3. MATERIALI E METODI
3.1 Campioni impiegati nell’esperimento
I campioni utilizzati nel nostro studio sono sieri prelevati da gatti specific-pathogen free (SPF) vaccinati e mock vaccinati. Il primo gruppo, costituito da 7 animali siglati GC, GD, GE, GF, GG, GH e GI, è stato sottoposto ad una vaccinazione di tipo prime-boost:
• Nel prime gli animali hanno ricevuto per via sottocutanea 3 inoculi costituiti ciascuno da 300 μg del plasmide pVIVO/GM-CSF esprimente l’Env di FIV. Tali inoculi sono stati effettuati ai tempi zero, 3 e 12 settimane.
• A 21 settimane dal tempo zero, il boost ha comportato il prelievo da ogni animale di linfociti T autologhi i quali sono stati trasdotti in vitro con l’Env di FIV e poi reinfusi intraperitonealmente nelle cavie.
I 2 gatti mock vaccinati, siglati GK e GL sono stati sottoposti alla medesima vaccinazione ma utilizzando nel prime 300 μg di plasmide pVIVO e nel boost linfociti trasdotti con particelle virali vuote (VLP).
• Trascorse 30 settimane dal primo inoculo del prime, la fase di challenge ha previsto l’infezione di tutte le cavie (vaccinati e mock vaccinati) con il ceppo virale Petaluma (FIV-Pet).
Nei sieri prelevati al momento del challenge è stata valutata la presenza di anticorpi neutralizzanti mediante test di neutralizzazione classici e di ultima generazione. Il primo metodo ha previsto l’impiego del virus wild-type e di una linea cellulare T-linfoide (MBM) mentre il secondo saggio è stato allestito con vettori FIV-derivati ed una popolazione cellulare di facile propagazione (CrFK CD134+).
3.2 Produzione particelle virali esprimenti luciferasi
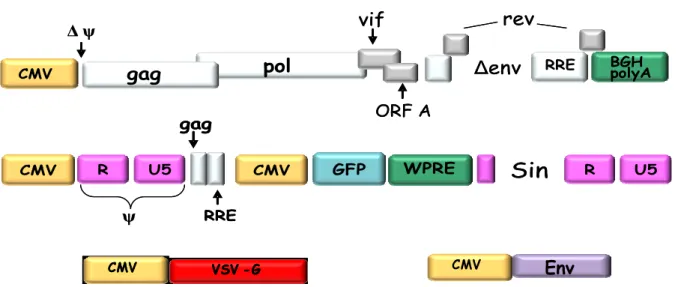
Per lo studio sono state prodotte particelle virali pseudotipizzate con la glicoproteina Env di FIV e veicolanti la luciferasi come gene reporter (FIV-Luc). Per produrre tali particelle, secondo il sistema split-component, sono stati utilizzati un plasmide detto
packaging che codifica le proteine strutturali ed enzimatiche, un costrutto envelope che esprime l’Env di FIV ed un plasmide (detto vettore) che veicola il gene reporter luciferasi. Quest’ultimo è stato appositamente costruito a partire dal costrutto LAW34GFP.
Sono state inoltre prodotte particelle virali pseudotipizzate con la glicoproteina G del virus della stomatiche vescicolare esprimenti sempre la luciferasi (VSV-G/Luc) impiegate come controllo positivo nel saggio di trasduzione (Fig. 3.1).
Figura 3.1: Plasmidi utilizzati nello studio, in ordine dall’alto in basso: costrutto packaging,
costrutto LAW34GFP e costrutti envelope.
3.2.1 Amplificazione gene luciferasi
La sequenza genica è stata amplificata a partire dal plasmide commerciale pGL3 (Promega, Milano, Italia) mediante PCR.
In particolare sono stati utilizzati primer senso (S) e antisenso (AS) provvisti di una coda aggiuntiva al 5’ contenente un sito di taglio rispettivamente per l’enzima di restrizione EcoRV e BglII (Fermentas, Milano, Italia): BglII S (5’ GGAAGATCTTCATGGAAGACGCCAAAAACATAAAGAAAGGC 3’) ed ECORV AS (5’ CCGGATATCTTACACGGCGATCTTTCCGCCCTTC 3’). Il frammento amplificato presenta una lunghezza di circa 1600 bp.
Per la PCR 1 μg di campione è stato aggiunto alla miscela di reazione costituita dai seguenti reagenti: tampone di reazione 10x (Tris-HCl 10mM, pH 0,9; KCl 50mM; Triton X-100 0,1%), MgCl2 1,5mM, primer S e AS 0,4μM, dNTP’s 20μM e 1U di Taq
DNA polimerasi (Polymed, Firenze, Italia). La miscela di reazione viene portata ad un
CMV R U5 CMV GFP WPRE
Sin
R U5RRE gag
ψ CMV
CMV RR U5U5 CMVCMV GFPGFP WPREWPRE
Sin
RR U5U5 RRE gag ψ CMV VSV -G CMV VSV -G CMV VSV -GVSV -G CMV VSV -G CMV VSV -G CMV VSV -GVSV -G rev vif Δ ψ ORF A Δenv gag pol CMV RRE BGHpolyA rev vif Δ ψ ORF A Δenv gag pol CMV CMV RRE BGHpolyA Env CMV EnvEnv CMV CMVvolume finale di 50 μl con acqua sterile. Per verificare l’assenza di contaminazioni è stato aggiunto un controllo negativo costituito da acqua in sostituzione del DNA.
La reazione di amplificazione è stata suddivisa in più fasi: • Una denaturazione iniziale del DNA per 2 minuti a 94°C • Una fase ripetuta 25 volte che prevede:
1. denaturazione a 94°C per 30’’
2. annealing a 60°C per 30’’
3. estensione a 72°C per 1 minuto e 45’’
• Estensione finale di 5-7 minuti a 72°C
L’avvenuta amplificazione del frammento è stata controllata su gel d’agarosio all’1% dopo corsa elettroforetica.
3.2.2 Digestione enzimatica
L’amplificato ed il costrutto LAW34GFP sono stati digeriti con gli opportuni enzimi di restrizione BglII ed EcoRV che hanno rispettivamente i seguenti siti di taglio: AGATCT e GATATC. Nella miscela di reazione il campione e gli enzimi sono stati aggiunti al Buffer TANGO 10× (Fermentas) portando ad un volume finale di 40 μl con aggiunta di acqua sterile. La reazione di digestione è stata mantenuta per 1 h e 30 minuti a 37°C e disattivata a 65°C per 10 minuti. Allo scopo di valutare l’efficienza della digestione è stato preparato in parallelo ad ogni digerito anche un controllo negativo con tutti i componenti della reazione escluso l’enzima.
Dopodichè l’inserto è stato purificato dai sali della digestione mediante estrazione da gel con il kit NucleoSpin (Macherey-Nagel, Düren, Germania), mentre il plasmide LAW34GFP è stato purificato mediante precipitazione con sodio acetato ed etanolo; in particolare è stato addizionato di acetato di sodio 3M a pH=5.5 ed etanolo assoluto, raffreddato a -20°C, nelle proporzioni, rispettivamente, di 0,1 e 3 volumi di plasmide. Dopo l’incubazione a -20°C overnight oppure a -80°C per 3 h, il DNA precipitato è stato centrifugato a 12000 rpm per 30 minuti a 4°C. Dopo aver eliminato il sovranatante, è stato effettuato un lavaggio con etanolo al 70% e successiva centrifugazione a 12000 rpm per 10 minuti a 4°C. Infine, il pellet formatosi è stato risospeso in 25 μl di acqua sterile.
La quantificazione dell’inserto, dopo estrazione da gel, e del vettore, dopo la precipitazione, è stata effettuata mediante corsa elettroforetica su gel d’agarosio all’1%.
3.2.3 Ligation
Prima del clonaggio il vettore è stato defosforilato con 1U di Shrimp Alkaline Phosphatase (SAP) (Promega) per ogni μg di DNA in Buffer SAP 1X (Promega). La reazione è stata mantenuta per 30 minuti a 37°C e portata a 65°C per 15 minuti per disattivare l’enzima. L’amplificato purificato e il vettore defosforilato sono stati utilizzati nella reazione di ligation.
La reazione è catalizzata dall’enzima T4 DNA ligasi (Fermentas) partendo da 100-200 ng di plasmide ed una quantità di inserto che può variare da un rapporto molare di 1:5 a 1:30 in base alle dimensioni dello stesso.
La miscela viene incubata overnight a 16°C, con il successivo innalzamento della temperatura a 65°C per 10 minuti per disattivare l’enzima.
3.2.4 Trasformazione
Il prodotto della ligation è stato usato per trasformare il ceppo Stable2 di E.Coli reso competente e conservato a -80°C in aliquote da 100 μl. La trasformazione è stata eseguita sotto cappa aggiungendo 10-100 ng di DNA plasmidico alle cellule batteriche e lasciando incubare la miscela in ghiaccio per 30 minuti. Dopodichè le cellule batteriche sono state sottoposte a shock termico tenendole a 42°C per 45’’-90’’, in modo da alterare la permeabilità della membrana plasmatica per favorire l’entrata del DNA, e quindi trasferite velocemente in ghiaccio per 2 minuti per permettere la chiusura dei pori cellulari.
Le cellule batteriche così trasformate sono state messe a crescere in 1 ml di terreno SOC (2% Triptone, 0,5 estratto di lievito, NaCl 10mM, KCl 2,5mM, MgCl2 10mM,
MgSO4 10mM e glucosio 20mM) e poste in agitazione per 1 h a 37°C.
Infine le cellule sono state seminate su piastre Petri contenenti terreno LB Agar (1% Triptone, 0,5% estratto di lievito, 1% NaCl a pH 7,0, 1,5% agar batteriologico) addizionato di ampicillina (50 mg/ml) per permettere la selezione delle cellule trasformate. Successivamente le piastre sono state incubate overnight a 37°C.
3.2.5 Screening delle colonie
Per verificare l’avvenuta inserzione del frammento nel vettore, le colonie ottenute dalla trasformazione sono state sottoposte a screening mediante PCR.
La miscela di reazione per la PCR viene preparata come descritto nel paragrafo 3.2.1, utilizzando i primer ScrEC S (5’ GGTATCAAGCAAGGATATGGG 3’) e ScrEC AS (5’ TTGATTGTCGACACTAGATATTC 3’). Tuttavia rispetto al protocollo descritto precedentemente, durante il primo step, la temperatura iniziale di 94°C viene mantenuta per 10 minuti per ottenere la lisi cellulare ed il rilascio del plasmide. Successivamente lo screening viene controllato su gel di agarosio all’1% e solo gli amplificati che hanno una lunghezza compatibile con quella dell’inserto sono considerati buoni.
3.2.6 Estrazione DNA plasmidico
Le colonie che risultano contenere l’inserto di interesse sono state messe a crescere a 37°C overnight in 3 ml di terreno liquido LB con l’aggiunta di 3 μl di ampicillina. La crescita batterica è stata centrifugata a 4°C per 5 minuti a 12000 rpm andando poi ad eliminare il surnatante. Il pellet è stato risospeso in 100 μl di soluzione I per la lisi della parete (glucosio 50mM, 25mM Tris HCl a pH 8,0, 10mM EDTA pH 8,0); successivamente, sono stati aggiunti 200 μl di soluzione II alcalina (0,2N NaOH, 1%SDS) e 150 μl di soluzione III neutralizzante (3M potassio acetato, 5M acido acetico). Dopo un’incubazione di 3-5 minuti in ghiaccio, il lisato batterico è stato centrifugato a 4°C per 5 minuti a 12000 rpm e il surnatante, contenente il DNA plasmidico, è stato trasferito in un nuovo tubino. Sotto cappa è stato aggiunto un volume di fenolo-cloroformio pari a quello del campione ed è stata effettuata una centrifuga (12000 rpm, 4°C per 5’) per andare ad eliminare le proteine.
La centrifugazione determina infatti la formazione di due fasi: il fenolo-cloroformio con le proteine nella parte inferiore del tubino e la parte superiore contenente il DNA. Quest’ultimo è stato prelevato e precipitato con l’aggiunta di 0,7 volumi di isopropanolo. Quindi è stata effettuata un’ulteriore centrifugazione di 15 minuti a 12000 rpm ed il pellet sottoforma di DNA è stato lavato con 150 μl di etanolo al 70%. A questo punto è stata effettuata un’ultima centrifugazione (12000 rpm, 4°C per 5’).
Il pellet è stato asciugato e risospeso in 20 μl di H2O contenente RNAsi (un decimo del
volume), la quale è stata lasciata agire per 30 minuti a 37°C per permettere la degradazione di tutto l’RNA presente.
Dopo l’estrazione il DNA è stato controllato su gel di agarosio all’1% e quantificato misurando con lo spettrofotometro la densità ottica alla lunghezza d’onda di 260 nm. Inoltre il DNA estratto è stato sottoposto ad un’ulteriore screening per digestione (§3.2.2) controllato mediante corsa elettroforetica su gel d’agarosio all’1%.
3.3 Linee cellulari utilizzate
3.3.1 MBM
Si tratta di una linea cellulare T-linfoide ottenuta nel nostro laboratorio da peripheral blood mononuclear cells (PBMC) di un gatto SPF negativo per tutti i retrovirus felini attualmente noti (Matteucci et al., 1995). Le MBM richiedono come fattori di crescita interleuchina-2 (IL-2) e concavalina A (ConA) e crescono in sospensione. Come i linfoblasti di primo isolamento, anche queste cellule sono sensibili all’infezione con diversi ceppi di FIV.
Il terreno di coltura in cui vengono mantenute le cellule MBM è RPMI 1640 Medium (Sigma, St Louis, USA) addizionato con il 10% di siero bovino fetale (FBS) (Sigma) inattivato a 56°C per 35 minuti, L-glutammina 2mM (Sigma), penicillina 1000U/ml (Sigma), 100 μg di streptomicina (Sigma) e l’1% di aminoacidi non essenziali (Sigma). Nel caso specifico delle MBM il terreno RPMI viene addizionato con 20 U/ml di IL-2 ricombinante umana (Roche, Mannheim, Germania) e, una volta a settimana con 5 μg/ml di ConA (Sigma). Due volte a settimana le cellule vengono contate e viene valutata la vitalità, previa colorazione con Trypan Blue.
3.3.2 CrFK (Crandell Feline Kidney fibroblast)
Sono cellule derivate da fibroblasti renali di gatto. Crescono in aderenza in terreno DMEM (Dulbecco’s Modified Eagle’s, Sigma D5546) con il 10% di FBS addizionato con l’1% di L-glutammina, l’1% di amminoacidi non essenziali, penicillina 1000U/ml e 100 μg di streptomicina. Ogni 3-4 giorni le cellule vengono esaminate al microscopio ottico e, se confluenti lavate con PBS (PBS 10X: 80 g di NaCl, 2 g di
KCl, 14,4 g di Na2HPO4, 2,4 g di KH2PO4, HCl a pH 7,4), tripsinizzate
(Tripsina/EDTA, Sigma) e messe di nuovo in coltura in terreno fresco alla concentrazione di 2×104 cellule/ml.
La linea cellulare CrFK CD134+, gentilmente fornita dal Dr. Brian Willett (Università
di Glasgow), è stata geneticamente modificata per esprimere stabilmente il recettore cellulare CD134 (Shimojima et al., 2004) e prevede lo stesso trattamento in coltura descritto precedentemente.
Tutte le colture cellulari sono mantenute a 37°C in atmosfera umidificata con il 5% di CO2.
3.4 Trasfezione con polietilenimmina (PEI)
Le particelle virali FIV-Luc e VSV-G/Luc sono state prodotte mediante cotrasfezione del costrutto di packaging, costrutto vettore e plasmide envelope nella linea cellulare CrFK. Ventiquattro ore prima della trasfezione 1,5 × 106 cellule sono state seminate in
piastre Petri.
La quantità totale di DNA utilizzato è di 35 μg distribuita in rapporto 1:2:4 fra i tre costrutti (envelope, packaging e costrutto vettore). Per l’allestimento della trasfezione sono state preparate 2 soluzioni: la prima è composta da 600 μl di NaCl2 150mM e 100
μl di PEI 1μ mentre la seconda contiene il DNA totale portato ad un volume di 700 μl con l’aggiunta di NaCl2 150mM. Le due soluzioni sono state mescolate ed incubate per
15 minuti a temperatura ambiente, dopodiché la miscela è stata distribuita goccia a goccia in modo uniforme sulle cellule da trasfettare. Trascorse 6-8 ore il terreno è stato sostituito con 10 ml di terreno fresco e le cellule sono state mantenute in coltura per circa 48 h durante le quali lo pseudovirus è rilasciato nel surnatante. A due giorni dalla trasfezione il surnatante delle cellule trasfettate è stato raccolto e chiarificato mediante centrifugazione a 1800 rpm per 10 minuti. Le particelle virali prodotte sono state conservate a -80°C in aliquote.
Per valutare l’efficienza di trasfezione le cellule trasfettate sono state analizzate mediante lettura al luminometro (§ 3.6) ed il background di fondo è stata eliminato grazie alla luminescenza emessa dalle sole cellule usate come controllo negativo (Kc).
3.5 Trasduzione
Il giorno precedente la trasduzione sono state seminate 2,5 × 104 cellule CrFK e CrFK
CD134+/pozzetto in una piastra da 96 pozzetti. La trasduzione è stata effettuata
aggiungendo alle cellule 100 μl di pseudovirus FIV-Luc o VSV-G/Luc ed incubando la piastra a 37°C per 6 ore permettendo così l’assorbimento delle particelle virali alle cellule. Dopodichè il terreno è stato sostituito con terreno fresco e le cellule sono state incubate nuovamente a 37°C per 48 h. L’efficienza di trasduzione è stata valutata mediante una contemporanea lettura al luminometro delle cellule trasdotte con FIV-Luc e delle cellule usate come controllo negativo (Kc). Inoltre è stato previsto anche un
controllo positivo in cui le cellule sono state trasdotte con lo pseudovirus VSV-G/Luc.
3.6 Rilevamento luminescenza
A 48 h dalla trasduzione il terreno è stato rimosso e le cellule sono state incubate, per circa 2 minuti, con 200 μl/pozzetto di soluzione Britelite Plus (Luminescence Reporter Gene Assay System, PerkinElmer, Waltham, USA), necessaria per la lisi cellulare e per fornire il substrato della luciferasi. Trascorsa l’incubazione il campione è stato trasferito in una specifica piastra da lettura che verrà posizionata nel luminometro (MicroBeta Trilux, PerkinElmer). Lo strumento permette di rilevare l’espressione di luciferasi (relative luciferase unit, RLU) mediante il software MicroBeta Windows Workstation.
3.7 Produzione e purificazione ceppo virale FIV-Pet
Il ceppo FIV usato nel nostro lavoro è l’isolato Petaluma il quale determina una rapida infezione sistemica ed una diminuzione consistente dei linfociti T CD4+ nei gatti infetti
(§ 1.7). FIV-Pet è stato propagato su colture cellulari della linea MBM. Per garantire l’integrità strutturale delle particelle infettanti, il virus è stato chiarificato da eventuali detriti cellulari mediante una centrifugazione a 1800 rpm per 10 minuti e successivamente è stato concentrato.
3.8 Titolazione virale
La titolazione dell’isolato FIV Pet è stata effettuata sulla linea cellulare MBM.
In una piastra da 96 pozzetti sono stati posti 100 μl/pozzetto di diluizioni scalari in base 10 (da 10-1 a 10-6) del virus da titolare; ogni diluizione è stata testata in triplicato e
la piastra è stata incubata per 1 h a 4°C. Dopodichè ogni diluizione è stata aggiunta a 105 cellule/pozzetto MBM seminate il giorno prima in una piastra da 96 pozzetti. La
piastra così preparata è stata incubata a 37°C per 5 h; al quarto giorno è stato cambiato metà terreno sostituendolo con terreno fresco addizionato con IL-2. A 8 giorni dall’infezione sono stati misurati i livelli dell’enzima virale trascrittasi inversa (RT) nei sovranatanti di coltura. Il titolo virale è il reciproco dell’ultima diluizione in cui la RT è positiva e si esprime come dosi infettanti il 50% delle colture (ID50).
3.9 Dosaggio dell’enzima RT
Il saggio della RT, basato sul metodo ELISA, viene allestito di routine in laboratorio per rilevare l’infezione da parte di FIV in colture cellulari, per quantizzare il virus prodotto e per caratterizzare parzialmente il virus. Nel test in vitro si sfrutta la capacità della RT virale, ove fosse presente nel campione, di sintetizzare un filamento di DNA usando come stampo l’RNA (polyA), in presenza di oligodT che fungono da primer, nucleotidi biotinilati e cationi bivalenti Mg2+ che risultano indispensabili per il
funzionamento dell’enzima. Inoltre, tale dipendenza, permette di rendere specifica la reazione e di poter discriminare tra Retrovirus appartenenti a generi diversi, in quanto, per esempio, FIV necessita di ioni Mg2+ mentre il virus della leucemia felina (FelV) di
ioni Mn2+.
Il giorno precedente l’esecuzione del test, è stato allestito un coating ponendo 50 μl/pozzetto di N-idrossisulfosuccinimide (Sulfo-NHS) 12,7mM (Pierce, Rockford, USA) e polyA (Pharmacia, Upsala, Svezia) alla concentrazione di 80 μg/ml insieme con 50 μl/pozzetto di 1-etil3-carbodiimide idrocloruro (EDC) 10mM (Pierce). La piastra così preparata è stata tenuta overnight a temperatura ambiente e successivamente posta a 4°C fino al momento di iniziare il saggio vero e proprio.
Sempre il giorno precedente il saggio, sono stati preparati anche i campioni, aggiungendovi, con un rapporto 3:2, PEG 6000 e NaCl 4M (rapporto 4:1 tra PEG e NaCl). I campioni così trattati sono stati incubati overnight a 4°C. Il giorno del test i campioni sono stati centrifugati a 10000 rpm per 20 minuti a 4°C. Il sovranatante di coltura cell-free ottenuto con la centrifugazione è stato rimosso mentre il precipitato è stato risospeso in 50 μl di tampone di lisi composto da Triton X-100 0,25%, ditiotreitolo (DDT) 12mM e Tris-HCl 50mM a pH 8. A questo punto la piastra ha subito i primi 6 lavaggi con un tampone costituito da Tris HCl 10mM a pH 7, NaCl 0,15mM, EDTA 1mM e Tween20 0,01%.
Dopodichè sono stati seminati 12 μl/pozzetto del campione lisato insieme a 50 μl/pozzetto di una miscela di reazione contenente Tris HCl 62,5mM a pH 7.8, MgCl2
6,25 gr/ml, KCl 1M, DTT 12mM, dTTP 1μM, dUTP-biotinilato 0,5μM e 150 μl oligodNTP (1,185 mg/ml). E’ stata quindi effettuata un’incubazione di 2 ore a 37°C per permettere all’enzima virale RT, se presente nel campione nella forma funzionalmente attiva, di sintetizzare DNA a partire dai nucleotidi biotinilati forniti. Dopo questa fase, la piastra è stata lavata 6 volte sempre con il suddetto tampone di lavaggio, quindi l’avvenuta polimerizzazione del DNA è stata rilevata aggiungendo 100 μl/pozzetto di extravidina perossidasi (Sigma) diluita 1:5000 in tampone TBSE (Tris-HCl a pH 7.5 10mM, NaCl 0,15mM, EDTA 1mM e siero albumina bovina-BSA 1%). Questa fase richiede solo 10 minuti di incubazione a 37°C.
L’eventuale presenza di RT è stata rilevata mediante una reazione colorimetrica sviluppata dalla miscela costituita da H2O e 3,3’,5,5’-tetrametil-benzidina (TMB
Substrate Kit, Pierce), in rapporto 1:1 (vol/vol), che funziona come substrato della perossidasi. Lo sviluppo di colore richiede dai 5 ai 7 minuti, passati i quali la reazione è stata bloccata con 50 μl/pozzetto di H2SO4 1N.
Secondo questo metodo, la quantità di RT è direttamente proporzionale all’intensità di colore sviluppata, letta come assorbanza nell’intervallo di lunghezza d’onda compreso tra 450 e 650 nm con il processore Behring ELISA II (Hoechst, Germania). Sono considerati positivi, i campioni che presentano un valore di densità ottica (D.O.) 5 volte maggiore della media dei valori dei controlli negativi o comunque maggiori di 0,05 nel caso in cui i controllo negativi presentino una D.O. uguale a zero
.
3.10 Saggio di neutralizzazione classico
I campioni di siero utilizzati sono stati scomplementati a 56°C per 30 minuti, diluiti 1:32, 1:64, 1:128 e 1:256 in terreno RPMI completo ed incubati con 10 ID50 di virus
FIV-Pet per 1 h a 4°C o 37°C. Ogni diluizione è stata testata in triplicato, trascorsa l’incubazione ogni diluizione (150 μl/pozzetto) è stata seminata su 105 cellule MBM e
dopo 4 h di incubazione a 37°C, il surnatante è stato rimosso e sostituito con terreno fresco addizionato con IL-2 e ConA. Dopo 3 giorni, è stato cambiato metà terreno e la coltura cellulare è stata mantenuta a 37°C fino al settimo giorno, quando viene determinata nel sovranatante di coltura l’attività della RT (§ 3.9) verificando così l’efficienza di neutralizzazione dei campioni analizzati.
3.11 Saggio di neutralizzazione di ultima generazione
Come sopra descritto i sieri sono stati scomplementati e diluiti 1:32, 1:64, 1:128 e 1:256 in terreno DMEM completo ed incubati con quantità fisse di particelle virali FIV-Luc per 1 h a 4°C o 37°C. Ogni diluizione è stata testata in triplicato; trascorsa l’incubazione ogni diluizione (100 μl/pozzetto) è stata seminata su 3 × 104 cellule
CrFK CD134+ e dopo 4 h di incubazione a 37°C, il surnatante è stato rimosso e
sostituito con terreno fresco. Trascorse 48 h è stato tolto il surnatante dalle cellule ed è stata effettuata lettura al luminometro (§ 3.6) per verificare l’efficienza di neutralizzazione dei campioni in esame. Oltre alle cellule non trasdotte utilizzate come controllo negativo (Kc) il test ha previsto l’incubazione delle cellule con sieri senza
attività neutralizzante (Ks-) e la trasduzione cellulare con particelle virali FIV-Luc
(controllo positivo).
L’attività neutralizzante, espressa come percentuale di riduzione dell’entrata virale, è stata calcolata mediante la seguente formula:
x 100