Capitolo 4
Miscele “bioartificiali” per applicazioni nella
tecnica di Selective Laser Sintering (SLS)
4.1 Introduzione
Il Capitolo 4 tratta la produzione e la caratterizzazione di “miscele bioartificiali” a base di PCL, la cui importanza nel settore dell’ingegneria tissutale, così come in quello delle plastiche eco-compatibili, è stata già discussa nel paragrafo 1.4 del Capitolo 1.
Il PCL è un materiale sintetico biodegradabile e biocompatibile approvato per usi biomedici. Esso possiede buone proprietà meccaniche, ed è soggetto a degradazione enzimatica attraverso l’idrolisi dei legami esterei, da parte di enzimi quali colesterolo esterasi, lipasi, carbossil-esterasi. Tuttavia, la velocità di degradazione del PCL è lenta a causa della sua natura idrofoba e semicristallina: il suo tempo di riassorbimento è superiore ai due anni. Inoltre, il PCL ha una temperatura di transizione vetrosa molto bassa (-60°C), per cui a temperatura ambiente è amorfo e presenta una buona permeabilità ai farmaci con basso peso molecolare nei sistemi di rilascio per usi biomedici. Alcune delle principale applicazioni del PCL nel settore biomedico includono la produzione di scaffold per la rigenerazione degli assoni, delle ossa e della pelle, così come i sistemi per il rilascio di farmaci. In questo lavoro, il PCL è stato miscelato con un polisaccaride scelto tra amido, gellano e destrano, allo scopo di trovare il sistema maggiormente promettente per impieghi nell’ingegneria tissutale. La scelta è caduta su amido, gellano e destrano, poiché essi sono polimeri naturali biodegradabili, facilmente degradati dall’attacco enzimatico, che presentano una buona compatibilità.
L’amido è un biopolimero presente in forma di minuti granuli nelle radici, nei semi e nel fusto di varie piante, come il granoturco, il riso, il malto, l’orzo e le patate. Esso è composto da amilosio, che è un polimero lineare a base di unità di D-glucosio unite tramite legami α-1,4 glicosidici, e amilopectina, che è un polimero altamente ramificato con un peso molecolare che può raggiungere decine di milioni. Le miscele a base di amido sono state usate in varie applicazioni biomediche, dato che esse offrono la possibilità di ottenere materiali con proprietà diverse semplicemente variando il componente sintetico, i metodi di processo, gli additivi, e materiali di rinforzo. Le possibili applicazioni coprono un’ampia gamma di campi, come gli scaffold per l’ingegneria tissutale, i cementi ossei, gli idrogeli per il rilascio controllato di farmaci e i sostituti ossei nel campo ortopedico.
Il gellano è un polisaccaride esocellulare anionico, secreto dal batterio Sphingomonas elodea, e composto da unità ripetenti tetrasaccaridi a base di glucosio, acido glucuronico, e residui di ramnosio nel rapporto 2:1:1, unite in una catena lineare ( [→3)-β-D-Glucosio-(1→4)-β-D-Acido
glucuronico-Il destrano è un polimero ad alto peso molecolare del D-glucosio prodotto da varie colture batteriche. Nel campo dei biomateriali viene adoperato come plasma expander mentre i derivati del destrano sono adoperati per la rigenerazione ossea [Berrada et al., 1994]. Miscele tra PLA e destrano, inoltre, sono state recentemente prodotte e utilizzate come scaffold [Cai et al., 2002; Bacakova et al., 2004].
Nella letteratura scientifica, si ritrovano diversi studi sulle miscele tra PCL e amido (c.f.r. Capitolo 1, paragrafo 1.4.1), mentre mancano ricerche sulle miscele PCL/gellano e PCL/destrano per usi biomedici.
Il contenuto innovativo del lavoro consiste anche nella tecnica con cui sono state effettuate microfabbricazioni, il Selective Laser Sintering (SLS). Il SLS è una tecnica di prototipazione rapida, basata sull’uso dell’energia riscaldante di un laser a CO2 per sinterizzare il materiale in forma di particelle, seguendo una precisa geometria fissata al computer tramite un programma di grafica [Ciardelli et al., 2004; Pham et al., 1998]. La tecnica permette di creare oggetti a partire da un modello disegnato tramite opportuno software di grafica, che viene successivamente scomposto in tanti strati bidimensionali, che sono poi sinterizzati in sequenza, in modo da rigenerare l’oggetto di partenza.
4.2 Parte sperimentale
4.2.1 Materiali
Sono stati adoperati un poli(ε-caprolattone) (PCL) commerciale fornito da Polysciences, Inc. con peso molecolare medio ponderale di 45,000 e tre biopolimeri: amido solubile ACS per analisi (A; Carlo Erba), destrano (D; Pharmacie Fine Chemicals) e gellano (GL; Fluka). L’amido usato ha una composizione pari al 90 % in peso di amilosio e al 10 % in peso di amilopectina. Tutti gli agenti chimici usati sono stati adoperati senza ulteriore purificazione.
4.2.2 Preparazione di microparticelle
Sono state preparate tre miscele contenenti il 90.9% in peso di PCL e il 9.1% in peso di amido, gellano o destrano, tramite tecnica di precipitazione da soluzione. Le miscele sono state identificate con le sigle: PCL/A, PCL/D, PCL/GL. Dapprima è stata preparata una soluzione al 5% (w/v) di PCL in dimetilsolfossido (DMSO; Ridel de Haen) a 50°C, poi uno dei biopolimeri è stato aggiunto alla soluzione, mantenendo il sistema in agitazione a 50°C per 2 h. Le soluzioni sono state precipitate in metanolo (Carlo Erba) e i precipitati prima sono stati recuperati per centrifugazione (Centurion, 6000 series) a 4000 rpm per 5 min, poi sono stati essiccati in stufa da vuoto a 37°C per tre settimane. I campioni essiccati sono stati ridotti in polvere tramite macinazione manuale, adoperando azoto liquido allo scopo di infragilire il materiale. Le polveri sono state poi setacciate attraverso setacci con dimensione della griglia di setacciatura pari a 125 µm.
Le microparticelle di PCL sono state ottenute sia per macinazione manuale e setacciatura, come nel caso delle miscele, sia attraverso un metodo di emulsione semplice olio in acqua (O/W). E’ stata dapprima preparata una soluzione di PCL al 5% (w/v) in cloroformio (60 ml), ed è stata gocciolata in 1000 ml di acqua demineralizzata, tenuta sotto agitazione. La risultante emulsione è stata omogeneizzata tramite omogeneizzatore ad alta velocità (Art. Miccra-D8, Falc Instruments) a 23500 rpm per 5 min, in un becher raffreddato esternamente con una camicia di acqua e ghiaccio. Il sistema è stato mantenuto in agitazione per 24 h a temperatura ambiente sotto cappa, per consentire l’evaporazione del solvente. Le microsfere sono state recuperate mediante centrifugazione a 4000 rpm per 5 min, ed essiccate a 35°C in stufa ventilata per una settimana. La dimensione media e la distribuzione delle dimensioni della microsfere è stata valutata tramite la tecnica di light scattering. Lo strumento utilizzato è stata l’apparecchiatura Malvern Instruments M6 che può misurare particelle con dimensioni pari a 2 nm – 5 µm.
4.2.3 Tecniche di caratterizzazione fisico-chimica delle micro-particelle
4.2.3.1 Analisi calorimetrica
L’analisi calorimetrica è stata effettuata adoperando lo strumento Perkin Elmer Pyris Diamond equipaggiato con un sistema di raffreddamento Perkin-Elmer Intracooler 2P. I campioni (5-10 mg) sono stati analizzati all’interno di capsule in alluminio. Le scansioni non isoterme sono state effettuate tra –20°C e +100°C a 10°C/min, sotto flusso d’azoto. La temperatura di fusione (Tm) e l’entalpia di
fusione (∆Hm) della fase cristallina sono state determinate tramite una seconda scansione, mentre la
temperatura di cristallizzazione (Tc) e l’entalpia di cristallizzazione (∆Hc) sono state ottenute dalla
scansione di raffreddamento. Il grado di cristallinità del PCL (X) è stato calcolato dall’entalpia di fusione del PCL, come percentuale dell’entalpia di fusione del materiale totalmente cristallino (139.5 J/g) [Pitt et al., 1981].
Il tempo di cristallizzazione (tc) è stato determinato mediante l’equazione:
tc = (Ti-Tf)/r (4.1)
dove Ti e Tf ed r indicano le temperature iniziali e finali di cristallizzazione e la velocità di
raffreddamento. Ti e Tf sono le temperature alle quali la frazione di materiale cristallizzato è pari
all’1% e al 99%, rispettivamente.
Le cinetiche di cristallizzazione isoterma sono state analizzate in base al seguente metodo. I campioni sono stati scaldati a 100°C, mantenuti a tale temperatura per 5 min in modo da eliminare la storia termica del campione, poi raffreddati velocemente (200 °C/min) alla temperatura di cristallizzazione prescelta (Tc = 38-48 °C) e mantenuti a tale temperatura fino al completamento della cristallizzazione
del materiale. A questo punto il materiale è stato scaldato direttamente da Tc a 100°C a 10°C/min per
analizzare il comportamento a fusione dei campioni cristallizzati in condizioni isoterme. L’entalpia di cristallizzazione per ogni campione e ad ogni Tc è stata registrata in funzione del tempo. Il tempo di
inizio di analisi è stato assunto come il tempo a cui la temperatura raggiunge il valore programmato Tc.
La frazione X(t) di materiale cristallizzato ad ogni tempo t è stata valutata mediante il rapporto tra l’area cristallizzata al tempo t e l’area totale. Il tempo di semicristallizzazione (t0.5), vale a dire il
tempo a cui il grado di cristallinità del campione è pari al 50%, è stato calcolato come il tempo a cui
X(t) = 0.5.
4.2.3.2 Analisi microscopica
La morfologia dei campioni è stata esaminata tramite microscopio ottico a luce polarizzata (Letz Ortholux II POL-BK), equipaggiato con una piastra riscaldante (Linkam, model THMSE 600). In questo esperimento, si sono ottenuti dei film analizzabili al microscopio a partire da polveri pressate e scaldate tra due vetrini a 100°C per 5 min. Poi i campioni sono stati raffreddati all’aria a temperatura ambiente ed esaminati al microscopio ad un ingrandimento di 32X. Sono state acquisite delle immagini tramite una telecamera (JVC TK-1085E video camera) e una scheda per l’acquisizione delle immagini (Pinnacle System miro VIDEO DC30), che sono state poi visualizzate tramite programma Adobe Premiere 5.1.
4.2.3.3 Analisi termogravimetrica
L’analisi termogravimetrica è stata effettuata tramite un’apparecchiatura Perkin-Elmer TGA 6 Thermogravimetric Analyser sotto atmosfera di azoto. Gli esperimenti sono stati condotti ad una velocità di scansione di 10°C/min nell’intervallo di temperatura 30-600°C.
4.2.3.5 Analisi all’infrarosso (FTIR)
L’analisi all’infrarosso in modalità di contatto (FTIR-ATR) è stata effettuata tramite apparecchiatura Perkin-Elmer Spectrum One Spectrometer, nell’intervallo di lunghezza d’onda pari a 4000-600 cm-1.
4.2.3.6 Analisi di microscopia elettronica a scansione (SEM)
La morfologia delle polveri è stata valutata tramite apparecchiatura SEM del tipo JEOL JSM 5600 LV. I campioni sono stati rivestiti con Au prima dell’analisi al SEM, tramite apparecchiatura Edwards Sputter Coater B150S.
4.2.4 Selective Laser Sintering (SLS)
L’apparecchiatura di SLS adoperata è un prototipo progettato e costruito al Dipartimento di Ingegneria Meccanica, Nucleare e della Produzione dell’Università di Pisa [Ciardelli et al., 2004]. La camera di lavoro dell’apparecchiatura è equipaggiata con una piattaforma in grado di traslare lungo l’asse verticale, e con un blocco laser che comprende il raggio laser e un sistema di specchi, che sono posizionati in modo tale da indirizzare il raggio con precisione. Il laser a CO2 adoperato (SYNRAD modello J48-5S) ha una potenza nominale di 50 W. L’apparecchiatura è collegata ad un computer, provvisto di un software per la guida del fascio laser (CADMARK, Quantasystem, s.r.l.). Gli esperimenti di sinterizzazione sono stati effettuati su polveri sia asciutte sia bagnate con acqua demineralizzata in modo da ottenere uno slurry. La Figura 4-1 mostra schematicamente il funzionamento dell’apparecchiatura di Selective Laser Sintering.
Figura 4-1. Schema del funzionamento dell’apparecchiatura per la sinterizzazione tramite laser (Selective Laser Sintering).
La procedura che è stata seguita per la sinterizzazione è la seguente: è stato dapprima distribuito e livellato uno strato di polvere o di slurry spesso 0.3 mm su un vetrino da microscopio, sistemato in un apposito alloggiamento (Figura 4-2) tale da facilitare la stesura di uno strato omogeneo. Tale alloggiamento è stato a sua volta posizionato sulla piattaforma in grado di traslare verticalmente (abbassata di una quantità corrispondente allo spessore dell’alloggiamento).
Figura 4-2. Schema dell’alloggiamento usato per il vetrino da microscopio (26×76×1 mm3) su cui
si stende la polvere o lo slurry adoperati per la sinterizzazione. La cava che ospita il vetrino è profonda 1.3 mm ed è provvista di un foro centrale per facilitare l’estrazione del vetrino, successivamente al processo di sinterizzazione.
Successivamente, aree specifiche dello strato sono state sinterizzate, seguendo la geometria fissata tramite il software CADMARK. Ad ogni esperimento, sono stati fissati due parametri: la potenza del fascio laser (P), espressa come percentuale della potenza nominale, e la velocità di scansione del laser (BS). I primi esperimenti sono stati condotti sulle polveri di PCL, tentando un’ampia varietà di combinazioni per i parametri di processo (P = 1-3 W; BS = 10-80 mm/s), in modo da trovare la combinazione ottimale. Gli esperimenti successivi sono stati effettuati sugli slurries, dapprima adoperando PCL puro e poi le miscele. Gli esperimenti sono stati effettuati alternativamente mantenendo fissa la potenza (2 W) e variando il parametro BS (5-50 mm/s) oppure mantenendo fisso BS a 5 mm/s e variando la potenza tra 1 e 3 W in modo da trovare la combinazione ottimale dei due parametri. Sono state sinterizzate strisce lunghe 10 mm da polvere asciutta di PCL (P = 2 W; BS = 80 mm/s) e scaffolds bidimensionali in forma di griglie con geometria quadrata (2 × 2 mm2) a partire da slurries (P = 2 W; BS = 5-20 mm/s). Sono state anche sinterizzate strutture con area 1 × 1 mm2 a
partire da paste a base di polisaccaridi puri e di PCL puro, adoperando gli stessi parametri di sinterizzazione usati per le miscele. Le strutture sono state essiccate a temperatura ambiente per una settimana, poi le particelle non sinterizzate sono state allontanate e gli oggetti sono stati esaminati al microscopio ottico (Olympus AX70).
4.2.4.1 Caratterizzazione fisico-chimica delle microstrutture sinterizzate
4.2.4.1.1 Analisi SEM
Sono state analizzate al SEM (JEOL JSM 5600 LV) la superficie superiore (cioè più vicina al blocco laser), quella a contatto con il vetrino e le superfici fratturate in azoto liquido delle strutture sinterizzate.
4.2.4.1.2 Misurazione dell’angolo di contatto
L’angolo di contatto statico della superficie superiore di una particolare struttura compatta sinterizzata al laser (con area di 20 × 20 mm2) è stato misurato a temperatura ambiente, adoperando una goccia
d’acqua demineralizzata di 5 µl in un goniometro telescopico (Ramè-Hart, Inc.). Il telescopio ha un ingrandimento pari a 23 ×. Per ogni angolo riportato, sono state eseguite almeno tre misurazioni sullo stesso campione ma in differenti posizioni della superficie analizzata.
4.2.4.1.4 Analisi all’infrarosso tramite mappatura chimica
Mappe dei segnali FTIR relativi a zone microscopiche dei campioni sinterizzati sono state ottenute tramite l’apparecchiatura Spectrum Spotlight FT-IR Imaging System (Perkin Elmer). In breve, si è ottenuta dapprima una mappa infrarossa di una zona di interesse, che è stata visualizzata come mappa colorata, i cui colori corrispondono a varie intensità di assorbanza. La mappa è costituita da uno spettro per pixel. Sono stati poi acquisiti vari spettri singoli da vari punti all’interno della mappa, il più rappresentativo dei quali è stato scelto come spettro medio. A questo punto, è stata elaborata una mappa di correlazione dell’area rispetto allo spettro medio.
4.2.5 Coltura di fibroblasti e successive caratterizzazioni
Gli scaffolds bidimensionali sinterizzati in forma di griglia sono stati preparati alla coltura cellulare secondo la procedura descritta al paragrafo 2.2.2.10 del Capitolo 2, dove si ritrovano anche i dettagli sulla coltura cellulare. Le strutture polimeriche sono state seminate con una sospensione cellulare di fibroblasti murini NIH-3T3 in pozzetti di polistirene da 24 celle. Le misure di adesione cellulare sono state effettuate su tre campioni ad ogni tempo (2, 4, 24 h). Le procedure di fissaggio cellulare e colorazione sono state descritte al paragrafo 2.2.2.10 del Capitolo 2. I campioni sono stati analizzati al microscopio ottico (Olympus AX 70). L’area di adesione cellulare di ogni substrato polimerico è stata calcolata considerando la struttura costituita da tanti parallelepipedi, quante sono le linee sinterizzate. Le dimensioni dei parallelepipedi usati come modello sono state scelte in base alle dimensioni reali delle microstrutture, che sono state ricavate da micrografie SEM. L’indice della densità cellulare è stato calcolato come rapporto tra l’area occupata dalle cellule aderite sul substrato e l’intera area del substrato. Per calcolare l’area occupata dalle cellule, è stato adoperato un software sviluppato in ambiente Matlab e basato su un processo di segmentazione. Sono state selezionate tre zone di ogni campione, e fotografate al microscopio ottico usando un ingrandimento 10 X. Per ogni foto, sono state scelte 5 aree per la misura. L’area della superficie dello scaffold e quella occupata dalle cellule sono state calcolate per ognuna delle 5 aree e poi mediate. La procedura è stata ripetuta per le altre due foto di uno dei tre campioni prelevati a un definito tempo di prova. I tre dati sono stati poi mediati. Si è proceduto in modo analogo per gli altri due campioni relativi a quel tempo di prova. I tre valori medi di adesione di ciascun campione sono stati a loro volta mediati, ricavando il valore medio della densità cellulare a quel tempo. La densità cellulare sulle strutture è stata paragonata a quella di un campione di riferimento (un film di gelatina di tipo A). La morfologia cellulare è stata studiata su ogni campione, tramite foto al microscopio ottico con ingrandimento pari a 40 X (Olympus AX 70).
4.3 Risultati e discussione
In questo paragrafo vengono riportati i risultati della caratterizzazione chimico-fisica delle miscele “bioartificiali” a base di PCL e di un polimero naturale tra gellano, destrano e amido. L’analisi calorimetrica è stata condotta sulle miscele, allo scopo di valutare le differenze nei parametri di cristallizzazione e di fusione del PCL, causate dalla presenza del polimero naturale. L’effetto del polimero naturale sulla struttura cristallina del PCL è stato approfondito tramite analisi WAXD. L’analisi calorimetrica è stata effettuata anche sui campioni sinterizzati delle miscele, al fine di valutare eventuali variazioni dei parametri termici, legate al processo cui sono sottoposti i materiali. L’analisi di cristallizzazione isoterma è stata utile per evidenziare il grado di interazione tra i due componenti in miscela e l’effetto del polimero naturale sui meccanismi di nucleazione e di crescita dei cristalli del polimero sintetico. L’analisi morfologica tramite microscopio ottico è stata un valido supporto all’analisi calorimetrica. L’analisi FTIR-ATR è stata condotta per evidenziare la presenza di interazioni tra i componenti delle miscele, mentre l’analisi IR-Chemical Imaging ha permesso di valutare il grado di omogeneità nella composizione delle miscele. L’analisi termogravimetrica è risultata funzionale all’utilizzo dei materiali nel processo di Selective Laser Sintering, che prevede l’applicazione di una fonte termica concentrata per la sinterizzazione del materiale nella geometria desiderata. Le polveri a base delle miscele “bioartificiali” e di PCL, destinate alla sinterizzazione, sono
per avere informazioni sulle loro dimensioni e caratteristiche geometriche. La morfologia delle strutture sinterizzate è stata analizzata e confrontata con quella delle polveri di partenza, per capire quali sono le dimensioni e la geometria ottimali delle polveri per realizzare una sinterizzazione accurata con il tipo di laser impiegato in questo lavoro. La misura dell’angolo di contatto dei campioni sinterizzati è stata utile per capire il loro grado di idrofilia e come questo possa variare con la composizione della miscela. Infine, le prove di adesione cellulare in vitro, adoperando fibroblasti murini, hanno permesso di stabilire il grado di biocompatibilità delle miscele “bioartificiali”.
4.3.1 Proprietà termiche delle miscele PCL/polisaccaride risultanti dall’analisi DSC e
dal supporto dell’analisi tramite microscopio ottico.
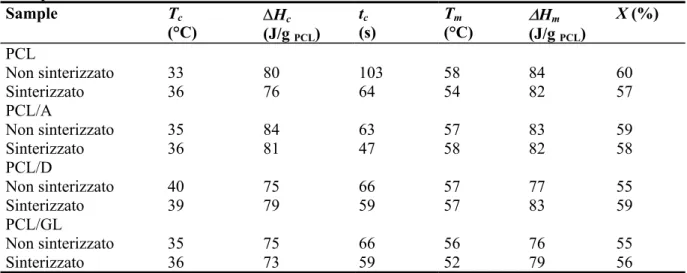
L’effetto del tipo di polisaccaride (amido, gellano, destrano) sulle proprietà termiche delle miscele a base di PCL è stato valutato tramite analisi DSC. Le Figure 4-3 a e 4-3 b mostrano rispettivamente le scansioni DSC di raffreddamento e di secondo riscaldamento per il PCL puro e per le miscele. I dati sono riportati in Tabella 4-1 e mostrano che l’introduzione del 9.1 % in peso di polisaccaride in miscela ha avuto un debole effetto sul grado di cristallinità del PCL e sulla temperatura di cristallizzazione. Questi effetti sono risultati più pronunciati per la miscela PCL/D. L’effetto nucleante del polimero naturale, suggerito dal piccolo incremento della Tc del PCL nelle prove non isoterme, è
stato confermato dall’analisi di cristallizzazione isoterma, che è stata effettuata nell’intervallo di temperatura 38-48 °C. Comportamenti simili sono stati riportati in letteratura per miscele incompatibili tra PCL e un polimero naturale [Rutot et al., 2001; Wu et al., 2003; Dubois et al., 2003; Duquesne et al., 2001].
Tabella 4-1. Dati DSC di cristallizzazione e di fusione per il PCL e per le miscele PCL/polisaccaridi. Sample Tc (°C) ∆H(J/g c PCL) tc (s) Tm (°C) ∆(J/g Hm PCL) X (%) PCL Non sinterizzato Sinterizzato 33 36 80 76 103 64 58 54 84 82 60 57 PCL/A Non sinterizzato Sinterizzato 35 36 84 81 63 47 57 58 83 82 59 58 PCL/D Non sinterizzato Sinterizzato 40 39 75 79 66 59 57 57 77 83 55 59 PCL/GL Non sinterizzato Sinterizzato 35 36 75 73 66 59 56 52 76 79 55 56
Figura 4-3. Scansioni DSC di raffreddamento (a) e secondo riscaldamento (b) per PCL puro e miscele (velocità di scansione: 10°C/min).
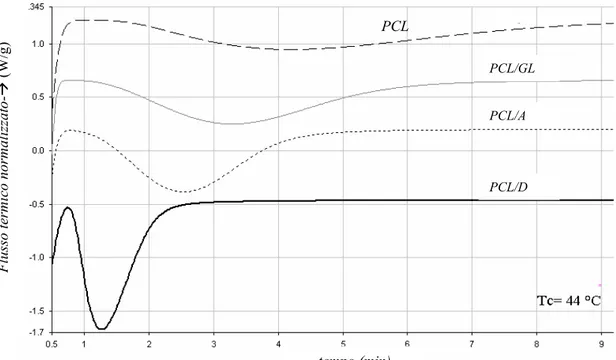
Per il PCL e per le miscele, è stato rilevato un solo picco di cristallizzazione isoterma per ogni TC,
come evidenziato dalla Figura 4-4 che mostra le curve di cristallizzazione isoterma per i vari campioni alla temperatura di 44°C. (a) (b) PCL/A PCL/GL PCL/A PCL/GL
Figura 4-4. Curve di cristallizzazione isoterma per il PCL e per le miscele a 44°C.
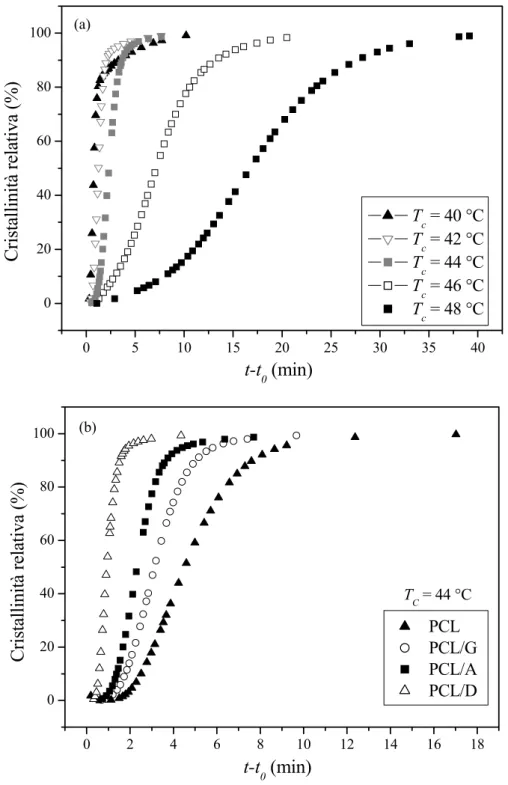
Le Figure 4-5 a e 4-5 b mostrano rispettivamente l’andamento della cristallinità relativa Xt in funzione
del tempo di cristallizzazione, per la miscela PCL/A cristallizzata a varie TC e per tutti i campioni
(PCL puro e miscele) ad una TC fissata. Le posizioni relative delle isoterme di cristallizzazione
dipendono da TC: una più elevata TC causa lo spostamento delle curve di Xt a tempi più lunghi. Al
contrario, per la composizione analizzata, la presenza del polisaccaride ha avuto l’effetto di accelerare la cinetica di cristallizzazione isoterma ad ogni TC.
PCL PCL/A PCL/GL PCL/D tempo (min) Fluss o termic o normalizzat o-Æ (W/ g)
Figura 4-5. Andamento della cristallinità relativa in funzione del tempo di cristallizzazione: (a) per campioni di PCL/A cristallizzati in condizioni isoterme nell’intervallo di temperatura 40-48 °C; (b) per campioni di PCL e di miscele cristallizzati a TC = 44°C. t0 è il tempo iniziale, vale a dire il tempo a cui la temperatura raggiunge il valore fissato TC.
La Figura 4-6 riporta i grafici del tempo di semi-cristallizzazione (t0.5) in funzione di TC per il PCL e
per le miscele. La velocità totale di cristallizzazione dei campioni ad ogni TC, calcolata come il
reciproco di t0.5, è risultata più elevata per le miscele rispetto che per il PCL. La velocità di
cristallizzazione ha mostrato il seguente ordine decrescente: PCL/D > PCL/A ≥ PCL/GL > PCL, nell’intervallo di TC studiato. 0 5 10 15 20 25 30 35 40 0 20 40 60 80 100 Tc = 40 °C Tc = 42 °C Tc = 44 °C Tc = 46 °C Tc = 48 °C
C
rista
llinità
re
la
tiva
(
%
)
t-t
0(min)
(a) 0 2 4 6 8 10 12 14 16 18 0 20 40 60 80 100 TC = 44 °Ct-t
0(min)
C
rista
llinità
re
la
tiva
(%
)
PCL PCL/G PCL/A PCL/D (b)Figura 4-6. Andamento del tempo di semicristallizzazione in funzione della temperatura di cristallizzazione isoterma per il PCL e per le miscele.
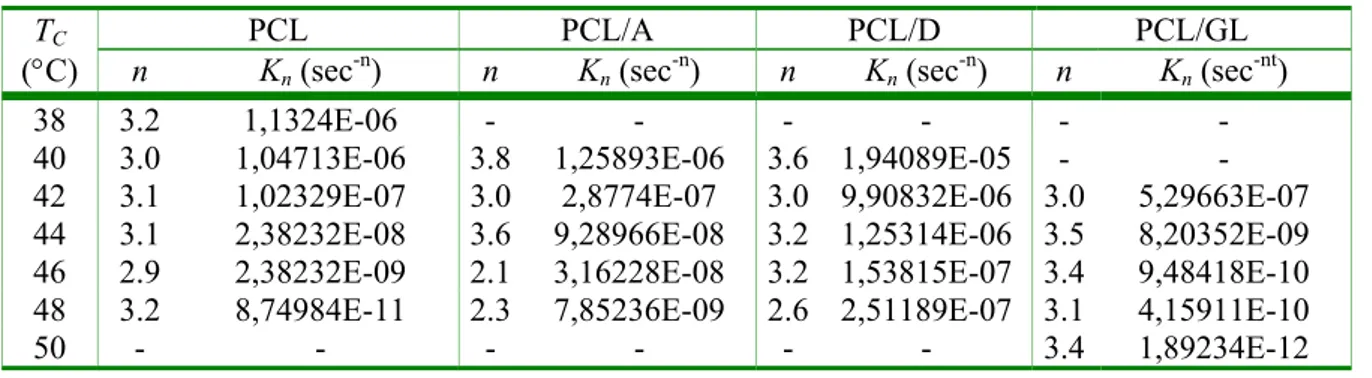
La cinetica di cristallizzazione dei campioni ad ogni TC è stata analizzata tramite l’equazione di
Avrami [Avrami, 1939]:
X(t) = 1 - exp(- Kn·tn) (4.2)
dove Kn è la costante cinetica e n l’esponente di Avrami, che dipendono dalla geometria di crescita e
dal tipo di nucleazione dei cristalli [Mandelkern, 1964]. I valori di Kn e n possono essere ottenuti dalle
intercette e dalle pendenze delle rette del log[-ln(1-X(t))] in funzione del log(t) per valori di X(t)<60-90%. Esempi di tali grafici sono riportati in Figura 4-7 per il PCL puro e per le miscele a varie temperature di cristallizzazione.
Tutti i grafici riportati in Figura 4-7 presentano un primo tratto lineare fino ad un valore di cristallinità relativa che dipende dalla temperatura di cristallizzazione e dal materiale, ed un secondo tratto lineare con pendenza inferiore per alti valori di X(t), che corrisponde ai fenomeni di cristallizzazione secondaria. I parametri del modello di Avrami per il PCL puro e per le miscele contenenti il 9.1 % in peso di polisaccaride sono riportati in Tabella 4-2.
308 310 312 314 316 318 320 0 5 10 15 20
t
0.5(min
)
T
C(K)
PCL PCL/A PCL/GL PCL/DTabella 4-2. Parametri di Avrami per il PCL e per le miscele a base di PCL
38 3.2 1,1324E-06 - - - -
40 3.0 1,04713E-06 3.8 1,25893E-06 3.6 1,94089E-05 - -
42 3.1 1,02329E-07 3.0 2,8774E-07 3.0 9,90832E-06 3.0 5,29663E-07
44 3.1 2,38232E-08 3.6 9,28966E-08 3.2 1,25314E-06 3.5 8,20352E-09
46 2.9 2,38232E-09 2.1 3,16228E-08 3.2 1,53815E-07 3.4 9,48418E-10
48 3.2 8,74984E-11 2.3 7,85236E-09 2.6 2,51189E-07 3.1 4,15911E-10
50 - - - - - - 3.4 1,89234E-12
Gli indici di Avrami, generalmente, hanno mostrato valori superiori a 3, indicativi di un meccanismo di crescita tridimensionale dei cristalli. In particolare, valori di n vicini a 3 hanno indicato fenomeni di nucleazione eterogenea atermica, mentre valori di n compresi tra 3 e 4 hanno indicato fenomeni di nucleazione mista (termica e atermica) [Mandelkern, 1964]. Per la miscela PCL/A, cristallizzata a 46-48°C, sono stati calcolati valori di n vicini a 2 che indicano un meccanismo di crescita bidimensionale dei cristalli [Mandelkern, 1964].
PCL PCL/A PCL/D PCL/GL
TC
Figura 4-7. Andamento del log[-ln(1-X(t))] in funzione di log(t) per le polveri di PCL (a) e delle miscele: (b) PCL/D 90.9/9.1; (c) PCL/A 90.9/9.1; (d) PCL/GL 90.9/9.1. 2.0 2.5 3.0 3.5 -1.5 -1.0 -0.5 0.0 0.5 1.0 TC=40°C TC=44°C TC=44°C TC=46°C TC=48°C PCL/GL 90.9/9.1 lo g(-ln (1-X(t ))) log[(t-t0)/s] 3.1 9.5 27.1 63.2 95.7 100 X(t ) 1.5 2.0 2.5 3.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 3.1 9.5 27.1 63.2 95.7 100 PCL/A 90.9/9.1 log (-ln(1 -X (t))) log[(t-t0)/s] TC=40°C TC=44°C TC=44°C TC=46°C TC=48°C X(t ) 1.0 1.5 2.0 2.5 3.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 TC=40°C TC=44°C TC=44°C TC=46°C TC=48°C PCL/D 90.9/9.1 lo g(-ln (1-X(t ))) log[(t-t0)/s] 3.1 9.5 27.1 63.2 95.7 100 X( t) 1.5 2.0 2.5 3.0 -1.5 -1.0 -0.5 0.0 0.5 1.0 TC=40°C TC=44°C TC=44°C TC=46°C TC=48°C 3.1 9.5 27.1 63.2 95.7 100 PCL log (-ln(1 -X(t ))) log[(t-t0)/s] X( t) (a) (b) (c) (d)
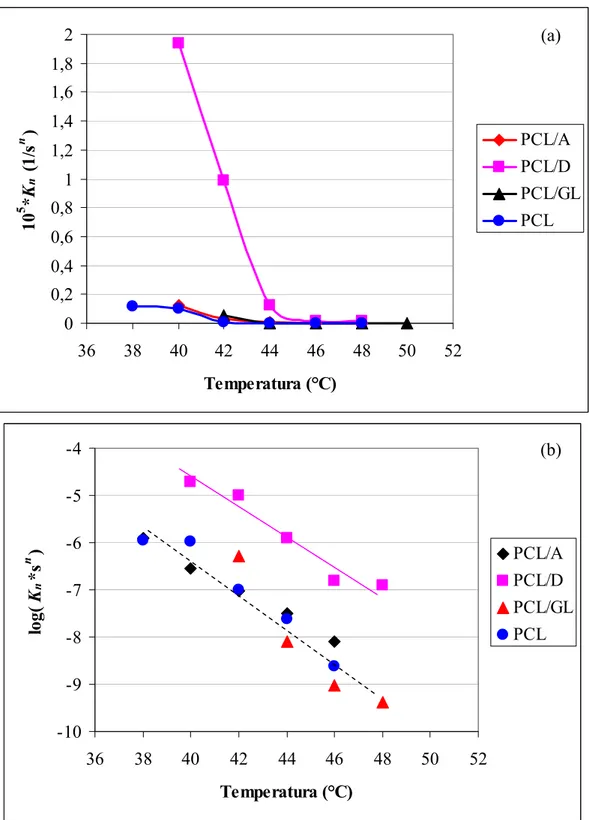
Le Figure 4-8 a riporta l’andamento della costante cinetica di Avrami per il PCL e per le miscele bioartificiali a base di PCL. La Figura 4-8 b mostra l’andamento del valore logaritmico della costante di Avrami in funzione della temperatura di cristallizzazione. La costante cinetica di Avrami, per ogni materiale, è risultata crescente al decrescere della temperatura di cristallizzazione, inoltre ha mostrato valori nettamente più elevati per la miscela PCL/D. Gli andamenti dei valori logaritmici della costante di Avrami sono risultati lineari e pressoché simili per le miscele PCL/A, PCL/GL e per il PCL puro.
0 0,2 0,4 0,6 0,8 1 1,2 1,4 1,6 1,8 2 36 38 40 42 44 46 48 50 52 Temperatura (°C) 10 5 *K n (1 /s n ) PCL/A PCL/D PCL/GL PCL -10 -9 -8 -7 -6 -5 -4 36 38 40 42 44 46 48 50 52 Temperatura (°C) log( K n *s n ) PCL/A PCL/D PCL/GL PCL
Figura 4-8. Andamento della costante cinetica di Avrami (a) e del valore logaritmico della costante cinetica di Avrami (b) per il PCL e per le miscele bioartificiali a base di PCL, in funzione della temperatura di cristallizzazione isoterma.
(a)
Le tracce di fusione dei campioni cristallizzati in condizione isoterma nell’intervallo 38-48 °C mostrano un singolo picco di fusione, che tende a spostarsi a temperatura più alta al crescere di TC. La
Figura 4-9 riporta il grado di cristallinità di tutti i campioni, registrato dalla scansione di riscaldamento successiva al mantenimento alla temperatura di cristallizzazione, in funzione della stessa TC. I gradi di
cristallinità di PCL, PCL/GL e PCL/A sono risultati simili, mentre quelli della miscela PCL/D hanno assunto valori più bassi. Il comportamento della miscela PCL/D può essere attribuito alla maggiore velocità di cristallizzazione della miscela PCL/D, che ha determinato la formazione di cristalli con dimensioni ridotte e un basso grado di perfezione.
36 38 40 42 44 46 48 50 0.40 0.45 0.50 0.55 0.60 0.65 0.70 PCL PCL/A PCL/GL PCL/D
G
rado di cris
tallin
ità (
X
)
T
c(°C)
Figura 4-9. Andamento del grado di cristallinità dei campioni di PCL e di miscele PCL/polisaccaride in funzione di Tc.
La Figura 4-10 mostra l’andamento delle temperature di fusione Tm, registrate nella scansione di
riscaldamento successiva alla fase di cristallizzazione isoterma, in funzione di TC, per ogni campione.
La temperatura di fusione all’equilibrio Tm0 del PCL e delle miscele a base di PCL è stata calcolata
sulla base della teoria di Hoffman e Weeks [Wunderlich, 1980], che stabilisce una relazione tra le temperature di fusione osservate (Tm) dopo ogni cristallizzazione isoterma a TC e la corrispondente TC
:
Tm = (1/2⋅β)⋅Tc + (1-1/2⋅β)⋅Tm0 (4.3)
Dove β è un fattore morfologico che correla lo spessore medio di lamella (l) dei cristalli allo spessore iniziale (l*) dei nuclei di crescita a Tc. La temperatura di fusione all’equilibrio è stata calcolata
dall’intersezione della retta Tm= Tc con la linea retta che approssima ai minimi quadrati i dati di Tm
riportati in grafico in funzione di TC. Nella Figura 4-10 sono riportate le rette di Hoffman-Weeks per
tutti i campioni.
310 320 330 340 330 332 334 336 338 340 342 PCL PCL/A PCL/GL PCL/D Tm=TC T m (K ) TC (K)
Figura 4-10. Andamento della temperatura di fusione registrata durante il riscaldamento a 10°C/min successivo alla fase di cristallizzazione isoterma, in funzione della temperatura di cristallizzazione isoterma.
La Tabella 4-3 riporta i valori di Tm0 e di β e il coefficiente di correlazione r2 dell’approssimazione ai
minimi quadrati. Il valore del parametro β è risultato maggiore di 1, in buon accordo con i risultati di
letteratura relativi a PCL e miscele a base di PCL [Leczano et al., 1986].
Tabella 4-3. Temperature di fusione all’equilibrio, parametri β e coefficiente di correlazione per il PCL e le miscele bioartificiali a base di PCL.
Campione Tm0 (°C) β r2
PCL 67 1.7 0.991
PCL/A 61 2.8 0.991
PCL/D 64 2.1 0.989
PCL/GL 65 1.6 0.989
Il valore Tm0 per le miscele PCL/GL e PCL/D ha subito una modesta variazione rispetto al caso del
PCL. Una maggiore variazione di Tm0 è stata invece riscontrata per la miscela PCL/A, risultato che ha
suggerito la presenza di maggiori interazioni tra i componenti di tale miscela.
Le osservazioni condotte al microscopio ottico su campioni in forma di film (ottenuti secondo la procedura indicata nella parte sperimentale) hanno evidenziato la presenza di sferuliti di 30-100 µm di diametro per il PCL, e di cristalliti di piccole dimensioni e di forma irregolare per le miscele (Figura 4-11). Queste osservazioni sono un’ulteriore conferma dell’effetto nucleante del polimero naturale sulla cristallizzazione del PCL, evidenziato dall’analisi calorimetrica: le miscele, infatti, hanno mostrato una morfologia che è tipica dei processi di cristallizzazione veloci.
Figura 4-11. Immagini al microscopio ottico per campioni sottili di film di PCL (a), PCL/D (b), PCL/A (c) e PCL/GL (d), ottenuti da particelle pressate tra due vetrini mantenuti alla temperatura di 100°C per 5 min, e successivamente raffreddati a temperatura ambiente. Le barre indicano 25 µm.
A questo punto, è possibile fornire un commento adeguato relativamente ai valori dei tempi totali di cristallizzazione dei materiali sinterizzati e non sinterizzati (tc), riportati in Tabella 4-1.
Le miscele hanno mostrato una maggiore velocità di cristallizzazione rispetto al PCL: ciò può essere attribuito sia all’effetto nucleante del polimero naturale sulla cristallizzazione del PCL, sia alla minore frazione di materiale cristallizzabile all’interno delle miscele.
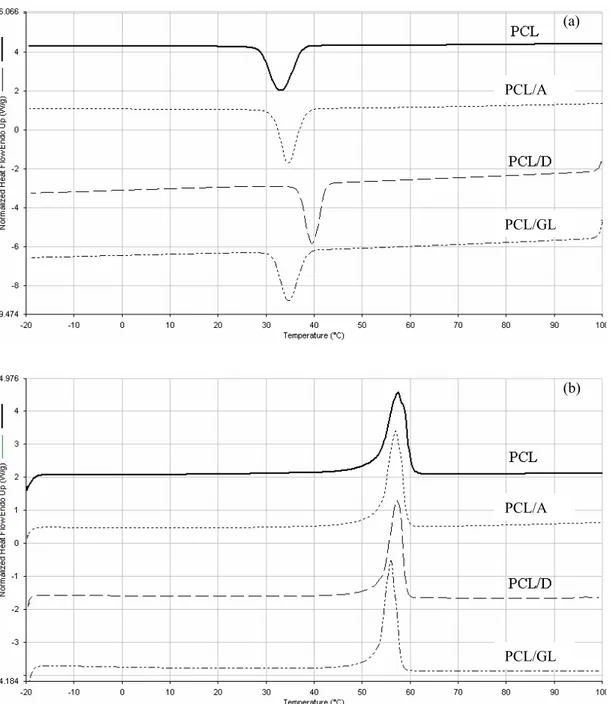
4.3.2 Analisi termogravimetrica (TGA)
Le curve relative all’analisi TGA dei polimeri naturali hanno evidenziato due fenomeni degradativi (Figura 4-12 a): la vaporizzazione dell’acqua contenuta nel campione nell’intervallo 70-100 °C, e la pirolisi del polimero la cui temperatura di massima velocità di degradazione (TD) è risultata pari a
250°C per il gellano, 320 °C per l’amido e 350 °C per il destrano. Le curve TGA del PCL hanno evidenziato un singolo evento degradativo, corrispondente alla pirolisi del polimero, la cui T vale
(a)
(b)
rispetto a quella registrata per i polisaccaridi puri. Al contrario, il PCL in miscela è degradato termicamente ad una temperatura più bassa di 15-25 °C rispetto a quella registrata per il PCL puro, probabilmente a causa delle interazioni tra il PCL e i prodotti di degradazione dei polimeri naturali. Per la miscela PCL/D, il picco di pirolisi è risultato più ampio e i fenomeni di degradazione sono iniziati ad una temperatura più bassa rispetto al PCL puro e alle altre miscele.
In conclusione, la stabilità termica delle miscele PCL/polisaccaride 90.9/9.1 (w/w) è risultata limitata dal tipo di polimero naturale miscelato e sufficientemente elevata da giustificare i nostri tentativi di sinterizzare le miscele tramite apparecchiatura SLS.
Figura 4-12. Curve termogravimetriche derivative (DTG) per (a) i componenti puri; (b) il PCL e le miscele.
4.3.3 Analisi di diffrazione ai raggi X (WAXD)
I grafici relativi all’analisi WAXD del PCL e delle miscele (Figura 4-13) evidenziano i loro picchi principali per valori di 2θ pari a 21.2°, 21.8° e 23.5°, che sono quelli tipici della cella unitaria
cristallina ortorombica [Hu et al., 1990]. L’intensità relativa dei picchi è risultata simile per tutti i campioni, evidenziando un grado di cristallinità simile per il PCL e per le miscele. Questo risultato non è contradditorio rispetto all’analisi DSC: l’analisi WAXD è stata effettuata sulle particelle così
(b)
(a)
APCL GL
come sono state prodotte, mentre i dati DSC commentati sono stati ricavati dalla seconda scansione di riscaldamento.
Figura 4-13. Spettri WAXD del PCL puro e delle miscele
2⋅θ
PCL/A
PCL PCL/D PCL/GL
4.3.4. Analisi FTIR-ATR
La Figura 4-14 mostra gli spettri FTIR-ATR dei componenti puri e delle miscele. Lo spettro del PCL ha mostrato bande caratteristiche a 2920 cm-1 e 2850 cm-1 associate alle vibrazioni di stretching dei gruppi CH2 e a 1756 cm-1 associate all’assorbimento del gruppo carbonile.
Lo spettro FTIR-ATR del gellano puro ha mostrato bande caratteristiche a 3382 cm-1 (stretching O-H),
a 1926 cm-1 (stretching C-H), a 1608 cm-1 (assorbimento dell’anione carbossilato) e a 1046 cm-1
(assorbimento dell’anello piranosidico).
Lo spettro del destrano ha mostrato una banda nell’intervallo di frequenze 3350 e 3200 cm-1, dovuta
alle vibrazioni di stretching O-H, un’altra banda a 2926 cm-1 tipica dello stretching C-H, e bande
parzialmente sovrapposte negli intervalli di frequenza 1200-800 e 1260-1000 cm-1 associate,
rispettivamente, agli assorbimenti del gruppo C-O-C e del gruppo alcolico C-O.
4000 3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 650. cm-1 %T PCL/D PCL/GL PCL/A PCL GL A D
Figura 4-14. Spettri FTIR-ATR del PCL puro e delle miscele
Lo spettro FTIR dell’amido ha mostrato bande caratteristiche a 3350-3200 cm-1 (stretching O-H), a
2850 e 2920 cm-1 (stretching C-H), a 1640 cm-1 (vibrazioni di bending O-H dell’acqua assorbita), a
1462 cm-1 (bending del gruppo CH
2), a 1445-1325 cm-1 (bending e wagging del C-H), a 1243-1205
cm-1 (bending del gruppo O-H), e a 960 cm-1 e a 1190 cm-1 (stretching C-O).
Gli spettri FTIR delle miscele hanno mostrato le tipiche bande di assorbimento rilevate per i componenti puri, e non si sono evidenziate variazioni nella frequenza delle bande caratteristiche, a causa della mancanza di forti interazioni tra i componenti delle miscele.
4.3.5. Caratteristiche degli scaffolds ottenuti mediante Selective Laser Sintering
La Figura 4-15 a-c mostra la morfologia delle polveri utilizzate per il processo di sinterizzazione. La macinazione manuale e la successiva setacciatura hanno prodotto polveri con geometria irregolare e disomogenea, superfici porose, e una distribuzione dimensionale ampia (< 125 µm). La Figura 4-15 d mostra invece la morfologia delle particelle ottenute tramite processo di emulsione semplice O/W: le particelle sono risultate sferiche, con una superficie liscia e una distribuzione dimensionale più stretta. L’analisi tramite light scattering ha mostrato che le particelle hanno un diametro D compreso tra 3 e
7.2 µm e che il 76% delle particelle ha un diametro inferiore a 4.6 µm (Figura 4-16 a-b). I risultati hanno permesso di individuare i valori dei diametri delle particelle, mediati sulle superfici e sui volumi, che valgono rispettivamente:
- d (3,2) = 4.4 µm (detto anche diametro di Sauter) - d (4,3) = 4.4 µm
Figura 4-15. Immagini SEM delle particelle usate per la sinterizzazione tramite laser: (a) PCL/D; (b) PCL/A; (c) PCL/GL; (d) PCL
(d)
(b)
(c)
(a)
0,00 20,00 40,00 60,00 80,00 100,00 0,00 2,00 4,00 6,00 8,00 D (micron) P erc en tu al e p ar ti cel le co n di am et ro < D (% ) 0,00 10,00 20,00 30,00 40,00 50,00 60,00 70,00 80,00 6,7 5,75 4,95 4,3 3,7 3,2 d (micron) P art ic el le con d iam et ro m ed io d (% )
Figura 4-16. Risultati dell’analisi tramite light scattering delle particelle di PCL ottenute per emulsione semplice: a) percentuale numerica cumulativa di particelle con diametro < D; b) percentuale numerica di particelle con diametro medio pari a d.
Il grado di precisione delle strutture sinterizzate è risultato influenzato dalla granulometria della polvere. La risoluzione verticale e l’accuratezza laterale della sinterizzazione può essere migliorata adoperando particelle di dimensioni inferiori rispetto allo spot del laser (< 50 µm), mentre una distribuzione stretta delle dimensioni delle particelle potrebbe garantire una maggiore omogeneità nel trasferimento termico, e quindi influenzare il processo di layer bonding nella sinterizzazione a più strati e il grado di uniformità delle dimensioni laterali delle piste sinterizzate.
I primi esperimenti sono stati condotti sulle polveri asciutte di PCL ottenute per macinazione manuale. La difficoltà principale riscontrata ha riguardato la possibilità di stendere uno strato micrometrico e compatto di polvere, a seguito delle piccole dimensioni e della geometria irregolare delle particelle di PCL. Sono state provate varie combinazioni dei parametri P e BS per sinterizzare semplici piste longitudinali bidimensionali. La combinazione di parametri ottimale è risultata P = 2W e BS = 80 mm/s. La densità di energia (ED), vale a dire l’energia laser applicata per unità di superficie, è
(a)
direttamente proporzionale al rapporto P/BS. Il valore di ED influenza varie proprietà, quali la rugosità superficiale, la densità, la resistenza a trazione, l’accuratezza dimensionale, la presenza di fenomeni di
curling e di cracking [Ho et al., 2003].
La scarsa adesione tra le polveri livellate e il vetrino di supporto unita alla bassa densità delle particelle nello strato deposto sono state la causa dei modesti risultati ottenuti in fase di sinterizzazione. Le strisce sinterizzate hanno mostrato una forma irregolare, e sono risultate interrotte in alcuni punti. La Figura 4-17 riporta un’immagine ottenuta tramite microscopio ottico di una striscia sinterizzata a partire da particelle di PCL prodotte per macinazione manuale.
Figura 4-17. Immagine al microscopio ottico di una striscia sinterizzata da polveri di PCL ottenute per macinazione (P = 2W; BS = 80 mm/s)
Come successivo tentativo, sono state utilizzate “paste” acquose di polvere di PCL e acqua demineralizzata. La presenza dell’acqua ha garantito sia una maggiore densità dello strato deposto sia una più stretta adesione tra le particelle e il vetrino. Sono stati effettuati molti esperimenti per trovare i parametri ottimali per la sinterizzazione, che alla fine sono risultati P = 2W e BS = 5 mm/s per le miscele e P = 2W e BS = 20 mm/s per il PCL puro. La presenza del polimero naturale, che presumibilmente ostacola il flusso del PCL fuso, ha reso necessaria una maggiore densità di energia
ED. Una maggiore densità di energia determina l’aumento della temperatura dello strato da
sinterizzare e, a sua volta, una temperatura più elevata decresce la viscosità del PCL fuso, favorendone il flusso [Rosa et al., 2004].
I parametri di sinterizzazione ottimali per le miscele PCL/polisaccaride 90.9/9.1 w/w sono risultati indipendenti dal tipo di polisaccaride impiegato (gellano, destrano o amido).
L’osservazione tramite microscopio ottico delle strutture sinterizzate a partire da slurries acquosi ha rivelato una riproduzione sufficientemente accurata, da parte del sistema di sinterizzazione, della corrispondente geometria prodotta con software di grafica CADMARK (strisce o griglie).
Successivamente, sono stati realizzati degli scaffold bidimensionali con una geometria a griglia con maglie quadrate. Per ogni maglia, la distanza tra gli assi delle strisce sinterizzate, che costituiscono i lati opposti del quadrato della maglia, è risultata di 2 mm. La Figura 4-18 riporta un’immagine, fotografata con una macchina fotografica digitale, di uno scaffold in forma di griglia, ottenuto da uno
slurry a base di PCL/GL. Tutte le griglie prodotte, delle quali la Figura 4-18 fornisce un esempio,
hanno mostrato una geometria regolare. Inoltre, l’osservazione delle griglie non ha permesso di individuare i segni più evidenti di una degradazione termica, che corrispondono all’assunzione di una colorazione giallo-marrone da parte dei materiali sinterizzati.
Le immagini SEM delle superfici superiori (cioè in contatto con l’aria) e inferiori (cioè in contatto con il vetrino) e delle sezioni fratturate in azoto liquido dei campioni sinterizzati sono riportate nelle Figure 4-19, 4-20 e 4-21.
Figura 4-19. Immagini SEM delle superfici superiori di campioni di: (a) PCL/D; (b) PCL/GL; (c) PCL/A; (d) PCL.
(a) (b)
(d) (c)
Figura 4-20. Immagini SEM della superficie inferiore di campioni sinterizzati: (a) PCL/D; (b) PCL/GL; (c) PCL/A; (d) PCL.
(a) (b)
(d) (c)
Figura 4-21. Immagini SEM della sezioni fratturate relative a campioni sinterizzati di: (a) PCL/D; (b) PCL.
La morfologia dei campioni di miscela sinterizzati ha mostrato variazioni nello spessore delle sezioni, seguendo un andamento caratteristico: le superfici superiori, esposte direttamente all’azione del laser, sono risultate generalmente lisce e regolari, mentre quelle a contatto con il vetro sono risultate porose e irregolari. La sezione ha evidenziato un buon grado di sinterizzazione fino ad una certa profondità e un’elevata porosità nella porzione di sezione vicina al vetrino di supporto. I pori possono essere dovuti a bolle di vapore intrappolate.
Al contrario, la morfologia delle strutture sinterizzate in PCL è risultata più omogenea. I risultati dell’analisi SEM hanno evidenziato che la morfologia delle strutture sinterizzate dipende strettamente dalla morfologia delle particelle di partenza.
Le superfici delle strutture sinterizzate al laser si sono rivelate idrofile con angoli di contatto (misurati sulla superficie superiore dei campioni sinterizzati) nell’intervallo 52°-66° (Tabella 4-4). Sulla base delle indicazioni di letteratura, l’idrofilia dei substrati per coltura cellulare, con angoli di contatto intermedi, favorisce l’adesione cellulare [Saltzman et al., 1997]. La bagnabilità superficiale è risultata crescente nell’ordine: PCL/A > PCL/D ≈ PCL > PCL/GL: sia possibili riduzioni del peso molecolare del PCL in fase di sinterizzazione, sia il tipo di polisaccaride miscelato hanno influenzato il grado di idrofilia dei campioni.
(a)
Tabella 4-4. Angoli di contatto per i campioni sinterizzati.
Sample Contact angle
PCL 58° ± 4
PCL/A 52° ± 4
PCL/D 66° ± 1
PCL/GL 62° ± 3
La Tabella 4-1, riportata nel paragrafo 4.4.1, riporta anche le proprietà termiche dei campioni sinterizzati, che, generalmente, sono risultate simili a quelle dei campioni non sinterizzati. L’eccezione principale è stata costituita dal PCL sinterizzato, che ha evidenziato una crescita di 3°C nella temperatura di cristallizzazione e una diminuzione di 4°C della temperatura di fusione, rispetto al PCL non processato. Queste variazioni possono essere attribuite a variazioni del peso molecolare del PCL durante la sinterizzazione. Le proprietà termiche delle miscele si sono mantenute sostanzialmente invariate con la sinterizzazione, fatta eccezione per la miscela PCL/GL, la cui Tm è diminuita di 4°C,
rispetto al caso delle polveri non sinterizzate.
La Tabella 4-1 ha mostrato una decrescita del tempo totale di cristallizzazione (tc) per i campioni
sinterizzati, rispetto ai materiali di partenza, particolarmente evidente per il PCL. Questo comportamento può essere attribuito a una ipotizzabile diminuzione del peso molecolare del PCL durante la sinterizzazione.
Slurries dei polisaccaridi sono state processate usando gli stessi parametri adoperati per le miscele. I
polisaccaridi puri processati al laser hanno modificato colore, passando dal bianco al marrone chiaro (amido e destrano) o al marrone scuro (gellano). Tuttavia, l’analisi FTIR-ATR dei campioni sinterizzati non ha mostrato segni di variazione della composizione chimica. A titolo di esempio, sono stati riportati in Figura 4-22 gli spettri FTIR-ATR dell’amido prima e dopo il trattamento di sinterizzazione, che non evidenziano variazioni di rilievo.
Questi risultati hanno dimostrato che i fenomeni di degradazione termica dei polisaccaridi dovuti all’azione del laser sono stati limitati alla parte superficiale dei campioni, maggiormente esposta al laser. I campioni di miscele e di PCL sinterizzati al laser (usando i parametri di sinterizzazione più severi, che sono quelli ottimali per le miscele) non sono anneriti superficialmente. Gli spettri FTIR-ATR dei campioni di miscela e di PCL, sinterizzati come indicato sopra, sono risultati analoghi a quelli dei rispettivi campioni non sinterizzati. Questi risultati hanno mostrato che la sinterizzazione non ha cambiato la struttura chimica dei materiali qui studiati. La miscelazione dei polisaccaridi con una percentuale elevata di un polimero resistente alle alte temperature (PCL), ha evitato che il componente meno resistente al calore (polimero naturale) subisse fenomeni di degradazione termica.
Figura 4-22. Confronto tra gli spettri FTIR-ATR dell’amido, prima e dopo la sinterizzazione.
I campioni sinterizzati al laser sono stati anche analizzati tramite mappatura chimica (IR-Chemical Imaging). Le Figure 4-23 a-c riportano, rispettivamente, la mappa spettrale relativa ad una zona di area 1×1 mm2, il suo spettro medio e la mappa di correlazione dello spettro medio all’interno della zona
analizzata, per un campione di PCL/A sinterizzato. Questo tipo di analisi è stata effettuata su più zone dello stesso campione, ed ha confermato l’omogenea distribuzione dei due componenti in miscela. La correlazione dello spettro medio infatti è risultata elevata ed uniforme (> 96%).
Le Figure 4-24 a-c mostrano la mappa spettrale, lo spettro medio e la mappa di correlazione rispetto allo spettro medio per un campione di PCL/GL. Anche in questo caso, la miscela è risultata piuttosto omogenea: la correlazione dello spettro medio è stata superiore al 98 %.
Le Figure 4-25 a-c mostrano la mappa spettrale, lo spettro medio e la mappa di correlazione rispetto allo spettro medio per un campione di PCL/D. La miscela PCL/D ha evidenziato zone a bassa correlazione rispetto allo spettro medio, causate dalla presenza di domini di destrano e attribuibili a fenomeni di segregazione di fase per la miscela PCL/D.
4000.0
3600 3200 2800 2400 2000 1800 1600 1400 1200 1000 800 650.0
%T
Amido (polvere)
Amido (sinterizzato: P=2 W; BS=5 mm/s)
Figura 4-23. (a) Mappa spettrale di una zona con area 1 mm × 1 mm di un campione di PCL/A
sinterizzato. (b) Spettro medio dell’area (a). (c) Mappa di correlazione dell’area (a) rispetto allo (c)
(b) (a)
Figura 4-24. (a) Mappa spettrale di una zona con area 1 mm × 1 mm di un campione di PCL/GL
sinterizzato. (b) Spettro medio dell’area (a). (c) Mappa di correlazione dell’area (a) rispetto allo spettro medio riportato in (b).
(a)
(b)
Figura 4-26. (a) Mappa spettrale di una zona con area 1 mm × 1 mm di un campione di PCL/D
sinterizzato. (b) Spettro medio dell’area (a). (c) Mappa di correlazione dell’area (a) rispetto allo (a)
(b)
4.3.6 Prove di adesione cellulare
La Figura 4-26 mostra le aree percentuali occupate dalle cellule sulle micro-strutture a base di PCL e di miscele, confrontate con la densità cellulare su uno strato di controllo di gelatina. Il rapporto tra la densità cellulare sulle strutture e quella sullo strato di gelatina ad ogni tempo di prova è un indice dell’efficienza di adesione cellulare [Ciardelli et al., 2004]. L’adesione cellulare sugli scaffold ha evidenziato una crescita nel tempo, seguendo l’ordine PCL/A > PCL/GL > PCL/D. L’introduzione dell’amido e del gellano ha determinato un notevole aumento dell’adesione cellulare, rispetto al caso del PCL puro. L’amido si è rivelato il polisaccaride più efficace tra quelli esaminati nell’aumentare la biocompatibilità del PCL in miscela. Le cellule hanno aderito sulle strutture di PCL/A ad una velocità leggermente inferiore rispetto a quella dello strato di controllo di gelatina, ma con un grado finale di adesione cellulare a 24 h, paragonabile a quello della gelatina. La velocità di adesione cellulare sulle strutture di PCL/GL è stata solo leggermente inferiore, rispetto a quella dello strato di controllo. Al contrario, le microstrutture di PCL/D hanno mostrato la più bassa velocità di adesione cellulare tra le miscele esaminate. L’introduzione del destrano, inoltre, ha determinato un aumento lieve della densità cellulare rispetto al PCL puro solo a tempi brevi, mentre dopo 24 h, l’adesione dei fibroblasti sugli
scaffold di PCL/D è risultata molto inferiore a quella del PCL puro. Questo comportamento può essere
motivato dal basso grado di compatibilità tra PCL e D, che ha causato la formazione di domini di D all’interno della matrice di PCL. Tuttavia, la letteratura scientifica riporta studi sulle miscele PLA/destrano, nei quali è stato evidenziato l’effetto benefico del destrano nell’accrescere la biocompatibilità del PLA, nonostante la bassa compatibilità tra i due componenti [Cai et al., 2002, 2003]. Una separazione di fase può essere causa di un debole miglioramento dell’adesione cellulare, mentre non basta a giustificarne una diminuzione. La spiegazione della bassa adesione cellulare della miscela PCL/D può essere ricercata, più verosimilmente, nell’arrangiamento delle macromolecole in miscela, che probabilmente è stato tale da sfavorire l’adesione cellulare. Un risultato simile è stato trovato da Barbucci et al. [Barbucci et al., 2003], nel caso di micropatterns superficiali di acido ialuronico o di solfato di acido ialuronico. Il solfato di acido ialuronico ha mostrato un arrangiamento molecolare “aperto”, che ha reso i suoi siti recettori disponibili per l’adesione cellulare. Al contrario, l’acido ialuronico ha evidenziato una struttura molecolare “chiusa”, che ha impedito l’adesione cellulare.
Le Figure 4-27 a-d mostrano rispettivamente le immagini ottenute tramite analisi al microscopio ottico dei fibroblasti che hanno aderito sulla gelatina, e sulle strutture di PCL/GL, PCL e PCL/D, dopo 24 h di prova. Il grado di adesione cellulare è variato con il tipo di campione, mentre la morfologia dei fibroblasti è risultata invariata.
0 20 40 60 80 100 120 2 4 24 Tempo (h) A re a o cc u p at a d alle c ellu le ( % ) PCL/A PCL/GL PCL/D PCL Gelatina
Figura 4-26. Andamento dell’area occupata dalle cellule, in funzione del tempo di prova, per il PCL e per le miscele. I dati riportati sono i valori medi, mentre le barre sono le deviazioni standard (n=45).
Figure 4-27. Immagini ottenute tramite microscopio ottico delle cellule aderite su a) PCL/A; b) PCL/GL; c) PCL; d) PCL/D dopo 24 h di prova. Le barre corrispondono a 40 µm.
(a) (b)
4.4 Conclusioni
Nel Capitolo 4 sono state studiate miscele “bioartificiali” tra PCL ed un polisaccaride (amido, gellano, destrano), contenenti il 9.1 % in peso di polimero naturale, e sono state utilizzate per la fabbricazione di scaffold sinterizzati tramite la tecnica di Selective Laser Sintering.
Fino ad oggi, le miscele “bioartificiali” sono state prodotte solo da pochi gruppi di ricerca nel mondo, tra cui il Gruppo Biomateriali dell’Università di Pisa: ogni nuovo studio, quindi, rappresenta un contributo importante per la ricerca scientifica. L’applicazione della tecnica di Selective Laser Sintering alle miscele “bioartificiali”, in particolare, rappresenta un aspetto innovativo, in quanto consente di allargare i possibili campi di impiego delle miscele “bioartificiali”, finora limitati quasi esclusivamente alle tecniche convenzionali di estrusione e di liofilizzazione. Vengono ora riportati e commentati i risultati più significativi dello studio effettuato. L’analisi delle proprietà fisico-chimiche delle miscele non ha rivelato la presenza di evidenti interazioni tra i componenti delle miscele “bioartificiali”: l’analisi FTIR non ha mostrato variazioni nella frequenza delle bande caratteristiche del PCL e dei materiali naturali. Solo l’analisi di Hoffman-Weeks, relativa al calcolo della temperatura di fusione all’equilibrio (Tm0) per le miscele tramite prove di cristallizzazione isoterma, ha mostrato
una lieve depressione della Tm0 rispetto al caso del PCL puro, che è la prova dell’esistenza di
interazioni tra i componenti delle miscele. Maggiori interazioni, in particolare, sono state registrate per la miscela PCL/A, che ha mostrato il massimo grado di depressione della temperatura di fusione all’equilibrio, rispetto al PCL puro. L’analisi della cinetica di cristallizzazione isoterma ha reso evidente l’effetto nucleante del polimero naturale sulla cristallizzazione del PCL. La cristallizzazione del PCL all’interno delle miscele è avvenuta tramite un processo di nucleazione eterogenea e ha prodotto un gran numero di piccoli cristalli dalle forme irregolari. La cristallizzazione del PCL puro, invece, ha portato alla formazione di sferuliti di dimensioni pari a 30-100 µm con contorni netti. La miscelazione del PCL con un polimero naturale, che degrada più rapidamente del PCL, ha diminuito il grado di cristallinità e le dimensioni dei cristalli di PCL, entrambi fattori che accelerano i processi degradativi. Anche se non si sono effettuate prove a riguardo, c’è da aspettarsi che i processi di degradazione in vivo e in vitro degli scaffolds bioartificiali siano più veloci di quelli delle strutture a base di PCL puro. L’analisi termogravimetrica è stata utile per capire la resistenza termica dei campioni prima del loro utilizzo nel processo di sinterizzazione: la degradabilità termica delle miscele è risultata leggermente inferiore rispetto al caso del PCL puro e influenzata dal tipo di polimero naturale miscelato.
Strutture a griglia sono state sinterizzate, dopo avere trovate le condizioni ottimali per il processo. Le strutture sinterizzate hanno mostrato proprietà termiche simili a quelle delle polveri di partenza: le uniche variazioni riscontrate sono state attribuite a eventuali diminuzioni del peso molecolare del PCL durante il processo di sinterizzazione. La miscelazione ha consentito di estendere il processo di sinterizzazione a sistemi contenenti i polimeri naturali amido, destrano e gellano, che, sinterizzati singolarmente, andrebbero soggetti a fenomeni di degradazione termica superficiale. L’analisi FTIR dei campioni sinterizzati non ha mostrato differenze rispetto ai campioni di partenza. L’analisi tramite mappatura chimica (IR-Chemical Imaging) dei campioni sinterizzati ha evidenziato una buona distribuzione dei due componenti per le miscele PCL/A e PCL/GL, mentre la miscela PCL/D ha mostrato la presenza di zone di segregazione di fase. Le superfici degli scaffold sono risultate idrofile, e in particolare dotate di una bagnabilità intermedia, capace di favorire l’adesione cellulare. Le strutture sinterizzate al laser hanno mostrato caratteristiche morfologiche dipendenti dalla morfologia delle particelle di partenza: polveri di forma sferica, con dimensioni sufficientemente uniformi hanno permesso di ottenere strutture omogenee, mentre particelle con forma irregolare e dimensioni varie non hanno consentito di ottenere strutture regolari. La miscelazione del PCL con amido e gellano ha permesso di ottenere materiali adatti a supportare l’adesione dei fibroblasti, mentre la miscelazione del destano con il PCL non ha aumentato la sua biocompatibilità. Le ragioni dei risultati di adesione cellulare poco soddisfacenti per la miscela PCL/D potrebbero essere ricercate in una disposizione delle macromolecole di destrano sulla superficie dei campioni sinterizzati poco adatta a favorire le interazioni con i recettori cellulari. L’adesione cellulare è un processo che dipende dal tipo di cellula seminata, dalla composizione chimica del materiale, e dalla morfologia dello scaffold (caratteristiche superficiali, topografia). Dopo avere individuato il materiale più adatto per un certo tipo di cellule, l’applicazione di una tecnica innovativa come il Selective Laser Sintering potrà permettere, in futuro, di ottenere l’architettura ottimale per la ricostruzione di un particolare tessuto.
4.5 Riferimenti bibliografici
1) Avrami, M. J. Chem. Phys. 1939, 7, 1103.
2) Bacakova, L; Filova, E.; Rypacek, F.; Svorcik, V.; Stary, V. Physiol. Res. 2004, 53 (suppl. 1), S35-S45.
3) Barbani, N.; Bertoni, F.; Ciardelli, G.; Cristallini, C.; Cascone, M. G.; Coluccio, M. L.; Giusti, P.
JABBs 2003, 1, 221.
4) Barbucci, R; Pasqui, D; Wirsen, A; Affrossman, S; Curtis, A; Tetto, C. J. Mater. Sci. Mater. Med.
2003, 14; 721-725.
5) Berrada, S.; Amedee, J.; Avramoglou, T.; Jozefonvicz, J.; Harmand F. J. Biomater. Sci. Polymer
Edn. 1994, 6, 211-222.
6) Cai, Q.; Wan, Y.; Bei, J.; Wang, S. Biomaterials 2002, 23, 4483-4492. 7) Cai, Q.; Wan, Y.; Bei, J.; Wang, S. Biomaterials 2003, 24, 3555-3562
8) Ciardelli, G.; Chiono, V.; Cristallini, C.; Barbani, N.; Ahluwalia, A.; Vozzi, G.; Previti, A.; Tantussi, G.; Giusti, P. J. Mater. Sci. Mater. Med. 2004, 15, 303-308.
9) Dubois, P.; Narayan, R. Macromol. Symp. 2003, 198, 233-243.
10) Duquesne, E.; Rutot, D.; Degee, P.; Dubois, P. Macromol. Symp. 2001, 175, 33-43. 11) Ho, H. C. H.; Cheung, W. L.; Gibson, I. Ind. Eng. Chem. Res. 2003, 42, 1850-1862. 12) Hu, H.; Dorset, D. L. Macromolecules 1990, 23, 4604-4607.
13) Leczano, E. G.; Coll, S. C.; Prolongo, M. G. Polymer 1996, 37 (16), 3603-3609. 14) Mandelkern, L. Crystallization of polymers. McGraw Hill, New York 1964.
15) Nunes, M. C.; Batista, P.; Raymundo, A.; Alves, M. M. ; Sousa, I. Colloids Surf. B: Biointerfaces
2003, 31, 21-29.
16) Ohkawa, K.; Kitagawa, T.; Yamamoto, H.; Macromol. Mater. Eng., 2004, 289, 33-40. 17) Pham, D. T.; Gault, R. S. Int. J. Mach. Tools Manuf. 1998, 38, 1257-1287.
18) Pitt, C. G.; Chasalow, F. I.; Hibionada, Y. M.; Klimas, D. M.; Schlinder, A. J. Appl. Polym. Sci.
1981, 26, 3779-3787.
19) Rosa, D. S.; Guedes, C. G. F.; Pedroso, A. G.; Calil, M. R. Mater. Sci. and Eng. C, 2004, 24, 659-662.
20) Rutot, D.; Duquesne, E.; Ydens, I.; Degee, P.; Dubois, P. Polym. Degrad. Stab. 2001, 73, 561-566. 21) Saltzman, W. M. In Principles of tissue engineering. Lanza R. P., Langer R., Chick W. L. Eds. R.
G. Landers Company: Georgetown TX, 1997; pp 225-246. 22) Wu, C. S. Polymer Degrad. Stab. 2003, 80, 127-134.
23) Wunderlich, B. Macromolecular Physics. Crystal melting. Wunderlich B. Ed; Academic Press, Inc: New York, 1980; Vol 3; pp 33-36.


![Figura 4-7. Andamento del log[-ln(1-X(t))] in funzione di log(t) per le polveri di PCL (a) e delle miscele: (b) PCL/D 90.9/9.1; (c) PCL/A 90.9/9.1; (d) PCL/GL 90.9/9.1](https://thumb-eu.123doks.com/thumbv2/123dokorg/7242512.79697/13.892.147.843.137.994/figura-andamento-funzione-polveri-miscele-pcl-pcl-pcl.webp)