1.1. La morte cellulare programmata: apoptosi
1.1.1. Apoptosi: un importante equilibrio cellulare
Le cellule hanno la capacità intrinseca di riuscire a gestire il delicato equilibrio tra sopravvivenza e morte: da una parte l’ingresso in divisione cellulare o la permanenza in uno stato di quiescenza, dall’altra l’apoptosi. In entrambi i casi, le cellule partecipano ativamente all’evento scelto. Per questo motivo, l’apoptosi è da considerarsi una manifestazione del funzionamento della cellula. Infati, alterazioni nel soosticato programma a poptotico o nei suoi meccanismi regolatori spostano, inevitabilmente, tale equilibrio verso un’instabilità talvolta dannosa per l’organismo che si manifesta soto f orma d i m alatie d e generative e tumorali.
1.1.1.1.
La scoperta e le diferenne con la necrosi
avanti il processo di morte; ed alla one, morendo, emete segnali che possono modiocare il comportamento delle cellule vicine, comprese quelle che la devono ingerire (per una revisione, (Arnoult, Akarid et al. 2002)).
La morte per necrosi (ogura 1.1), invece, avviene in seguito a gravi ed irreversibili danni patologici come ipossia, ipertermia, infezione virale, esposizione a vari agenti tossici o per l’atacco d el c omplemento ( Israels L ... 1 999). uesti e venti p ortano a d u na p erdita dell’integrità delle membrane degli organelli, incluso il nucleo, con conseguente rilascio del loro contenuto (ATP, proteasi e lisozimi) ono alla rotura dell’intera cellula e degradazione del DNA in modo aspecioco. Tale fenomeno, che la cellula subisce passivamente, innesca una risposta inoammatoria con rilascio di citochine da parte dei macrofagi.
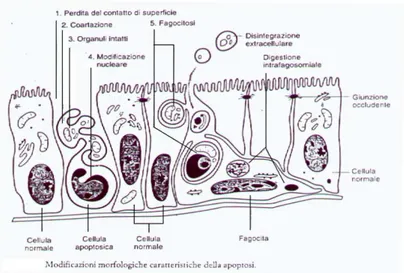
Come riassunto nella ogura 1.2, la serie di modiocazioni stereotipate che sono associate con il fenotipo dell’apoptosi vede le cellule ridurre il loro volume citosolico, condensarsi, perdere le specializzazioni di membrana (come i microvilli e le regioni specioche di contato con le altre cellule) e staccarsi dalle cellule vicine senza che ci sia una risposta inoammatoria, perchh a diferenza della necrosi, non avviene rilascio del contenuto citosolico nei tessuti circostanti.
Fig. 1.1. Principali eventi che caraterinnano la necrosi.
Una cellula che va incontro a necrosi a causa di gravi traumi subisce dapprima delle modiocazioni che sono ancora reversibili come la dilatazione di alcuni organuli; se, però, ai danni non può essere messo riparo si va incontro ad una perdita dell’integrità delle membrane degli organelli, compreso il nucleo, con
conseguente rilascio del loro contenuto (da “Patologia” vol.1, cap 3, (Wyllie 1992), Zanichelli Editore, 1995).
Fig. 1.2. Principali eventi che caraterinnano llapoptosi.
Come schematizzato, una cellula che va incontro ad apoptosi perde le specializzazioni di membrana e si stacca dalle altre. Tutavia, l’integrità degli organuli interni viene mantenuta almeno nelle fasi precoci del processo, mentre a livello nucleare avviene la disgregazione del/i nucleolo/i, la condensazione e la frammentazione della cromatina in porzioni di 180-200 paia di basi o multipli. La lunghezza di tali frammenti corrisponde a quella dei trati di DNA internucleosomale (.ranville, Carthy et al. 1998) ed il taglio è operato da endonucleasi, CAD (deossiribonucleasi ativata dalle caspasi) ative solo durante il processo apoptotico (Enari, Sakahira et al. 1998). La cromatina degradata si compata in granuli che si spostano verso la periferia del nucleo ed insieme ad altri frammenti di materiale nucleare raggiungono poi la membrana plasmatica, dove vengono circondati da evaginazioni della membrana stessa, fornendo alla cellula un aspeto simile a bolle (dall’inglese “blebbing”) che successivamente si staccano dalla cellula e vengono fagocitate dai macrofagi o da cellule vicine, senza che ci sia una reazione inoammatoria (da “Patologia” vol.1, cap 3, (Wyllie 1992), Zanichelli Editore, 1995).
Le principali carateristiche distintive tra le due forme di morte cellulare sono riassunte nella tabella 1.
(modiocata da (.ranville, Carthy et al. 1998)).
La prima evidenza dell’esistenza di un’informazione genetica specioca per il controllo della morte cellulare è stata fornita da esperimenti pionieristici sul nematode Caenorhabditis elegans (Ellis and Horvitz 1986), un metazoo la cui divergenza ologenetica precede quella umana di centinaia di milioni di anni. .li scienziati, studiando il suo sviluppo, si resero conto che il passaggio dalla forma embrionale a quella adulta prevedeva la perdita di esatamente 131 cellule. Tale processo doveva inevitabilmente prevedere un meccanismo di controllo altamente regolato. In seguito, grazie anche all’uso di mutanti genetici, è stato rivelato che la sopravvivenza o la morte delle cellule, durante il suo sviluppo, dipende dai prodoti di pochi geni; i principali sono: ced-3, ced-4, ced-9 e egl-1 (ced, dall’inglese cell death) (Meier, Finch et al. 2000).
L’apoptosi in C. elegans è divenuta modello di base per lo studio dello stesso fenomeno in altri sistemi cellulari. Nell’uomo e nel topo sono stati identiocati circa venti prodoti di geni che sono omologhi del repressore Ced-9 di morte cellulare e del suo antagonista Egl-1 (la famiglia
Si No
Necessità di ATP
Ativazione caspasi Degradazione non specioca
Proteine
Compromessi Cambiamento di permeabilità
Mitocondri
Condensazione, frammentazione e degradazione del DNA Distruto, degradazione di DNA
non specioca Nucleo
Intati Danneggiati
Bcl-2), come anche quatordici omologhi dell’esecutore Ced-3 (la famiglia delle caspasi) ed almeno un omologo di Ced-4 (Apaf-1 ovvero fatore apoptotico ativante le proteasi-1). Perciò, in contrasto al semplice paradigma di C. elegans, nelle cellule di mammifero, l’apoptosi può procedere lungo molteplici vie intracellulari molecolari. Per esempio, la via molecolare che porta all’apoptosi sarà addiritura diferente in un dato tipo di cellula, in risposta allo stesso segnale di morte, dipendendo dal particolare stato di diferenziazione della cellula. L’estensione della complessità del controllo molecolare della sopravvivenza e della morte della cellula non può essere pienamente apprezzato se non si prendono in considerazione i vari meccanismi epigenetici come lo splicing alternativo e modiocazioni p ost-traduzionali ( fosforilazioni, defosforilazioni o taglio proteolitico) che possono trasformare il prodoto di un dato gene sia in una proteina pro-apoptotica sia in una anti-apoptotica (per una revisione (Ameisen 2002)).
1.1.1.2. Apoptosi: signifcato biologico
implicata nella regolazione omeostatica dei tessuti, bilanciando il rapporto vita/morte di cellule danneggiate nell’adulto (Vaux, Haecker et al. 1994; .ranville, Carthy et al. 1998).
D’altra parte, è stata proposta la partecipazione di una difetosa regolazione dell’apoptosi nella patogenesi di molte malatie a ndando d al c ancro a lle m alatie c o me l ’ AIDS ( R udin a n d Thompson 1997) e ai disordini neurodegenerativi (per una revisione, (Ameisen 2002)) portando all’emergere di nuovi conceti r iguardo l ’intervento t erapeutico, o nalizzato a l la modulazione seletiva d ei m eccanismi c oinvolti n ella r egolazione d ella m orte e d ella sopravvivenza della cellula (Nicholson 2000).
Inone, l a m orte c ellulare p rogrammata p uò a nche e ssere p artecipe n el p rocesso d i invecchiamento regolato osiologicamente (Sastry and Rao 2000).
1.1.1.3. Llapoptosi nel sistema nervoso
Durante lo sviluppo del sistema nervoso circa la metà dei neuroni vanno incontro a fasi di regressione che terminano con la morte delle cellule stesse; questa grande perdita di cellule nervose è comune in molti tipi di neuroni di tuti gli invertebrati e sembra essersi evoluta come meccanismo adatativo durante lo sviluppo del sistema nervoso (Oppenheim 1991; Buss and Oppenheim 2004).
La sopravvivenza e lo sviluppo dei neuroni dei vertebrati dipende dalla produzione e dalla captazione di specioche neurotroone, tra le quali l’N.F (nerve grorth factor), il BDNF (brain derived neurotrophic factor) e le neurotroone ( NT)-3/4/5 l e q uali p romuovono l a
sopravvivenza e lo sviluppo specioco d ei n euroni ( Aloyz, B ammi e t a l. 1 998). I noltre, l o sviluppo delle cellule nervose richiede che queste ricevano segnali da altri neuroni con i quali instaurano appropriati contati sinaptici (Raf, Barres et al. 1993).
La mancanza di fatori di sopravvivenza può condurre alla morte; il senso di questo processo è che, tra il gran numero di neuroni prodoti durante la neurogenesi, soltanto la porzione di cellule che riceverà un adeguato supporto neurotrooco e c he i nstaurerà c orreti c o ntati, sopravvivrà e continuerà lo sviluppo mentre tute le altre cellule moriranno (Sastry and Rao 2000). D’altra parte, alcuni (Blaschke, Weiner et al. 1998) ritengono che l’apoptosi dei neuroblasti delle zone proliferative ventricolari rappresenti un fenomeno universale carateristico dello sviluppo del sistema nervoso e che sia una forma diversa da quella associata con la competizione sinaptica poichh tali neuroblasti non hanno ancora instaurato connessioni sinaptiche.
L’apoptosi sembra inoltre essere coinvolta in numerose malatie n eurodegenerative caraterizzate dalla perdita graduale di specioci gruppi di neuroni, tra le quali il morbo di Parkinson, la sclerosi laterale amiotrooca, la retinite pigmentosa, l’atrooa muscolare spinale ed il morbo di Alzheimer (Thompson 1995; ,uan and ,ankner 2000).
Una rapida descrizione di queste fasi viene riportata di seguito.
1.1.2.1.
Indunione
Numerosi fatori svolgono un ruolo chiave nell'induzione dell'apoptosi, fra questi per esempio il tratamento con TNF o farmaci citotossici, radiazioni ionizzanti e UV, shock da calore, ipossia, infezioni virali (per una revisione (Israels L... 1999)). Altri stimoli importanti sono la rimozione dei fatori di crescita presenti nel siero (Petmann and Henderson 1998) e la riduzione di disponibilità di molecole di ATP. Se il calo di ATP è massiccio ed improvviso la cellula muore per necrosi mentre se è pii moderato, muore per apoptosi (Leist, Single et al. 1997). L’ATP è inoltre necessario per l’ativazione d ella p rocaspasi-9 e q uindi p er l a condensazione e la frammentazione nucleare nelle fasi onali del processo (.ranville, Carthy et al. 1998)
.
1.1.2.2.
Esecunione
La fase di esecuzione sembra essere comune a tute o quasi le vie d’innesco ed è costituita da una serie di reazioni enzimatiche a cascata in un certo modo paragonabili a quelle del complemento o della coagulazione del sangue che, una volta innescate, procedono automaticamente portando inevitabilmente la cellula alla morte; è in questa fase che entrano in gioco le caspasi esecutrici.
Nei tessuti, i corpi apoptotici vengono rapidamente fagocitati dalle cellule circostanti e/o dai macrofagi e degradati all'interno dei lisosomi. Sono stati onora identiocati tre diversi sistemi mediante i quali i fagociti riconoscono i corpi apoptotici. A seconda del tipo cellulare e del fagocita interessato possono essere utilizzati il recetore delle asialoglicoproteine, quello della vitronectina (VnR)/CD36, o quello della fosfatidilserina (PS) (Savill and Fadok 2000), come riassunto in ogura 1.3. Si pensa che la scelta del meccanismo di riconoscimento usato dai fagociti sia infuenzato, oltre che dal tipo cellulare coinvolto, anche da stimoli provenienti dal microambiente. uale che sia la modalità con cui le cellule ed i corpi apoptotici vengono riconosciuti e fagocitati, il risultato onale è sempre quello di un'eliminazione pulita , senza sconvolgimento dell'architetura s truturale d e l t e ssuto e s e nza i n nesco d i u n p r ocesso inoammatorio.
Fig. 1.3. Riconoscimento di una cellula apoptotica da parte dei macroragi.
Recetore d elle a sialoglicoproteine: s ulle m embrane d i a lcuni t ipi c ellulari ( es. e patociti) d urante i l processo apoptotico vengono esposti alti livelli di glicoproteine desialilate che vengono riconosciute dalle cellule circostanti mediante il recetore delle asialoglicoproteine.
Recetore della vitronectina (VnR)/CD36: è il sistema pii complesso e meglio caraterizzato, utilizzato dai macrofagi derivati dai monociti. Al sistema di riconoscimento partecipano tre molecole: i) Trombospondina (TSP) molecola trimerica, adesiva, prodota e secreta dal macrofago, che funziona da ponte molecolare tra corpo apoptotico e complesso VnR/CD36; ii) VnR: presente sulle membrane del macrofago, è un eterodimero che appartiene alla famiglia delle integrine, molecole di membrana implicate nelle funzioni di adesione e ancoraggio cellulare; iii) CD36: molecola di superocie, tipicamente espressa dai monociti, dai macrofagi e dalle piastrine, la cui funzione osiologica non h ancora ben determinata. Recetore della fosfatidilserina (PS): normalmente i residui di PS sono situati sulla faccia citoplasmatica della membrana; nelle cellule apoptotiche l'asimmetria del foglieto l ipidico v iene p ersa i n s eguito all'ativazione di un enzima specioco ( scramblase ) che trasloca i residui di PS sulla faccia esterna della membrana (da (Savill and Fadok 2000).
1.1.3.
Le proteine coinvolte
A seconda dello stimolo apoptotico, il processo di morte prevede l’atuazione di proteine che possono essere raggruppate in due grandi famiglie distinte: quella delle caspasi e quella delle proteine Bcl-2 (B-cell lymphoma gene 2) (Breckenridge and Xue 2004; Ward, Kogel et al. 2004; Kim 2005; Lindsten, Zong et al. 2005; Shiozaki and Shi 2004).
1.1.3.1.
Le caspasi
Le caspasi appartengono ad una famiglia di cistein- proteasi citosoliche evolutivamente conservate, già a partire dal nematode C. Elegans ono all’uomo (Marks and Berg 1999). Esse rappresentano il cuore della messa in opera del “macchinario” apoptotico.
Le caspasi sono state classiocate in tre gruppi, i n b ase alla sequenza bersaglio (.orman, Orrenius et al. 1998; Strasser, O'Connor et al. 2000) ed al ruolo osiologico (Fuentes-Prior and Salvesen 2004):
.ruppo I (caspasi ativatrici di citochine): comprende le caspasi -1, -4, -5, -11, -12, -13, -14 ICE-simili, coinvolte in processi inoammatori ed apoptotici, le quali sono specioche per la sequenza Trp-.lu-Hist-Asp.
.ruppo II (caspasi iniziatrici dell'apoptosi): comprende le caspasi -6, -8, -9, -10 diretamente coinvolte nel processo apoptotico con speciocità per sequenze X-.lu-Hist-Asp/ X-.lu-Thr-Asp.
.ruppo III (caspasi efetrici dell'apoptosi): comprende le caspasi -2, -3, -7, CED-3-simili, diretamente coinvolte nel processo apoptotico, specioche per sequenze Asp-.lu-X-Asp.
Recentemente, nei mammiferi è stata identiocata a nche l a c aspasi-15, c he s embra e ssere implicata nell’ativazione sia di Bid che di caspasi-3 (Eckhart, Ballaun et al. 2005).
Le caspasi nella cellula si trovano come forme zimogene, con bassa atività biologica. Nella forma di procaspasi sono costituite da tre domini: un prodominio N-terminale, un dominio p20 di peso molecolare maggiore ed uno p10 di peso molecolare inferiore (Hengartner 2000).
L’ativazione avviene in conseguenza del taglio proteolitico tra il prodominio e p20 e tra p20 e p10. La forma ativa è costituita da due eterodimeri p20/p10 legati a dare un tetramero con due
procaspasi-8 e, la bassa atività biologica delle forme zimogene unite insieme, è suuciente ad ativarle reciprocamente;
- Ativazione per associazione con proteine regolatrici: è il caso della procaspasi-9 e della procaspasi-12.
Le caspasi agiscono modiocando processi biologici vitali per le cellule, provocandone la morte per apoptosi. Esse possono modulare l’atività di proteine regolatrici di meccanismi necessari alla sopravvivenza cellulare; inducono la degradazione del DNA in modo indireto (Enari, Sakahira et al. 1998) legandosi ad un inibitore (ICAD) normalmente legato ad una endonucleasi ativata da caspasi (CAD). La caspasi-3 scinde il legame ICAD-CAD, rendendo l’endonucleasi libera di agire. La CAD è responsabile della formazione di frammenti di DNA di 180 pb, o multipli interi, evidenziabili, mediante gel di agarosio, come un ladder nucleosomale. L’ativazione delle caspasi provoca cambiamenti morfologici nelle cellule. ueste assumono una forma tondeggiante con evaginazioni della membrana simili a bolle (blebbing). La perdita della normale strutura c ellulare è p resumibilmente c ausata d all’idrolisi d i p roteine citoscheletriche, come la gelsolina, l’actina e la laminina dell’involucro nucleare (Marks and Berg 1999).
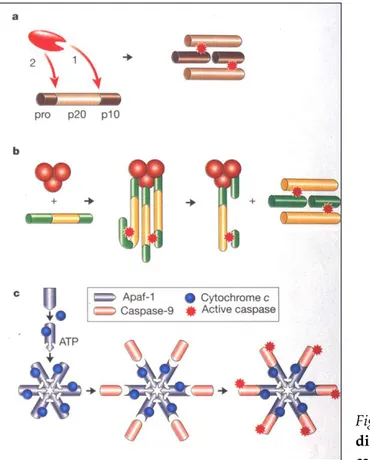
Fig. 1.4. Meccanismo
di ativanione d elle caspasi. Il meccanismo
di ativazione della caspasi include: taglio proteolitico mediato da altre caspasi (a); ativazione mediante contato (b); ativazione mediante un oloenzima (c). Ulteriori spiegazioni nel testo (Hengartner 2000).
1.1.3.2.
La ramiglia delle Bcl-2
Le proteine appartenenti alla famiglia di Bcl-2 assumono un ruolo importante nella regolazione della trasduzione del segnale apoptotico nelle cellule. Fino ad oggi sono stati identiocati 22 membri classiocati s ulla b ase d i s equenze -elica c onservate, conosciute come domini di
omologia Bcl-2 (BH) (Scorrano and Korsmeyer 2003). Per semplicità, sono stati distinti in due gruppi, in base ai tipi di domini che contengono. Come illustrato nella ogura 1.5, il primo gruppo, che comprende gli elementi che inibiscono l’apoptosi, come Bcl-2 e Bcl-XL, contiene quatro domini BH (da BH1 a BH4). Il secondo, che raccoglie gli elementi proapoptotici, è diviso a sua volta in due classi, proteine con tre domini BH (come Bax e Bak) e proteine con un solo dominio BH3 (Bad, Bid, Bim, Bik, Bmf) (Martin and Vuori 2004). La classe BH3 opera bloccando l’azione delle proteine anti-apoptotiche (Bcl-2, Bcl-XL) o ativando le proteine pro-apoptotiche Bax e Bak (Ward, Kogel et al. 2004).
Numerosi studi hanno rivelato che una sovraespressione di Bcl-2 aumenta la sopravvivenza dei neuroni simpatici in assenza di N.F (la deprivazione di N.F induce apoptosi) ed inoltre protegge dall’apoptosi indota d a r adiazioni i onizzanti, f armaci c hemioterapici, i possia e d infezioni virali. Proteine anti-apoptotiche come Bcl-2 possono impedire il rilascio del citocromo c oppure sequestrare la proteina Apaf-1, bloccando l’ativazione della procaspasi-9 (.ranville, Carthy et al. 1998; Fadeel, Zhivotovsky et al. 1999);(Kim 2005).
Fig. 1.5. Strutura dei domini dei membri della ramiglia di Bcl-2. Schema dei membri apoptotici ed antiapoptotici della famiglia di Bcl-2. Le posizioni dei domini BH sono solamente approssimative; TM: dominio transmembrana (modiocata da (Martin and Vuori 2004).
Le proteine di questa famiglia hanno una localizzazione dipendente dal ruolo svolto. L’elemento anti-apoptotico Bcl-2 è situato sopratuto s u lle m e mbrane d e gli o r ganelli ( r eticolo endoplasmatico, nucleo e la membrana esterna del mitocondrio). Bax, invece, è situata principalmente nel citoplasma, ancorata alle struture citoscheletriche (per una revisione, (Desagher and Martinou 2000)). .li stimoli apoptotici determinano la dimerizzazione di Bax
scoprendo il residuo C-terminale idrofobico che può quindi intercalarsi tra i lipidi di membrana (Suzuki, ,oule et al. 2000). Bax è struturalmente simile ad alcune tossine bateriche che formano dei pori ed è stato dimostrato che può formare dei canali in lisosomi. La sovraespressione di Bax o l’applicazione di Bax ai mitocondri isolati provoca rilascio di citocromo c dal mitocondrio anche in assenza di segnali apoptotici (Scorrano and Korsmeyer 2003). Bax e Bak possono agire sul reticolo endoplasmatico controllando l’omeostasi del calcio (Scorrano, Oakes et al. 2003).
1.1.4. Vie apoptotiche
L’apoptosi può essere indota d a m olteplici s timoli, s ia i ntra- c he e xtra-cellulari, i q uali ativano d iverse v ie (ogura 1 .6). La via estrinseca richiede la presenza di recetori n ella membrana citoplasmatica e coinvolge la caspasi-8; la intrinseca è incentrata sul ruolo svolto dai mitocondri e coinvolge la caspasi-9 (Hengartner 2000). Un’altra via è caraterizzata dalla ativazione della caspasi-2. Recentemente, sono state individuate vie che coinvolgono il reticolo endoplasmatico: una di queste è indipendente dall’azione delle caspasi, un’altra porta all’ativazione della caspasi-12 (ogura 1.9)
Fig 1.6. Schema delle vie che portano alllapoptosi. L’apoptosi può essere ativata da diversi segnali, in risposta ai quali vengono ativate le diverse vie (vedere testo). ueste sono dete (da sinistra a destra) estrinseca, intrinseca, dipendente da caspasi-2, indipendente da caspasi (da (Orrenius 2003).
(Nakagara, Zhu et al. 2000); (Orrenius 2003).
1.1.4.1.
Via estrinseca
Il legame ad un recetore (ogura 1.7), come ad esempio TNF ( tumor necrosis factor ), può determinare l’ativazione d ella c aspasi-8 e d i nnescare l a v ia e strinseca ( primo s chema d a sinistra in ogura 1.6). uesto è un processo complesso che richiede la trimerizzazione del recetore, reclutando il dominio di morte (DD, “Death Domain”) e due proteine adatatrici (TRADD e FADD/MORT1). La proteina FADD/MORT1, a sua volta, contiene un dominio che lega la procaspasi-8 ativandola (Hengartner 2000; Strasser, O'Connor et al. 2000).
La via mitocondriale (secondo schema da sinistra in ogura 1.6) è ativata in risposta sia ad insulti interni (come il danneggiamento del DNA) che a segnali extracellulari o alla mancanza di fatori di crescita: tuti i segnali elencati convergono sui mitocondri e conducono al rilascio del citocromo c, della proteina secondaria ativatrice di caspasi (SMAC), del fatore inducente l’apoptosi (AIF) che è legato alla condensazione della cromatina e della endonucleasi . la cui funzione non è stata ancora chiarita (Orrenius 2003). Implicati in questo processo sono i membri della famiglia Bcl-2 pro-apoptotici, in particolare Bax, Bak, Bid e Bim. uesti traslocano dal citosol (dove sono inativi) a gli o rganuli c ellulari d ove s ono a tivati v i a proteolisi, defosforilazione o altri meccanismi. Una volta localizzati sulla membrana mitocondriale esterna, i membri pro- competono con quelli anti-apoptotici nella formazione di canali atraverso cui verranno rilasciate le varie proteine.
Una volta rilasciato, il citocromo c si associa con la proteina fatore a tivante p r oteasi apoptotiche (Apaf)-1, con la procaspasi-9 e probabilmente con altre proteine, formando l’apoptosoma, complesso in grado di ativare l a c aspasi-9. L ’azione dell’apoptosoma viene antagonizzata da una famiglia di inibitori di caspasi chiamati inibitori dell’apoptosi (IAPs), ma questi, a loro volta, sono inibiti da SMAC/DIABLO e Htra/Omi (Breckenridge and Xue 2004). Il funzionamento dell’apoptosoma è regolato anche dalla oncoproteina pro-timosina- (Pro-T) e dal soppressore di tumore putativo associato alla proteina HLA-DR (PHAP) (Orrenius 2003).
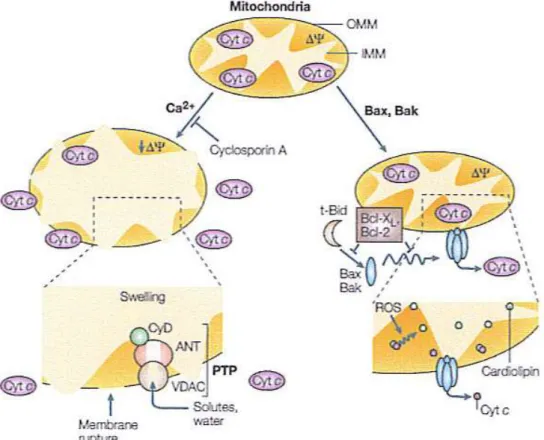
La ogura 1.8 illustra due dei probabili meccanismi con cui il citocromo c viene rilasciato nel citosol. Uno dei meccanismi coinvolge l’apertura di pori di permeabilità transitoria (PTP), costituiti da canali anionici voltaggio-dipendenti (VDCA), da traslocatore di nucleotidi di adenina (ANT), da cicloolina D (CyD), eprobabilmente da altre proteine. uesti PTP possono
lasciar passare soluti e molecole delle dimensioni di 1,5 kDa. L’apertura dei pori porta ad un rigonoamento della matrice, alla rotura della membrana mitocondriale esterna (OMM) e al rilascio del citocromo c (Cyt c) e di altre proteine già citate. Possono avvenire anche aperture transienti dei pori là dove una piccola frazione di mitocondri subisce una variazione di permeabilità in un dato momento. In questo caso, il rilascio delle proteine mitocondriali avviene senza rigonoamento di ampia entità o perdita del potenziale mitocondriale ((Δ) nell'intera popolazione (non mostrato). I pori transienti possono aprirsi in seguito alla diminuzione di potenziale di membrana mitocondriale. In entrambi i casi il processo è antagonizzato dalla ciclosporina A, che si lega a CyD e previene la formazione dei pori.
Un altro meccanismo di permeabilizzazione della membrana mitocondriale esterna coinvolge i membri della famiglia di proteine Bcl-2 (Bax, Bak e t-Bid). Il citocromo normalmente è legato alla membrana mitocondriale interna atraverso un fosfolipide anionico deto cardiolipina. La cardiolipina si trova soltanto nelle membrane mitocondriali e sopratuto, s e n o n esclusivamente, in quella interna. La dissociazone del citocromo c dalla cardiolipina è un importante primo passo per il rilascio del citocromo c nel citosol. Tale rilascio può essere stimolato dalla produzione di specie reative dell’ossigeno (ROS) così come dal legame del Ca2+ alla cardiolipina (Orrenius 2003).
Fig 1.8. Meccanismi di rilascio delle proteine dello spanio intermembrane del mitocondrio (da (Orrenius 2003)).
Recenti studi hanno dimostrato il coinvolgimento dei membri pro-apoptotici Bax e Bak nella regolazione del Ca2+ cellulare (Petronilli, Penzo et al. 2001; Scorrano, Oakes et al. 2003). Bax e Bak traslocano dal citosol al reticolo endoplasmatico (dove causano diminuzione delle scorte di Ca2+ del reticolo) ed ai mitocondri (nei quali aumentano la capacità di captare il Ca2+ e causano variazioni della permeabilità) (Petronilli, Penzo et al. 2001).
1.1.4.3.
Via dipendente da caspasi-2
L’ativazione da procaspasi-2 a caspasi-2 (secondo schema da destra in ogura 1.6), dovuta a danni del DNA, porta al rilascio di citocromo c e alla formazione dell’apoptosoma, in modo del tuto s imile a lla v ia i ntrinseca, c on m eccanismi n on d el t uto c h iari ( O rrenius 2 0 03; Philchenkov 2004).
1.1.4.5. Via del reticolo endoplasmatico
Il non correto funzionamento del sistema di ripiegamento delle proteine (UPR “unfolded protein response”) oppure le alterazioni dell'omeostasi del Ca2+ scatenano una serie di processi enzimatici che conducono il reticolo endoplasmatico ad uno stato di stress che, se si prolunga nel tempo, può condurre alla morte della cellula (Nakagara, Zhu et al. 2000; Rao, Peel et al. 2002). Lo stress del reticolo endoplasmatico induce l’ativazione d ella c aspasi-12. uest’ultima, ativata anche dalle calpaine, porta l’ativazione di caspasi-3 ed la successiva cascata di eventi apoptotici (Orrenius 2003).
Fig 1.9. Stress del reticolo endoplasmatico: cause e conseguenne. L’accumulo di proteine malfunzionanti stimola l’ativazione di alcune proteine del reticolo endoplasmatico (ER) da cui risultata una inibizione della traslocazione o una induzione dell’espressione genica. In risposta allo stress del reticolo endoplasmatico, le proteine malripiegate si legano alla chaperonina del reticolo endoplasmatico (Bip/.rp78), interrompendo la sua interazione con la chinasi Ire1 , che infuisce sulla regolazione genica. Ire1 può essere separata dalla membrana da una proteolisi limitata mediata da presenilina 1 (PS1). Una proteina omologa, Ire1 , idrolizza il RNA ribosomiale 28S, inibendo per tanto la traduzione. Inoltre, il fatore 6 a tivatore d e lla t r ascrizione AT F-6 e l a p r oteina c h inasi a t ivata da RN A (P E RK) so n o rilasciati dal RE e sono coinvolte nella ativazione d ella e spressione g enica e n ella t raduzione, rispetivamente. Fra i geni coinvolti si trovano quelli che aumentano il ripiegamento delle proteine nel lumen del RE, fra i quali BiP/.rp78, calreticulina e il fatore di trascrizione CHOP. Aumento di espressione di CHOP induce una diminuzione dell'espressione di Bcl2 e Bcl-XL. L'ativazione della procaspasi-12 avviene atraverso la formazione di un complesso con TRAF6 o Ire1 , o mediante l'idrolisi catalizzata dalla m-calpaina, che viene rilasciata dal complesso con il suo inibitore calpastatina. Il complesso Ire1- -TRAF2 può ativare la chinasi apoptotica ASK1 che porta all'ativazione della via della JNK, che può a sua volta condurre alla fosforilazione di Bcl-XL e Bcl2.BAP31 è una proteina associata a Bcl-2 che interagisce con la caspasi8, Bcl-XL e Bcl-2. uesta interazione è inibita da Spike, un altro componente della famiglia di BCl2 che contiene solo il dominio BH3. La caspasi-8 ativa può idrolizzare BAP31, il frammento N-terminale rimane nel RE e promuove apoptosi. Un canale ionico putativo è la proteina A4, la quale è stata identiocata come una proteina che si lega a BAP31. Variazioni nella concentrazione di calcio (le quatro frecce nel mezzo della ogura) p rovocano l'ativazione d i enzimi dipendenti del calcio (calpaine) e la produzione di ROS. La concentrazione luminale di calcio è controllata dalla Ca-ATPasi del RE e canali (recetore del ip3, recetore rianodinico), da proteine che sequestrano Ca2+ e Bcl2 (che possono regolare negativamente i livelli stazionari di Ca2+ (Orrenius 2003).
La via di traduzione del segnale apoptotico indipendente da caspasi (prima da destra in ogura 1.6) è mediata da granzime A (.rA) (Orrenius 2003). Dopo esser trasportato all’interno della cellula, atraverso p ori p rodoti d a p e rforina, . r A s c atena u n a v i a c a raterizzata da l la formazione di roture s ul D NA a s ingola e lica e d all’apparente m orfologia a poptotica (Orrenius 2003). La endonucleasi coinvolta nella formazione di tali danni al DNA è stata identiocata c ome D Nasi a tivata d a . r A ( . AAD), c o nosciuta a n che c o me N M 23-H1. L’atività di .AAD è inibita da specioci inibitori (I.AAD), conosciuti anche come complesso SET, il quale è collocato nel reticolo endoplasmatico. uesto complesso contiene un inibitore della proteina fosfatasi 2 (pp32), la proteina SET di assemblaggio del nucleosoma, HM.2 e Ape 1 (endonucleasi-1 apurinica). In questa via .rA idrolizza SET, HM.2 e Ape1, ma non pp32 e in questo modo .AAD viene rilasciata e ativata, e t rasloca a l n ucleo d ove i nduce l a frammentazione della catena di DNA (Orrenius 2003).
via estrinseca e quella intrinseca atraverso la proteina Bid, la quale dopo esser stata idrolizzata da caspasi-8, promuove il rilascio del citocromo c dal mitocondrio (via intrinseca) (Orrenius 2003).
L’ativazione della caspasi-8 e l’idrolisi di Bid si trovano anche in casi dove la via estrinseca non è ativata. A d e sempio, c iò a vviene n ell'apoptosi i ndota d a ll’1-metil-4-fenil-1,2,3,6-tetraidropiridina (MPTP) in cellule della substantia nigra. L’ativazione d i c aspasi-8 e l’idrolisi di Bid portano al rilascio del citocromo c e la successiva ativazione di caspasi-9, suggerendo che questo sia un probabile meccanismo di ampliocazione nella morte delle cellule dopaminergiche (Visranath, Wu et al. 2001).
Recenti ricerche, inoltre, hanno dimostrato che la via del reticolo endoplasmatico e quella del mitocondrio sono ampiamente interconnesse (Levine and Rabouille 2005). Infati, la proteina pro-apoptotica BAP31 risiede nel RE ed aumenta il segnale Ca2+ pro-apoptotico che passa dal RE al mitocondrio dopo che BAP31 è idrolizzata grazie all’azione della caspasi-8, localizzata nel mitocondrio (Breckenridge, Stomanovic et al. 2003; Chandra, Choy et al. 2004) (ogura 1.6). L’interdipendenza di questi compartimenti si basa sulla condivisione di regolatori dell’apoptosi. Un altro esempio è PACS 2, il quale si lega alla proteina Bid in seguito a uno stimolo apoptotico, e atraverso l’idrolisi mediata dalla caspasi-8, induce il rilascio di citocromo c. PACS 2 risponde, quindi, agli stimoli apoptotici traslocando dal reticolo endoplasmatico al mitocondrio (Simmen, Aslan et al. 2005).
AICA ribotide è un intermedio della biosintesi de novo delle purine ed è solitamente convertito in inosina-5’-monofosfato, il precursore di tute le purine (ogura 1.10). AICA riboside che deriva dall’idrolisi del ribotide non è rilevabile nelle cellule sane. Tutavia, la presenza di AICA riboside trifosfato è stata riportata negli eritrociti di pazienti afeti da sindrome di Lesch-Nyhan e di pazienti afeti da una sindrome causate da una aumentata atività della fosforibosil-pirofosfato (PRPP) sintetasi (Sidi and Mitchell 1985). In entrambe le sindromi è stata descrita un'accelerazione della sintesi de novo delle purine. L’accumulo di AICA riboside trifosfato è dovuto alla fosforilazione di AICA riboside. uesto viene generato dall’idrolisi dell' AICA ribotide, in quelle cellule dove c’è un aumento della sintesi de novo delle purine; una volta privo di carica è in grado di oltrepassare la membrana citoplasmatica ed esser captato dal circolo sanguigno (Sidi and Mitchell 1985). Recentemente, è stato dimostrato che la 5'-nucleotidasi ubiquitaria citosolica catalizza l’idrolisi dell' AICA riboside monofosfato ed è sovrativa negli eritrociti dei pazienti afeti da sindrome di Lesch-Nyhan (Pesi, Micheli et al. 2000). ueste osservazioni indicano che ci sono condizioni patologiche in cui AICA riboside può essere presente nel sangue come conseguenza di un disordine nel metabolismo delle purine. Sia la Lesch-Nyhan che un'elevata atività della PRPP sintasi sono associate a vari gradi di danni
Fig. 1.10. Metabolismo delle purine. (1) Ipoxantina-guanina fosforibosiltransferasi; (2) adenina fosforibosiltransferasi; (3) nucleoside monofosfato chinasi; (4) nucleoside difosfato chinasi; (5) adenosina chinasi; (6) 5’-nucleosidasi citosolica. Abbreviazioni: AMP, adenosina monofosfato; AS, adenosilsuccinato; IMP, inosina monofosfato; .MP, guanosina monofosfato; PRPP, 5’fosforibosil -pirofosfato; XMP, xantosina monofosfato; Z-riboside, 5-ammino-4-imidazolocarbossiammide riboside; ZMP, ZDP; ZTP, 5-ammino-4-imidazolocarbossiammide riboside 5’monofosfato, difosfato, trifosfato.
AICA riboside viene trasportato all’interno delle cellule atraverso d ei t rasportatori d i adenosina e viene fosforilato dalla adenosina chinasi (,oung, Radda et al. 1996): questo indica che può essere considerato un analogo dell’adenosina. Inoltre, il corrispetivo m onofosfato
PRPP
Z-riboside ZMP ZDP Z P
GMP XMP IMP AS AMP
guanina ipoxantina adenina
xantina acido urico (6) (3) (4) (1) (1) (2) (5)
esercita la sua azione all’interno delle cellule, dato che interagisce con i siti specioci per l’AMP (Muoio, Seefeld et al. 1999). Adenosina e AMP sono intermedi nel metabolismo dell’ATP e solitamente i loro livelli aumentano quando viene utilizzato l’ATP, in modo che svolgano diversi ruoli all’interno della cellula in risposta agli stress causati dalla deplezione energetica (Muoio, Seefeld et al. 1999).
La principale proteina ad essere studiata come sensore della regolazione del bilancio energetico cellulare è stata la proteina chinasi ativata da AMP (AMPK) (Kahn, Alquier et al. 2005) ed omologhi di AMPK sono stati identiocati in una grande varietà di eucarioti, da quelli pii primitivi, come .uardia lamblia e lievito (S. cerevisiae), vermi (C. elegans), inseti ( D. melanogaster), piante e mammiferi, suggerendone la primitiva origine e conservazione evolutiva (Hardie 2003; Carling 2004; Kahn, Alquier et al. 2005). Durante gli ultimi anni, è stato evidenziato che AMPK è coinvolta anche nella regolazione del bilancio energetico di tuto l’organismo, rispondendo a segnali ormonali e di nutrienti nel sistema nervoso centrale e nei tessuti periferici che regolano la richiesta di cibo ed il dispendio energetico (Kahn, Alquier et al. 2005).
1.3.1.
Strutura di AMPP
La proteina chinasi ativata d all’AMP ( AMPK), a ppartenente a lla f amiglia d elle serina/treonina chinasi (Chen, Heierhorst et al. 1999), è un complesso composto da una parte catalitica, subunità , ed una parte regolatoria, formata da altre due subunità, dete e (ogura 1.11). Di tute queste subunità ci sono diverse isoforme ( 1, 2; 1, 2; 1, 2, 3) e tale varietà fornisce 12 possibili combinazioni dell’assemblaggio dell’eterotrimero (Hardie 2005). La conservazione di questa strutura suggerisce che la formazione del complesso di tre subunità distinte sia un requisito essenziale per il funzionamento della chinasi. Nei mammiferi le subunità , e , con rispetivi pesi molecolari di ~63 kDa, ~40 kDa e ~38 kDa, sono codiocate da geni diversi. Il gene della subunità catalitica, isoforma 1, è stato localizzato sul
cromosoma 5p11, quello dell’isoforma 2, invece, è stato localizzato sul cromosoma 1p31. I geni delle subunità e sono stati localizzati sul cromosoma 12 (Mitchelhill 1997).
1.3.2. Ativanione di AMPP
Come suggerisce il nome, la AMPK è ativata a llostericamente d a 5 ’-AMP. Tutavia, i l complesso resta inativo a m eno che n on v enga f osforilato s u u n residuo d i t reonina, in posizione 172 (Thr-172), nel segmento di ativazione della subunità catalitica (Hardie 2005). Esistono altri tre modi atraverso cui l’AMP, una volta legato, può ativare il sistema: (i) convertendo la AMPK in un miglior substrato per la chinasi a monte, (ii) ativando diretamente quest’ultima, (iii) convertendo la AMPK in un peggior substrato per la proteina fosfatasi, inativatrice del complesso (Hardie 2003).
La chinasi necessaria alla fosforilazione di AMPK, chiamata inizialmente AMPK chinasi (AMPKK), è stata parzialmente puriocata d a e patociti d i r ato. u esto h a p o rtato a riconoscere che appartiene alla famiglia delle serina/treonina chinasi, e che era già stata scoperta come prodoto del gene mutato nella malatia autosomica recessiva umana di Peutz-Jeghers.
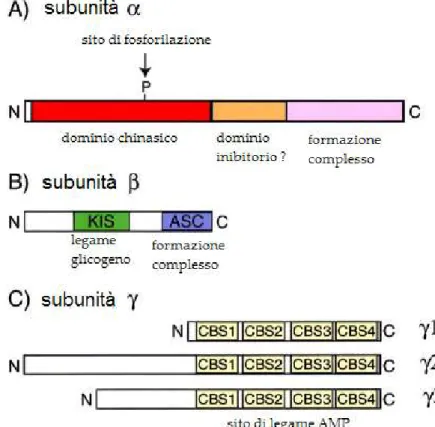
La subunità viene fosforilata in molteplici siti ma l’identità delle proteine chinasi che fosforilano questi residui o l’efeto della fosforilazione sull’atività della AMPK non è ancora conosciuto (Carling 2004). Ad essa viene riconosciuta la fondamentale funzione di impalcatura ed ancoraggio per le altre due subunità. La parte N-terminale, infati, contiene due domini deti ASC e KIS (kinase interaction sequence), i quali sono ben conservati in tuti gli eucarioti. Il primo, il dominio ASC, è richiesto per l’interazione con la subunità ; mentre al dominio KIS, che inizialmente sembrava dover ricoprire lo stesso ruolo ma con la subunità , è stata atribuita u na f unzione d el t uto d i versa ( H ardie 2 0 03). R e centi s t udi, i n fati, ha n no riportato che solo ASC è richiesto per la formazione del eterotrimero e che KIS potrebbe contenere le informazioni necessarie per il riconoscimento e legame del glicogeno (Hardie 2003). Infati, è stato visto che la subunità di AMK di tuti g li eucarioti contiene u n dominio di legame per i carboidrati che è relativo al dominio di enzimi che metabolizzano il punto di ramiocazione 1-6 in carboidrati con legami 1-4 come l’amido e glicogeno (Kahn, Alquier et al. 2005). Ciò suggerirebbe che AMPK sia diretamente sensibile alla quantità di glicogeno cellulare oltre che alla suuciente presenza di molecole energetiche. Per tale regione i domini KIS sono stati successivamente chiamati .BD (domini di legame del glicogeno).
In tute le specie, le subunità contengono quatro domini identici ripetuti in tandem deti CBS (cistationina-beta-sintetasi) o domini di Bateman (Carling 2004). La subunità della AMPK è provvista di due tandem di domini di Bateman. Ciascuno di questi domini lega una molecola di AMP o di ATP in un modo mutuamente esclusivo (Kahn, Alquier et al. 2005). proprio il legame dell’AMP ai domini CBS, della subunità , capace di promuovere l’ativazione allosterica della AMPK (Hardie 2003).
Tuti q uesti meccanismi, a ssicurano u na r isposta estremamente s ensibile a d o gni p iccolo aumento di AMP al di fuori di un’appropriata concentrazione. Poichh l’efeto ativazione è antagonizzato da alte concentrazioni di ATP, il sistema risponde non semplicemente ad un aumento dell’AMP ma all’aumento del rapporto AMP/ATP all’interno della cellula.
Fig 1.11. Schema dei domini di AMPP. Localizzazione del dominio catalitico sulla subunità (A); dominio di formazioni del complesso (ASC) e di legame al glicogeno sulla subunità (KIS) (B); domini di Bateman per il legame dell’AMP sulla subunità (C). Per ulteriori spiegazioni vedere testo (modiocato da (Hardie 2003).
La ogura 1.12 riassume i principali bersagli direti e indireti dell'AMPK. noto che nei vari tessuti (fegato, tessuto adiposo e muscolo), la AMPK fosforila enzimi chiave del metabolismo energetico come la acetil-CoA carbossilasi, la 3-idrossi-3-metilglutaril-CoA ridutasi, l a glicogeno sintetasi e la creatina chinasi, i quali controllano la sintesi degli acidi grassi, la sintesi del colesterolo, la sintesi di glicogeno e fosfocreatina rispetivamente. N el t essuto adiposo, la AMPK fosforila anche la lipasi mentre, nel muscolo, media la traslocazione dei trasportatori del glucosio (.LUT 4) atraverso u n m eccanismo c he d iferisce d a q u ello dall’insulina. La forma endoteliale della ossido nitrico sintetasi viene (anche essa) fosforilata ed ativata dalla AMPK (Hue, Beauloye et al. 2003); (Hardie 2003).
L’AMPK è in grado di regolare il trasporto di alcuni ioni. Ad esempio, inibisce il trasporto di Cl¯ atraverso CFTR (fatore regolatore trasmembrana obrosi ci stica) (vedere ogura 1.12) e il trasporto del Na+ nei polmoni agendo a livello apicale sul canale Na+ amiloride sensibile così come a livello basolaterale sulla pompa Na+/K+ ATPasica (Woollhead, Scot et al. 2005).
Fig. 1.12. Principali bersagli fsiologici e vie regolate da AMPP (Hardie, Scot et al. 2003). ACC, acetil CoA carbossilasi; HM.R. 3-idrossi-3-metilgutarilCoA; .PAT, glicerol fosfato acetil trasferasi; .S, glicogeno sintetasi; EF, fatore elongazione; mTOR, target di rapamicina di mammifero; HSL, ormone sensibile a lipasi; CFTR, regolatore di conduzione trasmembrana obrosi cistica; PFK2, 6-fosfo-2- fruto chinasi; eNOS, nitrossido sintasi endoteliale; IRS1. recetore insulina 1; .LUT, trasportatori glucosio; .6Pasi, glucosio-6-fosfatasi; PEPCK,
La traslocazione dei trasportatori per il glucosio è stata dimostrata in seguito ad ativazione di AMPK in diversi tipi cellulari. Ad esempio, la traslocazione di .LUT4 è stata dimostrata durante l’esercizio sia nel muscolo scheletrico che nel muscolo cardiaco (Coven, Hu et al. 2003), la traslocazione di .LUT 1 in condizioni di stress osmotico in epatociti di rato (Barnes, Ingram et al. 2002) e quella di .LUT3 in cellule renali quando la AMPK è ativata da NO (Cidad, Almeida et al. 2004).
AMPK è in grado di inibire anche la sintesi delle proteine, regolando la via TOR, con un meccanismo complesso, illustrato nella ogura 1.13. In particolare la AMPK fosforila la Thr 2446 di TOR impedendo la fosforilazione sulla Ser 2448 stimolata dai fatori di crescita. .li efeti di AMPK sull’espressione genica possono essere dovuti ad una fosforilazione di fatori di trascrizione oppure ad un efeto sulle importine che mediano il passaggio al nucleo (Hue, Beauloye et al. 2003).
Inoltre, Alba et al. (2004) hanno riportato la capacità della AMPK di inibire indiretamente l’atività dell’enzima NADPH ossidasi all’interno dei neutrooli umani riducendo il rilascio di specie reative dell’ossigeno (ROS), in particolare del radicale superossido (Alba, El Bekay et al. 2004). Ciò suggerisce che l’ativazione di AMPK in uno stato di bassa carica energetica cellulare riduce le difese dell’ospite scatenate da rilascio di ROS.
Fig. 1.13. Efeto antagonistico che la via della A MPP e d ella proteina chinasi B (PPBK
hanno sulla sintesi proteica. La via mTOR( = mammalian target of rapamycin) è ativata d a
amminoacidi e fatori di crescita, come I.F1; stimola la sintesi proteica e la crescita cellulare atraverso molteplici meccanismi, inclusi la fosforilazione della proteina chinasi S6K1 ed il fatore di inizio di traduzione 4E-BP1. I.F1 ativa la via che conduce dal substrato del recetore di insulina 1 (IRS1) all’ativazione della PKB, la quale fosforila TOR e TSC2. TSC1 e TSC2 formano un complesso. Le mutazioni di queste proteine causano la sclerosi tuberosa nell’uomo. La fosforilazione di TSC2 mediata da PKB inibisce TOR, probabilmente promuovendo l’atività .TPasi della proteina . Rheb. TSC2 è anche substrato da AMPK, la quale fosforila due siti diversi di quello fosforilato da PKB. E’ stato dimostrato che la fosforilazione mediata da AMPK è coinvolta nell’inibizione della crescita e nella protezione contro l’apoptosi indota da deprivazione di glucosio (Inoki, Zhu et al. 2003). PKB fosforila TOR nella Ser 2448 mentre AMPK fosforila la Thr 2446. E’ stato dimostrato che la fosforilazione di questi siti è mutuamente esclusiva (Hardie 2005).
1.3.4. Regolanione di AMPP
La AMPK può essere ativata d a s timoli o siologici c h e p roducono v a riazioni d e lla c a rica energetica cellulare e della disponibilità di riserve sia cellulari che dell’intero organismo. Per cui, si può dire che AMPK è ativata in condizioni di stress cellulare, durante l’esercizio e da ormoni secreti dal tessuto adiposo (ogura 1.14).
Inoltre, è stato dimostrato che, in diversi tipi cellule, può esser ativata anche da sostanze come l’AICA riboside (Sullivan, Brocklehurst et al. 1994; Corton, .illespie et al. 1995) e la metformina, un composto spesso utilizzato nella cura farmacologica del diabete di tipo II (Zou, Kirkpatrick et al. 2004). Recentemente è stato scoperto una sostanza, il composto C, in grado di inibire seletivamente AMPK (Kefas, Cai et al. 2004; Smith, Patil et al. 2005). .li efeti di AMPK possono essere studiati anche grazie all’uso di tecniche biomolecolari come vetori adenovirali, oligonucleotidi e small interferenig RNA con i quali vengono introdote nella cellula forme costitutivamente ative o dominanti negativi di AMPK, o viene implicata la sintesi dell'AMPK (Woods, Vertommen et al. 2003; Diraison, Motakis et al. 2004); (Pilon, Dallaire et al. 2004).
Recenti ricerche dimostrano, tutavia, che AMPK può essere ativata da una via indipendente dall’AMP (Birnbaum 2005). Tale via coinvolgerebbe il Ca2+ e Ca2+/calmodulina dipendente
Fig. 1.14. Probabili ruoli delllAMPP nel controllo del metabolismo energetico nelllintero
uesta via funziona anche in fetine di corteccia di rato, dove la depolarizzazione indota da K+, che provoca un aumento della concentrazione di Ca2+ senza modiocare i livelli dei nucleotidi di adenina, ativa AMPK (Harley, Pan et al. 2005).
1.3.4.1. Stress
La AMPK è ativata da una ampia varietà di stimoli cellulari che portano alla deplezione di ATP, inclusi veleni metabolici come inibitori del ciclo degli acidi tricarbossilici (arsenite), inibitori della catena respiratoria (antimicina A e azide) ed inibitori della sintesi di ATP (oligomicina) così come disaccoppianti della fosforilazione ossidativa (dinitrofenolo) (Kahn, Alquier et al. 2005). AMPK è anche ativata da stress patologici come la deprivazione di glucosio, ischemia (Hardie 2003; Russell 2003), ipossia, stress ossidativi, stress iperosmotico e tuti quegli stimoli che sono associati all’aumento del rapporto AMP:ATP (Fryer, Parbu-Patel et al. 2002; Hrang, Ha et al. 2005).
consumo dell’ATP ed ativa il metabolismo dei carboidrati ed acidi grassi per ripristinare i livelli di ATP nel muscolo. Inoltre, l'AMPK gioca anche un ruolo nella risposta adatativa del muscolo all'esercizio continuato alterando le riserve energetiche e l'espressione di geni sensibili all'esercizio. L’aumento dell’atività osica riduce il rischio di sviluppo di patologie insulina resistenti e diabete di tipo II nella popolazione umana (Hu, Manson et al. 2001) e l’ativazione di AMPK può portare ad efeti beneoci come l’aumento dell’ossidazione degli acidi grassi, biogenesi dei mitocondri e, probabilmente, la captazione di glucosio (Kahn, Alquier et al. 2005).
1.3.4.3.
Ormonale
stato recentemente dimostrato che AMPK è regolata anche da ormoni conosciuti come adipochine. uesti sono secreti dalle cellule adipose, che controllano il bilancio energetico in tuto l’organismo (Hardie 2005). La ogura 1.15 illustra i loro efeti sul tes suto adiposo e il fegato (dove si accumulano le riserve energetiche), l’ipotalamo (responsabile del controllo della fame e della sete) e il muscolo scheletrico (in cui la richiesta di substrati energetici è continua per lo svolgimento della contrazione). In ciascuno di questi tessuti gli ormoni sono in grado di modiocare l ’atività d e ll' A M PK e provocare una risposta atraverso e ssa. I n particolare, a livello dell’ipotalamo la AMPK viene stimolata da una diminuzione della concentrazione di glucosio e dall’ormone intestinale grelina. In entrambi i casi, la risposta è un aumento dell’assunzione di cibo; al contrario, la leptina, ormone segnale di scorta corporea di grassi, inibisce la AMPK nell’ipotalamo impedendo che altro cibo venga assunto. A livello del muscolo scheletrico, invece, la leptina e l’ormone adiponectina stimolano AMPK promuovendo l’ossidazione degli acidi grassi. Un efeto simile è svolto anche dall'adiponectina nel fegato
dove questa inibisce inoltre la gluconeogenesi; è stato dimostrato che la resistina ha efeti opposti alla adiponectina (Hardie 2005).
Di grande interesse è il potenziale ruolo che AMPK ha nel tratamento del diabete di tipo II (non dipendente dalla insulina). AMPK è infati ativata da composti b iguanidi come la metformina (Zhou, Myers et al. 2001); (Musi, Hirshman et al. 2002; El-Assaad, Buteau et al. 2003; Chan, Soltys et al. 2004), comunemente usata nel tratamento della suddeta patologia. Infati, la metformina è in grado di abbassare la produzione di glucosio (gluconeogenesi) nel fegato ed aumentare la captazione di glucosio nei muscoli (Zhou, Myers et al. 2001; Tankova 2003). Il meccanismo d’azione della metformina non è stato ancora chiarito. Sebbene sia stato dimostrato che la metformina è un inibitore del complesso I della catena respiratoria mitocondriale (Oren DJ 1995);(El-Mir, Nogueira et al. 2000) non è stata riscontrata una variazione del rapporto AMP/ATP che giustiochi l'ativazione dell'AMPK (Fryer, Parbu-Patel et al. 2002); (Harley, .adalla et al. 2002). Zou et al. hanno proposto un meccanismo d’azione della metformina che coinvolge la formazione di specie reative dell’azoto (ogura 1.16), che ativerebbero la AMPKK (Zou, Kirkpatrick et al. 2004). noto che l’AMPKK è ativata da specie reative dell’ossigeno (Hrang, Ha et al. 2005).
Fig. 1.16. Ipotesi di meccanismo di anione della metrormina nell' ativanione di AMPP. Studi su cellule endoteliali di bovino hanno portato al seguente modello: l’inibizione del complesso I della catena respiratoria mitocondriale da parte della metformina potrebbe generare O2-˙(radicale superossido), il
diferenziamento e sopravvivenza. Un numero crescente di risultati sperimentali suggeriscono che l’ativazione di AMPK può essere diretamente o indiretamente collegata ad efeti su questi processi.
Il ruolo che l’AMPK svolge nel controllo del destino cellulare non è chiaro; alcuni autori hanno correlato la sua ativazione con la sopravvivenza e altri con l’arresto della crescita o con la morte cellulare. Nelle tabelle 2 e 3 vengono riportati esempi, presenti in leteratura, sulla correlazione tra ativazione di AMPK e protezione o induzione dell’apoptosi.
La prima evidenza di un efeto anti-apoptotico di AMPK è stato descrito durante l’apoptosi indota da glucocorticoidi in timociti ( Stefanelli, Stanic et al. 1998). L’efeto pro tetivo è stato trovato anche nell’apoptosi indota da palmitato in astrociti, in cellule mesangiali e in cellule pancreatiche (Blazquez, .eelen et al. 2001; El-Assaad, Buteau et al. 2003; Mishra and Simonson 2005).
Negli astrociti il palmitato aumenta la sintesi ex-novo di cerammide e la stimolazione prolungata (48 ore) di AMPK con AICA riboside evita l’apoptosi. AICA riboside atenua l’induzione della serina palmitoiltrasferasi e della sintesi della cerammide ex-novo mediata dagli acidi grassi, senza avere efeti s u lla s i ntesi e l ’ ossidazione d e gli a c idi g r assi s t essi (Blazquez, .eelen et al. 2001).
In neuroni primari di ippocampo stimoli citotossici e deprivazione di glucosio ativano la AMPK e diminuiscono la vitalità mentre la preincubazione con AICA riboside riduce la morte cellulare. La somministrazione di oligonucleotidi antisenso delle subunità 1 e 2 di AMPK aumenta la morte neurale indota d a d eprivazione d i g lucosio e a bolisce l ’efeto neuroprotetivo di AICA riboside (Culmsee, Monnig et al. 2001).
Recentemente sono stati condoti e sperimenti c on c ellule c he e sprimono A MPK costitutivamente ativa o inativa. uesti esperimenti possono contribuire a chiarire il ruolo dell’AMPK nel controllo della sopravvivenza. L’incubazione di AICA riboside o la sovraespressione di AMPK costitutivamente ativa p revengono l ’apoptosi i ndota d a iperglicemia in cellule dell’endotelio della vena ombelicale (HUVECs) che è preceduta da un’inibizione dall’ossidazione degli acidi grassi, da una diminuzione del potenziale di membrana mitocondriale e dall’aumento degli acidi grassi esteriocati e della concentrazione di malonil-CoA (Ido, Carling et al. 2002). L’adiponectina sopprime l’apoptosi indota d a deprivazione di siero in cellule HUVEC mentre la trasfezione di AMPK dominante negativa provoca l’abolizione dell’efeto protetivo (Kobayashi, Ouchi et al. 2004). Inoltre, nei miociti di topo dopo un’ischemia, si trova una maggiore espressione di caspasi-3 e maggiore colorazione con TUNEL in cellule che esprimono un dominante negativo della subunità 2 di AMPK rispeto a cellule che esprimono la AMPK normale, suggerendo un ruolo protetivo (Russell, Li et al. 2004).
La tabella 2 riporta anche un esperimento in cui è stato usato AICA riboside come ativatore ma non è stata misurata l’atività di AMPK dagli Autori (El-Assaad, Buteau et al. 2003); non è scontato che l’efeto dell’AICA riboside sia sempre mediato da AMPK. Ad esempio, nelle cellule Jurkat (tabella 3) AICA riboside è in grado di entrare nelle cellule e di produrre apoptosi
Nelle cellule di carcinoma epatico umano AICA riboside ativa AMPK la quale catalizza la fosforilazione di p53 sulla Ser-15 e induce l’arresto della crescita (Imamura, Ogura et al. 2001). stato dimostrato che la deprivazione di glucosio o trasfezione con una forma costitutivamente ativa di AMPK producono un arresto del ciclo cellulare in fase .1 e fosforilazione di p53 sulla Ser-15 in obroblasti. E sperimenti d i m utazione d i q uesto r esiduo m ostrano c he q uesta fosforilazione è necessaria per l’arresto del ciclo cellulare. Inoltre, l’ativazione persistente di AMPK conduce ad una accelerazione della senescenza cellulare. uando cellule a basso passaggio sono trasfetate con AMPK costitutivamente ativa, esse manifestano un arresto prematuro mentre la trasfezione con un dominante negativo estende la durata della capacità replicativa delle cellule (Jones, Plas et al. 2005). anche noto che LKB1 è implicata nella soppressione della crescita di alcune linee cellulari di tumore umano (Hardie 2005) e può modiocare l’espressione dell’inibitore p21 Cdk mediata da p53 (Tiainen, Vaahtomeri et al. 2002). Recentemente inoltre, è stata trovata una disfunzione del ciclo cellulare dopo il silenziamento genico di lkb1 o di tre dei suoi bersagli putativi in Drosophila melanogaster (M. Betencourt-Dias and .lover 2004).
Tabella 2. Efeti protetivi delllativanione di AMPP.
timociti quiescenti
glucocorticoidi
AICA riboside
(Stefanelli, Stanic et al.1998)endoteliali (vena
ombelicale)
deprivazione siero
adiponectina
(Kobayashi, Ouchi et al.2004)pancreatiche di
topo
elevate conc.
glucosio e acidi
grassi saturi
AICA riboside
(El-Assaad, Buteau et al. 2003)
astrociti
palmitato
AICA riboside
(Blazquez, .eelen et al.2001)mesangiali
palmitato e acido
steartico
AICA riboside
(Mishra and Simonson2005)endoteliali (vena
ombelicale)
iperglicemia
AICA riboside,
CA-AMPK
(Ido, Carling et al. 2002)
Fibroblasti che
sovraesprimono
6PFK
deprivazione siero
AICA riboside
(Durante, .ueuning etal. 1999)miociti di topo
ischemia
riperfusione
insulto ischemico
(Russell, Li et al. 2004)colon, cellule di epatoma, cellule , cellule di neuroblastoma murino e umano, cellule ippocampali di topo. In alcuni di questi esempi non è stata misurata l’efetiva ativazione dell’AMPK e in altri il coinvolgimento dell’AMPK è stato studiato utilizzando la sua forma costitutivamente ativa. L o s tress o ssidativo i nduce a poptosi e a tivazione d i A M PK i n cellule di neuroblastoma murino. AICA riboside e CA-AMPK aumentano l’apoptosi indota da H2O2 (Jung, Lee et al. 2004).
In cellule la deprivazione di glucosio induce apoptosi e questo efeto è m i mato d a l tratamento con AICA riboside o CA-AMPK. L’ativazione dell’AMPK è accompagnata da ativazione di JNK, del fatore di trascrizione NF-FB e della caspasi-3 (Kefas, Cai et al. 2003). Inoltre, il tratamento con metformina induce ativazione di AMPK e aumento di apoptosi. Un’incubazione con il composto C inibisce l’ativazione della AMPK e l’efeto apoptotico della metformina (Kefas, Cai et al. 2004). L’ativazione di AMPK è stata riscontrata in fetine di ippocampo di topo dopo la deprivazione di glucosio e la ostruzione dell’arteria cerebrale mediale. Il danno ischemico è aumentato dagli ativatori di AMPK e ridoto dagli inibitori (McCullough, Zeng et al. 2005). uesti risultati contrastano con quelli otenuti durante l’ischemia o la deprivazione di glucosio in cellule ippocampali in coltura e in cardiomiociti (tabella 2) (Culmsee, Monnig et al. 2001; Russell, Li et al. 2004).
ipo di cellula
Stimolo
Ativatore di
AMPP
bibliografcoRirerimentolinfociti B
in B-CLL
AICA riboside
AICA riboside
(Campas, Lopez et al. 2003)
Jukart
riboside
AICA
AICA riboside
(Lopez, Santidrianet al. 2003)cancerogene di colon
5-FU
+
ginesteina
AICA riboside
(Hrang, Ha et al. 2005)
Neuro2a
neuroblastoma
H
2O
2AICA riboside,
CA-AMPK
(Jung, Lee et al.2004)Neuroblastoma
SH-S,5,
AICA riboside
AICA riboside
(Pesi, Micheli et al.2000)pancreatiche
deprivazione di
glucosio
AICA riboside
(Kefas, Cai et al.2003)pancreatiche
AICA riboside,
metformina
AICA riboside,
metfomina
(Kefas, Cai et al.2004)epatoma
AICA riboside
AICA riboside,
CA-AMPK
(Meisse, Van de Casteele et al. 2002)