Capitolo 7
MATERIALI E METODI
7.1 Sostanze utilizzate
La miscela batterica è stata fornita dal Centro Colture Sperimentali (Aosta, Italia), specializzato nella produzione di microrganismi destinati ad usi agricoli e di bonifiche ambientali. I substrati, i solventi e gli altri reagenti utilizzati sono stati acquistati dalla Sigma-Aldrich (Milano, Italia). Gli anticorpi primari sono stati acquistati dalla Daichi Pure Chemical Co. (Tokyo, Japan) e dalla Santa Cruz Biotechnology (Heidelberg,
Germany) -Italia), i reagenti
specifici per elettroforesi e blotting dalla Biorad Laboratories s.r.l. (Milano, Italia) o dalla Amersham.
7.2 Modello sperimentale
Lo studio è stato condotto su ratti maschi Wistar di tre mesi, stabulati in gabbie a temperatura ambiente con cicli luce/buio di 12 ore, 55% di umidità relativa, temperatura di 20°C ± 2°C, fornendo acqua e mangime ad libitum. La stabulazione e il sacrificio sono stati effettuati secondo le linee guida del Decreto Legislativo 27 gennaio 1992, n. 116
utilizzati a fini sperimentali o ad al (S.O. alla G.U. 18 Febbraio 1992, n. 40).
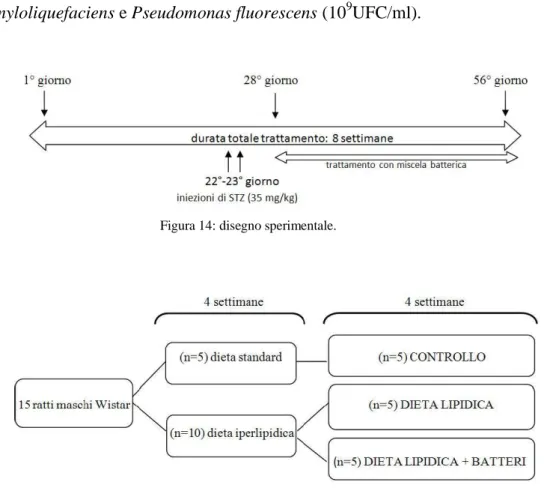
Il trattamento in vivo, articolato in due fasi, è durato 8 settimane (Figura 14) ed è stato effettuato su un totale di 15 ratti.
Nella prima fase (4 settimane) gli animali sono stati suddivisi in due gruppi:
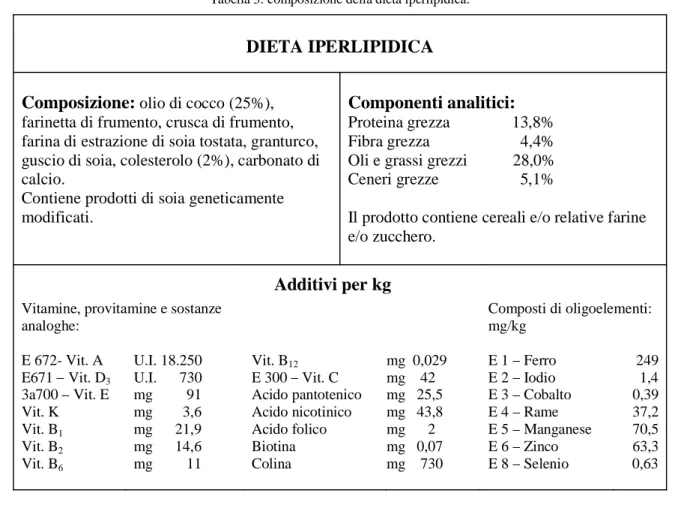
- gruppo di controllo alimentato con dieta standard (n=5), con 11% di energia derivante da grassi (4% della composizione) (Tabella 2);
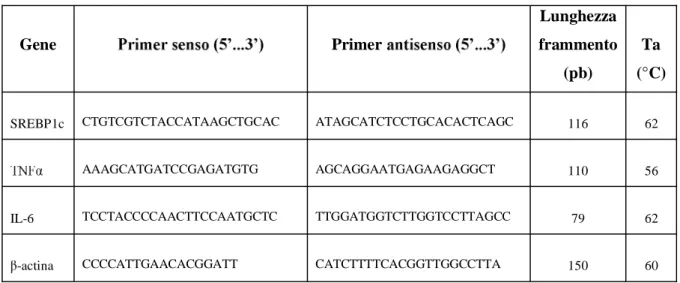
- gruppo alimentato con dieta iperlipidica (n=10), contenente il 55% di energia derivante da grassi (28% della composizione) (Tabella 3), e trattato con streptozotocina (35 mg/Kg) per due giorni consecutivi (22°-23° giorno di
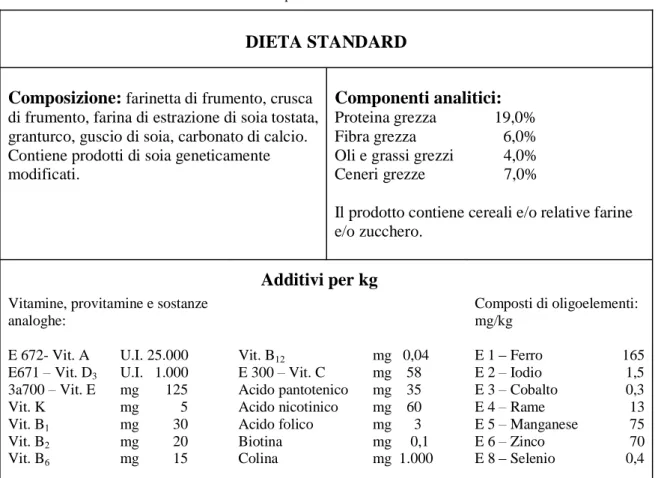
Al termine della prima fase, confermata la presenza di iperglicemia (glucosio >14 mmol/l), i 10 ratti appartenenti al secondo gruppo sono stati ulteriormente suddivisi in due gruppi (II e III) e sottoposti alla seconda fase di trattamento per 4 settimane.
I tre gruppi risultanti (Figura 15), sono stati così trattati:
I. CTR : gruppo di controllo alimentato con dieta standard (n=5), contenente
11% di energia derivante da grassi (Tabella 2);
II. IPD : gruppo alimentato con dieta iperlipidica (n=5), contenente il 55% di
energia derivante da grassi (Tabella 3);
III. IPD+B : gruppo alimentato con dieta iperlipidica (n=5), contenente il 55% di
energia derivante da grassi (Tabella 3) e contemporaneamente sottoposto a trattamento quotidiano per gavaggio con una miscela di Bacillus amyloliquefaciens e Pseudomonas fluorescens (109UFC/ml).
Figura 14: disegno sperimentale.
Figura 15: gruppi di trattamento.
La concentrazione della miscela di batteri somministrata quotidianamente ai ratti appartenenti al gruppo IPD+B è stata verificata mediante conta microbica presso il dipartimento di Scienze Vete
Durante il trattamento sono stati registrati il peso iniziale e finale degli animali, con i quali è stato calcolat
e la quantità di cibo assunto (Tabella 6). La sera antecedente il sacrificio, i ratti sono stati tenuti a digiuno. I ratti del gruppo III
trattamento batterico. Al momento del sacrificio a ciascun animale è stato effettuato, a livello cardiaco, un prelievo di sangue sul quale è stata poi eseguita la determinazione di alcuni parametri ematici. Dopo il sacrificio è stato asportato e pesato il fegato, per poterne calcolare il peso relativo (peso del fegato ale). Sono stati prelevati campioni di tessuto epatico, congelati in azoto liquido e conservati a
Una piccola porzione di tessuto epatico, destinata formalina 10%.
Tabella 2: composizione della dieta standard.
Ii
DIETA STANDARD
Composizione: farinetta di frumento, crusca di frumento, farina di estrazione di soia tostata, granturco, guscio di soia, carbonato di calcio. Contiene prodotti di soia geneticamente modificati.
Componenti analitici:
Proteina grezza 19,0% Fibra grezza 6,0% Oli e grassi grezzi 4,0% Ceneri grezze 7,0%
Il prodotto contiene cereali e/o relative farine e/o zucchero.
Vitamine, provitamine e sostanze analoghe: Additivi per kg Composti di oligoelementi: mg/kg E 672- Vit. A E671 Vit. D3 3a700 Vit. E Vit. K Vit. B1 Vit. B2 Vit. B6 U.I. 25.000 U.I. 1.000 mg 125 mg 5 mg 30 mg 20 mg 15 Vit. B12 E 300 Vit. C Acido pantotenico Acido nicotinico Acido folico Biotina Colina mg 0,04 mg 58 mg 35 mg 60 mg 3 mg 0,1 mg 1.000 E 1 Ferro E 2 Iodio E 3 Cobalto E 4 Rame E 5 Manganese E 6 Zinco E 8 Selenio 165 1,5 0,3 13 75 70 0,4
Tabella 3: composizione della dieta iperlipidica.
Ii
DIETA IPERLIPIDICA
Composizione: olio di cocco (25%), farinetta di frumento, crusca di frumento, farina di estrazione di soia tostata, granturco, guscio di soia, colesterolo (2%), carbonato di calcio.
Contiene prodotti di soia geneticamente modificati.
Componenti analitici:
Proteina grezza 13,8% Fibra grezza
4,4% Oli e grassi grezzi 28,0% Ceneri grezze 5,1%
Il prodotto contiene cereali e/o relative farine e/o zucchero.
Vitamine, provitamine e sostanze analoghe: Additivi per kg Composti di oligoelementi: mg/kg E 672- Vit. A E671 Vit. D3 3a700 Vit. E Vit. K Vit. B1 Vit. B2 Vit. B6 U.I. 18.250 U.I. 730 mg 91 mg 3,6 mg 21,9 mg 14,6 mg 11 Vit. B12 E 300 Vit. C Acido pantotenico Acido nicotinico Acido folico Biotina Colina mg 0,029 mg 42 mg 25,5 mg 43,8 mg 2 mg 0,07 mg 730 E 1 Ferro E 2 Iodio E 3 Cobalto E 4 Rame E 5 Manganese E 6 Zinco E 8 Selenio 249 1,4 0,39 37,2 70,5 63,3 0,63
Tabella 4: peso degli animali e assunzione di dieta.
peso medio iniziale (g) peso medio finale (g) assunzione media di dieta settimanale (g) controllo 202,20 ± 14,04 351,60 ± 47,78 138,43 dieta iperlipidica 192,00 ± 11,07 282,75 ± 35,57 148,59 dieta iperlipidica + batteri 215,80 ± 4,49 324,75 ± 15,41 155,59
7.3 Determinazione dei parametri ematici
ici (ALT, AST, trigliceridi, colesterolo totale, LDL, HDL, glucosio) è stata eseguita usando kit commerciali, presso i Laboratori della Fondazione Toscana Gabriele Monasterio (CNR - Pisa) dalla dott.ssa Maristella Maltinti.
7.4 Preparazione delle frazioni microsomiali e citosoliche
Il tessuto epatico, conservato a -80°C, è stato lavato con KCl 1,15% (p/v) e pesato. É stato successivamente omogenizzato in 4 volumi di tampone di omogenizzazione (KP 100mM pH 7,4 + KCl 1,15% (p/v) + EDTA 1mM) rispetto al peso del tessuto. uti a 10.000 g a 4°C per ottenere la precipitazione di nuclei e mitocondri. Il sovranatante, contenente la frazione microsomiale e quella citosolica, è stato recuperato e ulteriormente centrifugato a 33.000 rpm per 1 ora e 10 min a 4°C. Il sovranatante così ottenuto, contenente il citosol, è stato prelevato e conservato a -20°C. Il pellet, contenente i microsomi è stato lavato con tampone di omogeneizzazione, centrifugando per 1 ora a 33.000 rpm a 4°C, in modo da eliminare le proteine contaminanti non microsomiali. Il precipitato ottenuto è stato risospeso in 2 volumi di tampone di risospensione (KP 100 mM pH 7,4 + EDTA 0,1 mM) rispetto al peso iniziale del tessuto utilizzato, suddiviso in aliquote rapidamente congelate in azoto liquido e conservate a -80°C fino al loro utilizzo.
7.5 Determinazione del contenuto di proteine
Le proteine microsomiali e citosoliche sono state determinate come descritto da Lowry (1951)
del reagente di Folin-Ciocalteu (acido fosfotungstico-fosfomolibdico) che, in ambiente alcalino, ossida le sostanze contenenti Nei liquidi biologici questo reagente si lega prevalentemente alla tirosina, che contiene tale anello, e in misura minore anche a fenilalanina, triptofano e istidina. Quando viene utilizzato anche il reagente rameico, molecola proteica è in grado di reagire con il Folin, aumentando la sensibilità del metodo. Il complesso Cu-proteine che si forma riduce gli acidi fosfotungstico e fosfomolibdico a blu di molibdeno e blu di tungsteno. La
(Lowry O. H. et al., 1951).
7.6 Dosaggio del glutatione (reazione di Ellman)
-ditiobis-2--tio-2-nitrobenzoico (TNB), un composto di colore giallo rilevabile . Anche il glutatione ossidato, che viene ridotto dalla glutatione reduttasi, può contribuire alla formazione di TNB. La formazione di TNB è direttamente proporzionale alla concentrazione di glutatione totale (Ellmann G. L.,1959).
7.7 Perossidazione lipidica
La perossidazione lipidica è un processo che provoca danno cellulare e viene utilizzato come indicatore dello stress ossidativo nelle cellule e nei tessuti. La misurazione dei TBARS (Thiobarbituric Acid Reactive Substances) costituisce il principale saggio per il monitoraggio della perossidazione lipidica, che permette di valutare la concentrazione della malondialdeide (MDA), uno dei maggiori prodotti della perossidazione lipidica. In condizioni acide e ad elevate temperature (~100°C) la MDA prodotta reagisce con o 1:2 formando un addotto rilevabile (Mihara M. and Uchiyama M.,1978).
I risultati dei TBARS sono stati espressi in nmoli di MDA utilizzando la curva di taratura ottenuta mediante idrolisi d
soluzione 1% di acido solforico a 37°C per 15-20 minuti. Da tale soluzione madre sono state prelevate diverse quantità di MDA (0,25 nmoli, 2,5 nmoli, 5 nmoli e 10 nmoli) e fegato di ratto non trattato. Si è quindi proceduto alla reazione con TBA secondo il protocollo precedentemente descritto. Il bianco, ottenuto utilizzando il solo omogenato, è stato sottratto ai suddetti campioni.
7.8 Estrazione e quantificazione dei lipidi epatici
I lipidi sono stati estratti partendo direttamente dal tessuto epatico, secondo il metodo descritto da Folch et al. (1957), adattato al nostro sistema sperimentale. Campioni di 0,5 g di fegato sono stati omogeneizzati con 2 ml di acqua bidistillata e 2 ml di metanolo. Sul risultante omogenato sono state effettuate 3 successive fasi di estrazione con 2 ml di cloroformio; le frazioni di cloroformio riunite sono state lavate prima con 2 ml di KCl 1M e poi con 2 ml di acqua bidistillata. Dopo evaporazione sotto cappa e prolungato
essiccamento della soluzione di cloroformio sotto vuoto (fino a peso costante), è stato pesato e quantificato il contenuto in lipidi.
7.9 Dosaggio del citocromo P450
Il dosaggio del citocromo P450 si effettua su frazione microsomiale epatica mediante una misurazione spettrofotometrica che consiste nel registrare lo spettro tra 400 e 500 nm. A seguito di saturazione con monossido di carbonio, il CYP, nella sua forma ridotta con ditionito sodico, presenta un picco massimo di assorbimento a 450 nm che è proporzionale al suo contenuto nei microsomi (Omura T. and Sato R., 1964).
7.10 Attività enzimatiche
Etossicumatina-O-deetilasi (ECOD)
Questa attività, dipendente da molte isoforme di citocromo P450, è stata determinata su frazioni microsomiali epatiche. Permette la misurazione fluorimetrica della 7-idrossicumarina prodotta dalla deetilazione d i vari enzimi P450 (Ex: 390 nm, Em: 440 nm) (Aitio A., 1978). Questo protocollo consente di determinare valori di attività catalitiche maggiori di 0,2
Etossiresorufina-O-deetilasi (EROD)
Questa attività è stata eseguita su frazioni microsomiali epatiche e si basa sulla determinazione fluorimetrica della resorufina, che si forma in seguito alla deetilazione
R. A. et al., 1985).
Questo protocollo permette di rilevare valori di attività catalitiche maggiori di 0,2 pmol/min/mg prot.
Anilina idrossilasi (AnH)
- catalizzata essenzialmente dall
2E1, porta alla produzione del 4-ammino-fenolo che, trattato con fenolo in soluzione basica, produce il fenato corrispondente. La quantità di fenato presente in soluzione è
molecola ha il massimo assorbimento (Ko I. Y. et al., 1987). Questo protocollo permette di determinare valori di attività catalitiche maggiori di 30
p-Nitrofenolo idrossilasi (pNFH)
Il p-nitrofenolo viene idrossilato dal CYP2E1 in una reazione che produce un composto di colore giallo, il 4-nitrocatecolo, che presenta un picco di assorbimento massimo a 546 nm (Reinke L. A. and Moyer M. J., 1985). La misurazione del prodotto di reazione è
stata effettuata median .
Questo protocollo permette di determinare valori di attività maggiori di 0,1 pmol/(mg
Testosterone idrossilasi
Il testosterone viene ossidato in maniera regio- e stereo-selettiva da diverse isoforme di P450, quindi questo saggio viene utilizzato per determinare contemporaneamente
HPLC con colonna Supelcosil LC-18 (Amato G. et al., 1996) dei metaboliti che si
Per la determinazione sono stati utilizzati due tamponi:
2O (92,5%) + tetraidrofurano (7,5%);
L'analisi cromatografica è stata condotta ad una lunghezza d'onda di 254 nm.
utilizzato il corticosterone. Questo protocollo permette di determinare valori di attività catalitiche maggiori di
Benzilossichinolina debenzilasi (BQD)
La reazione di debenzilazione della benzilossichinolina è mediata dai CYP3A e porta alla produzione di 7-idrossichinolina, molecola che può essere quantificata fluorimetricamente (Ex: 410 nm, Em: 538 nm) (Stresser D. M. et al., 2000). Questo protocollo permette di determinare
Eme ossigenasi (HO-1)
-1 consiste nella rilevazione spettrofotometrica della bilirubina, prodotta a seguito della riduzione della biliverdina. La reazione richiede il substrato emina, una fonte di biliverdina reduttasi (3 mg di frazione citoplasmatica) e 2 mg di campione microsomiale contenente HO-1. La quantità di bilirubina formata è misurata
530 nm (Naughton P.
DT-diaforasi (NAD(P)H chinone ossidoreduttasi NQO1)
Questa attività si determina nella frazione citosolica epatica seguendo la riduzione del DCPIP (2,6-diclorofenolindofenolo) da parte del NADPH, che porta ad una
spettrofotometricamente a 630 nm.
Il coefficiente di estinzione molare è pari a 21,2 mM-1 cm-1 (Benson A. M. et al., 1980).
Catalasi
determinata spettrofotometricamente a 240 nm in funzione del tempo. Il coefficiente di estinzione molare è di 43,6 M-1 cm-1 (Aebi H., 1984).
Superossido dismutasi
La
(Marklund S. e Marklund G., 1974).
curva di taratura che è stata usata come riferimento per calcolare i ng di enzima.
Glutatione reduttasi
Questa attività richiede la presenza di GSSG e NADPH. La determinazione viene fatta allo spettrofotometro a 340 nm valutando il consumo di NADPH, la molecola che dona
. Il coefficiente di estinzione molare è pari a 6,22 mM-1 cm-1 (Wheeler C. R. et al., 1990).
Glutatione perossidasi
richiede la presenza di GSH, GSSG reduttasi, NADPH e terzialbutilidroperossido, che rappresenta il substrato di reazione. La misurazione viene fatta in
valutando il consumo del NADPH da parte della GSSG reduttasi, che riduce il GSSG consumato dalla GPx per ridurre il substrato. Il coefficiente di estinzione molare è pari a 6,22 mM-1 cm-1 (Badary O. A. et al., 2005).
7.11
e conservati a
-ata lavorando a temperatura ambiente.
campioni di fegato (circa 20 mg) sono stati omogeneizzati utilizzando il Tissue Lyser
- proseguita seguendo il protocollo
conservata a
-I campioni di RNA totale sono stati quantificati utilizzando il Nano-Drop , un nano spettrofotometro, che consente sia di determinare la concentrazione di RNA estratto che di valutare il grado di purezza del campione.
nda di 260 nm, mentre la purezza viene valutata sul rapporto di assorbimento tra 260/280 nm e 260/230 nm. Rapporti maggiori di 1,7 indicano un buon contenuto in acidi nucleici (principalmente RNA) rispetto al contenuto di proteine. Per poter procedere alla
I campioni sono stati preparati aggiungendo: -
-
-etidio)
- N
orizzontale, ad un voltaggio costante di 80 V per 30 minuti, utilizzando RNA buffer 1X come tampone di corsa. Alla fine della corsa le bande sono state visualizzate ponendo il gel su un transilluminatore UV.
RNA Il
reazione di retrotrascrizione sono stati effettuati in un unico passaggio, utilizzando il kit (Bio-Rad). In eppendorf è stata preparata una miscela di reazione per campione aggiungendo una soluzione di RNA del
, 16
iScript reverse transcriptase . Le eppendorf sono state incubate con i seguenti tempi e temperature: 5 minuti a 25°C, 30 minuti a 42°C, 5 minuti a 85°C, con holding a 4°C, e conservate in congelatore a -20°C.
Controlli utilizzati per la RT-PCR
Al fine di verificare l'efficacia del trattamento con la DNasi e poter escludere, quindi, la contaminazione da DNA genomico, che avrebbe potuto interferire con il cDNA nelle -e n-el r-ealizzar-e una mix in cui sono presenti tutti i reagenti tranne l'enzima (reverse-transcriptase). Le
- retrotrascrizione e corsa elettroforetica su
gel di agarosio. Per verificare l'avvenuta retrotrascrizione dell'RNA è stata effettuata su tutti i campioni una PCR con primers specifici per il gene housekeeping -actina.
Inoltre, per evidenziare eventuali contaminazioni da DNA nei reagenti necessari per l'amplificazione genica è stato fatto un controllo negativo per ogni reazione (bianco), in cui erano presenti tutti i componenti tranne il cDNA.
7.12 PCR real-time
La PCR real-time è una tecnica di amplificazione del DNA che, seguendo ad ogni ciclo real-time
numero di copie del frammento di DNA di interesse, permette di quantificarne accuratamente il contenuto iniziale. In questo studio sono stati analizzati attraverso
-6 e SREBP1c. La miscela di reazione è stata allestita aggiungendo:
- ,
- -Rad),
- -
- 2
-Rad) e la reazione è stata fatta
avvenir
Primer utilizzati
I primer utilizzati sono riportati in tabella 5, con le relative sequenze e temperature di annealing. I primer sono stati disegnati seguendo questa procedura:
- la sequenza nucleotidica dei trascritti dei geni d'interesse è stata ricercata in banca dati (GeneBank) e quindi scaricata;
- le varie sequenze sono state allineate e analizzate mediante il software CLUSTALW 1.61;
- sulla base delle sequenze specifiche sono stati scelti dei primer della lunghezza di circa 20 nucleotidi utilizzando il software OLIGO (disponibile in rete) ed utilizzando
evitare che i primer abbiano sequenze tali da potersi appaiare tra loro o formare strutture stabili (loops) al 3', riducendo così l'efficienza di amplificazione.
La temperatura di annealing (Ta) è specifica per ogni coppia di primer ed è stata calcolata con la formula: (2 °C per ogni A/T + 4 °C per ogni G/C) 5 °C. Inoltre, dopo aver stimato la Ta per ogni primer con la formula appena descritta, ogni coppia di primer è stata testata a differenti temperature per rilevare la migliore temperatura di annealing.
Tabella 5: primer utilizzati e loro caratteristiche.
Gene Primer Lunghezza frammento (pb) Ta (°C) SREBP1c CTGTCGTCTACCATAAGCTGCAC ATAGCATCTCCTGCACACTCAGC 116 62 AAAGCATGATCCGAGATGTG AGCAGGAATGAGAAGAGGCT 110 56
IL-6 TCCTACCCCAACTTCCAATGCTC TTGGATGGTCTTGGTCCTTAGCC 79 62 -actina CCCCATTGAACACGGATT CATCTTTTCACGGTTGGCCTTA 150 60
7.13 Elettroforesi (SDS-PAGE) e Western blotting
Elettroforesi
L'SDS-PAGE (Sodium Dodecyl Sulphate - PolyAcrylamide Gel Electrophoresis, elettroforesi su gel di poliacrilammide in presenza di sodio dodecil solfato) è una tecnica elettroforetica che permette l'analisi di estratti proteici. Durante l elettroforesi le molecole contenute nel campione vengono separate in base alle loro dimensioni ed è così possibile mettere in evidenza la presenza di una particolare molecola.
ttroforesi verticale su minigel di acrilammide al 10%. Dapprima sono stati preparati i campioni
Il sample buffer è un tampone di caricamento che contiene SDS (detergente anionico che si lega fortemente alle proteine e le denatura), blu di bromofenolo (colorante
tracciante ionizzabile che permette di se
-mercaptoetanolo (riduce i ponti disolfuro delle proteine e quindi ne destabilizza la struttura terziaria), glicerolo e saccarosio (servono a rendere più densa la soluzione del campione). La carica netta della proteina denaturata viene completamente mascherata dalle molecole di SDS, che le rende cariche negativamente.
I campioni da separare non vengono direttamente caricati sul gel di separazione, ma su un gel di caricamento che consente di impaccare le proteine contenute nel campione in una sottile banda, prima che queste entrino nel gel di separazione. Infatti, quando viene applicata la corrente, tutte le specie ioniche presenti devono migrare alla medesima
mentre quelle più grandi vengono rita bromofenolo raggiunge il fondo del gel.
) per circa 40 minuti (Laemli U. K., 1970; Sambrook J. and Russell D. W., 1989).
Western blotting
Questa tecnica consente il trasferimento del campione proteico dal gel ad appositi filtri di nitrocellulosa, sui quali successivamente potrà essere rilevata la proteina di interesse utilizzando un anticorpo specifico. Inizialmente le proteine, separate in base al loro peso
per il trasferimento, utilizzando corrente costante a 250 mA per 130 minuti circa (Towbin H. et al., 1979). In seguito è stato effettuato il bloccaggio dei siti aspecifici della membrana con una soluzione di latte al 5% in TTBS (NaCl 0,2 M, Tris 50 mM, pH 7,4, Tween 20%, H2O) per 2 ore. Al termine di questa operazione sono stati aggiunti
gli anticorpi primari e poi gli anticorpi secondari, che si sono legati al complesso antigene-anticorpo.
Per il rilevamento è stato utilizzato un metodo di sviluppo con chemioluminescenza (kit Super Signal West Pico Chemiluminent Substrate - Pierce).
Dosaggio dei P450 3A2, 2C11 e 2E1 mediante Western blotting
policlonali ratto (goat rat) sono stati diluiti in TTBS 1:3000 (anticorpo anti-2C11) o 1:500 (anticorpo anti-3A2 e anti-2E1) e lasciati in incubazione sulla membrana per tutta la notte in agitazione a 4°C. Gli anticorpi secondari anti-capra coniugati con la perossidasi sono stati diluiti 1:3000 o 1:2000 in TTBS e incubati per 60 minuti a temperatura ambiente.
7.14 Analisi istologica
Al momento del sacrificio degli animali, dal fegato di ciascun ratto è stata asportata una piccola porzione che è stata subito fissata in formalina 10%.
dei batteri che promuovono la degradazione
poiché preserva la struttura microscopica e submicroscopica del tessuto durante A questo punto ciascun pezzo di tessuto è stato diviso in due porzioni: una inclusa in paraffina, destinata alla colorazione con ematossilina-eosina, e una congelata a -20°C, destinata alla colorazione con oil red.
Inclusione dei campioni in paraffina
diafanizzazione dei tessuti, seguendo il protocollo di Mark et al., 2007. Nella fase di
diafanizzazione, att
i tessuti in xilene (3 passaggi). A questo punto, al fine di ottenere sezioni sottili da osservare al microscopio ottico, i campioni sono stati inclusi in paraffina fusa a 58°C (3 passaggi, ciascuno di 90 minuti) fino ad ottenere la sua completa penetrazione nel
metallici e ricoperti con altra paraffina liquida, lasciata raffreddare fino a completa solidificazione, per ottenere un blocco sezionabile al microtomo. Da ciascun campione è stato ricavato un piccolo cubo di paraffina che è stato montato sul microtomo e tagliato cuno contenente 10 sezioni, e lasciati asciugare su una piastra riscaldata per 12 ore. Al momento della colorazione, i vetrini sono stati sparaffinati immergendoli in xilolo per 15 minuti (2 passaggi), in etanolo assoluto per 5 minuti (2 passaggi), in etanolo 95% per 5 minuti, in etanolo 70% per 5 minuti. Le sezioni sono state fatte asciugare e si è provveduto alla colorazione.
Ematossilina-eosina
La colorazione ematossilina-eosina è importante per ottenere informazioni generali sulla -violaceo basico e come tale ha affinità per le sostanze acide. Va a legarsi alle componenti cellulari cariche negativamente, come acidi nucleidi, proteine di membrana, la mucina e le sostanze fondamentali dei connettivi ad alto tenore di mucopolisaccaridi, come la matrice
acido che colora in rosa la maggior parte delle struttura citoplasmatiche e, in generale,
I vetrini sono stati immersi nella soluzione di ematossilina per 5 minuti, sono stati lavati con acqua di fonte per 5 minuti e poi in acqua distillata per 1 minuto. Successivamente sono stati posti per 1 minuto nella soluzione di eosina e poi lavati in acqua distillata per pochi secondi. Le sezioni sono state disidratate nella serie ascendente degli alcoli immergendoli per 2-3 secondi in etanolo 70%, etanolo 90%, etanolo assoluto (2 passaggi) e infine in xilolo per 5 minuti.
I vetrini ancora bagnati sono stati chiusi con vetrini coprioggetto usando un opportuno collante (balsamo di Canada) e sono stati osservati al microscopio ottico.
Oil red
Questa colorazione, specifica per i lipidi, è stata eseguita a partire da campioni di tessuto epatico congelati. Il colorante utilizzato è la Oil Red O Solution 0,5% in
propilen glicole (Sigma), un colorante liposolubile che permette di evidenziare la dei tessuti.
Le sezioni istologiche criostato dopo aver fissato i
frammenti di tessuto su appositi supporti con PELCO
CryO-Z-INC.), una soluzione che consente l del campione e favorisce il taglio. Per ogni campione sono stati realizzati 2 vetrini, ciascuno contenente 10 sezioni dello Prima di procedere alla colorazione, i vetrini sono stati lasciati asciugare per 60 minuti a temperatura ambiente. Successivamente sono stati immersi in propilen glicole assoluto per 3 minuti a temperatura ambiente, incubati con la soluzione di Oil Red a 60°C in stufa per 10 minuti, immersi in propilen glicole 85% per 3 minuti a temperatura ambiente e infine lavati rapidamente in acqua bidistillata (2 passaggi). Sono stati inseriti in una soluzione di ematossilina per 30 secondi, lavati con acqua corrente per 2 minuti e subito chiusi con vetrini coprioggetto utilizzando glicerina.
è stata fatta al microscopio ottico.
7.15 Analisi statistica
si statistica è stata fatta mediante ANOVA ad una via con il test di Bonferroni, utilizzando il software GraphPad PRISM 4 (Camussi A. et al., 1990 Zanichelli). Gli intervalli di significatività sono i seguenti:
significativamente differente rispetto al gruppo di controllo *p<0,05 significativamente differente rispetto al gruppo di controllo **p<0,01 significativamente differente rispetto al gruppo di controllo ***p<0,001 significativamente differente rispetto al gruppo dieta iperlipidica °p<0,05 significativamente differente rispetto al gruppo dieta iperlipidica °°p<0,01 significativamente differente rispetto al gruppo dieta iperlipidica °°°p<0,001.