49
3. MATERIALI E METODI
3.1
Materiali
La selezione dei pazienti da includere nel presente progetto di tesi è stata effettuata utilizzando un database informatizzato contenente i dati di oltre 550 pazienti con carcinoma del colon-retto, genotipizzati presso il laboratorio di Anatomia Patologica dell’Azienda Ospedaliera Pisana nel corso del 2011. Il criterio d’inclusione nella casistica, oggetto di questo studio, è stata l’analisi mutazionale dei codoni 12, 13 e 61 del gene KRas e dell’esone 15 del gene BRAF effettuati sia sul prelievo di tessuto primitivo che su quello metastatico.
Da una prima analisi del database è emerso che la genotipizzazione dei suddetti geni risultava incompleta per una buona parte dei pazienti; inoltre, pur essendo in uno stadio avanzato della malattia, non tutti i pazienti disponevano sia di un prelievo di tessuto tumorale primitivo che di un prelievo di tessuto metastatico. Ho proseguito, quindi, nella ricerca dei casi da inserire nel mio studio, a partire dai pazienti che disponevano di un prelievo metastatico; in dettaglio, su 556 pazienti 124 presentavano una metastasi. Pertanto, ho verificato, tramite un software di gestione dei dati clinico-patologici (WinSap) in uso presso l’Anatomia Patologica 3, se per ciascuno di questi pazienti fosse disponibile negli archivi tissutali il prelievo di tessuto tumorale primitivo. Da questa verifica è risultato che 60 pazienti disponevano di un prelievo di tessuto tumorale primitivo. In definitiva, al fine di valutare quanto esposto precedentemente nello scopo di questo elaborato, sono stati inseriti in questa casistica 60 pazienti con una neoplasia primitiva e relativa metastasi.
50
Dopo questo primo momento di selezione dei pazienti, indispensabile nella progettazione di questo lavoro, ho proseguito con l’esecuzione dell’analisi mutazionale dei geni in esame mediante tecnologia di pirosequenziamento. In alcuni casi, è stato possibile utilizzare i dati sullo stato mutazionale prodotti precedentemente a scopo diagnostico, e dove necessario è stata completata la genotipizzazione; in altri casi l’analisi mutazionale è stata effettuata ex novo. In particolare sui 60 prelievi di tessuto tumorale primitivo l’analisi mutazionale dei geni presi in esame è stata condotta integralmente a partire dall’estrazione del DNA. Invece, tenuto conto dei risultati sullo stato mutazionale ottenuti a scopo diagnostico per il prelievo metastatico, ho eseguito in 17 casi l’analisi mutazionale del gene KRas codone 12-13, in 32 casi l’analisi mutazionale del gene KRas codone 61 e in 23 casi l’analisi mutazionale del codone 600 del gene BRAF.
51
3.2
Metodi
3.2.1
Organizzazione ed analisi dei dati
L’organizzazione dei dati, una volta eseguita la selezione dei pazienti, è avvenuta mediante l’ausilio di un software statistico StatSoft STATISTICA
8.0. Per una completa analisi dei dati ho inserito nel foglio di lavoro
informazioni riguardanti il sesso e l’età al momento dell’intervento di ciascun paziente. Ho ricercato e inserito, inoltre, dati circa le caratteristiche del materiale di partenza (biopsia, istologico), le diagnosi istologiche, la percentuale di cellule neoplastiche, eventuali analisi mutazionali eseguite in precedenza. Per le diagnosi istologiche, in particolare, ho chiesto il parere dell’anatomopatologo con lo scopo di unificare, dove possibile, le diverse forme di diagnosi in modo da agevolare l’analisi dei dati epidemiologici.
3.2.2
Estrazione e purificazione del DNA
L’estrazione del DNA è stata condotta su 60 prelievi di tessuto tumorale primitivo e 32 prelievi di tessuto tumorale metastatico.
Il tessuto tumorale da cui viene estratto il DNA proviene da interventi chirurgici di rimozione, di tutta o una parte, della massa tumorale o da interventi di biopsia; fissato in formalina e incluso in paraffina (FFPE,
formalin-fixed paraffin embedded), viene tagliato in fettine di circa 10 µm e
poste su vetrino. È necessario rimuovere completamente la paraffina che include il tessuto attraverso un processo chiamato “sparaffinatura” che consiste nell’immersione dei vetrini in xilolo. Dopo questo passaggio, occorre reidratare il tessuto immergendolo in soluzioni di etanolo a
52
concentrazione decrescente (99%, 95%, 70%, 50%) e infine in acqua. Il tessuto privo di paraffina viene dissecato, cioè “grattato” con un ago sottile in modo da rimuovere dalla superficie del vetrino la regione di tessuto che contiene la maggior concentrazione di cellule tumorali. Questa regione viene appositamente selezionata dall’anatomo-patologo, che, visualizzando al microscopio il corrispondente vetrino colorato (secondo la colorazione ematossilina-eosina), individua la parte che deve essere raschiata.
I passaggi successivi prevedono l’impiego di un kit commerciale, il QIAamp DNA mini kit (QIAGEN). La sezione di tessuto macrodissecata viene sottoposta a digestione a 56° C con proteinasi K, un enzima che digerisce le membrane delle cellule, compresa quella nucleare, e le proteine intracellulari. Il tempo di digestione minimo è di tre ore. Dopo la digestione con proteinasi K, viene aggiunto 200 µl di buffer AL e il campione viene sottoposto alla digestione finale a 70° C per dieci minuti. Successivamente all’aggiunta di 200 µl di etanolo (100%) il campione viene trasferito in apposite colonnine fornite dal kit per essere sottoposto ad una serie di centrifugazioni e lavaggi con buffer specifici. In particolar modo vengono utilizzati 500 µl di buffer AW1 e 500 µl di buffer AW2 in alternanza a centrifugazioni della durata di 1-2 minuti a velocità di 8000-13000 rpm. Il passaggio finale prevede l’eluizione del DNA utilizzando 30-35 µl di buffer AE, l’incubazione per un min e la centrifugazione a 8000 rpm; il DNA estratto può essere conservato a 4° C per un breve periodo o a -20° C per periodi più lunghi.
53
3.2.3 Quantificazione e normalizzazione del DNA
Prima di poter utilizzare il DNA per qualsiasi tipo di esperimento è necessario valutare la concentrazione degli acidi nucleici e la qualità del campione. La concentrazione di DNA dei campioni viene misurata attraverso un’analisi spettrofotometrica. Lo spettrofotometro è uno strumento analitico che fornisce il valore della concentrazione di una sostanza in soluzione. Il principio scientifico si basa sull’emissione di un fascio di luce incidente la soluzione. La lunghezza d’onda del fascio
di luce e la stessa assorbibile da molecole come gli acidi nucleici, 260nm. Quando la luce attraversa la soluzione contenente il DNA, fuoriesce ad un’intensità inferiore a quella incidente, perché una parte viene assorbita dal DNA stesso. Piccoli volumi
di campione, fino ad 1 μl, sono stati caricati su una apposita piastra di lettura che crea una colonna di liquido a diretto contatto con fibre ottiche; impostato il valore dello standard (bianco), è stato possibile effettuare la
lettura dell’assorbanza e quindi stimare direttamente la concentrazione del soluto. La qualità del DNA è stata verificata con la misurazione del rapporto A260/A280. Tale rapporto deve essere compreso tra 1,8 e 2; valori più alti indicano contaminazione da proteine e sostanze organiche. In seguito alla misura spettrofotometrica, una certa quantità di DNA estratto viene diluito in un volume predefinito al fine di raggiungere concentrazione di DNA pari a 20 ng/ml.
54
3.2.4
Pirosequenziamento
Il metodo
Il pirosequenziamento costituisce, oggi, una tecnica innovativa nell’ambito del sequenziamento genomico. Il principio del metodo si basa sul sequenziamento mediante sintesi e permette di caratterizzare a livello molecolare le alterazioni delle sequenze nucleotidiche. La tecnica consente il monitoraggio della sintesi di DNA mediante il rilevamento della bioluminescenza prodotta al termine di una cascata di reazioni enzimatiche.
Il processo può essere diviso in diversi passaggi: durante la reazione di pirosequenziamento, la DNA polimerasi catalizza l’incorporazione di un dNTP per volta all’interno del filamento di DNA se è complementare alla base del filamento stampo, secondo la seguente reazione:
(DNA)n + dNTP polimerasi (DNA)n+1 + PPi
Ogni evento di incorporazione è accompagnato dal rilascio del pirofosfato in quantità equimolare a quella del nucleotide incorporato. In presenza di adenosina-5’-fosfosolfato (APS), l’ATP solforilasi converte il PPi in ATP. L’ATP, a sua volta, guida la conversione, catalizzata dalla luciferasi, della luciferina in ossiluciferina con conseguente produzione di luce di intensità proporzionale all’ATP.L’intera reazione è schematizzata di seguito:
PPi + APS solforilasi ATP + luciferina + O2 luciferasi Luce + AMP +
55
Un enzima, l’apirasi, degrada continuamente tutti i dNTP che non vengono incorporati e l’ATP in eccesso secondo le reazioni seguenti:
dNT apirasi dNDP + dNMP + Pi
ATP apirasi ADP + AMP + Pi
Non appena la degradazione è completata viene aggiunto un altro dNTP, i
dNTP vengono aggiunti ciclicamente uno alla volta. Poiché il dATP è un substrato della luciferasi, al suo posto viene utilizzata l’adenosina-tio-trifosfato (dATPS) che viene incorporata dalla polimerasi ma non viene riconosciuta dalla luciferasi. Il segnale luminoso così generato viene rilevato da una camera fotosensibile (CCD Camera) e registrato in un apposito
pirogramma. La presenza del segnale ci conferma la presenza del nucleotide
nella specifica posizione della catena, mentre l'intensità del segnale è proporzionale al numero di ripetizioni della base lungo lo stesso filamento: un impulso doppio o triplo, per esempio, è indice dell'inglobamento nello stesso ciclo di 2 dNTPs (ripetizione della stessa base per 2 o 3 volte sul templato); viceversa un segnale nullo indica che il dNTP aggiunto in quel ciclo non è presente. In questo modo è possibile ottenere la sequenza nucleotidica d’interesse e valutare le eventuali alterazioni.
Per la reazione di pirosequenziamento sono stati utilizzati dei kits commerciali “Anti-EGFR MoAb response (BRAF e KRAS status)” della DIATECH certificati CE-IVD. La sensibilità del sistema per il codone 12 del gene KRas, intesa come percentuale minima di allele mutato rilevabile, è stata validata dalla DIATECH usando diluizioni seriali di DNAKRas mutato estratto dalla linea cellulare tumorale SW620 mescolato in diverse proporzioni con DNA KRas wild-type estratto dalla linea cellulare HT29. Per la validazione della sensibilità del sistema riguardo al codone 600 di BRAF
56
sono state usate diluizioni seriali di DNA BRAF mutato estratto dalla linea cellulare tumorale SKMEL8 mescolato in diverse proporzioni con DNA BRAF wild-type estratto dalla linea cellulare SW620. In entrambi i casi i risultati hanno indicato che il sistema può rilevare il 5% di cellule mutate. La specificità del kit è stata valutata testando, per ognuno dei tre test, campioni di tessuto tumorale e non, di diversa natura e con diverso status mutazionale, già caratterizzati presso il laboratorio di ricerca della ditta.
Tra i notevoli vantaggi di questa metodologia vi è quello di ottenere un’accurata quantificazione allelica che permette un controllo oggettivo della qualità dei dati ed un facile monitoraggio della performance del saggio. In dettaglio, viene ritenuto wild-type per i codoni 12, 13 e 61 del gene KRas un campione la cui frequenza dell’allele mutato, in specifiche posizioni della sequenza nucleotidica, è inferiore al 7%; questo indica che la frequenza dell’allele mutato è inferiore al limite di sensibilità del sistema inteso nel suo insieme (selezione materiale, estrazione, amplificazione, pirosequenziamento). Il campione presenta una mutazione nello specifico codone di KRas quando la frequenza allelica supera il 10%. Nell’intervallo compreso tra il 7 e il 10% il protocollo indica di ripetere l’amplificazione in doppio e sequenziare in doppio ognuno dei due replicati per un totale di 5 risultati utili. Se dopo tale ripetizione la frequenza allelica risulta essere compresa ancora una volta nell’intervallo del 7-10% il campione risulta indeterminato e andrebbe indagato con metodi alternativi. Si ritiene wild-type per l’esone 15 del gene BRAF un campione la cui frequenza allelica, nella specifica pozione, risulta essere inferiore al 3%; il campione presenta la mutazione quando la frequenza allelica è maggiore del 5%. Anche in questo caso, per situazioni che cadono nell’intervallo 3-5% va ripetuta l’analisi e valutato l’ulteriore risultato.
57
Amplificazione end-point
La reazione a catena della polimerasi (PCR) è una reazione mediante la quale una sequenza di acido nucleico può essere amplificata esponenzialmente in
vitro. Tutti i campioni di DNA estratti da tessuto primitivo e da metastasi
sono stati amplificati utilizzando il kit commerciale TaKaRa Ex TaqTM R-PCR Custom ed EvaGreenTM Dye; l’EvaGreen è una molecola fluorescente
che, aggiunta nella miscela di reazione, permette di visualizzare istante per istante l’andamento dell’amplificazione grazie alla possibilità dello strumento di rilevare le variazioni di fluorescenza ad una determinata fase del ciclo amplificativo stabilita dall’operatore. Tale processo, noto come Real-Time PCR, essendo computerizzato ha il vantaggio che ad ogni ciclo l’intensità di fluorescenza, in aumento, viene rilevata e visualizzata su un monitor come curva ad andamento esponenziale.
La miscela di reazione della PCR viene allestita sotto cappa a flusso laminare utilizzando materiale (provette e puntali) sterilizzato, e contiene:
un tampone di reazione contenente sali, per mantenere costanti le condizioni di pH;
ioni magnesio sotto forma di MgCl2, necessari per stabilizzare i nucleotidi. una miscela contenente i quattro deossinucleotidi (dNTP),
DNA Polimerasi,
una miscela di primers per ogni codone in esame acqua
Di seguito vengono riportate le quantità di reagenti utilizzati per ciascun campione:
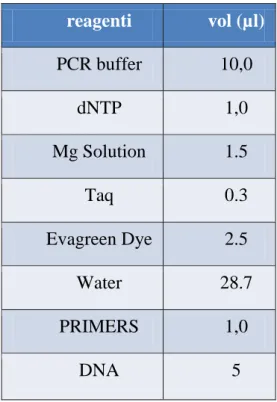
58 reagenti vol (µl) PCR buffer 10,0 dNTP 1,0 Mg Solution 1.5 Taq 0.3 Evagreen Dye 2.5 Water 28.7 PRIMERS 1,0 DNA 5
Tabella 2. Reagenti utilizzati nella reazione di amplificazione
La reazione di amplificazione avviene in un termociclatore, strumento in grado di variare la temperatura molto rapidamente. Infatti, il processo di amplificazione comprende tre fasi che si ripetono ciclicamente:
La fase di denaturazione, in cui la doppia elica di DNA si separa nelle due semieliche costituenti. La temperatura di denaturazione è stabilita in base alla lunghezza del frammento da denaturare e alla composizione in basi, da cui dipende l’energia necessaria per rompere i legami a idrogeno che tengono uniti i due filamenti.
La fase di annealing, in cui la temperatura raggiunta favorisce l’appaiamento specifico dei primers nelle regioni complementari di DNA. Durante questa fase si ha la formazione dei legami a idrogeno tra i primers e
59
le loro rispettive sequenze complementari sui filamenti denaturati. Solitamente tale temperatura di annealing è compresa tra 37-65°C e la fase di annealing ha una durata di circa 30 secondi. La temperatura rappresenta quindi un punto critico della PCR, in quanto da essa dipende sia la resa sia la specificità della reazione.
La fase di extension, in cui la DNA polimerasi catalizza la sintesi dei nuovi filamenti complementari a quelli di partenza. In particolare tale enzima utilizza come substrato per la sintesi del filamento ex novo i dideossinucleotidi (dNTP: deossinuclotide trifosfato) e procede in direzione 5’3’ dello stampo incorporando un dNTP alla volta all’estremità 3’-OH dei primers. La temperatura di extension è generalmente compresa tra 70-72°C, temperatura che dipende dal tipo di DNA polimerasi utilizzato in quanto ciascun enzima ha una sua temperatura ottimale alla quale polimerizza più velocemente.
La ripetizione ciclica di queste fasi permette un’amplificazione estremamente rapida del materiale genetico d’interesse. La regione amplificata è conosciuta con il termine di amplicone.
Per i codoni 12, 13 e 61 del gene KRas le condizioni del ciclo di amplificazione, ottimizzate sul Rotor-Gene TM 6000, sono le seguenti: 95°C per 20 secondi
57°C per 30 secondi 35 cicli 72°C per 30 secondi
Per l’esone 15 del gene BRAF le condizioni del ciclo di amplificazione, ottimizzate sul Rotor-Gene TM 6000, sono le seguenti:
60 95°C per 30 secondi
55°C per 30 secondi 40 cicli 72°C per 30 secondi
L’amplificazione dei codoni 12 e 13 del gene KRas è stata eseguita per: - 60 campioni di DNA estratto da tumore primitivo
- 17 campioni di DNA estratto da metastasi
L’amplificazione del codon 61 del gene KRas è stata eseguita per: - 60 campioni di DNA estratto da tumore primitivo
- 32 campioni di DNA estratto da metastasi
L’amplificazione dell’esone 15 del gene BRAF è stata eseguita per: - 60 campioni di DNA estratto da tumore primitivo
61
La reazione di pirosequenziamento
Per la reazione di pirosequenziamento è stato utilizzato il kit commerciale
Anti-EGFR MoAb response (BRAF status e KRAS status) della DIATECH.
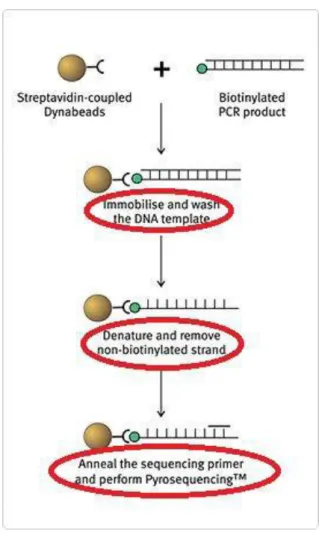
Ilprotocollo può essere schematizzato come segue:
Immobilizzazione dei prodotti di amplificazione biotinilati sulle biglie di sefarosio ricoperte da streptavidina (Streptavidin Sepharose TM High Performance) mediante agitazione per 15 minuti in una miscela di legame, la Binding Mix, che viene dispensata nei pozzeti di una Sample Preparation
Plate.
Allestimento della piastra per il sequenziamento. Questa fase prevede la preparazione di una miscela di annealing con i primers specifici che sarà dispensata nella piastra per il sequenziamento.
Denaturazione dei prodotti di amplificazione immobilizzati utilizzando il sistema accessorio PyroMark TM Vacuum Prep Workstation 220-240 V (Biotage AB )
Annealing dei primer di sequenziamento mediante l’incubazione della piastra di sequenziamento su un blocco termostatato a 80°C per 2 minuti a cui segue il raffreddamento della piastra per 5 minuti e l’alloggiamento della stessa nel pirosequenziatore.
Caricamento della cartuccia mediante dispensazione nelle posizioni predefinite dei PyroMark Gold Q96 Reagents (QIAGEN) rappresentati da enzima, substrato e nucleotidi.
Avvio della reazione di pirosequenziamento che avrà una durata di 20 minuti circa.
62
Figura 5. Fasi principali della reazione di pirosequenziamto
63
La reazione di pirosequenziamento dei codoni 12 e 13 del gene KRas è stata eseguita per:
- 59 casi di tessuto tumorale primitivo - 16 casi di tessuto tumorale metastatico
La reazione di pirosequenziamento del codone 61 del gene KRas è stata eseguita per:
- 58 casi di tumore primitivo - 29 casi di metastasi
La reazione di pirosequenziamento dell’esone 15 del gene BRAF è stata eseguita per:
- 59 casi di tumore primitivo - 22 casi di metastasi