1. INTRODUZIONE
1.1 Il problema del biofouling
Il termine inglese fouling, derivante dal verbo to foul, che significa letteralmente insudiciare, incrostare, indica l’insieme delle incrostazioni rilevabili su una superficie in contatto con un ambiente esterno. Più specificatamente si parla di biofouling, quando l’intero processo sia determinato dalla colonizzazione dinamica delle superfici sommerse, come ad esempio delle carene di navi, ad opera di numerosi organismi viventi uni e pluricellulari (quali batteri, molluschi o alghe) (Fig. 1.1).[1]
Figura 1.1– Esempio di fouling ad una chiglia di una nave
Il grado di biofouling sulla superficie di corpi sommersi dipende da diversi fattori, tra loro interagenti, come la natura chimica del materiale di rivestimento, la tensione superficiale dello stesso, l’abbondanza e la varietà degli organismi presenti nell’ambiente.[1]
Questa indesiderata colonizzazione ha un grave impatto, in particolare per l’industria navale, in quanto comporta il deterioramento delle superfici di scafi, pale ed eliche, arrecando problemi alle imbarcazioni, quali difficoltà nella manovrabilità, rallentamento e incremento dei consumi di carburante, con il conseguente aumento delle emissioni di gas serra.[2]
Ne conseguono, inoltre, le complicazioni dovute all’introduzione di specie marine in ambienti non autoctoni; il trasporto di questi organismi da una zona all’altra degli oceani porta all’inserimento di nuove specie in ecosistemi locali con danni gravissimi sul piano delle biodiversità.[3,4]
L’Organizzazione Marittima Internazionale ha stimato un aumento del 34% (dal 38 al 72%), dal 2000 al 2020, delle emissioni dovute ad un maggior consumo di carburante per condurre la stessa velocità di crociera, qualora non si compiano azioni correttive e vengano introdotte nuove tecnologie. [5]
E’ stato stimato che, complessivamente, i costi globali dei problemi connessi al biofouling sono dell’ordine dei 6,5 miliardi di dollari annui.[6]
Risulta dunque necessario lo sviluppo di nuove tecnologie e nuovi materiali per riuscire a contrastare o comunque limitare il problema del biofouling, particolarmente in ambiente marino.
2
1.2 Il processo biovegetativo
L’ambiente marino è caratterizzato da una vasta gamma di organismi, tra i quali sono presenti quelli responsabili del biofouling, diversificati sia secondo la zona oceanografica che per specie e dimensione. Essi vengono distinti in microfouling e macrofouling.
Il microfouling è formato da colonie cellulari di batteri, diatomee e protozoi; il macrofouling è costituito da molluschi e da organismi con esoscheletro (detto hard-fouling), ma anche da alghe pluricellulari e spugne (detto soft-fouling).[7]
L’attacco vegetativo è un fenomeno complesso ed è il risultato di una serie di processi la cui diversità ed estensione sono influenzate da una serie di fattori fisici, chimici e biologici nell’immediata vicinanza del substrato.
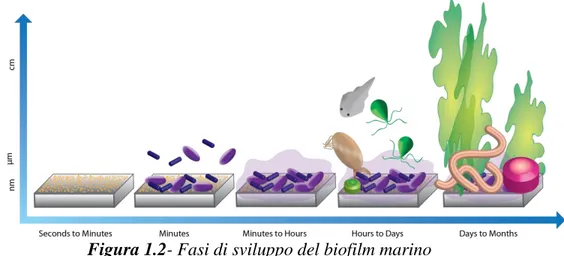
A prescindere dalle differenze legate all’ambiente ed al substrato, è possibile descrivere schematicamente, mediante 5 fasi, non necessariamente consecutive, gli eventi che si susseguono dinamicamente dopo l’immersione di un corpo solido in mare (Fig. 1.2).[1]
1. Entro pochi minuti dall’immersione del corpo in mare, inizia la deposizione di un film “condizionante” costituito prevalentemente da polimeri organici (quali polisaccaridi e proteine) e composti inorganici. Questo materiale organico si trova disciolto nell’acqua e deriva dalla decomposizione di organismi vegetali ed animali, ovvero dalla presenza degli “acidi umici” del mare. La formazione di questa pellicola di “condizionamento” provoca una modifica delle proprietà del substrato, quali la carica superficiale, le caratteristiche chimiche e le componenti dell’energia superficiale. Questo film ha un ruolo fondamentale in quanto nessun organismo né il particellato inorganico possono depositarsi prima che tale rivestimento sia completato. [8]
2. In un lasso di tempo che va da un minimo di 24 ore ad un massimo di alcuni giorni, questo substrato permette il verificarsi del processo di adesione batterica, avviando così la formazione del biofilm primario. Questo è costituito da organismi unicellulari, tra cui prevalgono batteri corti di forma bastoncellare, cui si aggiungono organismi foto sintetici, il cui numero diviene progressivamente dominante.
3. Si ha dunque la colonizzazione secondaria, ovvero la crescita batterica e la produzione dell’esopolimero. I microrganismi, presenti nell’acqua ed in grado di raggiungere l’interfaccia solida, utilizzano nutrienti reperibili per la loro crescita e moltiplicazione e si legano alla superficie, grazie alle proteine e i polisaccaridi da essi prodotti. Questa crescita batterica è accompagnata dalla formazione di sostanze polimeriche extracellulari (EPS) che portano alla genesi del biofilm microbico/batterico (ossia un biofilm in cui i batteri sono inglobati, detto anche
slime). Questo processo prosegue in maniera esponenziale fino al raggiungimento di
INTRODUZIONE
3
4. La crescita del biofilm microbico, anche grazie all’aumento dimensionale degli organismi filamentosi, si realizza non solo come aumento di spessore, ma è generalmente caratterizzata dalla formazione di vere e proprie appendici filamentose che agevolano la cattura e l’adesione di spore, microalghe, funghi e protozoi, oltre che di particelle inorganiche. [8] E’ a questo stadio, che solitamente si ottiene entro la fine della prima settimana dall’immersione, che avviene l’inizio della colonizzazione da parte di organismi pluricellulari.[9]
5. Entro due o tre settimane, si ha la crescita del macrofouling, tra cui tipicamente balani, mitili e macroalghe. Questo è il fenomeno che crea i maggiori problemi associati al biofouling marino.
Figura 1.2- Fasi di sviluppo del biofilm marino 1.3
Come affrontare il problema del biofouling
Il biofouling marino è considerato un notevole problema per la navigazione, per cui si è sempre cercata una possibile soluzione per preservare le navi dalle incrostazioni.
Esistono molte strategie per il controllo del biofouling, anche se il metodo più pratico ed immediato rimane quello di impedire l’insediamento degli organismi marini. In realtà questo risulta impossibile, per cui oggi giorno sono studiati dei rivestimenti antivegetativi non tossici che svolgano un’azione antifouling, e che al tempo stesso evitino di uccidere le specie adese senza rilasciare nell’ambiente sostanze inquinanti. L’interesse per questa tipologia di rivestimenti si è acceso di recente a seguito di legislazioni che hanno bandito dal commercio vernici a base di composti del tributilstagno (TBT), imponendo una valutazione più rigorosa dei biocidi attivi. Infatti il Comitato MECP (Marine Environment Protection Commitee) e l’organizzazione IMO (International Marine Organization) hanno approvato il piano che prevede l’eliminazione graduale delle vernici a base di metalli pesanti (tra cui Sn, Pb, As) entro Gennaio 2003 ed in maniera completa entro Gennaio 2008.[10,11]
I rivestimenti a rilascio del fouling (fouling-release) sono le alternative ad oggi più diffuse a livello commerciale: il loro sviluppo si basa su considerazioni sulle proprietà superficiali in generale e le loro relazioni con l’attacco dei microrganismi.[12]
L’obiettivo di tali vernici non è quello di prevenire il fouling, quanto quello di minimizzare le interazioni tra le molecole
4
biologiche extracellulari e la superficie solida, in modo tale che la cellula aderita venga rilasciata sotto l’azione di deboli forze di taglio, come quelle esercitate dal movimento della nave.[13]
1.4
Rivestimenti antivegetativi
Con il termine antivegetativo si intende quella classe di vernici aventi come obiettivo la prevenzione, riduzione e l’eliminazione della formazione di organismi marini sulle strutture immerse in acqua.
Durante la fine del 1970, gli sforzi di ricerca e sviluppo si sono concentrati principalmente sul successo del sistema copolimero auto-pulente a base di TBT. Tuttavia, a causa dell’impatto ambientale connesso con tali composti, nei primi anni ’80 sono stati sviluppati rivestimenti privi di stagno.[14] L’attuale strategia anti-fouling può essere divisa in due categorie:
i. Rivestimenti attivi chimicamente, che agiscono inibendo o limitando gli insediamenti degli organismi mediante composti attivi chimicamente;
ii. Rivestimenti non tossici, che tendono ad inibire l’attacco degli organismi o ne migliorano il rilascio senza coinvolgere reazioni chimiche.
Nel periodo di transizione fra i rivestimenti a base di stagno e i rivestimenti antifouling non tossici, sono stati rivendicati rivestimenti autopulenti chimicamente attivi privi di stagno come i rivestimenti più efficienti in circolazione. Il primo rivestimento fouling release non tossico è stato brevettato nel 1961; questo impiegava resine siliconiche reticolate come rivestimenti marini, ed è stato effettuato un intenso lavoro dal 1990 relativo allo sviluppo di rivestimenti sia siliconici sia fluorurati.[2,15]
1.4.1
Rivestimenti antivegetativi a base di biocidi
Le tecnologie antifouling chimicamente attive, che si basano su vernici a rilascio di composti attivi (detti biocidi), possono essere suddivise in tre categorie: a matrice solubile, a matrice insolubile e autopulenti.
i. Le vernici a matrice solubile, dette anche vernici erodibili, contengono biocidi miscelati con grandi porzioni di leganti a base di colofonia e di suoi derivati. Incorporano pigmenti tossici come ossidi di rame, ferro e zinco. A contatto con l’acqua di mare, i biocidi e i leganti solubili sono simultaneamente disciolti e rilasciati. Le vernici tradizionali a matrice solubile mantengono la loro azione antifouling per non più di 12-15 mesi.[16]
ii. Le vernici a matrice insolubile impiegano leganti ad alto peso molecolare che risultano essere insolubili in acqua di mare, come polimeri acrilici, vinilici, epossidici o gomme clorurate; piccole quantità di colofonia possono essere aggiunte.[13] Queste vernici sono anche dette hard-antifouling grazie alla loro buona resistenza meccanica e alla capacità di riuscire ad incorporare una grande quantità di agente tossico. Queste molecole, o particelle, attive, possono essere rilasciate gradualmente. Il legante non è solubile in acqua di mare, a differenza
INTRODUZIONE
5
dell’agente tossico che contiene, che viene rilasciato; l’acqua di mare si diffonde così attraverso i pori lasciati vuoti da quest’ultimo e va a sciogliere le prossime particelle di sostanza tossica. Tuttavia, dato che le particelle di sostanze tossiche esposte sono sempre più profonde nel film di vernice, la struttura è resa più rugosa e soggetta ad inglobare inquinanti marini; il rilascio dell’agente tossico va quindi diminuendo con il tempo, e la protezione tende a diventare inefficiente.[2] Per questa ragione l’efficienza di questa classe di vernici antifouling è stimata dai 12 ai 24 mesi, in base anche alla severità delle condizioni di esposizione della superficie.[16]
iii. Le vernici autopulenti sono a base di copolimeri acrilici o metacrilici, i quali sono facilmente idrolizzabili in acqua. Questi copolimeri miscelati con biocidi permettono di mantenere una superficie liscia e sono in grado di controllare il tasso di rilascio di biocida, mediante in controllo del tasso di erosione del legante.[17]
Gli organismi responsabili del fouling, che si attaccano sulla superficie del film di vernice, vengono eliminati simultaneamente alla matrice, che tende a decomporsi mediante idrolisi dei gruppi laterali della catena del copolimero.
I copolimeri (met)acrilici sono stati progettati per imitare il ben noto copolimero (met)acrilico organostannico impiegato nelle vernici a base di tributilstagno (TBT). I leganti privi di stagno impiegano gruppi esterei a base di rame, silicio, o zinco al posto del TBT.
Tali pitture subiscono il seguente meccanismo quando vengono immerse in acqua di mare:
L’acqua diffonde all’interno del rivestimento portando alla dissoluzione delle particelle del biocida. Dato che la matrice del copolimero è idrofoba, l’acqua è ostacolata nel penetrare all’interno del film. Pertanto, il mare riesce a malapena a riempire i pori creati dalle particelle di biocida solubile.[2]
Dato che i legami esterei sono idroliticamente instabili in condizioni leggermente basiche, come quelle che si ritrovano nell’ambiente marino, ha luogo una lenta e controllata idrolisi del rivestimento, reazione confinata a pochi micrometri dalla superficie.[2, 16]
Con il tempo, l’acqua marina dissolve più particelle di biocida, causando la crescita dell’area lisciviata e rendendo il film del copolimero più fragile e facilmente erodibile dall'acqua di mare, lasciando così una nuova area fresca del rivestimento scoperto per il successivo rilascio di molecole attive (effetto autopulente).[16]
Il tasso di dissoluzione del pigmento e quello di dissoluzione di ioni attraverso lo strato lisciviato diventa uguale al tasso di erosione del legante, raggiungendosi un valore così costante dello spessore dello strato
6
lisciviato. Questo spessore risulta notevolmente stabile e basso (10-20 µm) per tutta il tempo di vita del rivestimento.[13]
La dissoluzione è regolata a livello molecolare; infatti, il grado di rilascio di biocida è controllato dal grado di polimerizzazione e dal carattere idrofilo del copolimero legante, che dipende dalla porzione di gruppi idrolizzabili sulla catena del copolimero.[17] Tipicamente queste vernici autopulenti sono formulate per ottenere una velocità di erosione compresa fra 5-20 µm l’anno, che permette di estendere i tempi di ripristino fino a 5 anni.[16]
A seguito del divieto di prodotti a base di TBT in vernici antifouling, sono state sviluppate delle alternative contenenti elevate quantità di composti a base di rame, in quanto è circa dieci volte meno tossico del TBT. Co-biocidi sono stati utilizzati per migliorare le prestazioni antifouling dei rivestimenti a base di rame.[18]
La proprietà fondamentale di un buon biocida antifouling, in rispetto dell'ambiente, è che sia efficace nel prevenire sporcamento della superficie senza persistere nell’ambiente in concentrazioni maggiori di quelli che possono causare effetti dannosi.[19]
L’attenzione sempre più crescente nei confronti del carattere tossico dei composti biocidi in vernici antifouling, ha portato all’attuale regolamentazione e la conseguente necessaria approvazione per la loro applicazione.[2]
1.4.2 Rivestimenti non tossici
1.4.2.1
Superfici micro/nano strutturate
Studi condotti sull’influenza delle caratteristiche del substrato riguardo l’adesione degli organismi marini hanno messo in evidenza come questa risulti influenzata dalle imperfezioni nanoscopiche della superficie.[20] Infatti, una delle strategie impiegate per rivestimenti antifouling, è quella di limitare fisicamente l’adesione degli organismi marini tramite l’impiego di superfici micro/nano strutturate, prendendo spunto dal mondo naturale (“bioinspired surface design”).[21]
Per questo tipo di superfici è necessario manipolare le proprietà chimico-fisiche e meccaniche della superficie, per far sì che l’organismo non riesca ad aderire e percepisca la superficie come “non conveniente” per l’insediamento (Fig. 1.3).[22]
INTRODUZIONE
7
Figura 1.3- Confronto di immagini SEM fra (a) la superficie reale della pelle di squalo
(b) una superficie stampata in PDMS con microstruttura d’ispirazione ad essa.
I metodi più comuni impiegati per produrre superfici micro topografiche sono: il processo di ablazione laser, la fotolitografia, o per stampo e deposizione, e generalmente vengono applicati a substrati di polidimetilsilossani (PDMS), polivinilcloruri (PVC), o poliammidi.[2,23]
La maggiore difficoltà che ostacola il commercio di questi rivestimenti micro/nano strutturati, è dovuta al prezzo e l’impraticabilità su grandi navi.
1.4.2.2
Superfici fouling-release
I rivestimenti fouling-release sono rivestimenti privi di biocidi. L’idea generale di questo tipo di rivestimenti è quella di minimizzare le adesioni fra i microrganismi e la superficie, in modo da rendere possibile la rimozione del fouling mediante stress idrodinamico dovuto al movimento dell’imbarcazione o per semplice pulizia meccanica.[24]
Inoltre, la levigatezza, caratteristica di questi rivestimenti, permette loro di ridurre la resistenza al movimento della nave, con conseguente riduzione del consumo di carburante e di emissioni di gas serra (Fig. 1.4).
Figura 1.4- Esempio di rilascio di un generico organismo marino aderito su una superficie
fouling release.
In letteratura e su brevetti ritroviamo, come rivestimenti fouling release, due maggiori classi di polimeri esplicanti la caratteristica di rilascio di fouling, ossia matrici sia a base siliconica sia fluoropolimerica. [2]
8
Le specifiche proprietà fouling release sono generalmente correlate all’idrofobia e alla bassa energia della superficie, ma sono anche influenzate da altri parametri, quali la rugosità superficiale, il modulo elastico, e lo spessore del film depositato.
Le vendite dei rivestimenti fouling release sono aumentate in maniera significativa a seguito della convenzione dell'Organizzazione Marittima Internazionale nel 2003, e attualmente sono stimate attorno al 10% del volume delle imbarcazioni commerciali.[25]
Il tempo di vita stimato per questa classe di rivestimenti è dai 5 ai 10 anni di servizio, rendendo quindi questa tecnica meno costosa rispetto alle precedenti soluzioni illustrate come antifouling, considerando anche i benefici associati al risparmio di carburante per le imbarcazioni.
1.5
Proprietà delle vernici fouling release
I rivestimenti fouling release devono agire come barriera per evitare che si instaurino forti legami fra gli organismi e lo strato superficiale della chiglia. Le caratteristiche fondamentali che un materiale di questo tipo deve avere sono[13, 26]:
Un numero significativo di gruppi attivi in superficie che siano liberi di muoversi, conferendo così bassi valori di energia superficiale
Basso valore di energia interfacciale Basso valore di modulo elastico “Levigatezza” a livello molecolare Alta mobilità delle catene laterali attive Resistenza all’abrasione
Alta stabilità chimica e durata in acqua di mare
Dopo aver preso atto di tali fattori, che influenzano la bio-adesione, la ricerca scientifica si è spinta verso lo sviluppo di materiali che accomunano le proprietà di bassa energia superficiale e modulo elastico. Ad oggi, i polimeri conosciuti che maggiormente posseggono tali requisiti sono i siliconi ed i fluoropolimeri.
1.5.1 Tensione superficiale e modulo elastico
L’adesione e la crescita di organismi sulla superficie dipendono dalle energie interfacciali. Nel meccanismo di bioadesione intervengono parametri di tipo chimico, fisico e meccanico. Anche la bagnabilità è correlata con la bioadesione.[27]
Dal punto di vista termodinamico la tensione superficiale può essere definita come la quantità di lavoro necessario per aumentare l’estensione della superficie di un’unità mantenendo costante la temperatura del sistema. Questa risulta una proprietà che opera all’interfaccia tra il fluido in questione ed un materiale di altra natura, come un solido, un gas o un altro liquido con cui è immiscibile.
INTRODUZIONE
9
La tensione superficiale, e più in generale le proprietà di superficie, possono essere studiate tramite misure dell’angolo di contatto, ossia la valutazione dell’angolo formato da una goccia di liquido a contatto con una fase solida.[28]
1.5.1.1
Relazione fra angolo di contatto ed energia superficiale
E’ stato stabilito un modello termodinamico per descrivere l’adesione del microrganismo alla superficie, per cui l’energia libera di adesione (ΔGadh
) è legata alle energie interfacciali tra il substrato, l’organismo marino ed il liquido circostante:[29, 30]
(1) Dove γBS è l’energia interfacciale tra l’organismo responsabile del biofouling ed il substrato, γBL è l’energia interfacciale tra l’organismo responsabile del biofouling ed il liquido circostante, e γSL è l’energia interfacciale tra il substrato ed il liquido.
L’adesione dei batteri e delle spore dell’alga verde Ulva linza sono state spiegate tramite questo modello.[31] Tuttavia le notevoli difficoltà che si incontrano nel determinare tali valori di energia superficiale rendono l’equazione difficilmente praticabile.
Modelli descritti in letteratura affrontano il problema della determinazione delle energie interfacciali e delle energie superficiali mediante misure di bagnabilità tramite misure di angolo di contatto.
La teoria di Young descrive la condizione di equilibrio di una goccia di liquido, circondata dal proprio vapore, posta in contatto con una superficie liscia (Figura 1.5).[32]
Figura1.5- Rappresentazione di una goccia di liquido depositata su un substrato solido e le relative
tensioni superficiali. Teoria di Young
Nel caso in cui il liquido non bagni completamente la superficie solida, esso stabilisce un angolo di contatto θ, che dipende dal bilancio delle energie superficiali: la tensione superficiale del liquido γL, la tensione superficiale del solido γS, e la tensione interfacciale solido/liquido γSL, come segue:
10
(2) Quando il liquido impiegato è acqua, l’angolo di contatto dell’acqua (θw) è correlabile al carattere idrofilo/idrofobo della superficie solida; si possono presentare diverse situazioni:
Massima bagnabilità: il liquido si spande sulla superficie solida, θw= 0. Bagnabilità parziale: corrisponde ad una superficie idrofila, θw < 90° Bagnabilità scarsa: corrisponde ad una superficie idrofoba, θw > 90° Superfici superidrofobe, θw > 150°
In generale, minore è l’angolo di contatto che si forma fra superficie e solvente, maggiori sono le interazioni.[2]
Considerando l’equazione di Young per calcolare l’energia superficiale del solido γS, γSL e γL devono essere note. Owens-Wendt, Rabel e Kaeble [33] indicano che l’energia superficiale è uguale alla somma di una componente dispersiva ( ) e di una componente polare ( ):
(3) La prima componente ( ) riflette le interazioni di dispersione che si verificano all’interfaccia, mentre la seconda ( ) riflette le interazioni polari, induttive, a idrogeno, e acido-base. Questi parametri sono relazionati con γSL mediante l’equazione:
(4) Dove e sono i contributi delle forze dispersive e polari alla tensione superficiale γL del liquido bagnante impiegato. Combinando le equazioni 2 e 4, si ottiene:
(5) In questo modo è possibile calcolare e tramite la misura dell’angolo di contatto ottenuto mediante due liquidi bagnanti diversi sulla superficie del medesimo solido, in modo tale da avere un sistema di equazioni risolvibile.
L’utilizzo di questa teoria comporta che la tensione superficiale del solido si mantenga costante al variare della natura del liquido bagnante utilizzato. Sperimentalmente ciò non si verifica e al variare della coppia di liquidi a contatto con il solido, si ottengono valori differenti delle componenti polari ed apolari dell’energia superficiale totale.
INTRODUZIONE
11
Un ulteriore contributo alla teoria della tensione superficiale proviene da Good, van Oss e Chaudhury.[34, 35] Questi intraprendono un approccio acido-base per il calcolo dell’energia superficiale, assumendo la tensione superficiale come la somma di una componente apolare di Lifshitz-van der Waals e una componente polare acido-base di Lewis :
(6) La componente polare acido-base può essere ulteriormente suddivisa in sottocomponenti, utilizzando termini specifici per la componente acida di Lewis (γ+
), il carattere elettron accettore, e per quella basica (γ
-), il carattere elettron donatore, avendo perciò:
(7) Combinando queste equazioni con quella di Young, si ottiene la seguente equazione:
(8) I tre parametri caratterizzanti la tensione superficiale di un solido possono essere determinati misurando gli angoli di contatto all’equilibrio di almeno tre liquidi di cui siano note le componenti della tensione superficiale.[2, 29]
L’adesione degli organismi marini è storicamente relazionata alla tensione superficiale critica del substrato γC, introdotta da Zisman nel 1950.[36] Questa tensione rappresenta il valore critico di tensione superficiale del liquido (γL), al di sotto del quale esso si spanderebbe liberamente sul substrato (θ= 0°). γC viene determinato mediante misura di angolo di contatto di una serie di liquidi posti sulla superficie solida e tracciando il coseno dell’angolo in funzione della tensione superficiale del rispettivo liquido ed estrapolando a θ= 0° mediante una retta (Fig. 1.6).
12
Figura 1.6- Grafico di Zisman: andamento dell’angolo di contatto in funzione della tensione
superficiale del liquido
Il corrispettivo valore di tensione superficiale del cosθ= 1, è chiamato “tensione superficiale critica γC”, ed è tipica del solido. Una volta noto il valore di γC, si può stabilire a priori se un liquido bagni o meno una superficie solida confrontando questo valore con la tensione superficiale del liquido γL . Infatti un liquido avente una tensione superficiale γL minore rispetto al valore di γC del solido bagnerà completamente la superficie.
Tuttavia, l’utilizzo di tale parametro non è sufficiente per descrivere le interazioni che si sviluppano in superficie tra liquido e un substrato solido. Infatti il valore di γC risulta largamente dipendente dalla natura del liquido impiegato. Questa teoria si dimostra totalmente inefficiente, infatti, quando vengono considerati liquidi polari, capaci di generare per esempio interazioni tra le due fasi, con il risultato di una deviazione dalla linearità.[2] In letteratura, è stata stabilita una relazione empirica tra γC e la quantità relativa di materiale bioadeso, comunemente chiamata “curva di Baier” (Fig. 1.7).
INTRODUZIONE
13
Figura 1.7- Curva di Baier: relazione tra adesione e tensione superficiale critica per un substrato
solido
Il punto chiave fondamentale di questa curva è rappresentato dal minimo di adesione relativa a 22-24 mN∙m-1 che non cade in corrispondenza del più basso valore di tensione superficiale critica. Ciò implica che l’adesione non dipende soltanto dalla tensione superficiale. Un ruolo fondamentale viene anche esplicato dal modulo elastico (E) del substrato. Infatti l’adesione risulta direttamente proporzionale alla radice quadrata del prodotto tra il modulo elastico (E) e la tensione superficiale critica (γC) (Fig. 1.8).[37] Tali relazioni sono tuttavia puramente empiriche e non si può escludere che esistano altri parametri che possano influenzare l’adesione in modo sostanziale.
Figura 1.8- Relazione tra adesione e la radice quadrata della tensione superficiale e modulo elastico
Come si nota dal grafico, un basso valore di bioadesione coincide con un basso valore di modulo elastico perché la mobilità di questo tipo di superficie permette all’adesivo di attaccarsi meno fortemente riducendo così la forza necessaria per rompere il legame formatosi, e quindi per la sua rimozione.
14
In accordo a usta interpretazione, gli elastomeri siliconici, sono sfruttati come rivestimenti fouling-release. Questi presentando bassi valori di modulo elastico, il poli(dimetilsilossano) (PDMS) ha un valore di modulo elastico fra i più bassi nel campo dei polimeri (0,002 GPa), e energie superficiali di 20-26 mN/m, corrispondente al minimo valore di adesione per Baier, permettono di ottenere il comportamento un buon rilascio di fouling.[38]
1.5.2 Struttura chimica
Ovviamente, anche la struttura del polimero gioca un ruolo importante nei confronti della bioadesione.
Confrontiamo quindi le caratteristiche di polimeri più impiegati nelle formazioni di rivestimenti fouling-release: i polimeri siliconici, i fluoropolimeri e i polimeri ossietilenici.
1.5.2.1
Proprietà dei polimeri siliconici
I rivestimenti fouling-release sono principalmente composti da elastomeri di silicone contenenti gruppi organici ed inorganici. La catena principale è costituita da atomi di silicio e di ossigeno alternati. Ogni atomo di silicio è legato a due gruppi organici, mentre gli atomi di silicio terminali alla catena hanno un terzo gruppo, che può essere un sostituente idrossi, ammino, o alcossi. Nell’elastomero siliconico più comunemente impiegato nelle formulazioni di vernici antifouling, il PDMS, i gruppi laterali sono gruppi metilici, come illustrato in Figura 1.9.
Figura 1.9- Struttura chimica del PDMS
Questi polimeri di PDMS hanno elevati angoli di contatto con l’acqua, una bassa tensione superficiale critica, e un basso valore di modulo elastico.
Queste fondamentali proprietà sono connesse alle specifiche caratteristiche del legame silossanico,[39] che è la combinazione di una struttura flessibile e di gruppi laterali a bassa energia superficiale. La catena del silossano ha una flessibilità unica conferita da diversi parametri, quali la distanza di legame Si-O (1.65 Å), il legame silossanico planare (159°), la parziale natura ionica (non direzionale) del legame, e l’alternanza di gruppi bivalenti nello scheletro ottenendo una maggiore spaziatura tra i corrispondenti sostituenti del Si.[26] La
INTRODUZIONE
15
flessibilità unica di questi polimeri è evidenziata da temperature di transizione vetrosa molto basse, Tg attorno a -127°C.
Gli svantaggi maggiori di questo tipo di materiali sono le scarse proprietà meccaniche e la facilità con cui possono lacerarsi; ciò limita il loro campo di utilizzo o rende necessaria l’aggiunta di cariche rinforzanti.
Complessivamente, i siliconi vengono considerati materiali antiadesivi con proprietà migliori anche dei polimeri fluorurati. [27] Infatti, i rivestimenti siliconici hanno generalmente valori più alti, rispetto ai fluorurati, di energia superficiale e, quindi, determinano legami leggermente più forti con gli organismi del fouling; a causa però dei loro bassi moduli elastici si ha una migliore rimozione dello strato di fouling formatosi.[7]
1.5.2.2
Proprietà dei polimeri fluorurati
I polimeri fluorurati sono una classe particolare di polimeri con caratteristiche anche molto diverse dalle altre materie plastiche. La sostituzione di atomi di idrogeno con atomi di fluoro comporta evidenti cambiamenti nelle proprietà chimo-fisiche dei materiali. La natura del legame C-F, un legame covalente a elevata energia e altamente polarizzato, determina le caratteristiche di elevata stabilità termica e resistenza all’aggressione chimica.
Le principali caratteristiche dei fluoropolimeri possono essere riassunte in:
Anfifobia: sono materiali sia idrofobi che lipofobi, ovvero repellenti sia ai liquidi polari sia apolari;
Bassa adesione: dovuta alla bassa energia superficiale;
Resistenza al calore: resistono a più di 260°C per lunghi periodi; Resistenza all’invecchiamento e alle sollecitazioni dinamiche
Difficilmente degradabili: sono trasparenti ai raggi UV, resistenti all’ossidazione e all’attacco degli aggressivi chimici e dei microrganismi.
I fluoropolimeri sono ben conosciuti per la loro natura non polare, che conferisce caratteristiche idrofobe alle loro superfici e una tensione superficiale critica molto bassa, attorno a 10-20 mN/m.
Zisman[36] ha stabilito che l’energia superficiale dipende dall’intorno chimico e tende a diminuire in questo ordine:
-CH2 (36 mN/m) > -CH3 (30 mN/m) > -CF2 (23 mN/m) > -CF3 (15 mN/m)
Ne consegue, che l’introduzione di gruppi fluorurati nelle catene laterali delle macromolecole potrebbe essere un’ottima strategia per lo sviluppo di materiali impiegati come rivestimenti per il controllo del biofouling o bassa energia superficiale.
16
Nonostante le ottime proprietà chimico-fisiche, l’elevato costo e la scarsa solubilità in tutti i solventi classici limitano l’impiego di fluoro polimeri. Un efficace bilancio tra proprietà e prestazioni si può ottenere in polimeri parzialmente fluorurati in cui unità o segmenti fluorurati siano inseriti all’interno di catene idrocarburiche o come ramificazioni in catene laterali. L’inserimento, anche di sole poche unità di fluorurato, altera notevolmente le proprietà del polimero e in particolare quelle della superficie, rendendo il materiale più processabile e meno costoso.
Sebbene i rivestimenti fouling release a base di fluoropolimeri abbiano esibito una buona efficienza nella prevenzione e nel rilascio di organismi responsabili del biofouling, come per l’alga Ulva, questi risultano poco efficienti riguardo l’adesione delle diatomee.[40]
Lo scopo principale dello sviluppo di rivestimenti marini a rilascio di fouling è quello di riuscire a creare una superficie resistente a tutti i tipi di organismi responsabili del fenomeno. Sono state studiate superfici più polari, come per esempio rivestimenti contenenti frazioni di polietilenglicol (PEG) in combinazione con frazioni fluorurate. Si parla quindi di rivestimenti anfifilici, cioè contenenti unità idrofile in combinazione opportuna con unità idrofobe.
1.5.2.3
Caratteristiche dei polimeri ossietilenici
Le superfici di PEG sono conosciute per la loro resistenza all’adsorbimento delle proteine e all’adesione delle cellule; inoltre hanno anche mostrato resistenza all’insediamento e buon rilascio degli organismi marini responsabili del fouling.[40]
Il PEG è il polietere più conosciuto e diffuso commercialmente; è costituito da una catena flessibile formata da gruppi etilenici collegati da legami eterei, che generalmente termina con gruppi ossidrilici (Fig. 1.10).
Figura 1.10- Struttura del PEG
Le proprietà fisiche del PEG (come ad esempio la viscosità, la temperatura di cristallizzazione, ecc.) variano a seconda del peso molecolare, e quindi della lunghezza media della catena, mentre le proprietà chimiche sono quasi identiche al variare di questo. I pesi molecolari oscillano dai 300 ai 10 000 000 g/mol, passando rispettivamente da un liquido viscoso ad un solido ceroso ad un solido cristallino.
INTRODUZIONE
17
I rivestimenti a base di PEG, nonostante siano caratterizzati da un’energia superficiale relativamente alta (>43 mN/m), presentano bassa energia interfacciale con l’acqua (5 mN/m) [41], e una resistenza all’adesione data dalla repulsione sterica delle molecole di adesivo, causata dall’idratazione del PEG.[40]
Questo porta quindi a limitare il condizionamento e l’adesione dell’ EPS (sostanze polimeriche extracellulari) proteico sulla superficie.
1.5.3 Rivestimenti anfifilici micro/nano-strutturati
I rivestimenti anfifilici micro/nano-strutturati uniscono i benefici dei materiali idrofobi e di quelli idrofili. Diversi studi [31, 42] sulla colonizzazione delle diverse specie marine responsabili del fouling hanno dimostrato la preferenza per le superfici idrofobe di alcune specie, mentre altre tendono ad aderire con più forza su substrati idrofili.[43] Da queste diverse preferenze di colonizzazione nasce l’idea di sviluppare superfici anfifiliche, ossia superfici “ambigue” contenenti sia domini idrofili che domini idrofobi, in grado di “confondere” gli organismi e quindi di scoraggiarne l’adesione.
Rivestimenti di questo genere, attraverso micro/nano-separazioni di fase di copolimeri incompatibili, creano una superficie dinamica con locali variazioni chimiche e meccaniche.[31]
Per produrre questo tipo di rivestimenti esistono varie possibilità; la strada più seguita è quella di preparare copolimeri a blocchi anfifilici, i quali originano una superficie nano strutturata a causa della mutua incompatibilità dei singoli blocchi polimerici con distinte caratteristiche di idrofilia e idrofobia.[44]
Questi copolimeri a blocchi contengono un componente non polare, idrofobo, con bassa energia superficiale (ad esempio impiegano polimeri fluorurati o silossani) e un componente idrofilo con proprietà repellenti verso le proteine (ad esempio catene di PEG).
Spesso questi copolimeri sono caratterizzati da una catena principale stirenica o acrilica, alla quale sono legate le catene a base di PDMS/fluorurati/PEG (Fig. 1.11).
18
Figura 1.11- Struttura di un copolimero a blocchi anfifilico
L’auto-assemblaggio dei diversi blocchi produce una superficie complessa formata da domini di dimensioni micro-nanoscopiche. [45] Infatti quando il film polimerico è interfacciato con l’aria, il gruppo fluorurato tende a migrare in superficie, poiché caratterizzato da bassa energia superficiale, portando con sé la componente PEG. Viceversa, quando il rivestimento è immerso il acqua, la superficie tende ad arricchirsi di PEG, avendo questo una bassa energia interfacciale con tale liquido. Si ottiene così una superficie dinamica che tende a cambiare al variare delle condizioni ambientali che si presentano all’interfaccia. [41]
Figura 1.12- Meccanismo proposto per il riassestamento superficiale al momento dell’immersione in
acqua. Le sfere gialle rappresentano il fluoro, le rosse l’ossigeno, le celesti l’idrogeno e le grigie il carbonio.
INTRODUZIONE
19
Tali rivestimenti sono generalmente preparati da copolimeri a blocchi in cui il blocco silossanico [45-48] (PDMS) viene modificato con gruppi laterali fluorurati, ossietilenici oppure con entrambi ottenendo una struttura triblocco.[49, 50]
1.6
Fotopolimerizzazione
Il processo di polimerizzazione fotoindotto dei monomeri multifunzionali e di oligomeri è uno dei metodi più efficienti per produrre film a base di materiali polimerici.[51] Questi sistemi subiscono una rapida trasformazione del monomero liquido / oligomero nel polimero solido semplicemente mediante esposizione alle radiazioni UV in presenza di opportuni fotoiniziatori.[52] I fotoiniziatori svolgono un ruolo chiave nei sistemi ad UV generando le specie reattive, radicali liberi o ioni, in grado di iniziare la polimerizzazione dei monomeri multifunzionali e degli oligomeri. [53]
La tecnologia di fotopolimerizzazione da radiazione UV rappresenta una risposta concreta ed efficiente alla continua crescita dell’industria (in particolare per il rivestimento di superfici metalliche, di legno, per la verniciatura della plastica e per la produzione di compositi), in grado di ricercare e sviluppare metodologie produttive che comportino sia ridotti quantitativi di energia sia un minor impatto ambientale.[54]
Questa tecnologia permette di ottenere entrambi i vantaggi, in quanto consente la sostituzione di impianti di polimerizzazione, che hanno notevoli consumi energetici, e consente di ridurre le emissioni nocive, dato che generalmente la maggior parte dei prodotti fotosensibili sono privi di solvente e quindi risultano validi dal punto di vista ecologico.[55]
La radiazione UV è una radiazione elettromagnetica a lunghezze d´onda comprese tra 100 e 400 nm. La radiazione UV confina con la luce visibile, di lunghezza d´onda più corta, e con i raggi X, di lunghezza d’onda superiore (Fig. 1.12).
Figura 1.12- Spettro elettromagnetico
La fotopolimerizzazione risulta un metodo importante se si vogliono eseguire polimerizzazioni a basse temperature, alle quali la formazione dei radicali per via termica è pressoché trascurabile per la maggior parte dei comuni iniziatori.[56]
La polimerizzazione fotoiniziata si può avere in diversi modi, che possono comportare il trasferimento dell’energia raggiante assorbita da una molecola di una sostanza appositamente aggiunta al sistema o presente in tracce come impurezza (il fotosensibilizzatore) ad un’altra molecola con successiva formazione di radicali primari, oppure un’eccitazione diretta di una molecola (che può essere anche lo stesso monomero) con formazione da questa dei radicali per rottura di un legame. Radiazioni ultraviolette nel campo attorno ai 360 nm provocano ad
20
esempio (generalmente per fotolisi diretta) la formazione di radicali anche a basse temperature da perossidi e azocomposti, e di sostanze contenenti gruppi carbonilici (ed es. il benzoino), a volte troppo stabili per agire da iniziatori termici. La radiazione assorbita può anche essere in parte trasformata in calore, o riemessa sottoforma di una fluorescenza; il meccanismo globale, dall’assorbimento della radiazione fino alla formazione dei radicali primari, è quindi talvolta complicato e comporta diversi processi in competizione.
Il foto-iniziatore impiegato è il 2-idrossi-2-metilpropiofenone. I chetoni aromatici vengono impiegati grazie alla loro capacità di assorbire energia a lunghezza d’onda elevate (più basse energie), subendo scissione omolitica del legame in α al carbonile (Fig. 1.13). [59]
Figura 1.13- Meccanismo di scissione del legame
1.6.1 Meccanismo
La fotopolimerizzazione richiede un iniziale processo di formazione di radicali liberi primari, ottenuti per via fotochimica:
Inizio
I 2R· (dove I rappresenta l’iniziatore ed R· i radicali primari)
Il radicale così formato è in grado di reagire rapidamente con un monometro insaturo addizionandovisi e formando così un nuovo radicale generalmente più stabile del precedente che costituisce il centro attivo della polimerizzazione:
R· + M P1·
(dove M rappresenta il monomero e P1· il “primo” radicale della catena in accrescimento) Dopo l’inizio si avrà la successiva addizione del monomero sul “primo” radicale della catena in accrescimento e la catena si propagherà con spostamento del centro attivo radicalico all’estremità delle catene stesse; a questo stadio ne seguiranno rapidamente moltissimi altri a costruire una serie di reazioni di crescita:
O OH h O OH * O OH +
INTRODUZIONE 21 Propagazione P1· + M P2· --- Pi· + M Pi+1·
(dove i rappresenta il numero di unità monomeriche contenute nel radicale in accrescimento). La propagazione avviene molto rapidamente finché non avviene la terminazione della catena, con formazione del polimero inattivo e scomparsa di radicali. Ciò si può verificare per reazione diretta tra due radicali polimerici che possono dare luogo ad accoppiamento, formando, tramite un legame covalente, una sola molecola polimerica:
Termine
Pn· + Pm· Pn+m
oppure per disproporzionamento fra gli stessi tramite ad es. l’estrazione di idrogeno, formando due molecole polimeriche:
Pn· + Pm· Pn + Pm
La tendenza delle specie radicaliche alla terminazione è molto forte, ma è controbilanciata dalla loro bassissima concentrazione rispetto a quella del monomero.
Le catene polimeriche possono cessare di crescere anche mediante trasferimento del centro attivo da una macromolecola in accrescimento ad un’altra per reazione con una sostanza (trasferitore) volutamente aggiunta o no, che può anche essere il solvente, il monomero, il polimero ecc., con formazione di un nuovo radicale. Il trasferimento di catena permette la regolazione del peso molecolare del polimero senza bloccare la polimerizzazione, dato che per ogni catena che smette di crescere ne può nascere un’altra.[56]
Poiché nella polimerizzazione fotoindotta è coinvolto un meccanismo a radicali liberi, alle tipiche condizioni che governano la polimerizzazione termica si aggiungono altri fattori associati alla fotopolimerizzazione, fattori come la fonte di irraggiamento, la scelta delle lunghezze d’onda di irraggiamento, la distanza della sorgente dalla cella di reazione, il foto-iniziatore, il solvente, la presenza di ossigeno, la temperatura, ecc. [57]
Sistemi di rivestimento ottenuti mediante fotopolimerizzazione presentano diversi vantaggi quali: buon rapporto costo / prestazioni, basso consumo di energia, ad alta resistenza meccanica e chimica dei rivestimenti finali, assenza di composti organici volatili (senza solvente), e l'alta velocità del processo anche a temperatura ambiente.[58]
2. SCOPO DELLA TESI
La soluzione del problema dell’adesione, proliferazione e crescita della fauna e della flora sulle superfici immerse in mare o in acqua dolce riveste oggi un grande interesse in ambito tecnologico ed economico-ambientale. Inoltre, la crescente preoccupazione per i problemi di impatto ambientale connessi con l’uso indiscriminato dei rivestimenti antivegetativi, nonché la più rigida regolamentazione recentemente introdotta contro l’uso massiccio di agenti biocidi, hanno reso necessari la ricerca e lo sviluppo di materiali nuovi ed originali. Si richiede, infatti, che i rivestimenti antivegetativi della futura generazione siano non tossici, non si (bio)accumulino e non persis tano nei differenti ecosistemi, pur mantenendo un’efficiace prestazione biologica contro i più svariati organismi marini.
In questo contesto si inserisce una moderna linea di ricerca che mira alla progettazione e allo studio di rivestimenti protettivi in grado di inibire le interazioni con gli organismi vegetativi e di permettere il distacco di quelli eventualmente aderiti. L’adesione di tali organismi è un tipico fenomeno interfacciale che potrebbe essere controllato su scala micro-/nano-scopica, intervenendo sui meccanismi di interazione chimica, fisica e meccanica tra la struttura, la morfologia, la topografia della superficie e gli elementi sensibili di esplorazione dei siti di stanziamento e di secrezione dei collanti extra-cellulari degli agenti vegetativi.
Sebbene la notevole varietà degli organismi vegetativi e dei loro me ccanismi di attacco non lasci intravedere la possibilità di sviluppare un unico ed universale tipo di rivestimento antivegetativo, è ormai generalmente accettato che le nuove linee vincenti per combattere il processo biovegetativo coinvolgono l’opportuna combinazione di bassa energia superficiale e proprietà elastomeriche di basso modulo elastico. In particolare, superfici con caratteristiche anfifiliche, cioè che presentino proprietà chimiche e/o fisiche molteplici e tra loro contrastanti, sembrano offrire maggiori possibilità di ostacolare i processi di interazione fra substrato e sostanze organiche extracellulari secrete dai microrganismi.
Seguendo queste linee guida, nel presente lavoro di tesi abbiamo perseguito l’obiettivo principale di progettare e preparare nuovi film polimerici a base siliconica caratterizzati
24
da diversa filia/fobia e con potenziali proprietà di segregazione superficiale allo scopo di originare strutture molecolari di carattere “ambiguo”. Per raggiungere tale scopo abbiamo previsto di utilizzare una strategia semplice ed innovativa rispetto a quella generalmente descritta per la preparazione di film siliconici, potenzialmente applicabili come rivestimenti antivegetativi.[14] In particolare, volevamo reticolare la matrice polidimetilsilossanica tramite un meccanismo radicalico a catena invece che di policondensazione a stadi, in modo tale da evitare l’utilizzo di catalizzatori a base di metalli pesanti (quali composti di stagno e bismuto), ridurre le quantità di solvente usate nella preparazione dei film e condurre la reazione a temperatura ambiente senza necessità di successivi processi di finitura a più alte temperature.
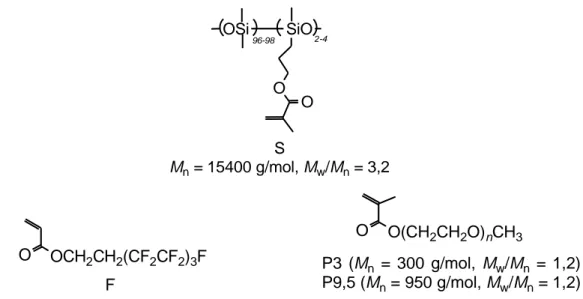
Per la preparazione dei film con le proprietà ricercate abbiamo scelto tre monomeri commerciali a base (met)acrilica contenenti una catena polidimetilsilossanica principale (S) e catene laterali perfluorurate (F) e ossietileniche (P3 e P9,5) ( Fig. 2.1).
S
F P3 (n= 5)
P9,5 (n= 20)
Figura 2.1- Formule di struttura dei monomeri S, F, P3 e P9,5.
Volevamo quindi preparare film derivanti dalla foto-polimerizzazione di miscele bi- e tri-componente caratterizzati da un diverso bilancio idrofobo/idrofilo determinato
SCOPO
25
variando il rapporto ponderale F/P3 (o F/P9,5) tra i due monomeri monofunzionali e da un basso modulo elastico ottenuto dalla reticolazione del macro -monomero polidimetilsilossanico poli-funzionale. In questo modo si intendeva produrre film di diversa natura anfifilica potenzialmente capaci di rispondere in modo specifico all’ambiente esterno, aria o acqua, inviando così messaggi diversificati ed “ambigui” in grado di confondere gli organismi biovegetativi a contatto con essi.
Un altro obiettivo era quello di verificare la bagnabilità dei film con liquidi di interrogazione di diversa polarità, in modo da poter calcolare dai valori di angolo di contatto (θ) misurati i corrispondenti valori di tensione superficiale sv da associare qualitativamente all’adesività dei film, e quindi alle loro capacità antivegetative. In questo ambito, lo studio di analisi chimiche della superficie mediante la spettroscopia fotoelettronica ai raggi X (XPS) ci avrebbe fornito utili informazioni riguardo alla reale composizione della superficie più esterna, particolarmente in relazione alla loro suscettibilità di riorganizzazione chimica e strutturale dopo immersione in acqua dei film.
Nello stadio finale della tesi abbiamo iniziato una valutazione preliminare delle prestazioni biologiche di alcuni dei rivestimenti preparati mediante saggi ecotossicologici utilizzando il batterio Vibrio fischeri, la microalga Dunaliella tertiolecta e il crostaceo Artemia salina. Infine, abbiamo avviato lo studio dello stanziamento e della rimozione della diatomea
Navicula salinicola e dell’adesione e del distacco del serpulide Ficopomatus enigmaticus, da
poter eventualmente correlare con la struttura chimica dei film da impiegare come rivestimenti protettivi antivegetativi in ambienti marini.
3. RISULTATI E DISCUSSIONE
In questo lavoro di tesi abbiamo progettato e messo a punto la preparazione di nuovi film siliconici reticolati caratterizzati da un’intrinseca natura anfifilica, potenzialmente utilizzabili come rivestimenti protettivi a bassa bagnabilità e ad azione antivegetativa e/o a rilascio di fouling. Di seguito descriveremo dettagliatamente la strategia seguita per la preparazione dei film, basata sulla foto-polimerizzazione di macro-monomeri (meta)acrilici mono- e poli-funzionali disponibili in commercio. Questa metodologia semplice e innovativa aveva il vantaggio di non richiedere significative quantità di solvente per la preparazione dei campioni e permetteva di evitare l’utilizzo di catalizzatori a base di metalli pesanti per la reticolazione della matrice silossanica. Successivamente presenteremo i risultati ottenuti nei nostri studi di caratterizzazione dei film prescelti con riferimento sia alle proprietà di massa (termiche e meccaniche) che di superficie (bagnabilità e composizione chimica). Infine, discuteremo le caratteristiche tossicologiche e le prestazioni biologiche che i film esibivano nei confronti di alcuni organismi marini.
3.1
Monomeri e omopolimeri
Per la preparazione dei film reticolati anfifilici sono stati individuati tre componenti essenziali, quali il copolimero (metacrilossipropil)metilsilossano-co-dimetilsilossano con un contenuto di unità metacriliche del 2 4% in moli (denominato S), ed i monomeri 1H,1H,2H,2H-perfluoroesiletil acrilato (denominato F) e polietilenglicol monometil etere metacrilato con due diversi pesi molecolari (Mn = 300 g/mol e Mn = 950 g/mol denominati rispettivamente P3 e P9,5) (Fig. 3.1).
28
Figura 3.1- Struttura chimica dei macro-monomeri (met)acrilici:
(metacrilossipropil)metilsilossano-co-dimetilsilossano (S), 1H,1H,2H,2H-perfluoroesiletil acrilato (F), polietilenglicol monometil etere metacrilato (P3 e P9,5).
In particolare, il primo rappresentava il reagente principale per la formazione dei film polimerici reticolati. Il contenuto di funzionalità metacriliche nel copolimero silossanico è stato scelto relativamente basso (2 4% in moli) in modo tale da generare una matrice con un basso grado di reticolazione e, quindi, un basso modulo elastico. Inoltre, la natura silossanica di questo macro-monomero era in grado di impartire caratteristiche idrofobe e lipofile all’intero sistema.
I monomeri F e P erano invece mono-funzionali: mentre F comprendeva una catena perfluorurata relativamente lunga capace di conferire proprietà di idrofobia e lipofobia assai spiccate, P conteneva una catena ossietilenica in grado di impartire un buon grado di idrofilia, oltre che lipofilia. Il carattere idrofilo poteva essere incrementato aumentando la lunghezza della catena polimerica laterale, passando da circa 5 (P3) a circa 20 (P9,5) unità ossietileniche (Tabella 3.6).
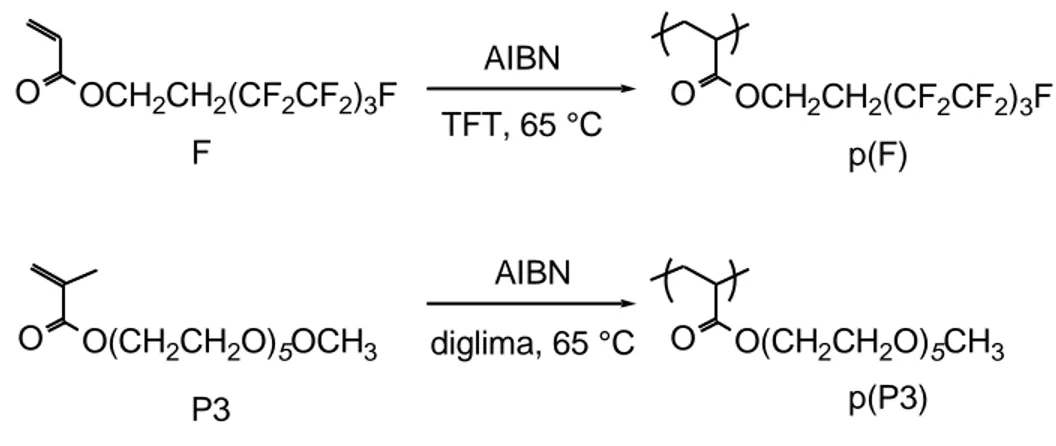
I monomeri F e P3 sono stati usati per preparare i corrispondenti omopolimeri p(F) e p(P3) allo scopo di utilizzarli come composti modello dei film delle diverse miscele polimeriche, soprattutto in relazione alle proprietà termiche e di bagnabilità superficiale.
Le reazioni di ompolimerizzazione sono state condotte secondo le classiche modalità della polimerizzazione a radicali liberi con ,’-azobis(isobutirronitrile) (AIBN) (rapporto
OSi SiO O 2-4 O 96-98 S Mn = 15400 g/mol, Mw/Mn = 3,2 O OCH2CH2(CF2CF2)3F F O O(CH2CH2O)nCH3 P3 (Mn = 300 g/mol, Mw/Mn = 1,2) P9,5 (Mn = 950 g/mol, Mw/Mn = 1,2)
RISULTATI E DISCUSSIONE
29
ponderale monomero/AIBN = 100:1), in soluzione di diglima o trifluorotoluene (TFT) a 65 °C per 65 ore (Fig. 3.2).
Figura 3.2- Sintesi degli omopolimeri p(F) e p(P3).
3.2
Preparazione dei film polimerici
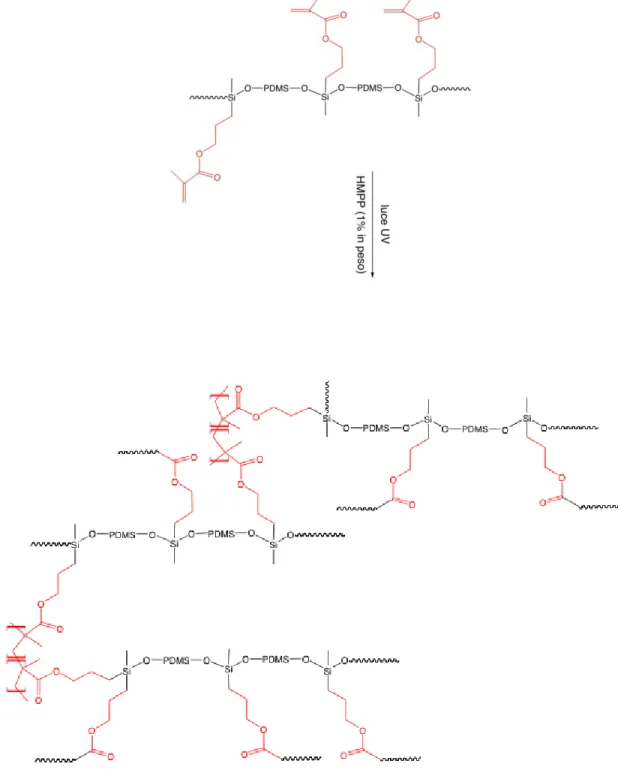
Il macro-monomero silossanico S è stato, inizialmente, utilizzato per la preparazione di film reticolati costituiti dalla sola matrice polidimetilsilossanica, di seguito denominati S100. La polimerizzazione di S è stata condotta in presenza di un foto-iniziatore (1% in peso) mediante irraggiamento con lampada UV (Fig. 3.3). La scelta dell’iniziatore è ricaduta sul 2-idrossi-2-metil propiofenone (HMPP) che, rispetto ad altri foto-iniziatori del tipo benzoino (vedi ¶ 1.6), aveva il vantaggio di essere solubile nel macro-monomero silossanico, non richiedendo quindi l’aggiunta di un solvente per ottenere un sistema miscelato in modo omogeneo.
O OCH2CH2(CF2CF2)3F F O OCH2CH2(CF2CF2)3F p(F) AIBN TFT, 65 °C O O(CH2CH2O)5OCH3 P3 AIBN diglima, 65 °C O O(CH2CH2O)5CH3 p(P3)
30
Figura 3.3- Schema di reticolazione del macro-monomero (metacrilossipropil)
metilsilossano-co-dimetilsilossano.
La conversione dei gruppi reattivi metacrilici in funzione del tempo di esposizione ai raggi UV è stata investigata mediante spettroscopia FT-IR, al fine di scegliere un tempo di reazione tale per cui si avesse una conversione pressoché totale delle funzioni metacriliche
RISULTATI E DISCUSSIONE
31
polimerizzabili. In Figura 3.4 è riportata la sovrapposizione degli spettri FT-IR registrati per la miscela S100 sottoposta ad irraggiamento per tempi crescenti, da un minimo di 20 ad un massimo di 40 minuti. In ogni caso, si riconoscevano i segnali caratteristici dello scheletro principale del polidimetilsilossano: 2969 cm1 ( CH del CH3), 1261 cm1 ( CH3 di SiCH3), 1088 cm1 e 1018 cm1 ( SiOSi) e 800 cm1 ( Si-C e CH3). Inoltre, era possibile osservare la banda di stiramento a circa 1720 cm1 del C=O dei gruppi esterei derivanti dalle funzionalità metacriliche delle catene laterali. Mentre i primi segnali FT-IR rimanevano nell’intervallo di tempo esaminato essenzialmente invariati per intensità e per frequenza di assorbimento, il secondo diminuiva di intensità e si spostava di frequenza da 1720 cm1 a 1735 cm1. Ciò era dovuto al procedere della polimerizzazione con conseguente perdita della coniugazione elettronica C=C-C=O nell’unità metacrilica. Tuttavia, un ingrandimento della regione spettrale compresa tra 1650 cm1 e 1500 cm1 ci permetteva di evidenziare significative differenze nella miscela prima e dopo l’irraggiamento. In particolare, si notava che, già dopo 20 minuti di esposizione alla lampada, il segnale a 1639 cm1, dovuto allo stiramento del doppio legame C=C, era completamente sparito ad indicare una conversione pressoché totale dei gruppi funzionali metacrilici. Gli spettri registrati a tempi di irraggiamento più lunghi rimanevano sostanzialmente invariati, presentando solo una leggera diminuzione dell’intensità della banda compresa tra 1640 e 1540 cm1
. Per queste ragioni, è stato scelto un tempo di esposizione intermedio di 30 minuti.
32 4000 3800 3600 3400 3200 3000 2800 2600 2400 2200 2000 1800 1600 1400 1200 1000 800 600 0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 A Numero d'onda (cm-1) S100 t0 S100 t20' S100 t30' S100 t40' 1520 1540 1560 1580 1600 1620 1640 1660 0,00 0,02 0,04 0,06 0,08 0,10 A Numero d'onda (cm-1) S100 t0 S100 t20' S100 t30' S100 t40'
Figura 3.4- a) Sovrapposizione degli spettri FT-IR della miscela S100 registrati per diversi tempi di
RISULTATI E DISCUSSIONE
33
Il macro-monomero polifunzionale S è stato, quindi, foto-polimerizzato alternativamente con il co-monomero ossietilenico P3 (o P9,5) o fluorurato F per dare film polimerici bi-componenti del tipo SxP3 (o SxP9,5) o SxF, dove x indica la percentuale in peso di S nella miscela (Tab. 3.1). Sono stati inoltre preparati film polimerici derivanti dalla polimerizzazione di miscele tri-componente costituite da S, P3 (o P9,5) e F di seguito denominate SxP3(o P9,5)Fm/n, dove x e m/n indicano, rispettivamente, la percentuale in peso di S nella miscela il rapporto ponderale P/F nella restante porzione (1 x), cioè 70/30 (7/3), 50/50 (5/5), 30/70 (3/7) (Tab. 3.2).
Una schematizzazione delle reazioni di fotopolimerizzazione per produrre i film reticolati a base di S, P e F viene riportata nella Figura 3.5.
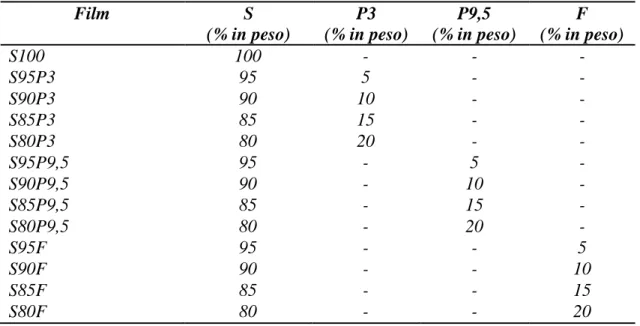
Tabella 3.1- Composizione delle miscele bi-componente
Film S (% in peso) P3 (% in peso) P9,5 (% in peso) F (% in peso) S100 100 - - - S95P3 95 5 - - S90P3 90 10 - - S85P3 85 15 - - S80P3 80 20 - - S95P9,5 95 - 5 - S90P9,5 90 - 10 - S85P9,5 85 - 15 - S80P9,5 80 - 20 - S95F 95 - - 5 S90F 90 - - 10 S85F 85 - - 15 S80F 80 - - 20
34
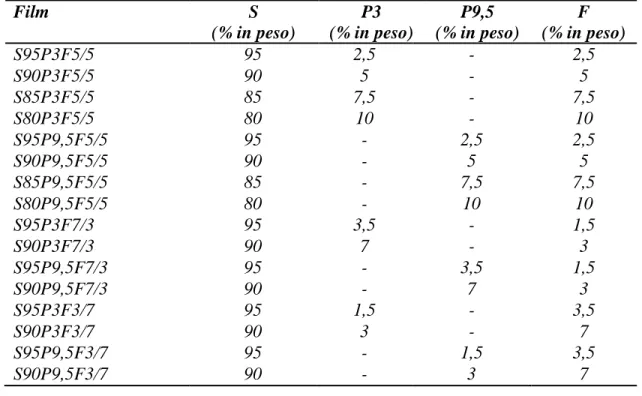
Tabella 3.2- Composizione delle miscele tri-componente
Film S (% in peso) P3 (% in peso) P9,5 (% in peso) F (% in peso) S95P3F5/5 95 2,5 - 2,5 S90P3F5/5 90 5 - 5 S85P3F5/5 85 7,5 - 7,5 S80P3F5/5 80 10 - 10 S95P9,5F5/5 95 - 2,5 2,5 S90P9,5F5/5 90 - 5 5 S85P9,5F5/5 85 - 7,5 7,5 S80P9,5F5/5 80 - 10 10 S95P3F7/3 95 3,5 - 1,5 S90P3F7/3 90 7 - 3 S95P9,5F7/3 95 - 3,5 1,5 S90P9,5F7/3 90 - 7 3 S95P3F3/7 95 1,5 - 3,5 S90P3F3/7 90 3 - 7 S95P9,5F3/7 95 - 1,5 3,5 S90P9,5F3/7 90 - 3 7
Figura 3.5- Rappresentazione schematica della foto-polimerizzazione di una miscela tri-componente
(in nero S, in verde: F e in blu: P3 o P9,5).
In generale, il contenuto dei co-monomeri ossietilenico e fluorurato è stato variato in un intervallo di composizione relativamente stretto (1,5 20% in peso) al fine di modulare l’anfifilia del sistema, pur cercando di mantenere le proprietà meccaniche elastomeriche della matrice silossanica. Le condizioni sperimentali adottate per la foto-polimerizzazione delle miscele erano del tutto simili a quelle utilizzate per S100, caratterizzate da un rapporto ponderale monomeri/iniziatore 100/1 ed un tempo di irraggiamento alla lampada UV di 30
RISULTATI E DISCUSSIONE
35
minuti. A causa della scarsa miscibilità tra i vari monomeri coinvolti, in particolare tra S e P3 (o P9,5) e tra F e P3 (o P9,5) è stata necessaria l’aggiunta di una piccola quantità di solvente (300 l/g di etil acetato) per ottenere miscele omogenee. In analogia con quanto precedentemente osservato per S100, l’analisi FT-IR confermava che 30 minuti di esposizione erano un tempo sufficientemente lungo per ottenere una reazione completa dei gruppi (met)acrilici (Fig. 3.6).
a) 3500 3000 2500 2000 1500 1000 500 0,0 0,5 1,0 1,5 2,0 2,5 A Numero d'onda (cm-1) S90P3 t0 S90P3 t30
36 b) 3500 3000 2500 2000 1500 1000 500 0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8 2,0 2,2 A Numero d'onda (cm-1) S90F t0 S90F t30 c) 3500 3000 2500 2000 1500 1000 500 0,0 0,5 1,0 1,5 2,0 2,5 3,0 A Numero d'onda (cm-1) S90P3F5/5 t0 S90P3F5/5 t30
Figura 3.6- Sovrapposizione degli spettri FT-IR registrati prima (t0) e dopo 30 minuti (t30’) di
RISULTATI E DISCUSSIONE
37
I film derivanti dalle miscele bi-componente SxP3 si presentavano opachi anche per contenuti percentuali piuttosto bassi di P3 del 5% in peso. Questo suggeriva che in seguito alla reazione di polimerizzazione si verificava una separazione di fase dovuta probabilmente all’incompatibilità chimica tra le catene silossaniche e quelle ossietileniche. L’aumento del contenuto di P3 ( 15% in peso) portava alla formazione di film con superfici macroscopicamente rugose (Fig 3.7 (a)). Questi fenomeni erano ancor più accentuati nel caso del sistema contenente il co-monomero P9,5, per il quale era possibile ottenere un film con una superficie apparentemente liscia solo per contenuti inferiori al 10% in peso (Fig 3.7 (b)). Diversamente, le miscele SxF originavano film perfettamente trasparenti e caratterizzati da una superficie liscia, per qualunque contenuto di F (Fig 3.7 (c)). Tuttavia, all’aumentare di F il film si irrigidiva diventando sempre più fragile e quindi difficile da maneggiare. Tale fragilità era indice di un minor carattere elastomerico del film della miscela rispetto a quello della sola matrice silossanica. Per queste ragioni, solo le miscele bi- e tri-componente contenenti percentuali ponderali di P3 (o P9,5) e F minori o uguali al 10% in peso sono state scelte per la preparazione dei rivestimenti da sottoporre alla valutazione biologica con diversi organismi vegetativi e su di esse è stata inoltre focalizzata la caratterizzazione termica, meccanica e di superficie.
(a) (b)
(c)
38
I campioni per le prove dinamico-meccaniche sono stati preparati per foto-reticolazione delle miscele corrispondenti in piastre di petri in PTFE. In questo modo si ottenevano film polimerici “free-standing” dello spessore di circa 1 mm. Per gli studi di bagnabilità e XPS e per le prove biologiche, i film sono stati preparati mediante deposizione della corrispondente miscela su vetrini porta-oggetto e successiva foto-reticolazione. I rivestimenti così ottenuti avevano spessori di 400 500 m. In tutti i casi, i film venivano essiccati a temperatura ambiente per 24 ore e ricotti a 120 °C per 12 ore al fine di favorire la migrazione all’interfaccia polimero-aria delle co-unità fluorurate a bassa energia superficiale.
Per evitare possibili fenomeni di delaminazione del rivestimento polimerico in seguito a prolungata esposizione all’acqua e a forze di taglio relativamente elevate, i supporti di vetro dei film da sottoporre alle prove biologiche avevano ricevuto preventivamente un opportuno trattamento che prevedeva: i) un lavaggio a 80 °C con soluzione piranha (H2O2/H2SO4 conc. 30/70 vol/vol) per ripristinare i gruppi ossidrilici sulla superficie e ii) funzionalizzazione con 3-(metacrilossipropil)trimetossisilano per introdurre i gruppi reattivi metacrilici ancorati alla superficie (Fig. 3.8). In seguito ad irraggiamento UV, queste funzionalità potevano quindi reagire con quelle dei co-monomeri nella miscela, legando covalentemente il film reticolato al substrato di vetro. Preme, tuttavia, sottolineare che questa tecnica di funzionalizzazione si è rivelata solo parzialmente efficace in quanto alcuni dei campioni sottoposti alle prove biologiche davano comunque luogo a delaminazione (vedi ¶ 3.7.2.1). Ciò era probabilmente dovuto ad un grado di funzionalizzazione della superficie troppo basso, che non consentiva un fissaggio forte e stabile del film al vetro.
.