69
3. PDK1 inhibitors
3-Phosphoinositide dependent protein kinase-1 (PDK1) is part of the PI3K/Akt pathway which regulates many growth factors and oncogenes. PDK1 signals to a number of proteins shown to be important in cancer, for example, S6K and Akt. PDK1 is reported to phosphorylate Akt on residue Thr308 thereby resulting in its activation. Catalytically active Akt promotes cancer progression through suppression of apoptosis and stimulation of proliferation, metabolism and angiogenesis. Consequently PDK1 inhibitors may be promising oncology drugs and several medicinal chemistry efforts aimed at generating potent PDK1 inhibitors have recently been published.
3.1. Bisindolylmaleimides
Bisindolylmaleimides (BIM, 3,4-di-1H-indol-3-yl-1H-pyrrole-2,5-dione) have been reported as scaffolds for various ATP-competitive PK inhibitors, and among them compounds with activity against PDK11.
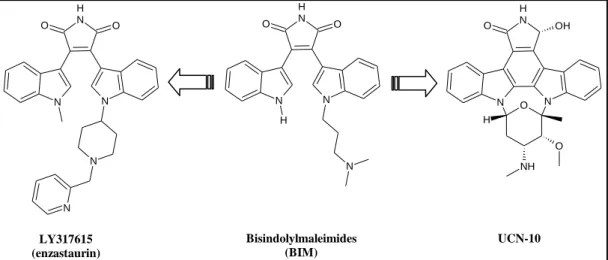
N N N H O O N N N N N H O O H N N N N H O H OH O O NH LY317615 (enzastaurin) Bisindolylmaleimides (BIM) UCN-10
Figure 3.1: Structure of BIM and analogs
However, this compound have been developed primarily as highly potent PKC inhibitors with IC50 values in the low nanomolar range, but they have also been
70 shown to block further PK such as MSK1, MAPKAPK1a, S6K1, Chk1, GSK-3b, and AMPK in a panel of 29 kinases2.
In 2005 researchers at Eli Lilly & Co. patented LY317615 (enzastaurin), which is useful for treating prostate cancer and Akt-mediated diseases3. Therein, enzastaurin was characterized as a dual inhibitor of PDK1 (EC50=370 nm) and p70S6K (EC50<500 nm). Recently, a phase II clinical study was published in which enzastaurin was demonstrated as a PKC inhibitor with therapeutic potential against large B-cell lymphoma4.
Compared with BIM , UCN-01 possesses a planar 5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-5-one scaffold (identical to the core of staurosporine) and showed much higher potency against PDK1 (IC50= 6 µm)5.
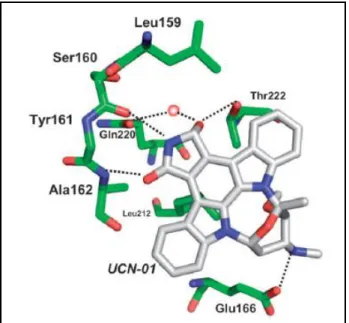
Figure 3.2:Binding mode of UCN-01 in the ATP binding pocket of PDK1
Analysis of the UCN-01–PDK1 complex relative to BIM–PDK1 complexes suggests a putative favourable energy conformation for planar UCN-01 (and staurosporine) in the ATP binding pocket of PDK16.
71
3.2. Indolinones
In 2006 researchers at Bayer Schering Pharma AG patented a class of indolinone derivatives as PDK1 inhibitors. These compounds possess the (3Z)-3-(1H-pyrrol-2-ylmethylene)-1,3-dihydro-2H-indol-2-one scaffold which is also the core structure of sunitinib, a multi-kinase inhibitor7.
Unsubstituted core 1 blocked Akt2 activation in the low micromolar range and was initially identified as a lead structure by high-throughput screening using a PDK1-mediated Akt2 activation assay (cAKT2)8. Compound 1 was determined to inhibit PDK1 in vitro with an IC50 value of 0.52 µM. It also blocked the activation of Akt in tumour cells. However, this compound (1) was nine-fold less potent against the highly homologous PKA (IC50=16 µM). The optimization of this lead structure resulted in potent single-digit nanomolar PDK1 inhibitors with selectivity over PKA (Table 3.1). N H R2 N H R1 O Compound R1 R2 IC50(nM)a 1 SO2NH2 H 290 2 SO2NH2 Me 67 3 SO2NH2 Et 14 4 SO2NH2 Ph 29 5 NHCONH2 H 18 6 NHCONH2 Et 3 7 NHCONH2 4-Pyr 10 8 (BX-517) NHCONH2 Me 5 9 OH Me 80
Table 3.1. Derivatives of 1 and their biological activity as cAkt2 inhibitors.
a
72 In particular, compound 8 (also referred to as BX-517) was reported to be a highly potent PDK1 inhibitor. This compound is 320-fold selective over PKA and 100-fold selective (or better) against a panel of seven Ser/Thr and Tyr kinases9.
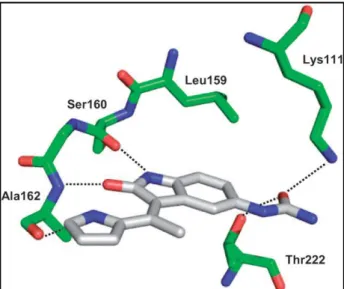
An X-ray crystallographic analysis of BX-517 in the ATP binding pocket of PDK1 disclosed the binding mode at the molecular level.
In particular the pyrrole–indolinone core is involved in three key H bonds. The indolinone nitrogen interacts with the Ser160; the indolinone oxygen accepts an H bond from Ala162. The pyrrole nitrogen addresses an H bond to the carbonyl group of Ala162.
The 5-urea group accepts an H bond directly from both the side chains of Lys111 and the hydroxyl function of Thr222.
Figure 3.3:Binding mode of BX-517 in the ATP binding pocket of PDK1
This Thr222 interaction may account for a degree of selectivity for PDK1, as it was also shown to be unique for the binding mode of UCN-01 in contrast to staurosporine in PDK15. The greater potency of BX-517 for PDK1 over the closely related PKA may be explained by the bulky gatekeeper Met120 in PKA. This residue is directed into the ATP binding pocket and thereby presumably causes clashes with the urea moiety of the inhibitor, in contrast to the less bulky Leu159 in PDK1 that provides space to accommodate BX-517. In line with this notion, BX-517 showed similar nanomolar activity in the PDK1 assay and in the PDK1-mediated Akt2 activation assay (IC50=5 nM), and it blocked Akt2 activation in cells with sub-micromolar potency. However, BX-517 has a poor ADME profile that includes a short half-life,
73 low metabolic stability, and poor solubility in aqueous media, thus prohibiting further development9.
The binding mode of BX-517 in PDK1 suggested substitution of the 4’-pyrrole moiety by hydrophilic groups, as the pyrrole moiety is situated in a solvent-accessible site of the ATP binding pocket. This position was therefore used to enhance water solubility and to optimise pharmacokinetic properties. Subsequently, medicinal chemistry efforts led to potent compounds (10–17, Table 3.2) in which 17 showed superior overall profiles in enzyme and cellular assays as well as improved pharmacokinetic properties9. N H N H NH O R N H2 O
Compound R cAkt2 IC50(nM)a p-Akt IC50(µM)b
10 H 20 3-10 11 Ph 21 - 12 O Me 19 0.01-0.1 13 OH O 45 >10 14 NH2 O 5 >1 15 NH O N Me Me 4 0.1-1.0 16 NH O N 4 1.0 17 N 8 0.1-1.0
Table 3.2. Second-generation indolinones and their biological activity as Akt2 inhibitors
a
74 However, 17 was only two-fold selective for PDK1 over PKA compared with the 320-fold selectivity of lead structure 1. No further profiling in a protein kinase panel of 18 or related compounds was reported9.
3.3. Pyridinonyl-PDK1 inhibitors
Researchers at Sunesis Pharmaceuticals/ Biogen Idec Inc. recently patented a series of substituted pyridinonyls as potent PDK1 inhibitors10. The concept of these inhibitors typically consists of using a flexible linker to tether 1-(3,4-difluorobenzyl)-2-oxo-1,2- dihydropyridine-3-carboxamide to an H bond donor (HD)/acceptor (HA) moiety that is presumably able to interact with Ala162 and Ser160 of the PDK1 hinge region.
Figure 3.4: Schematic representation of the concept of pyridinonyl PDK1 inhibitors. Typically
1-(3,4-difluorobenzyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (right) is tethered by a flexible linker (L) to a hinge binding moiety (HBM) bearing vicinal H bond donor (HD) and H bond acceptor (HA) groups.
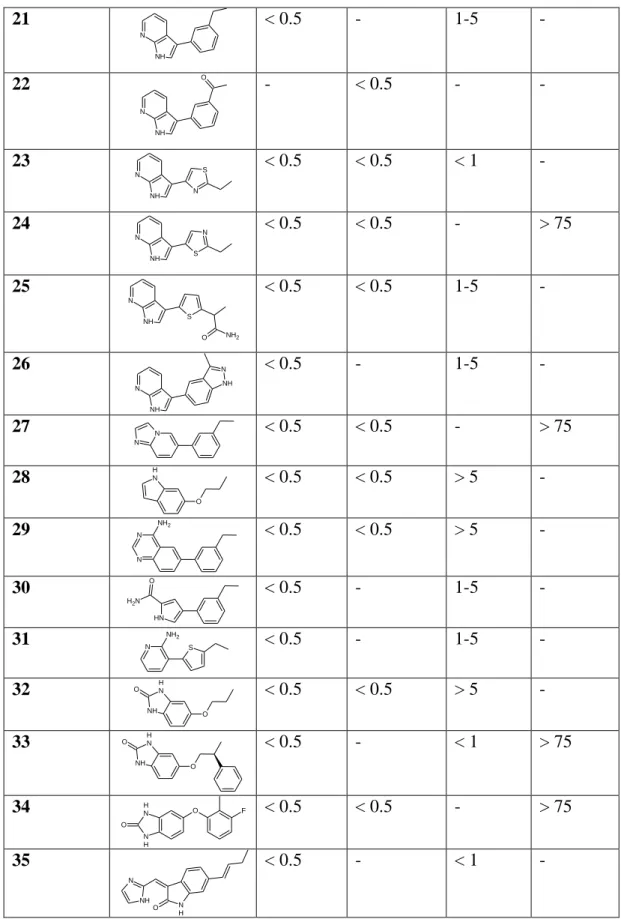
Examples of pyridinonyl inhibitors (18-35) and their biological properties are shown in Table 3.3. N O O N R H F F Compound R p-PDK1a IC50(µM) dp-PDK1b PC-3 Cell linec Westernd 18 N NH < 0.5 < 0.5 > 5 - 19 N NH < 0.5 < 0.5 - - 20 N N NH < 0.5 - 1-5 -
75 21 N NH < 0.5 - 1-5 - 22 N NH O - < 0.5 - - 23 N NH N S < 0.5 < 0.5 < 1 - 24 N NH S N < 0.5 < 0.5 - > 75 25 N NH S O NH2 < 0.5 < 0.5 1-5 - 26 N NH N NH < 0.5 - 1-5 - 27 N N < 0.5 < 0.5 - > 75 28 N H O < 0.5 < 0.5 > 5 - 29 N N NH2 < 0.5 < 0.5 > 5 - 30 N H O N H2 < 0.5 - 1-5 - 31 N NH2 S < 0.5 - 1-5 - 32 NH N H O O < 0.5 < 0.5 > 5 - 33 NH N H O O < 0.5 - < 1 > 75 34 NH O F N H O < 0.5 < 0.5 - > 75 35 O N H NH N < 0.5 - < 1 -
Table 3.3. Examples of 1-(3,4-difluorobenzyl)-2-oxo-1,2-dihydropyridine-3-carboxamides in the
inhibition of PDK1 and Akt.
a
Inhibition of phosphorylated PDK1. b Inhibition of dephosphorylated PDK1. c Inhibition of P308 Akt in PC-3 cells using an assay by MSD. d Decrease in cellular levels of Akt P308 in PC-3 cells by Western blot analysis.
76 Interestingly, to determine the biological activity of the compounds in this study, both a phosphorylated and a dephosphorylated PDK1 kinase assay, together with two different cellular techniques for measuring p-Akt (Thr308) phosphorylation were used (Table 3.3). The potent in vitro p-PDK1 inhibition elicited by compounds translates into significant decreases in p-Akt (Thr308) phosphorylation in cells detected by Western blotting. However, these compounds show a varied profile in terms of dephosphorylated PDK1 inhibition and p-Akt (Thr308) activity as determined by a phospho-Akt 308 ELISA technique11.
3.4. Imidazo[4,5-c]quinolines
Novartis AG patented in 2005 a class of substituted imidazo-[4,5-c]quinolines (Table 3.4) as potent PK inhibitors, particularly against PDK112.
N N N N R2 R1 R1 N Compound R1 R2 PDK1 IC50(nM)a 36 H H 34 37 H Me 110 38 H Et 327 39 Me H 150 40 Me Me 245 41 N N N N O Me Me N Me > 25000 42(NVP-BEZ235) N N N N Me Me N Me O Dual PI3K/mTOR inhibitor
Table 4. Examples of imidazo[4,5-c]quinolines and their biological activity as PDK1 inhibitors.
a
77 During the course of the medicinal chemistry programme the potentially metabolic weak methylcyano-moiety was replaced, leading to compound 40 showing decreased activity against PDK1. Further modifications of 36 by changing the imidazole ring to N-methylimidazolinone 41 resulted in a complete loss of PDK1 inhibition. Importantly, the compounds were tested in an in vitro PDK1 assay at an ATP concentration of 10 µM. The N-methylimidazolinone core can also be found in the dual PI3K/mTOR inhibitor 42 (also referred to as NVP-BEZ235, which recently entered phase I clinical trials)13.
3.5. Quinazolines
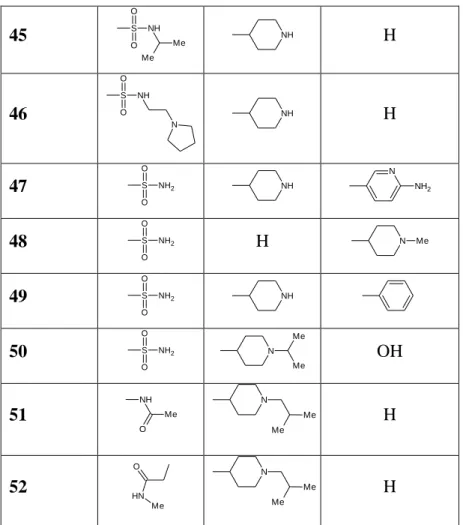
Novartis Vaccines & Diagnostics reported in 2007 on quinazoline derivatives (N-arylquinazolin-2-amines, scaffold 43) as inhibitors of PDK1, CDK1, and CDK214. Biologically active examples 43–52 of this compound class with IC50 values <5 µM in the PDK1 enzyme assay and EC50 values < 5 µM in a cell proliferation assay (A2780, PC-3, or PC-3MM cells) are presented in Table 3.5. Generally the N-arylquinazolin-2-amine scaffold is decorated with a combination of various functionalities including sulfonamides, piperidines, and aromatic moieties. Because no selectivity data concerning detailed IC50 values for PDK1 or other kinases were reported, it is not possible to reveal SAR.
N N N H O R2 R3 R1 Compond R1 R2 R3 43 S O O NH2 NH H 44 S O O N O N Me Me H
78 45 S O O NH Me Me NH H 46 S O O NH N NH H 47 S O O NH2 NH N NH2 48 S O O NH2 H N Me 49 S O O NH2 NH 50 S O O NH2 N Me Me OH 51 NH Me O N Me Me H 52 HN Me O N Me Me H
Table 3.5. Examples of N-arylquinazolin-2-amines with IC50 values < 5 µM against PDK1.
3.6. Dibenzo[c,f][2,7]naphthyridines
Researchers at Wyeth patented in 2007 a class of dibenzo[c,f]-[2,7]naphthyridines15 and in the same year published the discovery of this class of compounds as potent and selective PDK1 inhibitors16.
Compound 53 was identified by HTS as a lead structure with an IC50 value of 0.06 µM against PDK1, and the compound was shown to potently inhibit the growth of tumour cells.
The physicochemical properties of the poorly water-soluble lead 56 were improved by the introduction of functional groups in the side chain of the rigid dibenzo[c,f]-[2,7]naphthyridine scaffold (Table 3.6, 53–67).
79 N N R1 R2 R3 R5 R4 Compound R1 R2 R3 R4 R5 IC50(nM)a 53 NH2 Me NH2 OMe OMe 0.06 54 H H H OMe OMe 7.45 55 NH2 H NH2 OMe OMe 0.67 56 H Me NH2 OMe OMe 0.26 57 OH Me NH2 OMe OMe 0.04 58 Cl Me NH2 OMe OMe 2.12 59 NH2 Me NH2 F OMe 0.58 60 NH2 Me NH2 O N NH OMe 0.29 61 NH2 Me NH2 Me N N H Me OMe 2.49 62 NH 2 Me NH2 N H N Me Me OMe 0.97 63 NH2 Me NH2 N O O OMe 0.06 64 NH2 Me NH2 O N Me Me OMe 0.20 65 NH2 Me NH2 Me O Me OMe 1.18 66 NH2 Me NH2 O Cl OMe 0.04
67 OMe Me NH2 OMe OMe 0.05
Table 3.6. Examples of dibenzo [c,f] [2,7]naphthyridines and their biological activity as PDK1
inhibitors
a
The compounds were tested in an in vitro PDK1 assay at an ATP concentration of 10 µm.
80 Comparison between 64 (IC50= 0.2 µM, N-derivative) and 65 (IC50=1.18 µM, C-derivative) indicates a significant impact that a basic nitrogen or an alkyl chloride 66 (IC50=0.043 µM) situated in the side chain has on activity.
Replacement of the 3’-NH2 function (53 IC50=0.06 µm) by a 3’-methoxy group (67 IC50=0.05 µM) was tolerated.
3.7. PDK1 activators and modulators
These compounds do not target the ATP binding site, but instead target the regulatory hydrophobic motif pocket (HM, also referred to as PIF in the case of PDK1) and may have the ability to activate the enzyme allosterically. In PDK1, the HM/PIF pocket docks the HM of substrate kinases such as RSK, p70S6K, and SGK only when they are phosphorylated. This interaction not only provides docking for the substrates, but it also activates PDK1, in turn enabling ATP binding and phosphorylation of these substrate kinases17,18.
In 2004 researchers at Phosphosites GmbH reported small molecule as novel kinase regulators19. Subsequently, five compounds that possess the (3-oxo-1,3-diphenylpropyl) thioacetic acid scaffold have been published and were shown to be selective PDK1 modulators (Table 3.7)20.
R2 R1 S O O R4 R3 O * Compound R1 R2 R3 R4 PDK1a S6K1a PKAa 68 Cl H H H 234 109 100 69 Cl H H Me 109 109 109 70 Cl Cl H H 223 101 109 71 H Cl H H 110 93 91 72 Cl Cl Cl H 109 105 102
Table 3.7. Examples of (3-oxo-1,3-diphenylpropyl)thioacetic acid derivatives and their biological
activity as PDK1 modulators.
a
81 PDK1/PIF-mediated substrate docking of SGK and S6K is required to trigger their own phosphorylation. However, it is expected that an acidic moiety present in the PIF pocket would inhibit their binding and thus activation by PDK1. In line with this hypothesis, specific PDK1- catalyzed phosphorylation of S6K1 and SGK1 was blocked by 68 in an enzyme assay, as well as in HEK293 cells at a concentration of 200 µM.
Taken together, the (3-oxo-1,3-diphenylpropyl)thioacetic acid compounds are simultaneously activators of PDK1 and inhibitors of the PIF substrate kinases S6K1 and SGK1, and can therefore be considered as modulators of PDK1. Activation of S6K is induced by nutrients via the mTOR pathway, which triggers negative feedback loops that inhibit the insulin signaling pathway. These compounds could be useful in treating obesity-induced insulin resistance; however, the clinical use of these compounds remains to be evaluated.
3.8. Derivative of the natural compound
Recently has been identified one derivative of the natural compound inositol 1,3,4,5,6- pentakisphosphate (InsP5), named 2-O-Bn-InsP5, which possesses enhanced pro-apoptotic and anti-tumour activity compared to the parent molecule InsP5.
Figure 3.5:Derivative of the natural compound inositol 1,3,4,5,6- pentakisphosphate (InsP5).
Kinase profiling assays on almost 60 different kinases revealed that this compound potently and specifically inhibits PDK1 in vitro and the PDK1-dependent phosphorylation of Akt Thr308 in cell lines. This compound showed to be active also
82 in vivo assay21. These results are particularly important considering that this represents the first specific and selective PDK1 inhibitor and make it an interesting new molecule to use as a model to design novel specific PDK1 inhibitors. It has been proposed that inositol phosphates can bind PDK1 PH domain and retain this kinase in the cytosol, preventing Akt phosphorylation at Thr30821.
83
Reference chapter 3
1. C. Peifer, T. Stoiber, E. Unger, F. Totzke, C. Schächtele, D. Marmè, R. Brenk, G.
Klebe, D. Schollmeyer, G. Dannhardt. J. Med. Chem. 2006, 49, 1271–1281.
2. J. R. Graff, A. M. McNulty, K. R. Hanna, B. W. Konicek, R. L. Lynch, S. N.
Bailey, C. Banks, A. Capen, R. Goode, J. E. Lewis, L. Sams, K. L. Huss, R. M. Campbell, P. W. Iversen, B. L. Neubauer, T. J. Brown, L. Musib, S. Geeganage, D. Thornton, Cancer Res. 2005, 65, 7462–7469.
3. J. R. Graff (Eli Lilly and Co.), WO 2005/041953, 2005.
4. M. J. Robertson, B. S. Kahl, J. M. Vose, S. De Vos, M. Laughlin, P. J. Flynn, K.
Rowland, J. C. Cruz, S. L. Goldberg, L. Musib, C. Darstein, N. Enas, J. L. Kutok, J. C. Aster, D. Neuberg, K. J. Savage, A. LaCasce, D. Thornton, C. A. Slapak, M. A. Shipp, Clin. Exp. Metastasis J. Clin. Oncol. 2007, 25, 1741–1746.
5.D. Komander, G. S. Kular, J. Bain, M. Elliott, D. R. Alessi, D. M. F. Van Aalten, Biochem. J. 2003, 375, 255–262.
6. S. Bartlett, G. S. Beddard, R. M. Jackson, V. Kayser, C. Kilner, A. Leach, A.
Nelson, P. R. Oledzki, P. Parker, G. D. Reid, S. L. Warriner, J. Am. Chem. Soc.
2005, 127, 11699–11708.
7. S. Faivre, G. Demetri, W. Sargent, E. Raymond, Nature Rev. Drug Discovery 2007, 6, 734–745.
8. I. Islam, J. Bryant, Y. L. Chou, M. J. Kochanny, W. Lee, G. B. Phillips, H. Yu, M.
Adler, M. Whitlow, E. Ho, D. Lentz, M. A. Polokoff, B. Subramanyam, J. M. Wu, D. Zhu, R. I. Feldman, D. O. Arnaiz, Bioorg. Med. Chem. Lett. 2007, 17, 3814–3818.
9.I. Islam, G. Brown, J. Bryant, P. Hrvatin, M. J. Kochanny, G. B. Phillips, S. Yuan, M. Adler, M. Whitlow, D. Lentz, M. A. Polokoff, J. Wu, J. Shen, J. Walters, E. Ho, B. Subramanyam, D. Zhu, R. I. Feldman, D. O. Arnaiz, Bioorg. Med. Chem. Lett.
2007, 17, 3819–3825.
10. K. E. Lind, K. Cao, E. Y.-S. Lin, T. B. Thinh, B. T. Tangonan, D. A. Erlanson,
K. Guckian, R. L. Simmons, W.-C. Lee, L. Sun, S. Hansen, N. Pathan, L. Zhang (Sunesis Pharmaceuticals), WO 2008/005457, 2008.
11. S. M. Gowan, A. Hardcastle, A. E. Hallsworth, M. R. Valenti, L. J. K. Hunter, A.
K. De Haven Brandon, M. D. Garrett, F. Raynaud, P. Workman, W. Aherne, S. A. Eccles, Assay Drug Dev. Technol. 2007, 5, 391–401.
12. H.-G. Capraro, P. Furet, C. Garcia-Echeveria, F. Stauffer (Novartis AG), WO
2005/054238, 2005.
84
14. S. Ramurthy, X. Lin, S. Subramanian, A. C. Rico, X. M. Wang, R. Jain, J. M.
Murray, S. E. Bashman, R. L. Warne, W. Shu, Y. Zhou, J. Dove, M. Aikawa, P. Amiri, W. Wang, J. M. Jensen, A. S. Wagman, K. B. Pfister, S. C. Ng (Novartis Vaccines & Diagnostics, Inc.), WO 2007/117607, 2007.
15. A. Gopalsamy, M. Shi, K. Kutterer, K. T. Arndt (Wyeth), US 2007/0135429, 2007.
16. A. Gopalsamy, M. Shi, D. H. Boschelli, R. Williamson, A. Olland, Y. Hu, G.
Krishnamurthy, X. Han, K. Arndt, B. Guo, J. Med. Chem. 2007, 50, 5547–5549.
17. R. M. Biondi, P. C. F. Cheung, A. Casamayor, M. Deak, R. A. Currie, D. R.
Alessi, EMBO J. 2000, 19, 979–988.
18. R. M. Biondi, D. Komander, C. C. Thomas, J. M. Lizcano, M. Deak, D. R.
Alessi, D. M. F. Van Aalten, EMBO J. 2002, 21, 4219–4228.
19. R. Biondi, M. Engel (Phosphosites GmbH), EP 1486488A1, 2004.
20. M. Engel, V. Hindie, L. A. Lopez-Garcia, A. Stroba, F. Schaeffer, I. Adrian, J.
Imig, L. Idrissova, W. Nastainczyk, S. Zeuzem, P. M. Alzari, R. W. Hartmann, A. Piiper, R. M. Biondi, EMBO J. 2006, 25, 5469–5480.
21. Falasca, M.; Chiozzotto, D.; Godage, H.Y.; Mazzoletti, M.; Riley, A.M.; Previdi,



![Table 4. Examples of imidazo[4,5-c]quinolines and their biological activity as PDK1 inhibitors](https://thumb-eu.123doks.com/thumbv2/123dokorg/7569309.111422/8.892.233.726.541.1118/table-examples-imidazo-quinolines-biological-activity-pdk-inhibitors.webp)
![Table 3.6. Examples of dibenzo [c,f] [2,7]naphthyridines and their biological activity as PDK1 inhibitors](https://thumb-eu.123doks.com/thumbv2/123dokorg/7569309.111422/11.892.174.782.93.1075/table-examples-dibenzo-naphthyridines-biological-activity-pdk-inhibitors.webp)
