Capitolo 1
Metabolismo della Creatina e
Creatine Deficiency Syndromes
1.1 Metabolismo della Creatina
La sindrome da deficit della creatina (Cr) corrisponde ad un gruppo di errori congeniti del metabolismo di recente descrizione (fu Stöckler, nel 1994, a presentare il primo paziente affetto da un deficit metabolico di questa via), relativi sia alla sintesi che al trasporto della creatina, che hanno messo in luce il ruolo chiave di questa molecola, nonché consentito un approfondimento delle conoscenze in merito alle sue funzioni e al suo metabolismo [1] [2] [3].
Si tratta di tre difetti metabolici, due dei quali relativi alla sintesi della Cr (deficit degli enzimi AGAT e GAMT) ed uno al trasportatore (CRTR), che
I soggetti affetti da tali patologie, presentano segni e sintomi di riscontro frequente nell’ambito della clinica neuropsichiatrica dell’età evolutiva, come il ritardo mentale, i disturbi del linguaggio, l’epilessia, i disturbi del movimento e della sfera comunicativo-relazionale, elementi che spesso delineano quadri clinici che restano a lungo senza una diagnosi eziologica certa.
Da quanto emerso secondo i dati pubblicati ad oggi, si può sostenere che la diagnosi precoce e l’adeguato trattamento di questi errori metabolici consentano un sostanziale cambiamento della loro storia naturale.
Il fabbisogno giornaliero di creatina ammonta a circa 2 grammi: questi vengono per il 50% assunti con la dieta (in particolar modo con carne, pesce e proteine animali in genere) e per la restante quota prodotti per sintesi endogena, che avviene principalmente in fegato e pancreas. Tale molecola è poi accumulata, grazie ad un sistema di trasporto attivo, nelle cellule, in particolar modo in quelle a più alto fabbisogno energetico, come le fibre muscolari a rapida contrazione e le cellule nervose; qui può essere convertita in fosfocreatina (CrP) grazie ad uno dei 5 isoenzimi della Creatinkinasi (CK), situato nello spazio intermembrana dei mitocondri; viene così assicurato l’immagazzinamento di energia, convertibile in ATP mediante trasferimento di un gruppo fosforico all’ADP.
Infine, Cr e CrP sono degradate per via non-enzimatica a creatinina (Crn), che passa per diffusione passiva ai reni e viene escreta con le urine, in quantità direttamente proporzionale alla quota sintetizzata e alla concentrazione sierica di Cr. In un adulto sano, l’escrezione giornaliera di Crn varia da 1,5 a 2g e corrisponde ad un pool di 120g di Cr e CrP [4].
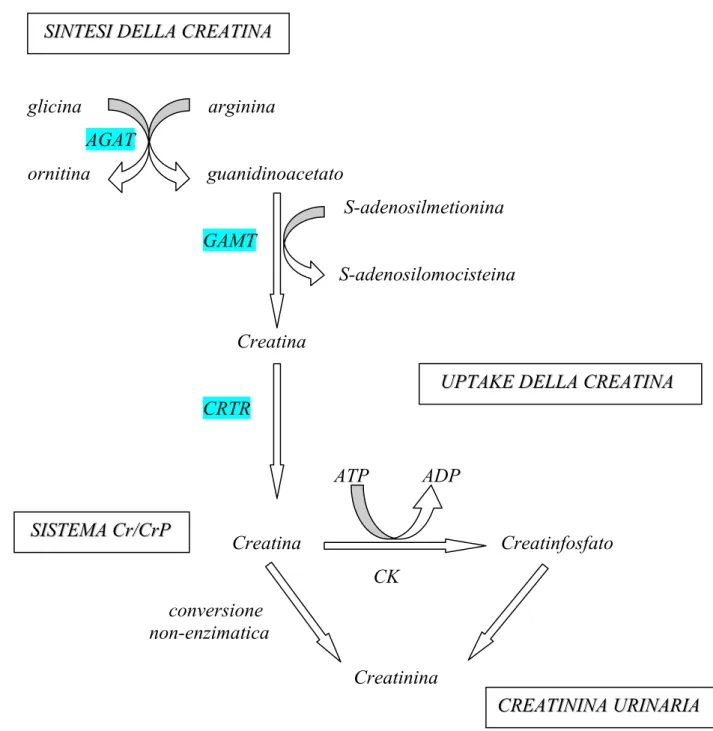
I substrati della reazione di sintesi della Cr sono arginina, glicina e metionina; gli enzimi catalizzatori la Arginina-Glicina-Amidinotransferasi (AGAT) e la Guanidinoacetato-Metiltransferasi (GAMT).
La prima tappa di tale via metabolica consiste nel trasferimento di un gruppo guanidinico dall’arginina alla glicina ad opera dell’enzima AGAT, da cui si ottiene acido guanidinoacetico (GAA) e ornitina. Tale tappa è autolimitantesi, in quanto inibita dal suo prodotto, l’ornitina e dalla Cr; viene invece indotta da tiroxina e ormone della crescita.
Il secondo step consente la formazione di Cr ed S-adenosil-omocisteina, catalizzata dall’enzima GAMT, che usa come substrati il GAA e l’S-adenosil-metionina [5].
La creatina così sintetizzata, raggiunge i tessuti ad alto fabbisogno energetico, ossia muscolo scheletrico e tessuto nervoso, grazie al suo specifico trasportatore transmenbrana (CT1), appartenente alla famiglia dei trasportatori Na-dipendenti.
glicina arginina AGAT ornitina guanidinoacetato S-adenosilmetionina GAMT S-adenosilomocisteina Creatina CRTR ATP ADP Creatina Creatinfosfato CK conversione non-enzimatica Creatinina
Fig. 1: Via metabolica della sintesi della creatina e sistema Cr/CrP
1.2 Creatine Deficiency Syndromes (CDS)
Il primo enzima della via metabolica della creatina è l’Arginina-Glicina-Amidinotransferasi (AGAT). Il gene codificante per l’enzima AGAT è localizzato sul cromosoma 15q15.3; l’AGAT è una proteina situata nello spazio intermembrana del mitocondrio: la localizzazione dell’enzima in questa sede
S SIINNTTEESSIIDDEELLLLAACCRREEAATTIINNAA U UPPTTAAKKEEDDEELLLLAACCRREEAATTIINNAA S SIISSTTEEMMAACCrr//CCrrPP C CRREEAATTIINNIINNAAUURRIINNAARRIIAA
permette la separazione della reazione della via metabolica della Cr da quelle che maggiormente utilizzano l’Arginina come substrato [2].
Il deficit di AGAT causa livelli molto bassi o assenti di Cr, Crn e GAA nei liquidi biologici e assenza del picco Cr/Crn alla 1H-MRS.
Il deficit di tale enzima è stato diagnosticato per la prima volta nel 2001, in due sorelle afferite all’IRCCS Stella Maris per ritardo mentale e del linguaggio associati a disturbi del comportamento e relazionali, nelle quali già un anno prima era stato riscontrato un importante deficit di Cr cerebrale, di cui non era però stata identificata la causa molecolare. Successivamente, la stessa diagnosi è stata posta per un cugino di secondo grado delle due bambine e per il loro fratello minore [6] [7].
Nonostante la limitata casistica in letteratura, la supplementazione di creatina monoidrato per os alla dose di 400 mg/Kg/die sembrava efficace nel ripristino della Cr cerebrale, per cui anche i nostri soggetti sono stati sottoposti a tale trattamento. Nel follow-up, monitorato con la 1H-MRS, si è avuto un recupero del picco di Cr/Crn del 90% nei tre bambini più grandi, consentendo negli anni la riduzione della dose terapeutica fino al raggiungimento della dose minima efficace. La terapia sostitutiva nel neonato, alla dose di 100 mg/Kg/die, dimostratasi efficace, ha confermato l’importanza di una diagnosi precoce, al fine di un intervento tempestivo che possa cambiare il corso naturale della malattia. A prova di ciò, il fatto che il soggetto presintomatico trattato ha uno sviluppo normale.
Il secondo enzima della via di sintesi della Cr è il Guanidinoacetato-Metiltransferasi (GAMT). Il gene che codifica per tale enzima è situato sul cromosoma 19p13.3 ed è composto da 6 esoni; corrisponde ad una proteina di 237 aminoacidi e ne sono state descritte, fino ad ora, 13 mutazioni e un polimorfismo [8].
Un difetto di questo enzima, trasmesso con modalità autosomica recessiva, comporta alte concentrazioni di GAA e bassi livelli di Cr in plasma, urine e liquor; anche in questo caso alla 1H-MRS si riscontra assenza del picco Cr/Crn. Il deficit di GAMT, descritto nel 1994 da Stöckler, è stato il primo errore congenito del metabolismo della Cr scoperto nell’uomo [3].
Lo spettro fenotipico di presentazione di questo disordine genetico, il più grave tra le CDS, varia da forme in cui predominano, in associazione al ritardo mentale, i segni extrapiramidali e/o piramidali e l’epilessia resistente al trattamento, ad altre in cui si riscontrano ritardo mentale meno importante ed epilessia trattabile. Le costanti di tale sindrome possono in ogni modo essere rintracciate nell’epilessia e nel ritardo globale dello sviluppo, compresa l’acquisizione del linguaggio [9] [10] [11].
La terapia del deficit di GAMT si è avvalsa di una supplementazione orale di Cr monoidrato ad alte dosi (350-2000) mg/Kg/die; se pure ciò consenta una normalizzazione dei livelli di creatina cerebrale, persiste nei pazienti trattati una sintomatologia neurologica ben più importante di quella riscontrata nei pazienti con deficit di AGAT. Ciò è da attribuirsi verosimilmente agli elevati livelli di GAA nei liquidi biologici, in particolar modo nel liquor, che spesso permangono
alti anche durante il trattamento. L’enzima AGAT è soggetto ad una retroinibizione da parte del prodotto finale della via metabolica, la Cr, ma la sua somministrazione ad alte dosi non riduce significativamente i livelli di GAA. Da ciò l’avanzare di strategie terapeutiche da affiancare alla reintegrazione di Cr quali:
- la somministrazione di ornitina esogena per indurre inibizione competitiva dell’AGAT;
- la restrizione dietetica di arginina, substrato dell’AGAT e precursore del GAA.
L’adozione di un approccio combinato, che utilizza tutte le soluzioni summenzionate, si concretizza in un rilevante miglioramento delle condizioni cliniche [12] [13].
Il quadro clinico dei deficit della sintesi della creatina viene modificato dal trattamento con efficacia variabile tra deficit di AGAT e GAMT; inoltre influisce sul risultato della terapia l’epoca della diagnosi: tanto più essa è precoce e tanto più tempestivamente viene intrapresa le reintegrazione di Cr, tanto migliori saranno i risultati ottenibili.
Il deficit del Trasportatore della creatina è il più frequente errore congenito del metabolismo della creatina, nonché una delle più comuni cause di ritardo mentale X-linked. Il gene per il trasportatore, detto SLC6A8 e conosciuto anche come CT1 o CRTR, si trova sul cromosoma Xq28, contiene 13 esoni e codifica
trasportatori Na-dipendenti, possiede 12 domini transmembrana. E’ stata dimostrata l’esistenza di un altro gene, detto CT2, sul cromosoma 16p11.1-16p.2, espresso però solo dal testicolo. I due geni presentano il 97% di omologia, ma il CT2 contiene un codone di stop prematuro nell’esone 4, il che suggerisce che codifichi per una proteina non funzionante o troncata.
Il primo deficit del trasportatore è stato descritto nel 2001, e da allora sono stati identificati più di 30 pazienti. Clinicamente, i soggetti affetti mostrano ritardo mentale di grado lieve-medio, epilessia, ritardo del linguaggio sia recettivo che espressivo, talvolta lieve ipotonia. Nei soggetti portatori è stato non infrequente il riscontro di disturbi dell’apprendimento e/o del comportamento [1].
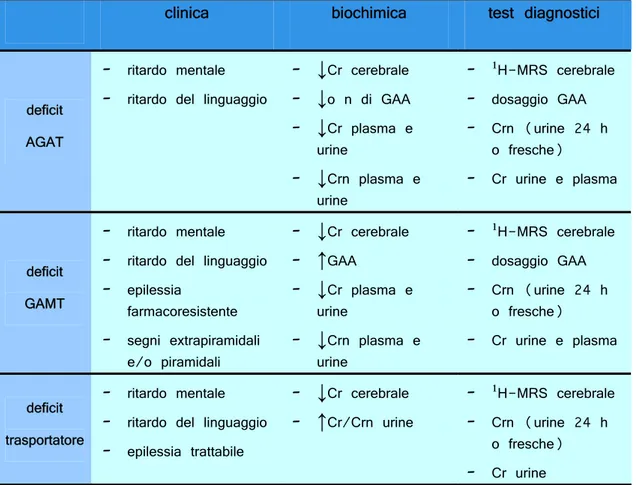
clinica
biochimica
test diagnostici
deficit AGAT
-
ritardo mentale-
ritardo del linguaggio-
↓Cr cerebrale-
↓o n di GAA-
↓Cr plasma e urine-
↓Crn plasma e urine-
¹H-MRS cerebrale-
dosaggio GAA-
Crn (urine 24 h o fresche)-
Cr urine e plasma deficit GAMT-
ritardo mentale-
ritardo del linguaggio-
epilessia farmacoresistente-
segni extrapiramidali e/o piramidali-
↓Cr cerebrale-
↑GAA-
↓Cr plasma e urine-
↓Crn plasma e urine-
¹H-MRS cerebrale-
dosaggio GAA-
Crn (urine 24 h o fresche)-
Cr urine e plasma deficit trasportatore-
ritardo mentale-
ritardo del linguaggio-
epilessia trattabile-
↓Cr cerebrale-
↑Cr/Crn urine-
¹H-MRS cerebrale-
Crn (urine 24 h o fresche)-
Cr urineTab. 1: Schema riassuntivo delle caratteristiche cliniche e biochimiche e dei test diagnostici nelle CDS
1.3 Strumenti diagnostici
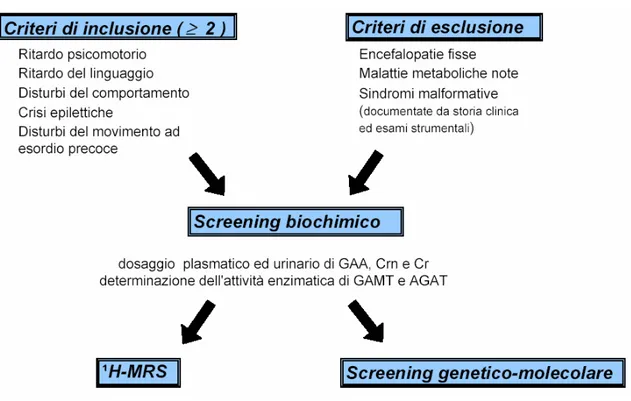
Nella diagnosi delle CDS, il primo imprescindibile passo è rappresentato dall’analisi della storia clinica del paziente, in cui rintracciare eventuali criteri di inclusione che possano indirizzare nella scelta dell’esecuzione delle indagini biochimiche (vedi algoritmo diagnostico Fig. 2). Queste saranno rappresentate, come trattato in precedenza, dai dosaggi plasmatici ed urinari di Cr, Crn e GAA, nonché dalla determinazione dell’attività dei due enzimi della via di sintesi della Cr, AGAT e GAMT.
Fig. 2: Algoritmo diagnostico dei deficit primari di creatina
Al momento della scoperta di uno dei deficit primari della creatina sulla base dei test biochimici, il ruolo della Risonanza Magnetica Spettroscopica (1H-MRS) consiste nell’evidenziare la presenza o l’assenza del picco di Cr cerebrale in vivo
Nella MRI le immagini vengono prodotte utilizzando il protone come nucleo soggetto al fenomeno di risonanza magnetica. Alcuni nuclei atomici, infatti, introdotti nel contesto di un campo magnetico uniforme, si rivelano capaci di assorbire energia elettromagnetica apportata dall’esterno, allorquando questa è erogata con opportuna frequenza. Questo assorbimento risonante rende conto della dizione del fenomeno di risonanza.
Nell’imaging si utilizza solo il segnale proveniente dall’eccitazione del nucleo dell’atomo di idrogeno dell’acqua. L’idrogeno è l’elemento più rappresentato nel nostro organismo, per cui consente di ottenere immagini con risoluzione spaziale nell’ordine dei millimetri [15].
Nella spettroscopia, al contrario, per evidenziare il segnale proveniente dalle specie molecolari, è necessario sopprimere selettivamente quello dell’H2O.
Questo espediente è necessario in quanto il campo magnetico che agisce sul nucleo di un atomo viene modificato dai campi prodotti dagli altri atomi presenti nella stessa molecola. Come conseguenza di questo effetto si ha il chemical shift (CS) o spostamento chimico, vale a dire uno spostamento della frequenza di risonanza del nucleo per ogni specie chimica di appartenenza. Il CS viene in genere espresso in parti per milione (ppm) che, riferendosi alla frequenza di risonanza di un composto di riferimento standard, normalizzano la distanza tra due righe spettrali al campo magnetico applicato. Nel caso dell’idrogeno e del carbonio il composto di riferimento è il tetrametilsilato.
In ambito clinico i nuclei usualmente studiati sono l’idrogeno (1H ) e il fosforo (31P) perché hanno un range di CS sufficientemente ampio da consentire la rilevazione di diverse specie chimiche [16] [17].
La 1H-MRS rappresenta la tecnica in genere utilizzata a scopo clinico in considerazione dell’alta concentrazione di idrogeno nelle strutture organiche e della sua elevata sensibilità. La 1H-MRS, inoltre, consente di rivelare numerosi metaboliti con diverso significato biochimico e fornisce il maggior numero di informazioni metaboliche, soprattutto dell’encefalo.
Lo spettro è costituito da un tracciato bidimensionale nel quale in ascissa è riportato il CS, in ordinata l’intensità del segnale. E’ composto da vari picchi di sostanze contenenti colina (Cho), creatina (Cr), N-acetil-aspartato (NAA), mioinositolo (mI), glutamina, glutammato (Glx) e, se presenti, da acido lattico (Lac) e da lipidi (Lip). La morfologia dello spettro protonico dipende dalle concentrazioni di tali metaboliti.
Lo spettro protonico si legge da destra verso sinistra: in uno normale il primo picco, quello più alto, è rappresentato dall’NAA e si localizza a 2.01 ppm. L’NAA è un metabolita molto importante dal punto di vista neuroradiologico perché numerose evidenze suggeriscono che esso sia presente solo all’interno della popolazione neuronale e che possa essere utilizzato come marker specifico del danno neuronale.
Il cluster successivo, costituito da piccoli picchi che risuonano tra 2.1-2.5 ppm, è rappresentato da molecole β e γ di Glx. Il glutammato è un neurotrasmettitore eccitatorio che ha un ruolo fondamentale nel metabolismo mitocondriale, nella neurotrasmissione, come precursore dell’Acido Gamma-Amino-Butirrico (GABA), come componente del Ciclo di Krebs, nella regolazione dei livelli di
Il picco successivo è quello della Cr e della CrP, misurate insieme a 3.03 ppm. È visibile un picco aggiuntivo a 3.94 ppm. Il segnale della Cr è il più stabile e per questo viene generalmente usato come riferimento interno nella valutazione delle alterazioni spettrali. La Cr ha un ruolo nel mantenimento dei sistemi energia-dipendenti nelle cellule dell’encefalo. La Creatinchinasi, infatti, converte la Cr in CrP utilizzando ATP e, quest’ultima, costituisce la riserva di fosfati ad alto contenuto energetico dei neuroni e provvede a mantenere costante il rapporto ATP/ADP. La Cr, quindi, può fornire informazioni sul metabolismo cellulare legato ai fosfati ed è considerata un aspecifico indicatore della densità cellulare. Questo metabolita, inoltre, ha assunto un ruolo ancora più importante con la scoperta degli errori congeniti del metabolismo della Cr che si manifestano con l’assenza del picco nello spettro.
Adiacente a quello della Cr troviamo il picco della Cho, localizzato a 3.22 ppm, che comprende composti contenenti Cho, come i fosfolipidi di membrana (fosfocolina e glicerofosfocolina) ed i relativi prodotti di degradazione. La Cho rappresenta un costituente del metabolismo fosfolipidico delle membrane cellulari e riflette il turnover di membrana.
Il picco situato a sinistra della colina è rappresentato dal mioinositolo (mI) a 3.56 ppm, che comprende mI e mI monofosfato. Questo segnale, recentemente, è stato attribuito allo scyllo-inositolo, forma isomerica dell’inositolo. Il mI è uno zucchero la cui concentrazione è elevata nelle cellule gliali ed è attualmente considerato un marker gliale.
Altro metabolita importante è l’acido lattico (Lac), metabolita terminale del metabolismo energetico, in particolare della glicolisi anaerobia, assente nello
spettro normale. Il picco del Lac si trova a 1.33 ppm. Quando presente, assume una particolare configurazione: è costituito da due picchi distinti risonanti in rapporto alle alterazioni del campo magnetico tra protoni adiacenti. Il picco è rilevabile in presenza di danno ischemico, quando la cellula non ha a disposizione sufficiente O2 per le sue necessità e quindi attiva il percorso
anaerobico della glicolisi con conseguente accumulo di lattato (shift fosforilazione ossidativa/glicolisi anaerobia).
Anche il picco dei Lipidi (Lip) è di norma assente; quando presenti, i lipidi producono picchi a 0.8-1.2-1.5-6.0 ppm, comprendenti i protoni metilici, metilenici, allelici e vinilici di acidi grassi non saturi. Questi metaboliti rappresentano segni di degradazione cellulare.
L’analisi quantitativa o semiquantitativa delle variazioni in ampiezza delle singole componenti spettrali, offre un possibile criterio di caratterizzazione dei tessuti in condizioni patologiche.
Di solito si ricorre alla modalità che utilizza un riferimento interno, spesso la Cr, o uno esterno, cioè l’area sana o, in sua assenza, campioni a concentrazione nota localizzati all’interno della bobina. Nel determinare se i rapporti sono normali, essi dovranno essere confrontati con rapporti normali per l’età del soggetto in esame e per la regione presa in considerazione.