22
CAPITOLO 2
METODI
2.1 PHASE 3.0PHASE3.092 è integrato in MAESTRO, l’interfaccia grafica (GUI) di Schrödinger, ed è un programma versatile per la costruzione di farmacofori, allineamento strutture, predizione di attività e ricerca in un database di composti.
Dato un set di molecole con alta affinità per un target proteico, PHASE identifica ipotesi farmacoforiche comuni, attraverso un campionamento conformazionale e un range di tecniche di scoring. Ogni ipotesi è accompagnata da un set di conformazioni allineate che suggeriscono il giusto modo con cui le molecole potrebbero interagire.
Le ipotesi trovate possono essere combinate con le loro attività in modo tale da creare modelli 3D QSAR che riassumono tutti gli aspetti della loro struttura molecolare, in relazione all’attività.
Questo programma supporta lo sviluppo SAR, e la scoperta, l’ottimizzazione e lo sviluppo di nuovi lead.
2.1.1 SVILUPPO DI UN MODELLO FARMACOFORICO
Lo sviluppo di un modello farmacoforico si basa sull’identificazione di comuni ipotesi farmacoforiche derivate sulla base di un set di ligandi attivi. Un’ipotesi farmacoforica corrisponde ad un arrangiamento spaziale di caratteristiche chimiche comuni a due o più ligandi attivi, con la quale si propone di spiegare le interazioni chiave coinvolte nel legame ligando-proteina.
I ligandi inattivi possono essere usati per eliminare ipotesi che non provvedono ad una buona spiegazione dell’attività solo sulla base del farmacoforo. Più in dettaglio il set di ligandi attivi determina una serie di modelli farmacoforici ai quali è assegnato uno score iniziale e le molecole inattive possono essere usate successivamente per verificare il grado con il quale i modelli distinguono i ligandi attivi da quelli inattivi.
23
Ogni ipotesi identificata da PHASE è ottenuta in base alla sovrapposizione tra i ligandi attivi e le caratteristiche associate a questa ipotesi. I descrittori o “features” che il programma identifica nei vari ligandi sono: Acceptors (A), Donor (D), Hydrophobic (H), Negative (N), Positive, Rings (R), ma è possibile crearne anche nuove.
Le ipotesi migliori possono essere usate per la ricerca, in un database 3D, di nuove molecole potenzialmente attive o per creare un modello 3D QSAR.
Le fasi principali che descrivono lo sviluppo di modelli farmacoforici sono:
a. preparazione dei ligandi b. creazione del sito
c. ricerca di farmacofori comuni
d. Analizzare le ipotesi attraverso funzioni di score e. Costruire modelli di 3D-QSAR
Digitare Maestro da shell, e cliccare su:
Applications>>>Phase>>>Develop Pharmacophore Model
Con questo comando si apre l’interfaccia grafica con la quale è possibile eseguire le fasi precedentemente elencate.
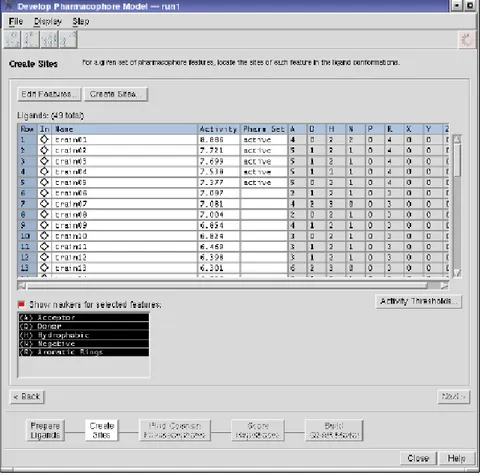
a. preparazione dei ligandi (figura 1)
Cliccando su From file… si aggiunge il file contenente ligandi attivi e non attivi, il quale elenco con relativa attività viene visualizzato nella tabella sottostante al pulsante.
Per poter visualizzare nella colonna phase set la voce “active” o “inactive” occorre cliccare su Activity Thresholds e impostare numericamente la soglia di attività e inattività dei composti.
24 Figura 1: Preparazione dei ligandi
Nel PHASE vengono usati ligandi su cui è già stata fatta l’analisi conformazionale, e quindi sono già stati create tutte le conformazioni possibili per ogni ligando (figura 1), oppure si introducono le molecole costruite su MAESTRO e l’analisi conformazionale viene fatta successivamente direttamente con PHASE (figura 2). Nel primo caso la procedura è la seguente: Con MAESTRO creare un unico file dove sono riportati tutti i ligandi
con le loro attività e salvarlo con estensione .mae;
Creare il DATABASE tramite il comando da Shell: $SCHRODINGER/utilities/phasedb_manage -db
/home/simone/db_lig –
new -mae lig.mae -confs false
fai l’analisi conformazionale del database:
$SCHRODINGER/utilities/phasedb_confsites -JOB lig_cs -db /home/simone/db_lig -confs all -sample thorough -amide trans -max 200
25
Converti il DATABASE in file con estensione .mae:
$SCHRODINGER/utilities/phasedb_export -db /home/laura/db_lig omae ligandi_cs.mae
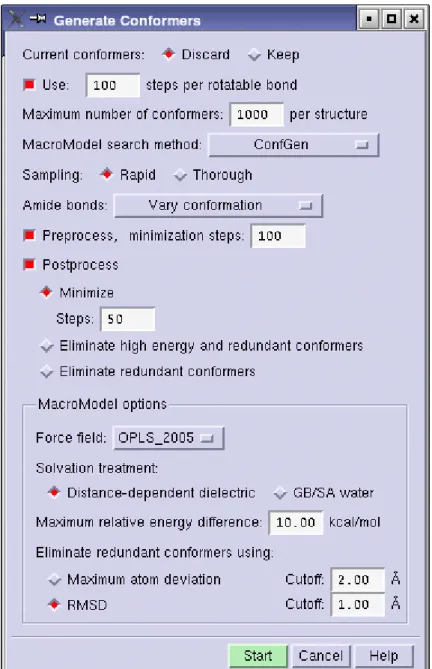
Nel secondo caso invece l’analisi conformazionale viene fatta da PHASE cliccando sul pulsante Generate conformers… (figura1), il quale apre una finestra di dialogo in cui possiamo impostare tutti i parametri da noi desiderati ( figura 2).
26 b. creazione del sito (figura 3)
Questa fase consente di identificare il sito farmacoforico per ogni conformazione di ciascun ligando, cliccando su Create Sites... Si apre la seguente finestra:
Figura 3: Pannello della creazione del sito.
Qui sono visibili le principali features che PHASE riconosce per la prima struttura conformazionale di ciascun ligando. Cliccando su Edit features… è possibile far comparire nuove features esistenti o crearne direttamente nuove.
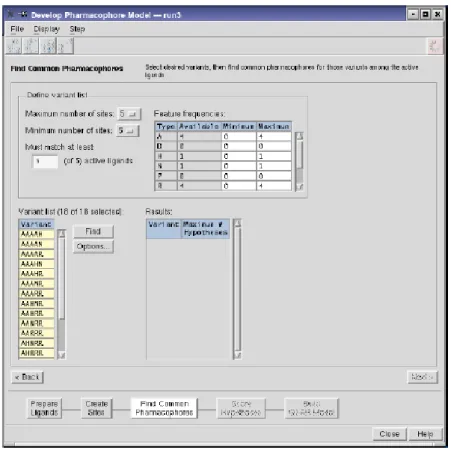
27 c. ricerca di un farmacoforo comune (figura 4)
Si apre la seguente finestra:
Figura 4: Pannello della ricerca dei farmaco fori comuni a piu molecole.
Cliccando su Find, PHASE esamina i farmacofori di tutte le conformazioni dei ligandi attivi e raggruppa tutti quei farmacofori che contengono stesse features con simili arrangiamenti spaziali. Di default, il programma identifica 5 modelli farmacoforici contenenti cinque siti. Il numero di siti può essere cambiato con un valore compreso tra 3 e 7 inclusi.
d. Analizzare le ipotesi attraverso funzioni di score (figura 5)
In questa fase, PHASE esamina ciascun farmacoforo e applica una procedura di scoring per identificare il farmacoforo che mostra il miglior allineamento con i ligandi attivi. Questo farmacoforo andrà a costituire
28
l’ipotesi farmacoforica per spiegare il modo con cui i ligandi attivi si legano al recettore.
Il processo di scoring misura la capacità dei vari ligandi di allinearsi ad una determinata ipotesi e viene calcolato attraverso tre vie:
1. Score di allineamento : deviazione RMSD tra la posizione dei siti farmacoforici della conformazione esaminata e la posizione dei siti nell’ipotesi faramacoforica;
2. Score vettoriale: media tra il coseno individuato tra i vettori delle features della struttura esaminata e quelli dell’ipotesi;
3. Score del volume: sovrapposizione delle interazioni di van der Walls degli atomi di ogni coppia di strutture, ad eccezione degli atomi di idrogeno.
Il punteggio finale è dato ad ogni ipotesi secondo la seguente equazione:
S=WsiteSsite+Wvec Svec+Wvol Svol+WselSsel+Wmrew-WE∆E+WactA
dove:
Ssite= 1 – Salign ⁄Calign
Wsite : score del peso del sito faramacoforico, definibile dall’utente. Di default è
1.0;
Salign: score di allineamento, deviazione RMS tra la posizione dei siti
farmacoforici della conformazione esaminata e la posizione dei siti nell’ipotesi faramacoforica;
Calign: cutoff di allineamento, definibile dall’utente. Di default è 1.2;
Wvec: score dell’importanza del vettore, definibile dall’utente. Di default è 1.0;
Svec : score vettoriale: media tra il coseno individuato tra i vettori delle features
della struttura esaminata e quelli dell’ipotesi;
Wvol : score del volume del sito farmacoforico, definibile dall’utente. Di default
29
Svol : rapporto del volume occupato dalla struttura esaminata e il rispettivo
conformero rispetto al volume totale (volume occupato da entrambi).
Il volume è applicato durante il secondo step, infatti alcuni hits vengono scartati a questo livello proprio per questo motivo.
Ssel : Score selettivo: è una stima empirica della rarità dell’ipotesi.
Wsel : Score del peso della selettività dell’ipotesi.
Wmrew :Se scegli di abbinare meno del numero totale dei ligandi attivi scelti,
puoi sperare di assegnare punteggio più alto all’ipotesi che se abbinassi un maggiore numero di ligandi attivi scelti. Il risultato arriva in questa forma laddove Wrew è regolabile dall’utente (1.di default)e m è il numero dei ligandi attivi che si abbinano all’ipotesi, meno uno. Se Wrew è incrementato molto oltre 1.0, bisogna curarsi di non renderlo troppo grande, altrimenti dominerebbe totalmente il risultato della funzione. Per esempio, se si hanno dieci attivi e Wrew è 1.4, questo contributo al punteggio può avere valore di 32. Gli altri termini hanno un massimo valore d 1.0.
WE∆E : è la penalizzazione introdotta per le ipotesi che hanno il ligando di
riferimento con un elevata energia relativa ad un conformero a più bassa energia che non può quindi essere un buon modello di legame con il recettore, a causa dell’elevato costo in termini di energia92
.
WactA : si possono penalizzare le ipotesi per le quali l’attività del legante
referente è più bassa della più alta attività, aggiungendo un multiplo dell’attività del legante referente al risultato, Wact A, dove A è l’attività.
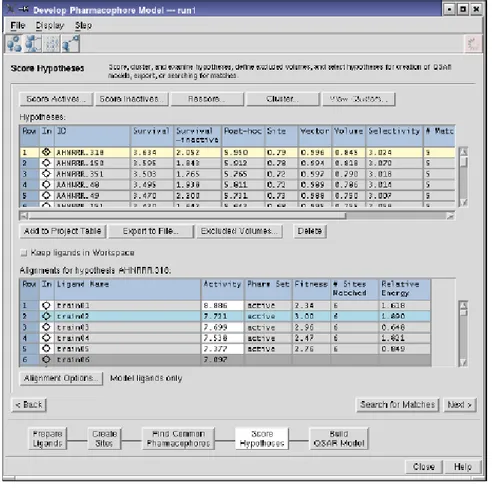
Il calcolo dello Score viene eseguita cliccando su Score Actives…, che assegna un punteggio in base al grado di allineamento dell’ipotesi farmacoforica con i ligandi attivi e Score Inactives.. che invece si basa sul grado di allineamento del modello con i ligandi inattivi.
Nel pannello mostrato in figura 5 sono visibili due tabelle; la prima contenente l’elenco delle ipotesi farmacoforiche trovate e la seconda il set di ligandi. Nella seconda tabella abbiamo tre tipi di colorazioni:
30
1. Grigio scuro: ligandi non presi in considerazione in quella determinata ipotesi
2. Grigio chiaro: ligandi presi in considerazione in quella ipotesi 3. Blu : ligando con fitness score perfetto, cioè ligando di riferimento
per quella determinata ipotesi ovvero ligando che si allinea perfettamente a quella determinata ipotesi.
Figura 5: Pannello delle ipotesi create, per ogni ipotesi è associato un
punteggio ed esiste una molecola di riferimento con miglior allineamento evidenziata di celeste.
e. Costruire modelli di 3D-QSAR (figura 6)
Dopo aver creato le Ipotesi cliccando sul tasto Next… arriviamo alla finestra di dialogo per costruire il modello 3D-QSAR.
31
1. Tabella dove sono riportate tutte le ipotesi scelte
2. Tabella dove sono riportati tutti i ligandi (sono in colore grigio scuro perché il modello 3D-QSAR non è ancora stato costruito). La prima operazione da fare è quella di scegliere il training set ed il test set e questo può essere fatto in due modi:
Selezionare in modo random le molecole che costituiranno il training set ed il test set cliccando su Apply… (si può scegliere solo la % delle molecole che costituiranno il training set).
Cliccare in corrispondenza del ligando all’interno della tabella nella colonna QSAR set ed è possibile scegliere in modo manuale per ciascun ligando se questo fa parte del training set, del test set, o di nessuno dei due.
32
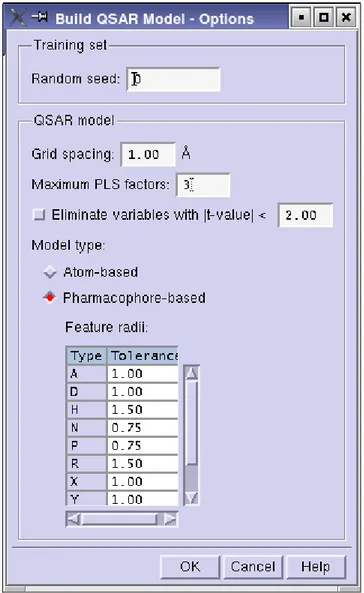
Cliccando sul tasto Options… si apre una finestra di dialogo dove è possibile impostare le opzioni per la costruzione del modello 3D-QSAR ( figura 7).
Figura 7: Pannello delle opzioni per costruire il
modello 3D-QSAR
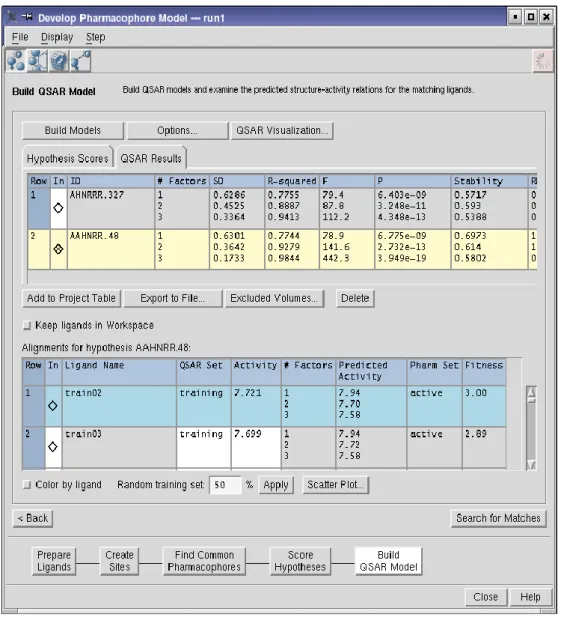
Selezionate le Opzioni (figura 7) cliccare sul tasto Build Models e verrà costruito il modello di QSAR. I dati del modello QSAR per ciascuna ipotesi verranno visualizzati nella finestra QSAR Results (figura 8) su cui basterà cliccare per farla apparire , e nella tabella sottostante ci saranno i risultati
33
dell’allineamento delle molecole associate a quella determinata ipotesi( figura 8).
Figura 8: Pannello del modello 3D-QSAR costruito, sopra ci sono i modelli
3D-QSAR, sotto ci sono i ligandi allineati a quel modello.
Una volta costruito il modello QSAR è possibile esportare le ipotesi e le molecole allineate alle ipotesi, visualizzarle graficamente.
E’ possibile visualizzare graficamente anche il modello QSAR cliccando su QSAR Visualization… (in blu vengono visualizzate le regioni che danno un contributo positivo ed in rosso le regioni che danno un contributo negativo).
34
2.1.2 PARAMETRI IMPOSTATI PER COSTRUIRE IL
MODELLO 3D-QSAR
Phase>>>Prepare Ligands>>>Activity Thresholds… Active if activity above = 8,377
Inactive if activity below = 6,000 Find Common Pharmacophores
Maximun number of sites = 5 Minimum number of site = 5 Build QSAR model >>> Options
Atom-based
2.2 GRID
GRID93 è una procedura computazionale che è in grado di trovare dei siti favorevoli di interazione tra una macromolecola (target) e piccoli gruppi atomici (probes) che mimano i gruppi funzionali più comuni posseduti dai farmaci. Per siti favorevoli d’interazione si intendono le regioni all’interno o in prossimità della macromolecola studiata le cui le proprietà chimiche, spaziali ed energetiche, sinergicamente favoriscono l’interazione con particolari gruppi atomici come gruppi carbossilici, amminici, etc…
Per l’individuazione di specifiche regioni di interazione, GRID include un elevato numero di probes che comprende tutti i più comuni gruppi funzionali organici, alifatici o aromatici, unitamente ad acqua, cationi ed anioni.
GRID può distinguere tra siti di legame anche quando i probes sono molto simili e questa specificità può aiutare nel comprendere relazioni attività-struttura o selettività di interazioni farmaco-recettore. Tali informazioni possono anche essere utilizzate per progettare nuovi agenti terapeutici o per aumentare la selettività di interazione di composti noti.
35
Nel programma GRID l’energia di interazione viene calcolata come somma totale di tutte le interazioni elettrostatiche, di Lennard-Jones e di legami ad idrogeno del probe con la struttura target, usando una funzione dielettrica che dipende dalla posizione per modulare le forti interazioni elettrostatiche fra i centri carichi. Gli atomi della molecola target sono considerati in posizione fissa o variabile durante il calcolo delle interazioni energetiche, mentre i movimenti degli atomi di idrogeno e dei lon pairs sono sempre pienamente considerati. L’energia di interazione per un probe che si trova in un punto dello spazio (coordinate x, y, z) è data da:
E = ELJ + Eel + Ehb
Il programma genera una griglia tridimensionale intorno alla molecola ed il calcolo viene ripetuto con il probes ad ogni successivo punto della griglia finché un valore di energia è stato assegnato ad ogni punto.
Se gli atomi della molecola target sono mantenuti fissi, i contributi all’energia totale corrispondenti all’energia di Van der Waals e a quella columbiana sono le classiche equazioni riportate in letteratura. Il contributo dovuto al legame ad idrogeno è invece peculiare. Non ci sono infatti equazioni generali per descrivere l’energia dei legami ad idrogeno, ma sono state sviluppate diverse regole empiriche per lo studio della distribuzione e della geometria dei legami ad idrogeno basate sullo studio statistico effettuato su banche dati cristallografiche.
In GRID la funzione scelta per modellare i differenti tipi di legame ad idrogeno è la seguente:
Ehb = ErEtEp
Questa complessa funzione multiparametrica tiene conto della distanza tra atomo accettore e donatore, della geometria e ibridazione degli orbitali coinvolti nei legami ad idrogeno, e della dipendenza angolare dell’energia del legame ad idrogeno. Er, infatti, descrive la dipendenza dell’energia del legame ad idrogeno dalla distanza tra gli atomi donatore ed accettare; Et descrive la mobilità degli idrogeni o degli orbitali coinvolti nel legame ad idrogeno; Ep, infine, descrive la dipendenza angolare dell’energia di legame a idrogeno.
36 2.3 GREATER
GREATER94 è l’interfaccia grafica usata dall’utente per eseguire i programmi del “GRID package”. Attraverso GREATER, l’utente può importare strutture molecolari, calcolare i campi di interazione molecolari e osservare o esportare i risultati. Questa interfaccia, inoltre, è integrata con il GVIEW che aiuta l’utente a visualizzare simultaneamente le strutture del target ed i risultati del calcolo.
Il primo stadio di un calcolo di GRID93 è quello di importare uno o più target molecolari.
Le strutture devono essere provviste di un formato 3D che sia supportato da GREATER (.pdb, .mol2, .sdf). GREATER supporta anche il formato .kout prodotto dal programma GRIN.
Per lavorare con un solo target si utilizza il comando Targets>>>Add Single Target
Se invece vogliamo lavorare con un gruppo di molecole usiamo il comando
Targets>>>Add Multiple Targets
Si apre una finestra di dialogo nella quale compaiono due opzioni:
Single file per importare un singolo file contenente le strutture di più targets.
Multiple files or listfiles per importare una lista di file ognuno dei quali descrive una singola struttura.
Una volta selezionato il tasto OK, GREATER usa il programma GRIN per convertire ciascun file nel formato .kout usato da GRID. Questo lavoro viene eseguito in background. Se la conversione è avvenuta correttamente appare un messaggio “ready” per ciascun file convertito.
I targets che sono stati importati sono mostrati nella finestra principale di GREATER e sono separati l’uno dall’altro da una linea. Le informazioni riportate per ciascun target sono:
37
molecule name in cui deve essere inserito il nome della molecola.
status questo campo mostra se tutto il lavoro preparatorio eseguito su ogni target è stato svolto correttamente. Se vi sono stati dei problemi la linea di status si colora in giallo (GRIN warning) o in rosso (GRIN errors).
charges carica totale sulla molecola, calcolata da GRIN.
activity i valori di attività possono essere editati in questo apposito spazio o importati da un file di testo attraverso il comando
Fields>>>Import Activity list
filename nome del file che contiene tutte le informazioni relative alla struttura del target.
Per calcolare i campi di GRID l’utente deve:
selezionare uno o più probes che può scegliere da un menu di GREATER;
definire i parametri di calcolo;
definire la posizione e la grandezza della griglia all’interno della quale viene svolto il calcolo (GRID Box).
Per scegliere i probes si utilizza il comando:
Probes>>>Choose probes
una finestra di dialogo mostra due tabelle che rappresentano le due categorie di probes: probes a singolo atomo o probes multi-atomo.
L’utente può anche definire un nuovo probe con il comando Probes>>>Define new probe
Le direttive usate da GRID che l’utente può scegliere sono raggruppate in quattro categorie:
1. “box”: definisce alcune caratteristiche della griglia che saranno utilizzate per il calcolo, come l’apertura in Amstrong o il numero di piani per Amstrong;
38
3. output: definisce come i risultati del calcolo saranno scritti nei file di output GRIDLONT e GRIDKONT;
4. advanced: usato per definire alcuni versatili e avanzati aspetti. Una volta che sono state importate le molecole e sono stati scelti i probes il calcolo viene lanciato con il comando
Compute>>>Run
Quando il calcolo è terminato è possibile visualizzare i campi di interazione molecolare e salvarli con i comandi
Target>>>View fields File>>>Save computation
2.3.1.PARAMETRI USATI PER COSTRUIRE I MODELLI 3D-QSAR Probes C3 , O
Output>>>LIST LIST-2 Run>>> Advanced Joint
2.4 GOLPE
GOLPE95 è un programma che viene utilizzato con lo scopo di costruire un modello 3D-QSAR. Quando il programma viene lanciato appare una finestra dove possiamo individuare 8 menu: file, pretreatment, modeling, X-selection, list, plot, utilities, help.
La procedura che GOLPE offre per la costruzione del modello 3D-QSAR può essere divisa in varie fasi:
Importazione dei dati; Pretrattamento dei dati;
Costruzione di un modello PLS iniziale e sua validazione; Selezione e rimozione delle variabili;
Costruzione di un nuovo modello PLS e sua validazione; Interpretazione del modello.
39
2.4.1 IMPORTAZIONE DEI DATI
Il programma permette di importare i campi di interazione molecolare da Sybyl, Grid e Spartan.Nel nostro lavoro abbiamo importato i dati da GRID93 cliccando su:
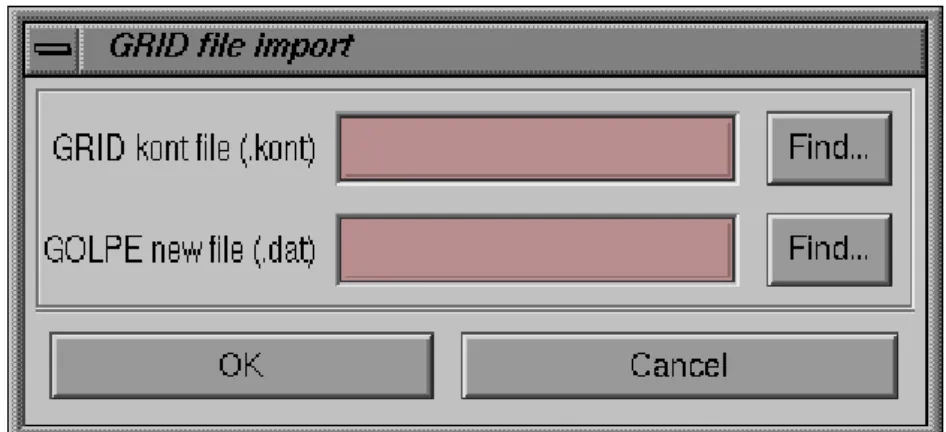
File>>>Import Fields>>>GRID-Kont
Con questo comando il programma permette di prendere direttamente il file binario GRID.KONT senza prima processarlo; si apre la seguente finestra di dialogo (figura 9)
:
Figura 9: Pannello per importare file in Golpe
Si clicca sul Find… superiore e si va a cercare nell’opportuna directory il file GRID.KONT. La stessa directory deve anche contenere le strutture dei target nel formato .kout. Poi si clicca sul Find… inferiore e si salva il file come .dat. Cliccando OK si genera il file GOLPE/SIMCA che è il file input standard di GOLPE. Il file può successivamente essere letto selezionando
File>>>Open data File
A questo punto GOLPE lista i nomi e le attività dei composti contenuti nel file. Nei casi in cui il file non contenga già i valori delle attività, essi possono essere aggiunti a questo punto
40 File>>>Import Activity
Il comando permette di importare il file contenente le attività in formato ASCII. Generalmente i valori sperimentali di attività vengono convertiti in scala logaritmica.
2.4.2 PRETRATTAMENTO DEI DATI
Il pretrattamento dei dati è uno step molto importante in un’analisi 3D-QSAR. Il comando
Pretreatment>>>ClassicPretreatment
offre un semplice modo per applicare un pretrattamento di base sul file di input. Il comando
Pretreatment>>>AdvancedPretreatment
offre un’alternativa migliore ed è stato specificatamente ideato per lavorare con i campi di GRID93.
Appare una finestra che mostra le principali caratteristiche dei dati e offre numerose opzioni:
minimum cutoff il programma permette di applicare un valore di cutoff minimo diverso per ciascuna variabile. I valori più bassi del minimo cutoff vengono sostituiti nel data file dal valore del cutoff selezionato.
In uno studio 3D-QSAR applicare un cutoff minimo non è comune come applicare un cutoff massimo ed è quindi consigliata la sua applicazione soltanto se l’utente lo ritiene indispensabile ai fini della ricerca.
La finestra di dialogo lavora in modo interattivo: appena si seleziona un cutoff il programma mostra sulla destra il numero di valori che verrà troncato applicando quello specifico cutoff. Per selezionare il valore, il cursore sulla scala proposta può essere spostato con il mouse oppure con l’uso delle frecce destra e sinistra sulla tastiera, in modo da avere piccoli spostamenti. Il tasto 2D-Plot mostra un istogramma della
41
distribuzione dei valori di X ed il tasto Profile l’andamento dei dati X. Entrambi i plots possono essere utili nella selezione dell’appropriato valore di minimo cutoff.
maximum cutoff anche per il massimo cutoff, come per il minimo, si può applicare un valore diverso per ogni tipo di variabile. I valori più alti di questo valore massimo sono poi sostituiti nel data file dal valore del cutoff.
Definire un appropriato cutoff è di grande importanza in uno studio 3D-QSAR.
I dati provenienti dall’analisi di GRID sono generalmente ottenuti da calcoli di meccanica molecolare. L’energia di interazione calcolata nei punti di GRID sulla superficie di Van der Waals delle molecole ha un valore positivo (repulsivo) molto elevato. Questo ha senso da un punto di vista puramente teorico ma occorre tener presente che quando costruiamo il modello alti valori andranno ad incrementare artificialmente la varianza di un gruppo di variabili che, in realtà, contengono poca informazione. Per non avere effetti indesiderati i valori di energia positiva troppo alti devono essere troncati.
Secondo alcuni autori il valore ottimale di massimo cutoff è 30 Kcal/Mol, secondo altri è 5 Kcal/Mol. Un’altra ragionevole possibilità è troncare i valori positivi in modo da avere una distribuzione totale più simmetrica. Ad esempio, se il minimo valore di energia del data file è – 10 Kcal/Mol, è conveniente troncare i valori positivi a 10 Kcal/Mol. I tasti 2DPlot e Profile forniscono un consistente aiuto nella scelta, in analogia a quanto visto per il minimum cutoff.
zeroing values i valori vicini a zero, tendono a introdurre rumore di fondo nel data file ed è quindi appropriato sostituire questi valori con 0.000. L’utente può scegliere se azzerare i valori positivi, negativi o entrambi e può scegliere il valore critico assoluto. Le variabili che hanno un valore assoluto più basso del valore critico vengono sostituite da 0.000.
42
min.sd cutoff le variabili con una bassa deviazione standard (sd) mostrano una varianza molto piccola nel data file. L’utente può allora scegliere un valore minimo di sd: le sd inferiori a questo valore minimo saranno convertite in variabili inattive.
n-level vars le variabili “N-level“ hanno un piccolo valore e inoltre presentano una cattiva distribuzione degli oggetti. Sono pericolose perché obbligano il modello a spiegare più della varianza di un gruppo di oggetti con un’alta influenza, generando risultati falsi e fuorvianti. In accordo al numero di livello e alla distribuzione dei valori noi si possono distinguere tre tipi di variabili N-level:
2-levels variables ovvero variabili che presentano solo due valori in tutto il data file, uno dei quali appare soltanto in 1, 2 o 3 oggetti; 3-levels variables ovvero variabili che presentano solo 3 valori in
tutto il data file, due dei quali appaiono solo per 1 o 2 oggetti;
4-levels variables, ovvero variabili che presentano solo 4 valori in tutto il data file.
E’ necessario precisare che non tutte le variabili N-level sono ugualmente pericolose e, di conseguenza, dovranno essere trattate differentemente, secondo il loro livello di pericolosità. Le 2-level sono estremamente pericolose ed è opportuno rimuoverle dal data file in ogni caso. Le 3-level e le 4-level, invece, non hanno una grande influenza sul modello ed è l’utente che decide se eliminarle o meno. Tuttavia, dopo che il modello finale è stato ottenuto, è sempre consigliabile verificare la posizione delle variabili N-level nel modello.
Una volta impostate opportunamente tutti i parametri si clicca sul tasto Save per salvare le modifiche e nella finestra di dialogo salviamo le modifiche come file .dat nell’opportuna directory.
2.4.3 COSTRUZIONE E VALIDAZIONE DEL MODELLO PLS INIZIALE Per generare il modello PLS si usa il comando
43
Per costruire il modello sono utilizzati tutti gli oggetti contenuti nel file. Naturalmente la stringa di comando nel menu modeling non è applicabile se il data file non contiene i valori della variabile Y (ovvero i valori di attività).
Il numero delle variabili latenti calcolate può essere selezionato dall’utente. Il calcolo richiede un tempo variabile da alcuni secondi a qualche minuto in base al numero delle variabili X e Y e al numero degli oggetti.
Dopo di che GOLPE mostra il risultato della PLS nella finestra principale. Esempio:
Partial Least Squares (PLS) 15 objects 449 X-var 1 Y-var Y1 components XVarExp XAccum SDEC r2
0 0.0000 0.0000 1.0675 0.0000 1 18.7309 18.7309 0.5703 0.7146 2 12.7664 31.4973 0.4179 0.8468 3 19.7530 51.2503 0.3586 0.8871 4 10.4417 61.6920 0.3052 0.9183 5 14.4762 76.1682 0.2760 0.9331
Per ciascuna componente sono mostrati: XvarExp, Xaccum, SDEC, il coefficiente di correlazione (r2).
Per valutare la “bontà” del modello ottenuto è necessaria la sua validazione.
Il metodo di validazione del modello PLS è uno degli aspetti più importanti di GOLPE. Naturalmente questo comando può essere eseguito soltanto dopo aver costruito un modello PLS.
Per eseguire la validazione si esegue:
Modeling>>>Validate PLS model
Appare una finestra nella quale occorre scegliere:
Max-dimensionality permette di selezionare la massima dimensionalità del modello PLS da validare.
44
Validation mode permette di selezionare il metodo di crossvalidation da seguire: LeaveOneOut, LeaveTwoOut, Random Groups o Specific Groups.
Number of SDEP questa opzione si attiva solo quando abbiamo scelto il Random Groups come Validation mode. Indica il numero delle volte che sarà ripetuta la procedura. Il default è 20 volte.
Number of groups questa opzione si attiva soltanto quando abbiamo scelto Random groups o Specific Groups come validation mode. Specifica il numero di gruppi nei quali gli oggetti del data file saranno splittati. E’ consigliabile usare 5 gruppi quando gli oggetti sono 20 o più ed un minor numero di gruppi quando il numero degli oggetti è più piccolo.
Una volta che il calcolo è terminato GOLPE mostra nelle finestra principale i risultati della validazione. Esempio :
PLS Model Validation - 5 Random Groups 20 SDEP-calc Y1 components SDEP SDEV(sdep) q2 0 1.1599 0.0417 -0.1807 1 0.9637 0.0592 0.1850 2 0.9217 0.0888 0.2544 3 0.8738 0.1087 0.3300 4 0.8607 0.0933 0.3498 5 0.8639 0.0732 0.3451
Per ciascun componente sono riportati: la deviazione standard degli errori di predizione (SDEP), SDEV, il coefficiente di correlazione predittiva (q2):
dove:
Y= valore sperimentale Y’= valore predetto
45
Y= valore medio
N= numero degli oggetti
Il modo migliore per esaminare le informazioni date dalla PLS è quello di analizzare i “plot” ottenuti:
2d T-U scores plot rappresenta gli oggetti nello spazio delle X-scores (T) contro le Y-scores (U). Da questo plot l’utente può avere una chiara idea della correlazione tra X e Y ottenuta nel modello per ciascuna variabile latente. Il plot della prima variabile latente è quello che contiene più informazioni ed esprime le principali relazioni tra le attività e i descrittori strutturali. Può essere di aiuto per identificare gli oggetti “solitari”. Generalmente tali oggetti non correlano con la prima componente mentre vengono fittati dalla seconda o dalla terza LV ed appaiono nel T-U scores plot come oggetti completamente distinti dagli altri, comportamento tipico degli oggetti non significativi.
2d e 3d loading plots rappresentano le variabili originali nello spazio delle variabili latenti (P). Il loading di una singola variabile indica quanto questa partecipa nel definire la LV. Le variabili che contribuiscono poco a LV hanno un piccolo valore di loading e sono collocate al centro del plot.
2d e 3d weight plots rappresentano le variabili originali nello spazio dei contributi o “pesi” (W). I pesi rappresentano i coefficienti che moltiplicati per le X riescono a spiegare al meglio le Y. Le variabili che presentano un alto valore di “weight” sono importanti per spiegare la Y. GRID-plot di loading e weights in uno studio 3D-QSAR le variabili X sono ottenute dall’analisi di GRID condotta sul set di molecole. Il GRID-plot dei loading aiuta ad identificare le aree dello spazio che contribuiscono di più a spiegare le variabili latenti. Il grid plot dei pesi aiuta ad identificare le aree dello spazio che contribuiscono di più a spiegare la Y, cioè l’attività.
46
valori negativi (zone celesti) mettono in luce aree in cui un’interazione favorevole è correlata con un aumento dell’attività e un’interazione repulsiva è correlata con un decremento dell’attività; valori positivi (zone gialle) mettono in luce le aree in cui
un’interazione favorevole porta ad un decremento dell’attività mentre un’interazione repulsiva porta ad un aumento dell’attività.
2.4.4 SELEZIONE E RIMOZIONE DELLE VARIABILI
Le procedure di selezione delle variabili valutano gli effetti di singole variabili sull’abilità predittiva del modello così da determinare quali variabili sono rilevanti per il problema studiato. Nel primo step dell’analisi un piccolo numero di variabili sono estratte da una grande quantità di informazioni ridondanti. Dopo ciascuna procedura di selezione delle variabili, una lista di tutte le variabili non selezionate è memorizzata sul disco: queste variabili possono essere rimosse dal file in qualsiasi momento usando il comando:
Pretreatment>>>Delete unselected var.s
GOLPE offre due approcci di selezione delle variabili: D-optimal preselection e F.Factorial selection. Quando il numero delle variabili non è molto elevato (meno di 1000) è appropriato applicare immediatamente la F.Factorial selection mentre la D-optimal preselection è richiesta quando le variabili X sono qualche migliaio. Non è possibile applicare la F.Factorial selection se le variabili sono più di 4000.
2.4.4.1 GRUPPI DI VARIABILI
Nei problemi 3D-QSAR i cambiamenti nei valori delle variabili generate dai campi di GRID sono prodotti da alcune variazioni strutturali nelle molecole della serie.
E’ chiaro che le differenze strutturali tra le diverse molecole non si riflettono in una singola variabile, ma piuttosto in un gruppo di variabili. Per questo motivo le differenze strutturali sono descritte meglio da gruppi di
47
variabili che non da singole variabili. GOLPE offre la possibilità di generare gruppi che possono essere usati nella procedura di selezione delle variabili. Ci sono due buone ragioni che supportano l’uso dei gruppi di variabili:
1. Per valutare l’effetto delle variabili sulla predittività è concettualmente più corretto testare contemporaneamente tutte le variabili che contengono le stesse informazioni nello stesso istante; altrimenti può accadere che l’effetto della variabile individuale non sia valutato correttamente in quanto, quando questa è rimossa dalla matrice X, alcune variabili con le stesse informazioni sono ancora contenute nella matrice stessa.
2. Quando numerose variabili sono considerate tutte insieme includono nell’analisi anche informazioni sulla posizione 3D di queste variabili. Informazioni che vanno perdute se consideriamo una sola variabile. Esistono numerosi modi con i quali le variabili X possono essere raggruppate.
L’algoritmo di gruppi SRD sviluppato da GOLPE segue questa procedura: le variabili che contengono maggiori informazioni e che si chiamano “seed” vengono estratte dal data set. Successivamente, ciascuna variabile X è assegnata alla “seed” più vicina (si misura la distanza euclidea nello spazio 3D). Le variabili che si trovano distanti più di un certo cutoff dalla “seed” più vicina vengono assegnate ad un gruppo “fittizio” e rimosse dall’analisi. I gruppi così generati sono chiamati “Voronoi polyhedra” e possono essere usati direttamente; si può anche decidere di far collassare i gruppi vicini tra loro in un unico gruppo.
Il comando da utilizzare per generare i gruppi con la procedura appena descritta è
X-selection>>>SRD Appare una finestra dove l’utente potrà scegliere:
Max. dimensionality permette di selezionare la massima dimensionalità del modello PLS da validare.
48
Vars. Parameters si seleziona lo spazio dove viene svolta la procedura di selezione delle “seeds”. E’ possibile scegliere tra PCA loadings, PLS loading o PLS partial weights.
Number of seeds numero totale di variabili con le quali la procedura costruisce i Voronoi polyhedra. Come default viene assegnato il più piccolo di questi tre valori: 10% del numero di punti di grid, e 3000 variabili (la metà del numero di variabili attive).
Critical distance rappresenta la distanza entro la quale una data variabile X viene assegnata al gruppo della seed più vicina. Le distanze sono misurate come reali distanze euclidee nello spazio di grid intorno alle molecole. La distanza di default è 1.0 Å: con questa distanza la procedura lavora bene se il numero di seeds è alto, se invece questo è piccolo è preferibile aumentare la distanza critica.
Collapse groups selezionando yes si ottiene il collassamento dei Voronoi polyhedra.
Collapsing distance i gruppi che presentano tra loro una distanza inferiore a questa vengono collassati.
2.4.4.2 F.FACTORIAL SELECTION
La procedura GOLPE “Generating Optimal Linear PLS Estimations” è un metodo per eliminare variabili ed aumentare la predittività del modello PLS. I modelli ottenuti usando soltanto le variabili selezionate da GOLPE sono infatti più predittivi del modello PLS su tutte le variabili. Per fare la selezione delle variabili GOLPE costruisce un grande numero di “modelli ridotti” simili ai modelli completi ma con un minor numero di variabili. La predittività di ciascun modello è valutata usando la cross-validation e GOLPE mette in relazione la predittività del modello con la presenza o l’assenza di ciascuna variabile X in base ai valori ottenuti.
49
In questo processo, uno dei problemi principali è quello di trovare il modo più efficiente per valutare l’effetto individuale di ciascuna variabile sulla predittività del modello.
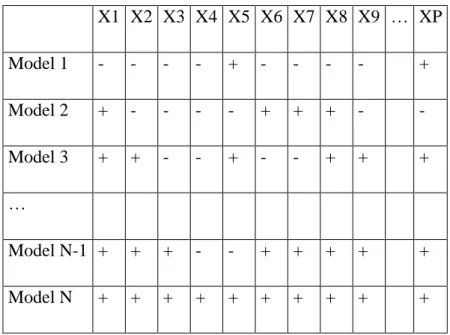
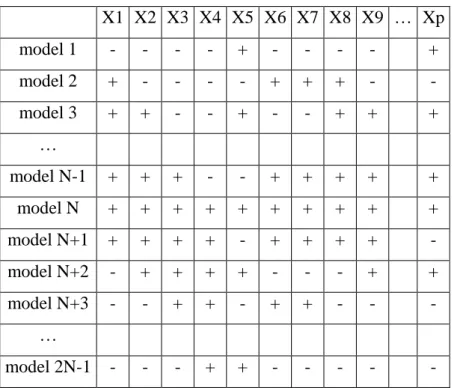
La strategia usata da GOLPE è quella di creare una “design matrix” seguendo uno schema FFD (Fractional Factorial Design) (figura 10). In questa matrice ciascun modello è una combinazione di variabili dalle quali si costruisce un modello PLS ridotto e se ne misura la predittività. La matrice contiene tante colonne quante sono le variabili. Ciascuna variabile può assumere valore positivo, se in quel modello la variabile X è utilizzata, o negativo se in quel modello la variabile X non è utilizzata.
X1 X2 X3 X4 X5 X6 X7 X8 X9 … XP Model 1 - - - - + - - - - + Model 2 + - - - - + + + - - Model 3 + + - - + - - + + + … Model N-1 + + + - - + + + + + Model N + + + + + + + + + +
Figura 10: Esempio di matrice usata da GOLPE per costruire un modello di PLS
ridotto.
Quando gli N modelli sono stati ottenuti ed è stato calcolato il valore di SDEP per ciascuno, sarà possibile quantificare come la presenza o l’assenza di una data variabile influenzi il valore di SDEP;
E = (SDEP+ - SDEP_) dove:
50
SDEP+ = valore medio di SDEP per tutti i modelli che includono la variabile
SDEP_ = valore medio di SDEP per tutti i modelli che non includono la variabile
N = numero totale dei modelli.
Sfortunatamente, nel FFD gli effetti principali delle variabili più significative sono confusi con alcuni dei loro termini di interazione e quindi si corre il rischio di selezionare come rilevanti variabili che in realtà non lo sono. Per minimizzare il rischio GOLPE introduce nella “design matrix” alcune variabili “fantasma”, cioè variabili che appaiono nella design matrix ma che non hanno corrispondenza nella matrice X. L’effetto di queste variabili è poi confrontato con quello delle variabili reali sulla base di uno Student-t misurato con un limite di confidenza del 95%.
Il confronto è svolto come segue.
Si calcola l’effetto medio delle variabili fantasma secondo la formula:
dove:
ED = effetto della variabile fantasma D
ND = numero di variabili fantasma incluse nella “design matrix” T critical = T critico per il 95% di confidenza
L’effetto di ciascuna variabile (E) è successivamente confrontato con l’effetto medio delle variabili “fantasma”. Da questo confronto le variabili sono classificate in: fisse, escluse o incerte. Se E<V, la variabile è incerta. Se E>V con E<0 allora la variabile provoca un decremento dell’SDEP (quindi un aumento della predittività), la variabile è fissa. Se E<V con E>0, la variabile aumenta l’SDEP e diminuisce la predittività; di conseguenza la variabile verrà esclusa.
Una volta fatta la selezione delle variabili quelle che sono risultate “escluse” vengono rimosse dal modello. Il metodo permette di scegliere se
51
eliminare o meno le variabili incerte; è comunque consigliabile eliminarle in ogni caso.
Il comando per avviare la FFD Selection è:
X-selection>>>F.Factorial Selection
I parametri da definire sono:
Use grouping of variables opzione attiva soltanto se i gruppi di variabili sono stati precedentemente generati. Selezionando ON, la selezione delle variabili viene eseguita sui gruppi di variabili anziché sulle variabili individuali. Questo significa che quello che viene valutato non è l’effetto sul potere predittivo dato da un singola ed isolata variabile ma l’effetto prodotto da un insieme di più variabili appartenenti ad un singolo gruppo.
Retain uncertain variables se si seleziona l’opzione ON le variabili “incerte” saranno rimosse dal data file.
Fold-over design attivare questa opzione significa ripetere tutte le combinazioni della “design matrix” dopo aver cambiato i segni (figura11). X1 X2 X3 X4 X5 X6 X7 X8 X9 … Xp model 1 - - - - + - - - - + model 2 + - - - - + + + - - model 3 + + - - + - - + + + … model N-1 + + + - - + + + + + model N + + + + + + + + + + model N+1 + + + + - + + + + - model N+2 - + + + + - - - + + model N+3 - - + + - + + - - - … model 2N-1 - - - + + - - - - -
52
Questo nuovo disegno ha come scopo principale quello di evitare che il modello confonda l’effetto di una variabile singola con l’effetto dovuto all’interazione tra le variabili. Questa procedura richiederà un tempo doppio rispetto a quella standard.
Combinations/variables ratio scala che controlla il numero delle righe della matrice “design”. E’ possibile calcolare il numero di modelli PLS che saranno testati come la più piccola potenza di 2 che dia un valore più alto del prodotto del numero delle variabili moltiplicato per il valore mostrato sulla scala.
Esempio, se sono presenti 500 variabili e il valore sulla scala è 2 (default), il numero di modelli è 1024 (2¹º); se invece il numero sulla scala è 3 i modelli sono 2048 cioè 2¹¹.
Dumnies permette di selezionare la % di variabili fittizie da introdurre nella matrice. Fissare il valore al 20% rappresenta una buona scelta nella maggior parte dei casi.
2.4.5 INTERPRETAZIONE DEL MODELLO
Una volta che abbiamo costruito un modello con soddisfacenti capacità predittive dobbiamo interpretarlo in modo da poter rispondere alle seguenti domande:
perché il composto più attivo è così attivo?
in che modo è possibile modificare la struttura delle molecole in modo da aumentarne l’attività?
Domande alle quali occorre rispondere in termini chimici e non in termini matematici o statistici. Interpretare un modello 3D-QSAR significa osservare diversi GRID plots nei quali l’informazione chemiometrica ottenuta dal modello è rappresentata nello spazio reale intorno alle molecole. Il plot di base per l’interpretazione è il PLS Coefficients: in questo plot è rappresentata l’importanza relativa di ciascuna posizione intorno alle molecole per spiegare l’attività:
53 Plot>>>Grid Plot>>PLS Coefficients
I valori qui rappresentati riassumono i contributi correttamente pesati di ciascuna componente principale. Occorre prestare molta attenzione, poichè il segno del coefficiente può indurre in errore.
Per le regioni CELESTI, un’interazione favorevole (negativa) aumenta l’attività mentre un’interazione sfavorevole (positiva) diminuisce l’attività.
Per le regioni GIALLE, un’interazione favorevole (negativa) diminuisce l’attività mentre un’interazione sfavorevole (positiva) aumenta l’attività.
Per comprendere meglio l’effetto di questi coefficienti sui valori di attività assegnati dal modello agli oggetti si può usare un secondo grafico: l’”activity contribution plot”, ottenibile con la sequenza dei comandi:
Plot>>>Grid Plot>>Activity Contributions
Il plot activity contribution è diverso per ogni composto ed è il risultato della moltiplicazione tra i valori dei coefficienti e i valori veri del campo per questo oggetto.
2.4.6 PARAMETRI USATI PER COSTRUIRE I MODELLI 3D-QSAR
Pretreatment>>>AdvancedPretreatment
Zeroing values: 0,06 Kcal/mol Min.sd cutoff: 0,10
n-lavel vars: 2-level
Modeling>>>Validate PLS model
Max dimensionality: 5
Validation mode: Random Group Num. of sdep: 20,0
54 X-selection >>> SRD
Number of seeds: 10% delle variabili attive Critical distance: 2,5 Å
Collapsing distance: 4 Å
X-selection>>>F.Factorial Selection
Use grouping of variables: attivo Retain uncertain variables: attivo Fold-over design: inattivo
Combinations/variables ratio: 2,0 % of dumnies: 20 %
Validation mode: Random Group Num. of SDEP: 20
Recalculate weights: yes
2.5 OMEGA 2
OMEGA 296 è un programma utilizzato per effettuare velocemente analisi conformazionale di ligandi. Tale programma inizia il processo di analisi dei torsionali esaminando il grafico molecolare e determinando i legami che possono essere liberamente ruotati. La selezione finale si basa sulla distanza RMS degli eteroatomi.
Ad ogni legame ruotabile viene poi assegnata una lista di possibili angoli diedri. L’attuale meccanismo per tale assegnazione si basa sulla corrispondenza SMART; anche se vi sono strategie alternative che hanno basi sia sperimentali che teoriche.
Le strutture intere sono assemblate combinando i frammenti a più bassa energia fino a che non termina l’analisi. Condizioni che determinano l’arresto del calcolo includono un limite sul numero totale di conformeri che possono essere creati, l’esaurimento della lista di frammenti o il superamento, da parte della somma delle energie dei frammenti, della finestra energetica della struttura del minimo globale.
Per essere accettato nella selezione finale un conformero deve avere un RMSD rispetto a tutti gli altri membri della selezione che non supera un certo valore limite definito dall’utente.
55
2.5.1 INTERFACCIA DELLA COMMAND LINE
Se si avvia OMEGA 2 senza argomenti si ottiene:
Si ottiene una descrizione dell’interfaccia della command line con l’opzione –help:
che dà il seguente output
Se si vogliono vedere tutte le opzioni della command line usare il comando –help all:
Per lanciare il calcolo di OMEGA 2 è necessario digitare da shell il comando come nell’esempio:
omega -in drugs.smi -out drugs.oeb.gz –fraglib /usr/local/openeye/data/omega2/fraglib.oeb.gz
2.5.2 PARAMETRI RICHIESTI
-in file contenente l’input che deve essere analizzato da OMEGA. -out file in cui saranno scritte le conformazioni generate da OMEGA.
-fraglib l’argomento è un file OEBinary V2 contenente le coordinate dei frammenti aciclici pre-costruiti e le conformazioni multiple di sistemi ciclici.
56
2.5.3 FORMATI DEI FILES
OMEGA può leggere e scrivere una gran varietà di formati, che sono interpretati automaticamente dal suffisso del nome del file.
File type Extension
SMILES .smi .ism .can .smi.gz .ism.gz .can.gz
SDF .sdf .mol .sdf.gz .mol.gz
SKC .skc .skc.gz
CDK .cdk .cdk.gz
MOL2 .mol2 .mol2.gz
PDB .pdb .ent .pdb.gz .ent.gz
MacroModel .mmod .mmod.gz
OEBinary v2 .oeb .oeb.gz
Old OEBinary .bin
2.5.4 PARAMETRI OPZIONALI INPUT/OUTPUT
-param l’argomento di questa opzione è il nome del file contenente i parametri di
controllo. Tutti i parametri necessari per l’esecuzione del programma possono essere forniti dal file dei parametri di controllo, sebbene qualsiasi stringa data esplicitamente nella command line sostituirà le opzioni che si trovano nel file.
OMEGA crea un nuovo file dei parametri con l’intero set dei parametri usati dopo ogni calcolo. Il nome del file scritto è dato dalla combinazione del prefisso di base con l’estensione .parm.
57
-prefix l’argomento definisce il prefisso che deve essere usato per i files di
informazione e di dati creati da OMEGA.
2.5.5 PARAMETRI PER L’ANALISI TORSIONALE
-maxconfs stabilisce il numero massimo di conformazioni che devono essere generate.
I conformeri vengono formati secondo un ordine energetico dei frammenti costituenti. Il valore di default in molti casi supera il numero di conformeri che sarebbe necessario, in modo da selezionare il miglior insieme dal punto di vista dell’RMSD e dei criteri energetici. Il valore di default è 400.
-rms determina il minimo Root Mean Square (RMS) cartesian distance al di sotto del quale due conformeri vengono duplicati. L’RMS viene calcolato dopo la sovrapposizione, in modo da poter avere la reale distanza minima tra i due conformeri. Se il valore di -rms viene abbassato OMEGA può creare gruppi che contengono più conformeri con una forma simile. Valori più alti portano a gruppi più piccoli con conformeri aventi forme diverse.
Il valore di default è 0,8.
2.5.6 FILES PARAM
I files param non sono altro che files contenenti un parametro per ogni riga. Ogni esecuzione di OMEGA crea un file param chiamato prefix.parm che contiene tutti i parametri usati da OMEGA e che può essere usato nei successivi lanci del programma con l’opzione -param.
2.5.7 PARAMETRI USATI PER L’ANALISI CONFORMAZIONALE
maxconfs è stato fissato a un valore pari a 10.000. rms è stato impostato a 0,5.
58 2.6 ROCS 2.2
ROCS97, Rapid Overlay of Chemical Structure, è stato sviluppato per attuare un’analisi in larga scala di database 3D attraverso il metodo della sovrapposizione in modo da trovare composti non intuitivamente simili che possono essere successivamente valutati per il processo di drug discovery.
ROCS è un metodo di sovrapposizione basato sulla forma; le molecole vengono allineate secondo un processo di ottimizzazione del corpo rigido che massimizza il volume sovrapposto. Il programma prende in considerazione solo gli eteroatomi dei ligandi ed ignora gli idrogeni.
Dato che il volume e la forma in questo contesto sono altamente correlati, la procedura di massimizzazione del volume sovrapponibile è un eccellente metodo per avere un’idea circa la somiglianza delle forme.
Sebbene il programma sia principalmente un metodo shape-based, l’utente può specificare di includere nel processo di sovrapposizione e di analisi della somiglianza considerazioni riguardanti la chimica, in modo da facilitare l’identificazione di quei composti che sono simili sia per la forma che dal punto di vista elettrostatico.
La sovrapposizione molecolare ha avuto un impatto limitato nell’analisi di database 3D a causa della bassa velocità (1-2 molecole/secondo) dei precedenti metodi; ROCS invece può effettuare l’allineamento alla velocità di 600-800 conformeri per secondo. L’unione di screening riguardanti forma e chimica rende ROCS uno strumento di incredibile potenza per l’analisi di grandi database 3D.
2.6.1 TEORIA
Due entità hanno la stessa forma se il loro volume corrisponde esattamente. Più il volume è differente tanto più sarà diversa la forma.
Il volume è un campo scalare, ovvero una funzione che ha un singolo valore numerico in ogni punto dello spazio.
59 Due oggetti non possono avere la stessa forma se il loro volume non è lo stesso; due oggetti possono però avere lo stesso volume ma non la stessa forma.
È possibile scrivere una precisa definizione di similitudine della forma:
dove f e g sono differenti funzioni caratteristiche. Se l’integrale è zero allora f e g sono realmente le stesse funzioni e quindi corrispondono alla stessa forma. Maggiore è l’integrale maggiore sarà la differenza tra le forme descritte da f e g.
Il tipo di uguaglianza descritta in S1 è indicata come metrica L1. Un altro tipo di metrica è la L2:
Questo integrale rappresenta quello standard utilizzato per definire l’uguaglianza di forma. Il vantaggio principale tra S1 e S2 è che quest’ultima non coinvolge la
funzione valore assoluto, che non è analitica. Moltiplicando i termini nell’integrale si ottiene:
cioè l’equazione fondamentale per il confronto della forma. Si può riscrivere come:
I termini I rappresentano il self-volume sovrapposto di ogni entità (molecola), mentre il termine O è la sovrapposizione tra le due funzioni. È necessario calcolare questi tre termini per comparare le forme dei due campi. I termini I sono indipendenti dall’orientamento ma non è così per il termine O.
Trovare l’orientamento che massimizza O, e quindi minimizza Sf,g equivale a
60 Il coefficiente di Tanimoto può essere descritto come la combinazione di I ed O secondo l’equazione:
Una misura alternativa della forma è il coefficiente di Tversky, che può essere descritto dall’equazione:
Generalmente α+β=1. ROCS calcola due valori di Tversky, uno in cui è la molecola query ad avere il pre-fattore α, nell’altro è la molecola del database ad avere il pre-fattore α..
2.6.2 PREPARAZIONE DEI FILES DI INPUT
File del Database
L’uso più comune di ROCS riguarda la sovrapposizione di grandi quantità di molecole (dbase file) su una molecola di riferimento (query). Il formato più comune per il file dbase è il multi-conformer OEBinary, creato con il programma OMEGA 2, anche se è possibile usare altri tipi di formato (SDF, MOL2 e PDB).
Un altro tipo di file è riconosciuto come bdase, ovvero un file il cui nome termina con .lst o .list che non è altro che una lista di files di molecole, una per riga.
File Query
Il secondo input richiesto da ROCS è un file contenente una o più molecole usate come query. ROCS leggerà le molecole dal file dbase e cercherà di sovrapporle alla molecola di riferimento.
ROCS può anche usare una griglia al posto della molecola come file di riferimento.
61
2.6.3 INTERFACCIA DELLA COMMAND LINE
Si ottiene una descrizione dell’interfaccia della command line lanciando rocs con l’opzione –help:
che darà il seguente output:
Per lanciare il calcolo di ROCS digitare da shell il comando come nel seguente esempio:
rocs –dbase input.oeb.gz –query riferimento.mol2 dove input.oeb.gz è il file di input con le molecole che devono essere allineate, mentre il file riferimento.mol2 rappresenta la molecola di riferimento sulla quale effettuare la sovrapposizione.
2.6.4 PARAMETRI RICHIESTI
-query (-ref) file contenente molecole o una singola griglia da usare come riferimento
I formati disponibili per la molecola query includono:
File type Extension
SDF .sdf .mol .sdf.gz .mol.gz
MOL2 .mol2 .mol2.gz
PDB .pdb .ent .pdb.gz .ent.gz
62 OEBinary v2 .oeb .oeb.gz
Old OEBinary .bin
mentre se la query è una griglia i formati possibili sono: Grid File type Extension
Grasp .phi
OpenEye .grd
CCP4 .map .ccp4
X-PLOR .xplor .xplmap
-dbase (-fit) file contenente una o più molecole 3D da sovrapporre con la query. I
formati supportati sono gli stessi del file query (non le griglie), oltre al formato ROCS DB ( .rocsdb) e il file list ( .lst o .list).
2.6.5 PARAMETRI OPZIONALI
-param definisce il file dei parametri di controllo. Il file dei parametri scritto dopo
ogni lancio di ROCS può essere usato con l’opzione -param nei successivi calcoli di ROCS. Ogni comando dato esplicitamente dalla command line sostituirà il comando trovato nel file specificato con il parametro -param.
2.6.6 OPZIONI GENERALI DELL’OUTPUT
-prefix prefisso usato per il nome dei file di output. Di default è usato il prefisso
ROCS.
-rankby definisce il punteggio da usare per classificare gli hits. È possibile scegliere
tra: tanimoto, tverskyd, tverskyq, scaledcolor, combo e overlap. Per default è impostato tanimoto.
63
-maxhits (M) numero massimo di hits che vengono riportati; questa opzione fa sì che
il programma termini di lavorare appena vi sono M molecole nella hitlist.
2.6.7 OPZIONI RIGUARDANTI IL COLORE
-chemff nome del force field per il colore, che può essere uno di quelli già costruiti
(ImplicitMillsDean o ExplicitMillsDean) oppure uno di quelli definiti dall’utente.
-optchem per scegliere di usare le forze e i gradienti del force field del colore come
parte dell’ottimizzazione della sovrapposizione.
2.6.8 FILE REPORT
Il file report di ROCS appare come una tabella con i campi descritti sotto. Per visualizzare correttamente il file in una shell occorre usare l’applicazione align nella command line.
Name nome del database di molecole; se il database contiene molecole
multi-conformere l’indice dello specifico conformero segue il nome della molecola.
ShapeQuery nome della molecola di riferimento; se la query è una molecola con più
conformeri al nome della molecola segue l’indice dello specifico conformero.
Rank classificazione numerica della lista di hits, basata sul metodo di punteggio che è
stato scelto di applicare. Se non sono state fatte modifiche si basa solamente su Tanimoto, ma può essere modificato dalla command line con il parametro –rankby; se non viene usata alcuna hitlist nel calcolo questo campo avrà come valore 0 (zero).
ShapeTanimoto questa colonna dà il valore della forma di Tanimoto; equivale a un
valore compreso tra 0 e 1.
Tversky(d) è calcolato per la molecola del file dbase con l’equazione riportata di
seguito, dove beta = 0,95.
Tversky(q) è calcolato con l’equazione precedente per la molecola query, con alfa =
64
ScaledColor viene calcolato un valore “graduato” del colore considerando il valore
del punteggio reale di un hits e dividendolo per il punteggio del colore della molecola query contro se stessa. Si tratta di un valore compreso tra 0 e 1.
ComboScore fornisce un punteggio che include sia la forma che il colore; ha un
valore compreso tra 0 e 2 ed è usato per classificare la hitlist quando dalla command line si specifica il parametro –rankby combo.
SubTan ulteriore punteggio che può essere calcolato specificando l’argomento di –
subtan. Non è compreso come default. Subtan è definito prendendo la posizione della molecola di riferimento e del database dopo la sovrapposizione finale e rimuovendo tutti gli atomi della molecola del file dbase che distano dagli atomi della query più di 1,5 Å. Si calcola poi la forma di Tanimoto usando queste due strutture ed il coefficiente di Tanimoto è salvato come SubTan.
Overlap rappresenta il valore assoluto del volume sovrapposto tra la query e la
molecola del database. Tale valore è dato in unità arbitrarie ed è più utile quando viene usata come query una griglia.
2.6.9 COLOR FORCE FIELD
Il force field chimico può essere usato per misurare la complementarietà chimica e perfezionare la sovrapposizione basata sulla forma in base alla somiglianza chimica. Tale force field è composto da regole che determinano i centri chimici più regole che determinano come tali centri interagiscono.
In ROCS esistono due color force field: ImplicitMillsDean ed ExplicitMillsDean. Entrambe definiscono sei TYPE simili, ovvero donatori di legame a idrogeno, accettori di legami a idrogeno, idrofobici, anioni, cationi e anelli. È raccomandato l’uso del force field ImplicitMillsDean.
ImplicitMillsDean include un semplice modello pKa che considera pH=7; definisce cationi, anioni, donatori e accetori in modo tale che è assegnato loro il giusto valore indipendentemente dallo stato di protonazione nel file dbase o query.
ExplicitMillsDean non include il modello pKa ed interpreta lo stato di protonazione e di carica di ogni molecola esattamente come è indicato nel dbase.
65
2.6.10 PARAMETRI USATI
chemff è stato usato il force field ImplicitMillsDean. optchem è stato impostato come true.
rankby si è scelto l’opzione combo.