56
2. PI3K/PTEN/mTOR pathway inhibitors
PI3K-PTEN-mTOR pathway plays a critical role in proliferation and survival in tumor cells and is also linked with resistance to radiotherapy, chemotherapy, endocrine therapy and novel anticancer therapies. In this setting, this pathway seems to bring together all the characteristics of a good target for the treatment of cancer.
Figure 2.0: Therapeutic targeting of the Akt pathway including Akt itself as well as its upstream
regulators and downstream effectors
2.1 RTK Inhibitors
Among RTKs, EGFR and Her2/Neu/ErbB2 are frequently altered in human cancer and primarily activate Akt as target for anticancer drug discovery the PI3K/Akt pathway. Two major approaches have been used to target the ErbB family, that is, small molecule tyrosine kinase inhibitors and humanized antibodies against the receptor extracellular domains1. In general, antibodies bind to the extracellular
57 domain of the receptors, inhibiting their activation by ligand, and promoting receptor internalization and downregulation, whereas small molecules competitively inhibit ATP binding to the receptor, thereby hindering autophosphorylation and kinase activation. At present, the most advanced of the newer therapies in clinical development are anti-EGFR monoclonal antibody IMC-C225 (cetuximab, Erbitux; Imclone), anti-ErbB2 and the reversible small-molecule inhibitors of EGFR, ZD-1839 (gefitinib, Iressa; AstraZeneca) and OSI-774 (erlotinib, Tarceva; OSI Pharmaceuticals). N N O O N O HN F Cl N N O O O N H O ZD-1839 OSI-774
Figure 2.1: Structure of RTK inhibitors
Both ZD-1839 and OSI-774 have been through phase I and phase II trials. Promising single-agent clinical antitumor activity has been reported in advanced NSCLC, head and neck cancer and prostate carcinoma2,3.
2.2 PI3K Inhibitors
Due to the high sequence homology between the four class I PI3K catalytic domains and the conserved topology of the ATP-binding site across the human kinome, the design of small-molecule PI3K inhibitors that exhibit a sufficient level of isoform selectivity (δ and/or γ vs. α and β) and crosskinase selectivity to enable oral treatment of chronic inflammatory diseases presents a formidable challenge4.
2.2.1 Inhibitors derivatives from natural products
The steroidal furan wortmannin was isolated in 1957 from Penicillium
wortmanni, and its structure was fully elucidated through x-ray crystallography by
researchers at Sandoz laboratories. Although the potent anti-inflammatory properties of compound had been known for some time, it was not until 1993 that its molecular target was determined to be PI3K5.
58 Further biochemical studies revealed that wortmannin functions as a mechanism-based inhibitor by forming an irreversible covalent adduct between C-20 of its strained, electrophilic furan ring and a lysine residue in PI3K that is normally involved in the phosphate transfer reaction.
O O O O O O O O H
Figure 2.2: Structure of Wortmannin
In the case of PI3Kγ, this residue is Lys833, and the covalent interaction can be clearly observed in the cocrystal structure of compound bound to porcine PI3Kγ. Several oxygen atoms in the steroidal skeleton also engage the enzyme through hydrogen bonds: the ketone oxygen of the D-ring interacts with Val882 in the hinge region of the ATP pocket; the A-ring carbonyl interacts with the Ser806 hydroxyl group; and both the B-ring ketone and the furan oxygen interact with Asp964. Since all of these residues are conserved within the class I PI3K family, it is not surprising that wortmannin is unable to discriminate between the four isozymes (IC50s at 50 μM
ATP: PI3K α = 4.0 nM; β = 0.7 nM; δ = 4.1 nM; γ = 9.0 nM). At higher concentrations wortmannin also crossreacts with myosin light-chain kinase (MLCK) and PI4K, as well as several PI3K-related kinases, such as mTOR, DNA-PK, ATM, and ATR. Although compound and related analogues have helped to define the role of PI3K in numerous intracellular signaling pathways, their development as viable therapeutic agents has been limited by poor aqueous solubility and stability (through hydrolytic ring opening of its furan), as well as high in vivo toxicity6.
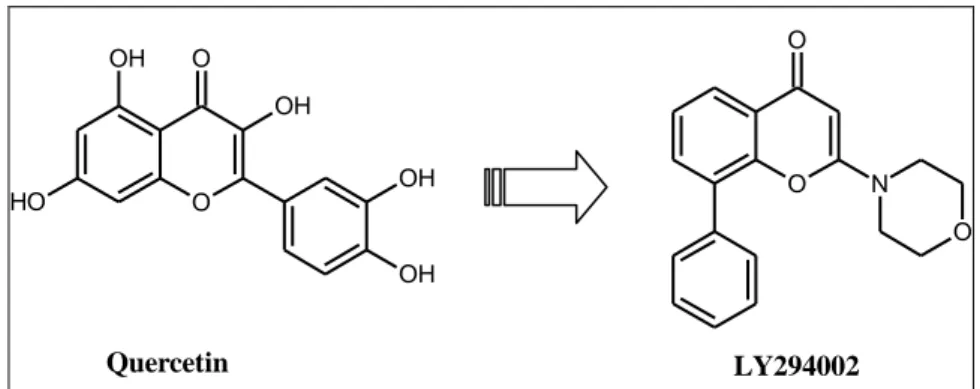
The naturally occurring flavanoid quercetin is a pan-kinase inhibitor with moderate affinity for PI3K (50 μM ATP; PI3Kα IC50 = 3.8 μM)7.
Researchers at Lilly explored substitution around the chromone core of quercetin and identified a morpholine moiety as a suitable replacement for the catechol ring. The lead compound from this study, LY294002, exhibits similar PI3K inhibition (20μM ATP; PI3Kα IC50 = 1.4 μM) as quercetin but also demonstrates significantly
59 improved cross-reactivity against off-target kinases (i.e. 50 μM of LY294002 does not inhibit PI4K, PKC, PKA, MAPK, EGFR, or c-Src).
O O N O O O OH O H OH OH OH Quercetin LY294002
Figure 2.3: Natural product PI3K inhibitors (Quercetin) and related analogue(LY294002)
Due to its chemical stability and non-covalent mechanism of action, LY294002 is often used in place of wortmannin to investigate the role of PI3K in various cellular processes. However, the results of these experiments should be carefully analyzed since recent studies have revealed that the overall selectivity of LY294002 towards class I PI3Ks, PI3K-related kinases, and numerous protein kinases may not be as high as originally though8.
In addition to steroid and flavanoid, the alkaloid staurosporine is a natural-product inhibitor of PI3K (Kd for PI3Kγ = 290 nM). A cocrystal structure of staurosporine bound to human PI3Kγ revealed that the lactam of the oxindole mimics the adenine ring of ATP by forming a two-point interaction with the backbone of Val882 in the hinge region of the enzyme. Unfortunately, this mode of binding is also accommodated by numerous protein kinases9.
N N N H O O N H O
60
2.2.2 PI3K δ inhibitors
Researchers at ICOS have disclosed a series of quinazolinone PI3Kδ-selective inhibitors that bear structural characteristics reminiscent of the xanthine alkaloids. A well characterized compound from this series10, IC87114 exhibits high
in vitro selectivity (100- to 1000-fold) for PI3Kδ over the other PI3K isozymes (IC50s
at 10 μM ATP: PI3K δ = 0.13 μM; α > 200 μM; β = 16 μM; γ = 61 μM), does not inhibit several related lipid kinases (IC50 > 100 μM for class II and class III PI3Ks,
PI4Ks, PIKKs, PIPKs), and is remarkably clean against a panel of 36 diverse protein kinases (<20% inh at 10 μM). This unique selectivity profile has enabled researchers to utilize IC87114 as a tool for deciphering the role of PI3Kδ in leukocytes11.
N N N N O CH3 CH3 O R N N N N N N N H2 O R: H Theophylline R: CH3 Caffeine IC87144 Figure 2.5: Quinazolinones and related PI3K δ inhibitors
2.2.3 PI3K γ inhibitors
Boehringer Ingelheim has focused their efforts on tricyclic 2-aminothiazoles12 and most recently disclosed a series of thiazolyl-dihydro-indazoles (1) with activity against PI3Kγ (IC50s < 600 nM)13.
61 N S N N NH O Cl NH O N Compound 1
Figure 2.6: Structure of 2-Aminothiazole PI3Kγ inhibitors.
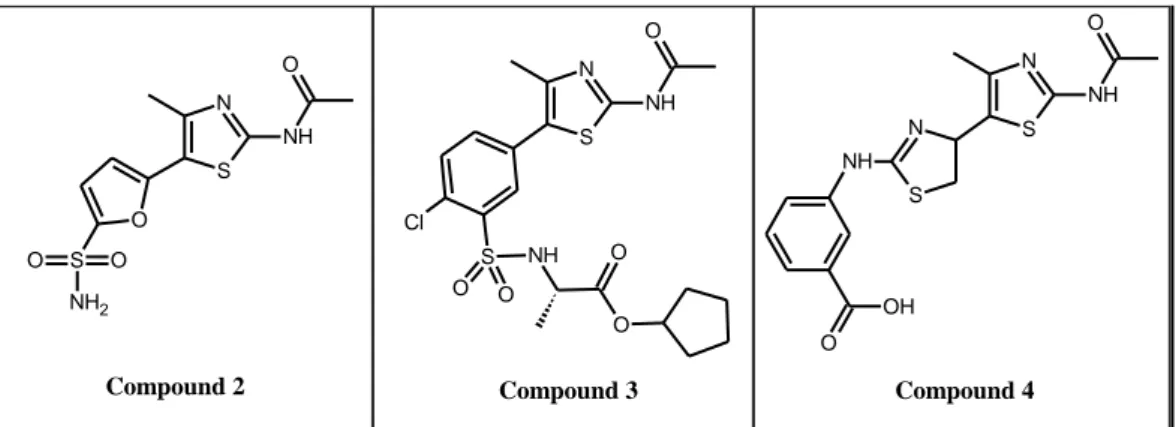
Additional 2-amidothiazoles have been reported by Vertex (2)14 and Chroma (3)15 with activities in a PI3Kγ enzymatic assay of IC50 < 0.10 μM and 0.10 < IC50 < 1.0
μM, respectively. Serono pharmaceutical16
has also described aminothiazoles as PI3Kγ inhibitors, of which bis-thiazole 4 is shown as a representative example.
N S O NH O S O O NH2 N S NH O Cl S NH O O O O N S NH O N S NH O OH
Compound 2 Compound 3 Compound 4
Figure 2.7: Structure of 2-Aminothiazole PI3Kγ inhibitors (Compound 2, 3, 4)
2.3 mTOR Inhibitors
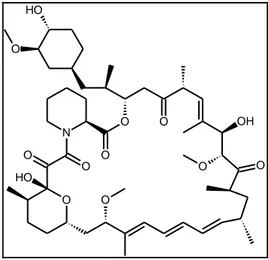
The only specific small molecule inhibitors of mTOR reported to date are rapamycin and its derivatives. Rapamycin, an allosteric inhibitor of mTOR, is a white crystalline solid that is insoluble in aqueous solutions, a property that has prevented development of a parenteral formulation17.
62 O O O N O O H O O O H O O OH O O
Figure 2.8: Structure of Rapamycin
Rapamycin was approved by the U.S. Food and Drug Administration in the 1990s as an immunosuppressant for use following renal transplantation. Rapamycin induces G1-arrest and/or delays in cell cycle transition, and in some cell lines, leads to apoptosis. The growth inhibitory effect of rapamycin is partially mediated by suppression of cap-dependent and 5’-TOP-dependent translation18, but it also inhibits cdk activation and accelerates the turnover of cyclin D1.
However, the poor chemical stability of rapamycin has limited its clinical use, and led to the development of three synthetic analogues (rapalogs) with superior pharmacokinetic properties.
2.3.1 Analogs of Rapamycin
A number of clinical trials with the rapalogs have now been completed. CCI-779/temsirolimus (Wyeth) is a prodrug that is metabolized to rapamycin. It inhibits the growth of a wide range of cancer types and was assessed in phase I/II trials with escalating doses to determine the maximum tolerated dose level in solid tumors, such as RCC (renal cell carcinoma), NSCLC, glioblastoma, prostate, breast, and pancreatic carcinomas. Synergistic anti-tumour activity was also detected when Temsirolimus was used in combination with conventional therapies (including cisplatin and gemcitabine) or other targeted therapies19.
63 O O O N O O H O O O O O OH O O OH O H O
Figure 2.9: Structure of CCI-779/temsirolimus
RAD-001/everolimus (Novartis) is a hydroxyethyl ether derivative of rapamycin suitable for oral administration. It showed activity against several human cancer in
vitro and tumours in animal models together with anti-angiogenic properties because
of its ability to inhibit proliferation of human vascular endothelial cells. Interestingly everolimus showed inhibitory activity towards human tumour xenografts models even when the cell line was insensitive to the drug in vitro. Some activity was also observed in hematologic malignancies but only modest activity has been reported for non-small cell lung cancer patients failing chemotherapy or epidermal growth factor receptor (EGFR)-targeted treatment or patients with recurrent or metastatic breast cancer20. O O O N O O H O O O O O OH O O OH O O O N O O H O O O O O OH O O P O RAD-001/everolimus AP23573/deforolimus
64 AP23573/ deforolimus (Ariad) derives from the substitution of the C-43 secondary alcohol moiety of the cyclohexyl group of rapamycin with phosphonate and phosphinate groups. It has been developed for both oral and intravenous administration and inhibits the proliferation of several human cancer cells in vitro and tumour xenografts in conjunction with cytotoxic agents21. It is currently in Phase III22.
2.3.2 mTORC1/mTORC2 dual inhibitors (TORCdIs)
A new generation of mTOR-specific kinase inhibitors has emerged from screening and drug discovery efforts directed toward the kinase active site of mTOR. Because they block the activity of both mTOR complexes they are commonly called mTORC1/mTORC2 dual inhibitors.
The pyrazolopyrimidine compounds PP242 and PP30 are selective inhibitors of mTOR kinase23. PP242 N N N N NH2 NH O H N N N N NH2 NH O N S PP30
Figure 2.11: Structure of pyrazolopyrimidine compounds PP242 and PP30
Besides being more effective than rapamycin in achieving cytoreduction and apoptosis in leukemia and multiple myeloma cells, perhaps the most striking thing about PP242 was its effect or lack thereof on the immune system24. At therapeutic doses in leukemia models PP242 produces much weaker immunosuppression than either rapamycin or PI103, which could translate into a better therapeutic ratio in the clinic25.
65 Although these inhibitors exhibit antiproliferative activity against several cancer cell lines and a glioma xenograft model, the poor pharmacological properties prevented their further clinical development. Recently, another compound, WYE132 was reported to show single-agent anticancer activity in tumor models of brain, breast, lung and renal cancer.
WAY001 WYE132 N N N N O H N O N H N N N N N O NH NH O O O Figure 2.12: Structure of compounds WAY001 and WYE132
The data from preclinical pharmacology supported its usefulness as an anticancer agent and provided a strong rationale for clinical development26.
2.4 mTOR/PI3K dual inhibitors
Several mTOR/PI3K dual inhibitors (TPdIs) have been developed. The development of these agents has benefited from previous endeavors with PI3K-selective inhibitors. Through kinase selectivity profiling and cellular assays, it was discovered that several of these PI3K inhibitors are in fact TPdIs, exemplified by PI103 and NVPBEZ235. As a founding member of the TPdI series, PI103 was discovered using HTS followed by a medicinal chemistry campaign27.
66 N N N O O N OH PI103 N N N N O N NVPBEZ235
Figure 2.13: Structure of mTOR/PI3K dual inhibitors : PI103 and NVPBEZ235
PI103 has not been evaluated in humans because of its poor in vivo pharmacokinetics (PK), it is now widely used as a chemical probe for the PI3K/mTOR pathway and as a lead compound for generating other PI3K and mTOR inhibitors28.
Several TPdIs have entered early-stage clinical trials that yielded incomplete but promising results. NVPBEZ235, generated by structure-based design, is an orally bioavailable TPdI. It was reported to inhibit tumor growth in many preclinical models, including prostate, breast, pancreatic, and renal cancers, glioblastoma, multiple myeloma, leukemia and sarcomas, and it enhanced the antitumor activity of several other cancer drugs such as vincristine and doxorubicin. NVPBEZ235 has appropriate pharmacological features that allowed it to enter Phase I/II clinical trials in patients with advanced solid tumors, including breast cancer29.
GSK2126458 is another TPdI with a low picomolar IC50 with excellent antitumor
activity. It has a sustained pharmacodynamic (PD) effect at very low circulating drug levels. It is now in a Phase I, open-label, dose-escalation study in subjects with solid tumors or lymphoma30. N N N N NH S O O O F F GSK1226458
67
Reference chapter 2
1. Chen JS, Lan K and Hung MC. Drug Resist. Updat. 2003, 6, 129–136.
2. Mitsiades CS, Mitsiades N and Koutsilieris M. Curr. Cancer Drug Targets. 2004,
4, 235–256.
3. Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S and Arteaga CL. Cancer Res. 2002, 62, 4132–4141.
4. Sundstrom. T. J.; Anderson, A. C.; Wright, D. L. Org. Biomol. Chem. 2009, 7,
840-850.
5. Wipf, P.; Halter, R. J. Org. Biomol. Chem. 2005, 3, 2053-2061.
6. Yano, H.; Nakanishi, S.; Kimura, K.; Hanai, N.; Saitoh, Y.; Fukui, Y.; Nonomura,
Y.; Matsuda, Y. J. Med. Chem., 1996, 39 (25), 5021–5024.
7. Matter, W. F.; Brown, R. F.; Vlahos, C. J. Biochem. Biophys. Res. Commun. 1992,
31; 186(2):624-31.
8. Gharbi, S. I.; Zvelebil, M. J.; Shuttleworth, S. J.; Hancox, T.; Saghir, N.; Timms,
J. F.; Waterfield, M. D. Biochem. J. 2007, 15, 404(1): 15-21.
9. Michael K. Ameriks and Jennifer D. Venable. Curr. Top. Med. Chem. 2009; 9(8):
738-53
10. Sadhu, C.; Dick, K.; Treiberg, J.; Sowell, C. G.; Kesicki, E. A.; Oliver, A.
Quinazolinone derivatives as inhibitors of human phosphatidylinositol 3-kinase delta. WO01081346, 2001.
11. Puri, K. D.; Doggett, T. A.; Douangpanya, J.; Hou, Y.; Tino, W. T.; Wilson, T.;
Graf, T.; Clayton, E.; Turner, M.; Hayflick, J. S.; Diacovo, T. G. Immunobiol. 2004, 103, 3448-3456.
12. Brandl, T.; Maier U.; Hoffmann, M.; Scheuerer, S.; Joergensen, A. T.; Pautsch,
A.; Breitfelder, S.; Grauert, M.; Hoenke, C.; Erb, K.; Pieper, M.; Pragst I. Thiazolyl-dihydroquinazolines. WO2007115929, 2007.
13. Grauert, M.; Maier, U.; Hoffman, M.; Scheuerer, S.; Joergensen, A. T.; Pautsch,
A.; Brandl, T.; Hoenke, C.; Breitfelder, S.; Klaus, E.; Pieper, M.; Pragst, I. Thiazolyl-dihydro-indazole. US20090093474, 2009.
14. Wang, T.; Green, J.; Cornebise, M.; Ledford, B.; Parsons, J.; Tanner, A.;
Westcott, J. Inhibitors of phosphatidylinositol 3-kinase. WO2008027584, 2008.
15. Moffat, D. C. F.; Davies, S.; Alesso, S. M.; Launay, D. F. M.: WO2007129048, 2007.
68
16. Quattropani, A.; Pomel, V.; Rueckle, T.; Grippi-Vallotton, T. Thiazole
derivatives and use thereof. WO2007082956, 2007.
17. Vignot S, Faivre S, Aguirre D and Raymond E. Ann Oncol. 2005; 16: 525-537.
18. Hay N and Sonenberg N. Genes Dev. 2004; 18: 1926-1945.
19. Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, Leister C,
Korth-Bradley J, Hanauske A and Armand JP. J Clin Oncol. 2004; 22: 2336-2347.
20. Houghton, P.J. Everolimus. Clin. Cancer Res., 2010, 16(5), 1368-1372.
21. Tsang, C.K.; Qi, H.; Liu, L.F.; Zheng, X.F. Drug Discov Today, 2007, 12(3-4),
112-124.
22. Dancey, J. Nat. Rev. Clin. Oncol., 2010, 7(4), 209-219.
23. Apsel, B. et al. Nat. Chem. Biol. 2008, 4, 691–699.
24. Hoang, B. et al. Blood. 2010, 116, 4560–4568.
25. Janes, M.R. et al. Nat. Med. 2010, 16, 205–213.
26. Yu, K. et al. Cancer Res. 2010, 70, 621–631.
27. Workman, P. et al. Nat. Biotechnol. 2006, 24, 794–796.
28. Raynaud, F.I. et al. Mol. Cancer Ther. 2009. 8, 1725–1738.
29. Maira, S.M. et al. Mol. Cancer Ther. 2008, 7, 1851–1863.