e ai ponti radiali: l’interazione tra coppia centrale e ponti radiali sembra costituire la chiave regolatrice dell’attività della dineina, tanto che le ciglia prive di queste strutture risultano completamente immobili.
1.3 - CARATTERISTICHE CLINICHE DELLA DISCINESIA CILIARE PRIMARIA Nella DCP la disfunzione delle ciglia dell’epitelio respiratorio provoca il ristagno del muco nelle vie aeree, cui consegue il ricorrere di infezioni il cui esito principale è l’inevitabile e progressivo decadimento della funzione respiratoria, talora anche di grado severo, con impatto rilevante sulla qualità della vita dei pazienti affetti [31]. Di conseguenza oggi si ritiene che sia necessario giungere alla diagnosi il più precocemente possibile, in modo da intraprendere quanto prima i trattamenti necessari per rallentare l’evoluzione della patologia. Nonostante ciò, questa condizione non solo viene ancora diagnosticata poco, ma viene anche diagnosticata tardi: infatti, nel 2010 una Task Force dell’European Respiratory Society (ERS) ha raccolto i dati di più di mille soggetti affetti da DCP di età inferiore a 20 anni e afferenti a 223 Centri di 26 Paesi europei, mettendo in evidenza come la malattia sia solitamente diagnosticata intorno ai 5,3 anni (3,5 anni in presenza di SVI e 5,8 anni in assenza di SVI), con una lunga coda di diagnosi poste addirittura in epoca adulta [17]. Il ritardo della diagnosi di DCP dipende in parte dal fatto che questa condizione è ancora poco conosciuta e che i test necessari per giungere alla diagnosi sono costosi e richiedono un elevato grado di expertise [17, 32]. Inoltre, la presentazione clinica della DCP, quando non si accompagni al SVI, è di solito aspecifica anche se, a volte, la diagnosi è ritardata nonostante la presenza di alcuni segni tipici già nei primi anni di vita [12, 33].
Le caratteristiche cliniche della DCP variano da soggetto a soggetto, ma per ciascuna epoca della vita si possono individuare dei segni / sintomi più specifici, che devono farne porre il sospetto (“pointers”) [3, 34]. Pointers generici di DCP sono la familiarità per questa condizione o altra ciliopatia, la consanguineità tra i genitori e la presenza di un difetto della lateralità (eterotassia) [35, 36]. Quest’ultimo dato può essere rilevato già in epoca prenatale mediante ecografia: il 50% dei soggetti con DCP ha, infatti, un SVI, mentre il 6% ha un situs ambiguus, ovvero un qualche grado di asimmetria sinistro / destra degli organi toraco-addominali, anche discordante da organo a organo [37, 38]. Inoltre, se da un lato è noto che i soggetti con DCP ed eterotassia hanno un rischio aumentato di 200 volte di avere anche una cardiopatia congenita rispetto alla popolazione generale con eterotassia [37], dall’altro è stata rilevata un’elevata prevalenza di anomalie delle ciglia respiratorie nei soggetti con cardiopatia congenita, ciò a dimostrazione del fatto che le ciglia svolgono probabilmente anche un ruolo
nell’organogenesi del cuore [39]. Di conseguenza, si consiglia di sottoporre tutti i neonati con SVI alle indagini diagnostiche per la DCP e di considerarne la possibilità nei soggetti con cardiopatia congenita, specialmente se associata ad eterotassia [34].
I meccanismi responsabili dell’associazione tra DCP ed eterotassia sono stati illustrati mediante numerosi studi sperimentali su modelli murini che hanno permesso di dimostrare la presenza, nel nodo embrionale, di particolari ciglia mobili (caratterizzate da una configurazione 9+0) in grado di determinare precocemente un flusso sinistrorso di fattori di trascrizione che, a sua volta, determina la corretta disposizione degli organi nelle prime fasi dello sviluppo embrionale. Inoltre, in alcuni di questi studi è stato inattivato un gene codificante la dineina, dimostrando la successiva comparsa di un’alterazione del movimento delle ciglia nodali a seguito della quale il situs di ogni organo asimmetrico (ovvero la disposizione preferenziale in un lato) diventa casuale [40]. In soggetti con DCP in cui la mutazione patogenetica non riguardi la dineina, ma una componente della coppia centrale, la probabilità di presentare eterotassia è, invece, sovrapponibile a quella della popolazione generale, dal momento che le ciglia nodali sono sprovviste della coppia centrale.
Per quanto riguarda il periodo neonatale, i bambini affetti da DCP possono essere asintomatici alla nascita, ma in più del 75% dei casi presentano un distress respiratorio senza causa apparente [41] probabilmente dovuto alla difficoltosa rimozione del liquido amniotico dalle vie aeree conseguente all’alterata funzione ciliare. La presenza di distress respiratorio senza causa apparente in un neonato a termine, eventualmente associato ad alterazioni radiologiche (in particolare ad atelettasie), deve, quindi, far porre il sospetto di DCP [42]. Inoltre, sin dai primi giorni di vita i bambini possono presentare rinorrea cui successivamente si associa anche una tipica tosse catarrale, che, nel tempo, diventa quotidiana ed è seguita dall’espettorazione di muco.
Nelle epoche successive i bambini con DCP tendono a presentare infezioni ricorrenti a carico delle vie aeree superiori (riniti ed otiti acute) ed inferiori (bronchiti ostruttive, broncopolmoniti) con il successivo instaurarsi di un quadro di bronchite cronica che nel giro di qualche anno può determinare lo sviluppo di bronchiectasie distribuite bilateralmente e più frequentemente localizzate a livello del lobo medio e dei lobi inferiori [43 - 45], mentre i lobi superiori sono relativamente risparmiati, a differenza di quanto avviene nella FC [46].
Parallelamente all’interessamento delle vie aeree inferiori, l’impegno del distretto nasale conduce rapidamente a un quadro di sinusite cronica spesso associata a cefalea ricorrente e dimostrabile con l’opacamento dei seni paranasali alla tomografia computerizzata (TC). Tale esame permette anche di studiare le dimensioni dei seni paranasali in relazione all’età del soggetto: a questo proposito il nostro gruppo ha recentemente dimostrato che l’ipoplasia e/o l’agenesia dei seni paranasali (in particolare dei seni frontali e dei seni sfenoidali) sono manifestazioni caratteristiche della DCP [47].
Inoltre, l’interessamento dell’orecchio medio, legato all’alterato trasporto muco-ciliare a livello delle tube di Eustachio, è caratterizzato da un’otite media cronica con deficit uditivo di tipo trasmissivo e da episodi ricorrenti di otite media acuta con perforazione del timpano. In questi soggetti il posizionamento di drenaggi transtimpanici non è risolutivo ed è, anzi, seguito da otorrea prolungata. Studi recenti hanno messo in evidenza come le problematiche relative all’orecchio tendano, però, a ridursi per frequenza e gravità a partire dall’adolescenza [48, 49].
Nell’età adulta, oltre alle manifestazioni presenti nelle epoche precedenti, si possono riscontrare una poliposi nasale e una riduzione della capacità riproduttiva. A questo proposito, nei soggetti di sesso maschile si può rilevare un quadro di infertilità (con preservata possibilità di riprodursi tramite fertilizzazione in vitro) poiché le alterazioni dell’assonema ciliare possono riflettersi anche su quello spermatico alterando la motilità degli spermatozoi (astenozoospermia) oppure sulle ciglia dei dotti deferenti ostacolando la progressione degli spermatozoi (azoospermia) [6], mentre nelle femmine l’alterazione delle ciglia a livello delle tube di Falloppio può esitare in subfertilità con aumentato rischio di gravidanza ectopica [50].
Infine, è necessario ricordare che l’alterazione genetica delle ciglia respiratorie può riflettersi anche sulle ciglia presenti in altri distretti corporei [15] quali, ad esempio, l’epitelio olfattivo (anosmia), l’endotelio corneale (cheratocono) [51], l’ependima (idrocefalo dovuto al rallentamento della circolazione liquorale) [52], l’epitelio retinico (retinite pigmentosa) [53] e il blastema renale (policistosi renale) [54].
Per quanto riguarda la storia naturale della malattia, non sono ancora disponibili evidenze sufficienti per descriverla con sicurezza. Vecchi testi su casistiche pediatriche descrivevano la DCP come una malattia a prognosi relativamente benigna: con l’accrescersi delle conoscenze e delle casistiche, invece, è stato dimostrato che le cose non stanno affatto così [2], dal momento
che in questi soggetti la funzione respiratoria può essere alterata precocemente, con comparsa di pattern ostruttivi alle prove di funzionalità respiratoria (PFR) già in età pediatrica [55] con una
marcata riduzione di tutti i parametri spirometrici (FEV1, FVC, PEF, FEV1/FVC) e un aumento
del Volume Residuo (RV) e dell’indice di Motley (RV/TLC) alla pletismografia corporea [56, 57]. Un recente studio longitudinale ha valutato il peggioramento funzionale in un gruppo di
pazienti con DCP seguiti in follow-up per trent’anni, documentando un decremento del FEV1
dello 0,8% su base annuale (superiore a quello della popolazione generale, ma inferiore al 3,6% della FC) [58]. In questo stesso studio è stato dimostrato che, a 10 anni dalla diagnosi, nel 57%
dei pazienti la funzione rimane stabile, nel 34% dei pazienti si assiste a un declino del FEV1 >
10% e nel 10% ad un miglioramento del FEV1 > 10%.
A dimostrazione del fatto che si tratta di una condizione più grave di quanto ritenuto in passato, sono stati pubblicati alcuni case reports relativi a pazienti che, nel tempo, hanno sviluppato un’insufficienza respiratoria cronica con ippocratismo digitale (digital clubbing), ipertensione polmonare e cor pulmonale; in alcuni casi si è addirittura reso necessario il trapianto di polmone [59].
Come nella FC, un ruolo fondamentale nel determinare la maggiore o minore velocità del declino funzionale è svolto dalle infezioni batteriche: nell’espettorato o nell’aspirato faringeo dei soggetti con DCP sono, infatti, di frequente riscontro l’Haemophilus influenzae, lo Staphylococcus aureus e lo Streptococcus pneumoniae, ma è possibile riscontrare anche lo Pseudomonas aeruginosa, il quale sostiene un’infezione cronica nei teenagers e nei giovani adulti (più tardivamente rispetto alla FC). Talora, anche se più di rado rispetto alla FC, vengono isolati i Micobatteri Non Tubercolari [2]. Se non trattate prontamente ed adeguatamente, tali infezioni possono determinare un netto peggioramento della funzione respiratoria.
In conclusione, per intraprendere il percorso diagnostico della DCP è necessario innanzitutto porne il sospetto. Tra le numerose manifestazioni cliniche descritte è necessario ricordarne quattro che, se presenti in numero di almeno due, rendono il sospetto di DCP estremamente fondato: si tratta del distress respiratorio neonatale (1), dell’interessamento precoce delle vie aeree superiori con rinorrea e otiti ricorrenti (2), dell’interessamento precoce delle vie aeree inferiori caratterizzato da tosse catarrale persistente (3) e della presenza di un difetto della lateralità (4) [42, 60].
1.4 - DISCINESIA CILIARE PRIMARIA: METODI DIAGNOSTICI
Una volta posto il ragionevole sospetto di DCP, dopo aver escluso le altre condizioni compatibili con il quadro clinico (la FC, le immunodeficienze, il deficit di α-1-antitripsina) è necessario inviare il paziente presso un Centro specializzato che si occupi della diagnosi di questa condizione, dal momento che le indagini necessarie richiedono strumentazioni costose, personale qualificato e molto tempo.
L’unico test di screening considerato affidabile per la DCP è la misurazione dell’Ossido Nitrico nasale, mentre, per quanto riguarda i test diagnostici propriamente detti, le raccomandazioni europee, pubblicate nel 2009, suggeriscono di eseguire sempre sia lo studio della funzione sia quello dell’ultrastruttura ciliare, mentre le colture delle cellule ciliate e l’immunofluorescenza restano appannaggio dell’ambito della ricerca o dei casi di difficile diagnosi [34]. L’approccio diagnostico ottimale prevede quindi l’impiego della misurazione dell’Ossido Nitrico nasale, dell’analisi della funzione ciliare e dello studio morfometrico dell’ultrastruttura ciliare [32]: se queste tre indagini sono positive la diagnosi è certa, mentre se esse risultano negative e il sospetto clinico è debole, la diagnosi è estremamente improbabile. Nessuno di questi test è in grado, da solo, di escludere o confermare la diagnosi.
Infine, la ricerca delle mutazioni genetiche è considerata ancora oggi un test di conferma. Alla complessa genetica della DCP è dedicato il capitolo II di questa tesi.
1.4.a - L’Ossido Nitrico nasale: il test di screening della DCP
Fino a qualche anno fa, quale test di screening per la DCP erano molto utilizzati i test per la
valutazione del trasporto muco-ciliare in vivo effettuati misurando il tempo di transito di una
particella di saccarina nel rinofaringe (test alla saccarina) [61] o di particelle colloidali marcate con Tecnezio-99 nelle vie aeree inferiori: questi metodi sono oggi considerati obsoleti, poiché gravati da bassa specificità e bassa sensibilità.
Attualmente, l’unico test di screening ritenuto valido per la DCP è la misurazione dell’Ossido Nitrico nasale (nNO): nel 1994, infatti, è stato dimostrato che nella DCP le concentrazioni nasali di questo gas sono estremamente ridotte [62] e, successivamente, numerosi studi hanno dimostrato che la sua misurazione rappresenta un test di screening
affidabile e sensibile per la DCP, sebbene poco specifico, dal momento che livelli ridotti di nNO possono essere rilevati anche nella FC, nella panbronchiolite e in altre condizioni, così come sono stati descritti casi di DCP con valori di nNO perfettamente nella norma [63]. L’Ossido Nitrico è prodotto fisiologicamente dalla NO-sintetasi a livello del corpuscolo basale delle ciglia dell’epitelio respiratorio, in particolare a livello dei seni paranasali, e svolge un ruolo importante nella modulazione dell’attività ciliare, nella vasodilatazione e nella difesa immunitaria. I meccanismi per i quali nella DCP l’nNO è ridotto non sono ancora noti: tra le ipotesi attualmente in studio sono la ridotta produzione dell’NO a causa di una ridotta espressione dell’NO-sintetasi, l’aumentato metabolismo dell’NO da parte dei batteri intrappolati nel muco e la ridotta diffusione del gas a causa dell’ostruzione degli osti dei seni paranasali [64]. Quale che sia la causa, valori ridotti di nNO supportano la diagnosi di DCP e confortano gli operatori sanitari nel percorso diagnostico intrapreso: tale test viene, quindi, universalmente ritenuto l’esame di prima linea da eseguire nel sospetto di DCP [65].
La misurazione dell’nNO è molto semplice e consiste nel posizionare, dopo accurata pulizia nasale, un’oliva a livello di una narice, tramite la quale l’aria viene aspirata dallo strumento che la analizzerà in tempo reale; per garantire l’isolamento della cavità nasale viene chiesto al paziente di soffiare contro una resistenza di modo che il palato molle si sollevi. Recentemente è stato dimostrato che l’nNO può essere efficacemente misurato anche nel bambino non collaborante mediante la tecnica del tidal breathing o del breath hold [65]. Gli strumenti a disposizione per questo test sono piuttosto costosi (in particolare quelli a chemiluminescenza), ma di recente sono stati sviluppati strumenti portatili dotati di sensori elettrochimici che sembrano garantire una buona affidabilità e che sono decisamente più economici [66].
Sebbene siano disponibili linee guida internazionali relative alla misurazione dell’nNO [67], i vari Centri utilizzano strumenti e metodiche diverse, per cui, ad oggi, non esiste un metodo standardizzato del tutto condiviso, così come non esiste un cut-off universalmente riconosciuto, per cui attualmente si considerano patologici valori di nNO inferiori a 105 - 290 ppb [47, 68] o valori di produzione di nNO al minuto inferiori a 77 nl/min [69]. Inoltre, non sono ancora disponibili valori di cut-off età-specifici, sebbene sia noto da tempo che anche nella popolazione generale nei bambini piccoli l’Ossido Nitrico nasale può essere fisiologicamente ridotto (si osserva un progressivo incremento soprattutto nei primi 5 anni di vita).
1.4.b - L’analisi dell’attività ciliare in vitro
Per eseguire l’analisi dell’attività ciliare in vitro le cellule ciliate vengono prelevate a livello del turbinato nasale inferiore mediante la tecnica del brushing nasale, utilizzando uno spazzolino citologico, come descritto da Rutland e Cole: la procedura, sebbene fastidiosa per il paziente, è semplice, poco invasiva e non richiede anestesia [70, 71].
Dopo aver eseguito il brushing, i campioni prelevati vengono messi in sospensione in un mezzo di coltura, quindi centrifugati a 1.000 giri per 2 minuti allo scopo di ottenere la sedimentazione del campione che verrà prelevato con una pipetta Pasteur e posto su un vetrino per l’immediata valutazione tramite microscopio ottico dotato di obiettivo ad immersione e di contrasto interferenziale di fase secondo Nomarsky. Il microscopio è connesso ad una videocamera digitale che consente la ripresa in tempo reale ad elevato ingrandimento delle immagini, che vengono inviate ad un elaboratore digitale che consente di migliorare la qualità di visualizzazione aumentando le dimensioni delle ciglia sul monitor da 5 - 8 µm a 3 - 5 cm [72]. Le immagini vengono quindi archiviate mediante un videoregistratore digitale ad alta velocità (25 frames al secondo) oppure tramite software computerizzati dedicati, per essere successivamente analizzate a velocità ridotta in modo da poter quantificare la frequenza del battito ciliare e la presenza di movimenti patologici e di eventuali alterazioni morfologiche ciliari [73, 74].
Nella DCP si osservano solitamente valori di frequenza del battito ciliare estremamente ridotti (< 6 Hz) se non addirittura la presenza esclusiva di ciglia immobili. L’analisi della sola frequenza del battito ciliare, però, non è sufficiente per escludere o confermare la diagnosi: sono stati, infatti, descritti casi di DCP con frequenza del battito ciliare normale ma pattern di movimento patologico e quindi inefficace, così come casi caratterizzati da battito ciliare ipercinetico e altrettanto inefficace. E’ quindi fondamentale eseguire l’analisi della frequenza
del battito ciliare insieme all’analisi del pattern del movimento ciliare [74]. Attualmente si
ritiene che i pattern di movimento ciliare patologico siano i seguenti [21, 46]:
- assenza di battito ciliare (ciglia immobili) o frequenza del battito ciliare estremamente
- battito ciliare discinetico (frequenza del battito ciliare ridotta o normale, ma con attività ciliare disorganizzata, inefficace o con movimenti circolari)
- battito ciliare ipercinetico (per lo più associato a movimenti rigidi, con scarsa escursione, simili a un tremolio)
La presenza di alterazioni del movimento ciliare viene giudicata significativa se è osservata in almeno il 40% dei campi microscopici osservati.
Per quanto riguarda gli aspetti morfologici, assume importanza il riscontro di ciglia ispessite e di aree di disepitelizzazione (ciglia diradate, sottili o raccorciate) che suggeriscono la presenza di una flogosi in atto: se il quadro di flogosi è particolarmente evidente, ciò permette di considerare le alterazioni del movimento ciliare di natura verosimilmente secondaria, rimandando il definitivo inquadramento diagnostico ad un successivo prelievo nasale eseguito a distanza di tempo ed in benessere clinico. L’attività ciliare può essere, infatti, alterata dall’esposizione ad agenti fisici o chimici (danno ossidativo, danno da pollutanti) e biologici (Virus Respiratorio Sinciziale, Adenovirus, Haemophilus influenzae, Chlamydia pneumoniae) e, di conseguenza, è sempre preferibile eseguire il prelievo quando il paziente è in buona salute, ovvero a distanza di tempo da un’infezione respiratoria [75, 76].
1.4.c - L’esame morfometrico ultrastrutturale delle ciglia dell’apparato respiratorio
Parte delle cellule prelevate tramite brushing nasale può essere utilizzata per l’esame dell’ultrastruttura ciliare al TEM: il campione viene fissato e quindi sezionato in sezioni semifini che vengono colorate con blu di toluidina e osservate al microscopio ottico per orientare l’operatore nella scelta del campo migliore da osservare al TEM. Il campo migliore è quello in cui, in sezione trasversale, le ciglia sembrano essere orientate il più possibile, poiché le sezioni utili per lo studio ultrastrutturale sono solo quelle che interessano l’assonema nella sua porzione intermedia, in quanto in quella prossimale e in quella distale sono fisiologicamente presenti alcune modificazioni strutturali che ne ostacolano la valutazione.
Le principali anomalie ultrastrutturali osservabili nella DCP interessano i bracci di dineina oppure il complesso centrale (coppia centrale e strutture ad essa collegate). In particolare si possono osservare le seguenti alterazioni [13, 77]:
- assenza dei bracci esterni ed interni di dineina;
- riduzione > 50% del numero dei bracci esterni ed interni di dineina;
- assenza isolata dei bracci esterni di dineina;
- accorciamento dei bracci esterni di dineina e possibile assenza di quelli interni;
- assenza dei ponti radiali e/o della guaina centrale con conseguente decentramento dei
microtubuli centrali (radial spoke defect);
- assenza o accorciamento dei microtubuli centrali e dislocamento di una coppia periferica
verso il centro (transposition defect), talora associato ad assenza dei bracci interni;
- assenza dei microtubuli centrali eventualmente associata ad assenza dei bracci interni di
dineina;
- alterazioni dei corpuscoli basali (in questo caso le cellule ciliate possono essere sostituite
da cellule provviste di lunghi microvilli);
- assenza dei legami di nexina e/o dei ponti radiali e/o dei bracci interni di dineina, con
conseguente perdita della disposizione circolare delle coppie di microtubuli e “disorganizzazione” dell’assonema (disorganized axonemes).
Inoltre, in una percentuale stimabile tra il 10 e il 25% dei casi, l’ultrastruttura ciliare risulta perfettamente normale [13], il che suggerisce che lo studio dell’ultrastruttura al TEM non debba essere più ritenuto, come era in passato, l’esame gold standard per la diagnosi di DCP e che, invece, debba essere sempre associato all’analisi del movimento ciliare [34]. Tuttavia, nel 2012 è stata introdotta la tecnica della tomografia elettronica applicata allo studio dell’ultrastruttura ciliare, che ha permesso di studiare l’intero assonema in 3D e di documentare, in alcuni casi di DCP, specifiche alterazioni submicroscopiche non visualizzabili al TEM [13, 77]: è lecito supporre, quindi, che, anche nei casi in cui l’ultrastruttura risulta normale, potrebbe essere possibile, in futuro, descrivere alterazioni ultrastrutturali submicroscopiche non rilevabili con la strumentazione ad oggi disponibile.
Un altro fattore da tenere in considerazione è la recente dimostrazione del fatto che la maggior parte dei deficit isolati del braccio interno di dineina possono essere reversibili: ne consegue la necessità di eseguire una rivalutazione della diagnosi nei soggetti in cui essa sia stata posta solo su questa base. A tal proposito bisogna ricordare come la corretta
visualizzazione dei bracci di dineina, e in particolare di quelli interni, sia particolarmente difficile da ottenere e quindi passibile d’errore [78].
Allo scopo di evitare di osservare lesioni ultrastrutturali secondarie dovute al danno epiteliale causato dall’infezione e dall’infiammazione, anche in questo caso è essenziale eseguire lo studio ultrastrutturale a distanza da eventuali episodi infettivi. Le lesioni secondarie, strettamente correlate allo stato di flogosi, sono presenti anche nel 10% dei soggetti sani ed in percentuale maggiore in vari tipi di patologia acquisita, sia acuta che cronica, dell’apparato respiratorio [79]. Il pattern di queste lesioni, che possono presentarsi isolate o variamente associate nello stesso assonema e che sono focali e transitorie (nella DCP le lesioni sono invece specifiche, universali e permanenti), è sostanzialmente diverso da quello rilevato nelle forme congenite ed è rappresentato da:
- swollen cilia, ovvero ciglia con aumento della matrice citoplasmatica;
- assonemi multipli (compound cilia), ovvero ciglia con più di un assonema completo o incompleto;
- cilia intracitoplasmatiche, cioè ciglia che si proiettano in larghe cavità intracellulari;
- alterazioni a carico delle coppie periferiche (una o più coppie periferiche assenti o
aggiuntive o dislocate, uno o più microtubuli periferici assenti o aggiuntivi);
- alterazioni delle coppie o dei microtubuli centrali (coppia o microtubulo centrale assenti
o aggiuntivi);
- microtubuli incompleti.
Le ciglia osservate al TEM vengono fotografate a ingrandimenti variabili e i dati qualitativi e quantitativi relativi a ciascun paziente vengono raccolti in un’apposita tabella: per ritenere l’esame significativo è necessario che vengano studiati almeno 100 assonemi adeguatamente sezionati e appartenenti ad almeno 10 cellule non adiacenti, derivanti da campioni diversi dello stesso epitelio [70].
1.4.d - Le colture cellulari e l’immunofluorescenza
L’iter diagnostico della DCP è un percorso lungo e irto di insidie, che non sempre permette di giungere a una diagnosi certa e definitiva. In questi casi, laddove esista un forte sospetto
clinico, è necessario ripetere gli accertamenti a distanza di tempo e nelle migliori condizioni cliniche possibili. Inoltre, nei laboratori dove è possibile predisporle, possono essere di grande aiuto anche le colture cellulari [34]: le cellule ciliate, infatti, una volta prelevate possono essere poste in un terreno di coltura adeguato ed osservate per tre settimane, eseguendo periodicamente la sostituzione del mezzo di coltura. Con tale metodica è possibile valutare in un ambiente privo di noxae patogene le tappe della ciliogenesi, ovvero il processo di rigenerazione delle ciglia. La ciliogenesi inizia già durante la prima settimana di messa in coltura, mentre dopo 2-3 settimane le cellule ciliate danno vita a sferoidi mantenendo la loro polarità: se la ciliogenesi, l’orientamento ciliare e l’attività ciliare sono efficienti, gli sferoidi sono in grado di ruotare su se stessi di 360 gradi e/o di migrare e/o di rimuovere i detriti [80, 81]. Esistono varie tecniche per ottenere le colture delle cellule ciliate, alcune particolarmente complesse e costose: il nostro gruppo ha recentemente messo a punto una metodica semplificata che richiede la sola messa in coltura in sospensione delle cellule ciliate prelevate mediante brushing nasale [82]. Questa tecnica permette di discriminare le alterazioni congenite da quelle secondarie già dopo 5 giorni di coltura valutando la rotazione e la migrazione degli sferoidi, la loro capacità di rimuovere i detriti e il pattern del movimento ciliare [83]. Ne consegue che le colture cellulari sono senza dubbio una tecnica promettente ed affidabile, sebbene ancora priva della standardizzazione necessaria per entrare a far parte dell’iter diagnostico di routine della DCP.
Infine, in alcuni centri di ricerca è stata applicata con successo la tecnica dell’immunofluorescenza allo scopo di evidenziare al microscopio a fluorescenza la presenza e la distribuzione delle proteine ciliari lungo tutta la lunghezza del ciglio [84, 85]. Questa tecnica, impiegata soprattutto negli studi di genetica, sta migliorando la nostra comprensione dei meccanismi patogenetici responsabili della DCP ma, essendo molto costosa, resta appannaggio dei centri di ricerca più avanzati.
1.5 - APPROCCIO TERAPEUTICO E FOLLOW-UP DEL PAZIENTE CON DCP Sebbene le manifestazioni cliniche della DCP siano ormai ben note, sono ancora pochissimi gli studi relativi ai trattamenti da attuare in questi pazienti (si tratta prevalentemente di case reports) e non sono ancora disponibili dati relativi a trials clinici. Le prime raccomandazioni internazionali evidence-based su questa condizione risalgono al 2009 [34], ma per quanto concerne il trattamento anch’esse sono basate sui pochi dati disponibili e su estrapolazioni tratte dalle raccomandazioni relative alla FC, nonostante il fatto che si tratti di un approccio scorretto, dal momento che le due patologie differiscono per fisiopatologia [34, 86].
Ad oggi non sono disponibili terapie specifiche in grado di correggere la disfunzione ciliare che è alla base delle manifestazioni della DCP: l’intervento terapeutico deve essere, quindi, finalizzato a rimuovere le secrezioni dalle vie aeree in quanto il loro ristagno favorisce la moltiplicazione di germi, i quali, a loro volta, stimolano una risposta infiammatoria da parte dell’ospite determinando il rilascio di enzimi proteolitici che danneggiano la mucosa dell’apparato respiratorio. Il ripetersi di questi processi infettivi è responsabile del decadimento della funzione respiratoria e quindi della progressione della malattia. Inoltre, l'accumulo delle secrezioni incrementa la resistenza al flusso aereo e il lavoro respiratorio, divenendo potenzialmente causa di iperinsufflazione e/o atelectasia e di alterazioni del rapporto ventilazione / perfusione.
A livello delle fosse nasali le secrezioni possono essere rimosse mediante irrigazioni nasali con soluzioni saline erogate con una leggera pressione alternativamente nelle due narici del bambino mantenuto in posizione ortostatica: le secrezioni, rese più fluide dall’irrigazione, devono essere successivamente rimosse con un apposito aspiratore se il bambino non è in grado di eliminarle spontaneamente soffiando correttamente il naso (una narice per volta, premendo la controlaterale, a bocca chiusa, senza soffiare energicamente onde evitare la disseminazione delle secrezioni nei seni paranasali o nelle tube di Eustachio). Metodi alternativi sono rappresentati dall’impiego di appositi strumenti che consentono di eseguire docce nasali micronizzate oppure di eseguire l’irrigazione della cavità nasali a caduta, favorendo il drenaggio anteriore delle secrezioni. Inoltre, l’uso di steroidi topici nasali dopo l’irrigazione può favorire la risoluzione della rinosinusite.
Per quanto riguarda le vie aeree inferiori, è di fondamentale importanza il ricorso quotidiano alla fisioterapia respiratoria allo scopo di favorire la clearance delle secrezioni [87]: utile è l'impiego del drenaggio posturale in cui, per mobilizzare e trasportare le secrezioni, viene sfruttata la forza di gravità ponendo il bambino in diverse posizioni e facilitando lo scollamento delle secrezioni con percussioni e vibrazioni esercitate sulla parete toracica.
Per mobilizzare le secrezioni a livello dei distretti bronchiali prossimali si può utilizzare la
tosse volontaria: numerosi colpi di tosse eseguiti in sequenza sono in grado di drenare porzioni
sempre più distali delle vie aeree, determinandone una progressiva “spremitura”, perché il punto di egual pressione, ovvero il punto in cui la pressione all'esterno del bronco (pressione pleurica) eguaglia quella vigente all'interno (pressione pleurica + pressione elastica), si sposta verso la periferia col procedere dell’espirazione, e con esso il punto di collabimento. La tosse non è in grado di drenare le piccole vie aeree poiché in periferia la loro superficie in sezione supera di gran lunga quella delle prime diramazioni bronchiali ed il flusso dell’aria, da cui dipende il potere espulsivo della tosse, è inversamente proporzionale a tale sezione.
Nel bambino più grande può essere applicata con successo anche la tecnica dell’espirazione
forzata, in cui si fa eseguire un’inspirazione a volume corrente ed un’espirazione forzata
attraverso la glottide aperta o semichiusa (huffing): tale tecnica utilizza i flussi aerei espiratori per mobilizzare e trasportare le secrezioni, ma, anche in questo caso, l’efficacia della rimozione delle secrezioni si riduce progressivamente dalle vie aeree centrali a quelle periferiche.
Di fondamentale importanza è l’impiego della PEP (Positive Expiratory Pressure) Mask, che permette di espirare attivamente contro resistenza fino alla capacità funzionale residua in modo da ottenere un prolungamento della fase espiratoria. Si ritiene che tale manovra possa determinare un’onda pressoria di ritorno in grado di mantenere pervie le vie aeree instabili: si comprende quindi perché la PEP Mask trovi impiego nei pazienti con malattia polmonare di tipo ostruttivo complicata da instabilità delle pareti delle vie aeree (ovvero in presenza di bronchiectasie o broncomalacie). Eseguire la tosse volontaria dopo l’impiego della PEP Mask favorisce l’espettorazione, anche perché tale strumento facilita la mobilizzazione espiratoria del muco mediante l'incremento di flussi aerei collaterali verso distretti precedentemente non ventilati per la presenza di secrezioni [88].
Alla fisioterapia deve essere associata un’adeguata terapia farmacologica: innanzitutto è fondamentale somministrare un broncodilatatore (β2-agonista) per via inalatoria prima di ogni seduta fisioterapica allo scopo di aumentarne l’efficacia. Inoltre i β2-agonisti e le xantine sono in grado di stimolare il battito ciliare e di incrementare la clearance muco-ciliare (per lo meno in vitro) e l’ipratropium bromuro, farmaco anticolinergico in grado di ridurre le secrezioni delle vie aeree superiori ed inferiori, viene utilizzato allo stesso scopo.
Per quanto riguarda gli steroidi inalatori, va considerato il fatto che l’esame dell’espettorato di questi pazienti, così come nella FC, dimostra la presenza di un’infiammazione tipicamente neutrofilica, ma con un livello di IL-8 tre volte più alto nei pazienti con DCP [3]: sebbene non siano disponibili ulteriori studi al riguardo, è ragionevole pensare che gli steroidi inalatori possano, quindi, avere un qualche ruolo nella terapia di questi pazienti.
Per quanto riguarda il controllo delle infezioni, è importante ricordare che nella DCP esse sono per lo più sostenute dall’Haemophilus influenzae, dallo Staphylococcus aureus e dallo Streptococcus pneumoniae, anche se nell’espettorato di questi pazienti vengono talvolta riscontrati anche lo Pseudomonas aeruginosa e i Micobatteri non tubercolari [2]. Ai primi segni di peggioramento dei sintomi respiratori o di deterioramento della funzione respiratoria, è quindi necessario intraprendere una terapia antibiotica per via generale ad alte dosi, meglio se sulla base di un antibiogramma [89]. Se il ricorso agli antibiotici è frequente, l’impiego di una terapia antibiotica profilattica può essere presa in considerazione (in particolare con macrolidi).
Nei casi in cui venga isolato lo Pseudomonas aeruginosa è necessario tentare una terapia eradicante simile a quelle utilizzate nei pazienti con FC, mediante aminoglicosidi eventualmente associati a β-lattamici, per os o per via endovenosa. Se il tentativo di eradicazione non sortisse effetti, è necessario ricorrere alla somministrazione periodica della tobramicina per via inalatoria (tramite nebulizzazione oppure tramite inalazione di polvere secca): questa via garantisce, infatti, di raggiungere concentrazioni più elevate di farmaco a livello della mucosa respiratoria rispetto alla via parenterale. Per il trattamento dell’infezione cronica da Pseudomonas accanto alla tobramicina sono state recentemente introdotte formulazioni per via inalatoria anche dell’aztreonam e della colistina, cui presto si assocerà anche la ciprofloxacina [90].
Nella DCP i sedativi della tosse devono essere ovviamente sconsigliati, mentre i mucolitici (ad esempio l’N-acetilcisteina) potrebbero avere un ruolo nella terapia di questi pazienti, così come l’inalazione della soluzione ipertonica, che si è dimostrata efficace nel migliorare la funzione respiratoria nei soggetti adulti con bronchiectasie non-FC. L’efficacia della deossiribonucleasi umana ricombinante (rhDNAasi), il cui utilizzo è routinario nella FC, non è stata invece verificata nella DCP, anche se in alcuni case reports sembra aver determinato un miglioramento delle condizioni respiratorie anche in questi pazienti [91]. Questo enzima ricombinante è, infatti, in grado di frammentare il DNA extracellulare (polianione viscoso liberato nel processo di degenerazione dei leucociti che si accumulano in risposta all'infezione) che è presente in concentrazioni elevate nelle secrezioni purulente: in vitro, l’enzima idrolizza il DNA presente nell'escreato dei pazienti e in tal modo ne riduce la visco-elasticità in modo consistente.
Un’ulteriore protezione nei riguardi delle infezioni può essere ottenuta tramite la somministrazione di vaccini, in particolare quelli contro la Bordetella pertussis, l’Haemophilus influenzae di tipo b, lo Streptococcus pneumoniae e il virus dell’Influenza A. Inoltre, molta attenzione dovrà essere rivolta ad evitare l’esposizione al fumo di tabacco e agli altri irritanti in grado di danneggiare la mucosa delle vie aeree e di stimolare la secrezione di muco, così come dovrà essere incoraggiato il ricorso all’esercizio fisico regolare, dal momento che esso si è rivelato efficace quanto e più dei β2-agonisti nel dilatare le vie aeree [92].
Talora i pazienti con DCP devono ricorrere alla chirurgia: la polipectomia nasale e la
chirurgia endoscopica funzionale sui seni paranasali (FESS) possono essere impiegate in
pazienti con infezioni dei distretti superiori che non rispondano alla terapia antibiotica [93]. In pazienti con bronchiectasie localizzate o atelettasie ritenute fonte di infezioni croniche altrimenti non risolvibili o di febbricola persistente o emottisi, che non rispondano ad una terapia medica aggressiva, può essere presa in considerazione la lobectomia [42]. In casi particolarmente gravi, per fortuna rari, si può arrivare ad un peggioramento della funzione respiratoria tale da rendere necessario il trapianto polmonare [59].
In considerazione della grande eterogeneità genetica della DCP la realizzazione di una terapia genica sembra un progetto alquanto futuribile. In realtà, alcuni gruppi hanno già realizzato esperimenti in vitro in questo senso: il gruppo francese di Bouvagnet ha, infatti,
eseguito un tentativo di terapia genica su cellule prelevate da un paziente con DCP caratterizzata da eterozigosi composta nel gene DNAI1 (assenza dei bracci esterni di dineina e conseguente immobilità ciliare). Le cellule, poste in coltura, venivano trasfettate mediante un lentivirus con il gene DNAI1 corretto: l’esperimento ha dimostrato che la trasfezione avviene correttamente e che il gene viene espresso dalle cellule trattate, che riprendono successivamente a produrre correttamente i bracci esterni di dineina e, quindi, a presentare un battito ciliare normale [94].
Più recentemente un esperimento analogo è stato eseguito su cellule murine mutate nel gene Dnaic1 (analogo del gene DNAI1), dimostrando che la terapia genica funziona in vitro sia su cellule indifferenziate sia su cellule già specializzate. Inoltre, il lentivirus con il gene corretto è stato somministrato ai topi per via nasale dimostrando l’efficacia della terapia anche in vivo, sebbene la quota di gene trasfettato sia fortemente limitata dalla presenza di rinite [95]. Ulteriori studi saranno necessari per dimostrare se la terapia genica possa essere eseguita in vivo anche sull’uomo, quale sia la sua efficacia e la sua durata nel tempo e, soprattutto, quali siano i suoi effetti collaterali.
Infine, per quanto riguarda il follow-up, anche in questo caso le raccomandazioni internazionali si basano su estrapolazioni tratte dalle raccomandazioni relative alla FC [34]. In generale è sicuramente utile valutare le condizioni del paziente periodicamente (almeno ogni 3 - 4 mesi): durante la visita è necessario eseguire un accurato esame obiettivo onde rilevare tempestivamente i segni di un’eventuale infezione respiratoria acuta e, in quell’occasione, sarà anche opportuno eseguire le misurazioni auxologiche e le prove di funzionalità respiratoria, in quanto l’esame spirometrico e/o l’esame pletismografico (a seconda del grado di collaborazione del paziente) permettono non solo di rilevare un transitorio peggioramento o miglioramento, ma anche di valutare l’andamento nel tempo delle condizioni respiratorie del paziente.
Nei pazienti con DCP è importante eseguire la coltura dell’espettorato o dell’aspirato faringeo (dopo 5 - 7 giorni di sospensione degli antibiotici) ogniqualvolta si presenti un improvviso peggioramento della funzione respiratoria e comunque almeno una volta al mese: ciò permette di intervenire tempestivamente in caso di infezione con una terapia mirata sulla base dell’antibiogramma.
Utile è anche effettuare periodicamente una visita otorinolaringoiatrica ed un esame audiometrico per identificare precocemente eventuali deficit dell’udito: ciò è particolarmente importante in età pediatrica, allo scopo di prevenire l’insorgenza di deficit del linguaggio legati all’ipoacusia.
Infine, non è necessario eseguire periodicamente la radiografia del torace, che deve essere riservata alle esacerbazioni infettive nel sospetto di complicanze, quali un versamento pleurico. Più utile può essere l’HRCT (High Resolution Computed Tomography) del torace, che permette di definire l’estensione delle bronchiectasie e di monitorarne la progressione nel tempo. Tuttavia, questo esame è gravato dai rischi biologici relativi all’esposizione alle radiazioni ionizzanti e, di conseguenza, non può essere utilizzato di routine nel follow-up di questi pazienti, a maggior ragione se in età pediatrica. Per superare questo limite si ricorre sempre più spesso alla risonanza magnetica, che si è dimostrata altrettanto affidabile nella valutazione della severità e dell’estensione delle bronchiectasie [96].