Capitolo 2
Materiali e Metodi
2.1 Materiali
Tutte le soluzioni utilizzate in questo lavoro di tesi sono state preparate con acqua deionizzata attraverso un sistema di purificazione Millipore S.A. 670120 Mosheim. I reagenti impiegati erano di grado analitico e non hanno subito ulteriori purificazioni. Le misure di pH sono state eseguite con un pH-metro della Metrohom (Herisau, Switzerland) corredato di un elettrodo a vetro combinato.
Al fine di lavorare in condizioni vicine a quelle fisiologiche il pH è stato mantenuto ad un valore pari a 7.0 tramite cacodilato di sodio (NaCac = (CH3)2AsO2Na) 10-2 M e la forza ionica
desiderata è stata raggiunta con aggiunte opportune di cloruro di sodio, magnesio o nichel. Le soluzioni stock di NaCac, cloruro di sodio (NaCl), cloruro di magnesio (MgCl2) e cloruro di
nichel (NiCl2) sono state preparate per pesata a partire dal prodotto commerciale solido
disciolto in acqua bideionizzata.
2.1.1 Polinucleotidi
Il poli(A)∙poli(U) e il poli(U) (sali di sodio liofilizzati, Sigma) sono stati sciolti in acqua bideionizzata. Le soluzioni stock dei polinucleotidi sono state standardizzate spettrofotometricamente usando il coefficiente di estinzione molare proprio di ognuno di essi: per il poli(A)∙poli(U) ε = 14900 M-1cm-1 a 260 nm, pH = 7, I = 0.10 M (NaCl) e per il poli(U) ε = 8900 M-1cm-1 a 260 nm, pH = 7, I = 0.10 M (NaCl) (Appendice I).
Le soluzioni di tripla elica di RNA, poli(A)∙2poli(U), sono state preparate miscelando quantità equimolari di poli(A)∙poli(U) e poli(U) nella soluzione tampone (NaCl 0.1 M, NaCac 0.01 M, pH = 7.0) e lasciando riposare il tutto per almeno 24 ore al buio e a temperatura ambiente. Le concentrazioni delle soluzioni di polinucleotidi sono espresse in molarità di singole basi (sb) per i polinucleotidi in singola elica, in molarità di coppie di basi (bp) per quelli in doppia elica
o in molarità di triplette di basi (bt) nel caso delle triple eliche. La concentrazione molare di polinucleotide viene indicata come CP. Tutte le soluzioni di lavoro, ottenute per diluizione
della soluzione stock, vengono conservate alla temperatura di 4°C.
2.1.2 Coralina
La struttura chimica del legante impiegato, la Coralina, è riportata in Figura 1.14 del Capitolo 1. La soluzione stock di Coralina è stata preparata sciogliendo una quantità nota del suo cloruro puro al 99.9% (Sigma-Aldrich) in acqua bideionizzata. Anche in questo caso le soluzioni di lavoro sono state ottenute per diluizione della soluzione stock di partenza. Sia il solido sia le soluzioni vengono conservate a 4°C al riparo dalla luce, per evitare eventuali effetti fotolitici. Le soluzioni di Coralina sono state preparate frequentemente perché questo colorante tende a decomporsi in soluzione acquosa. La loro concentrazione molare (CD) è
stata determinata sulla base della pesata diretta, utilizzando PM = 417.89.
2.2 Metodi
2.2.1 Misure spettrofotometriche
L’assorbimento di una radiazione UV o visibile è riconducibile all’eccitazione degli elettroni esterni di una molecola. Ogni specie molecolare è in grado di assorbire frequenze caratteristiche della radiazione elettromagnetica. La quantità di radiazione assorbita viene espressa dall’assorbanza (A), che è legata alla concentrazione del campione secondo la legge di Lambert-Beer
A = εbC 2.1
dove ε è il coefficiente di estinzione molare (in M-1 cm-1), b è il cammino ottico (in cm) e C è la concentrazione molare del campione.
Le misure spettrofotometriche sono state effettuate usando due spettrofometri diversi.
Il primo è uno spettrofotometro UV-vis a doppio raggio Perkin-Elmer Lambda 35 (Figura 2.1), dotato di lettura digitale fino al millesimo di assorbanza, che utilizza come sorgenti di
radiazione una lampada al tungsteno per la regione del visibile e una lampada allo Xenon (Hamamatsu) per la regione dell’ultravioletto.
Figura 2.1 Schema a blocchi di uno spettrofotometro UV-vis a doppio raggio.
Un monocromatore permette di selezionare una banda di lunghezze d’onda con la precisione di 1 nm rispetto a quella impostata. Il segnale proveniente dal campione e quello proveniente dal riferimento vengono inviati ad un rivelatore costituto da un fotodiodo. Il rivelatore è collegato con un sistema computerizzato di registrazione dei dati.
Le soluzioni sono contenute in cellette di quarzo con cammino ottico di 1cm e munite di tappo a tenuta; esse vengono mantenute alla temperatura desiderata mediante un bagno termostatico ad acqua avente la precisione di ± 0.1°C. Le misure di assorbanza sono relative rispetto all’assorbimento della cella contenente il riferimento, ovvero il solvente utilizzato per preparare le soluzioni. Tale strumento è disponibile presso il Dipartimento di Chimica dell’Università di Pisa ed è stato utilizzato per la quantificazione dei polinucleotidi e per le analisi cinetiche.
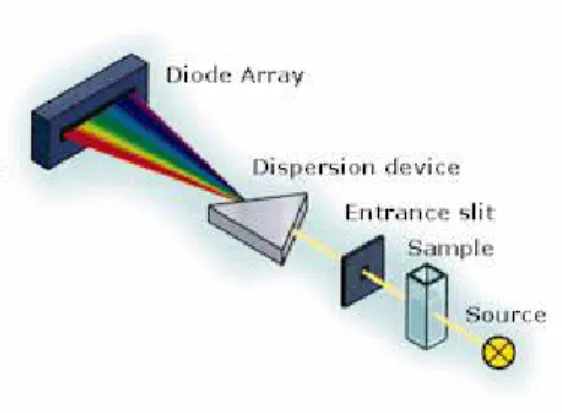
Il secondo spettrofotometro, Hewlett-Packard 8452A (Agilent Technologies), è invece a singolo raggio e sfrutta un campo di lunghezze d’onda tra 190 e 1100nm, ha fenditura da 1nm, la rivelazione è a diodo array e le sorgenti di luce sono lampade a Deuterio e Tungsteno. Questo strumento è assai più semplice rispetto ad uno spettrofotometro a doppio raggio non essendo dotato di monocromatore (Figura 2.2).
Figura 2.2 Schema dello spettrofotometro a diodo array utilizzato.
Il portacuvette è termostatato con sistema Peltier, modello HP-89090, con una precisione di ± 0.1°C. In questo strumento l’array ha 1024 elementi. La luce dispersa da un prisma in tutte le lunghezze d’onda arriva al diode array ed è misurata così simultaneamente, ovvero l’acquisizione dei dati avviene in parallelo per tutti i fotodiodi. L’elevata velocità di acquisizione dei dati è uno dei fattori che maggiormente lo discostano dalla strumentazione classica a doppio raggio. Con questo strumento sono state eseguite le misure di melting che saranno discusse in seguito. Lo strumento si trova presso il Dipartimento di Chimica Fisica dell´Università di Burgos in Spagna, dove ho avuto l’opportunità di lavorare per cinque mesi durante il mio internato di tesi.
2.2.2 Misure spettrofluorimetriche
La fluorimetria è una tecnica molto utile per studiare l’interazione di piccole molecole con acidi nucleici e per osservare i cambiamenti conformazionali che possono accompagnare tale processo. L’emissione di fluorescenza risulta infatti molto più sensibile all’intorno molecolare delle specie fluorescenti di quanto non lo sia l’assorbimento della luce.
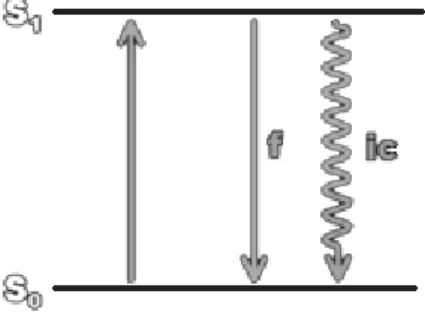
La spettrofluorimetria è una tecnica spettroscopica che consiste nell´eccitare la molecola di interesse con una radiazione monocromatica; successivamente, la molecola eccitata decade radiativamente mantenendo lo stesso stato di spin, emettendo una radiazione di fluorescenza. Gli elettroni raggiungono lo stato elettronico eccitato (assorbimento) in tempi brevi (10-15 s) e si distribuiscono tra i vari livelli eccitati. Il ritorno allo stato fondamentale può avvenire attraverso vari cammini descritti di seguito. Il processo di eccitazione dal livello S0 al livello
rivolta verso l’alto. Il processo di disattivazione dello stato eccitato (S1→ S0) è rappresentato
da due frecce rivolte verso il basso. La freccia ondulata rappresenta una disattivazione non radiativa detta conversione interna (IC) mentre la freccia continua rappresenta la disattivazione per fluorescenza (f).
Figura 2.3 Diagramma di Jablonski che illustra il fenomeno della fluorescenza (f).
La sensibilità molto elevata dei metodi fluorimetrici è dovuta al fatto che l’intensità di fluorescenza può essere aumentata incrementando la potenza della sorgente. Ciò non è invece possibile nel caso delle misure di assorbimento in quanto, sebbene possa essere aumentata l´intensità della sorgente I0, automaticamente viene prodotto un proporzionale aumento
dell’intensità emergente I e questo lascia invariata la trasmittanza, la quale è espressa come rapporto I/I0.
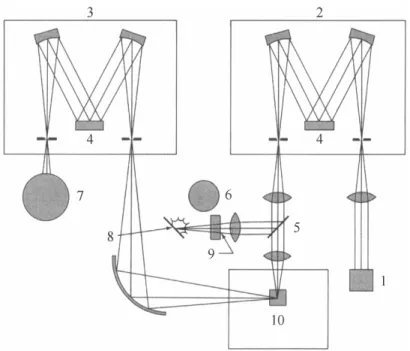
La radiazione di fluorescenza, emessa perpendicolarmente alla radiazione di eccitazione, viene inviata ad uno monocromatore con reticolo mobile il quale permette di registrare lo spettro di emissione. Lo strumento possiede in realtà due monocromatori: il primo è posizionato tra la lampada e la cella di misura e serve a selezionare la lunghezza d’onda d’eccitazione; il secondo è posizionato tra la cella e il rivelatore di fluorescenza e serve a selezionare la lunghezza d’onda di emissione. Il secondo monocromatore raccoglie la radiazione di fluorescenza emessa a 90° rispetto alla direzione del fascio di luce incidente, in modo tale da minimizzare le radiazioni diffuse provenienti dalla sorgente. La radiazione in uscita dal monocromatore arriva al detector (fotomoltiplicatore) che la trasforma in segnale elettrico. Questo, modulato in funzione della lunghezza d´onda, fornisce lo spettro di emissione. La Figura 2.4 mostra lo schema interno di uno spettrofluorimetro.
Figura 2.4 Schema interno di uno spettrofluorimetro: 1) lampada 2) monocromatore di eccitazione 3)
monocromatore di emissione 4) reticolo 5) divisore del fascio 6) tubo fotomoltiplicatore di riferimento 7) tubo fotomoltiplicatore del campione 8) riflettore totale 9) cella di compensazione dell’assorbanza 10) compartimento del campione
La fluorescenza è, come accenato sopra, una tecnica molto più sensibile dell’assorbanza; infatti permette di lavorare con soluzioni la cui concentrazione è dell’ordine di 10-7-10-8 M. Questo fatto è di fondamentale importanza quando si debbano studiare sistemi contenenti specie con tendenza all’auto-aggregazione, oppure sistemi biologici pei i quali le quantità disponibili sono sempre molto piccole.
L’intensità di fluorescenza dipende dall’intensità della radiazione eccitante, dalla quantità di molecole fluorescenti e dalla resa quantica di fluorescenza, che è una proprietà caratteristica di ogni fluoroforo e pertanto può essere considerata un analogo del coefficiente di estinzione molare. La legge che lega l’intensità di fluorescenza alla concentrazione assume, per basse concentrazioni di fluoroforo, una forma molto simile alla legge di Lambert-Beer (equazione 2.2).
F = ּC 2.2
In questa equazione F indica l’intensità di fluorescenza del fluoroforo di concentrazione molare C, mentre φ è il parametro ottico corrispondente, nella fluorescenza, al coefficiente nell´assorbanza (Appendice II).
In questo lavoro è stato utilizzato uno spettrofluorimetro Perkin Elmer LS55, dotato di lampada allo Xenon e lettura digitale fino al millesimo d’intensità di fluorescenza.
Il sistema polinucleotide/Coralina è stato studiato mediante titolazioni spettrofluorimetriche, nelle quali si aggiungono volumi crescenti di polinucleotide a concentrazione nota ad un volume noto di colorante a concentrazione nota, entrambi nelle opportune condizioni di pH e di forza ionica. Per quanto riguarda invece lo studio del sistema poli(A)∙poli(U)/Coralina al variare della concentrazione di ioni metallici in un primo set di esperimenti si aggiungono volumi crescenti di soluzioni a concentrazione nota di cloruro metallico ad una soluzione contenente un rapporto ben preciso di poli(A)∙poli(U)/Coralina. In un altro set di esperimenti, invece, una soluzione di Coralina in presenza di un dato cloruro metallico viene titolata con una soluzione di poli(A)∙poli(U) con la stessa concentrazione di cloruro. I cloruri utillizzati sono NaCl, MgCl2 e NiCl2.
Le aggiunte di titolante sono state eseguite con una microsiringa Hamilton azionata da una microvite, sistema che permette l’aggiunta di piccolissimi volumi con un’elevata precisione. Infatti un giro di microsiringa corrisponde a 8.3 µL e, essendo suddiviso in 50 parti, si ha che il volume minimo aggiunto è pari a 0.166 µL. Anche in questo caso le soluzioni erano contenute in una cella di quarzo con tappo a tenuta e termostatata alla temperatura di 25°C con la precisione di ± 0.1°C.
2.2.3 Curve di fusione (melting) del polinucleotide
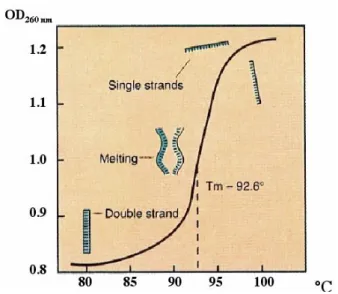
Le prove di denaturazione termica permettono di stabilire la temperatura di “fusione”, ovvero di separazione delle catene di un polinucleotide, oppure di misurare la temperatura della transizione elica-gomitolo. Tali prove vengono effettuate incrementando la temperatura di una soluzione contenente polinucleotide o complesso in concentrazioni opportune, e seguendo la variazione di assorbanza prodotta alla lunghezza d’onda di 260 nm. Le curve ottenute con questo procedimento hanno una forma sigmoidale e sono caratterizzate da un unico salto se il polinucleotide ha una struttura di doppia elica (Figura 2.5), oppure da due salti se ha una struttura di tripla elica. I punti di flesso di tali curve indicano la temperatura alla quale metà del polinucleotide ha subito la transizione, ovvero la temperatura di melting, indicata con Tm.
Per determinare con maggior precisione la temperatura di melting delle diverse transizioni è stata usata la curva che corrisponde alla derivata prima della curva di melting. Nel presente lavoro le curve di denaturazione termica sono state registrate con lo spettrofotometro a singolo raggio descritto precedentemente. Il programma che è stato utilizzato per questo tipo
di misure è basato su un gradiente di temperatura di 0.2°C al min ed un tempo di stabilizzazione di 1 min per ogni step.
Per quanto riguarda il sistema polinucleotide/cationi metallici, viene tenuta fissa la concentrazione di polinucleotide al valore di 1.7×10−5M e variata di volta in volta la concentrazione di catione. Per il sistema polinucleotide/Coralina la concentrazione di colorante è stata mantenuta costante al valore di 1.7×10−5 M mentre la concentrazione di polinucleotide è variata in modo da ottenere il rapporto CD/CP desiderato. Prima di avviare
l’incremento di temperatura la soluzione polinucleotide/Colorante ha riposato per circa due ore.
Figura 2.5 Esempio di curva di melting del DNA in doppia elica.
2.2.4 Dicroismo circolare
Il dicroismo circolare (CD) è una tecnica spettroscopica basata sulla capacità delle molecole chirali, ossia che presentano due configurazioni (levogira L e destrogira R), di interagire in modo diverso con la luce polarizzata circolarmente. Questa tecnica, capace di distinguere le differenze tra due forme enantiomeriche, è altamente sensibile alle caratteristiche tridimensionali delle molecole, cioè alla loro conformazione (Woody, 1995) e può dare informazioni, per esempio, anche sulla specifica orientazione di un farmaco relativamente ad un polinucleotide con il quale interagisce.
Molti leganti possiedono cromofori achirali e perciò non mostrano CD in soluzione; nonostante questo, quando queste molecole si legano all’elica dell’acido nucleico, che è una
struttura chirale, acquistano un CD indotto (Lyng et al., 1991 e 1992) che è caratteristico della loro interazione con il polinucleotide (Fasman, 1996). I cambiamenti nello spettro di dicroismo circolare di un polinucleotide in seguito all’interazione con un colorante possono essere utilizzati per determinare le costanti di equilibrio della reazione di binding e inoltre forniscono indicazioni qualitative circa la natura dell’interazione. La tecnica di dicroismo circolare fornisce quindi informazioni sulla struttura secondaria di biopolimeri, quali acidi nucleici e proteine e sul legame di piccole molecole con questo tipo di macromolecole.
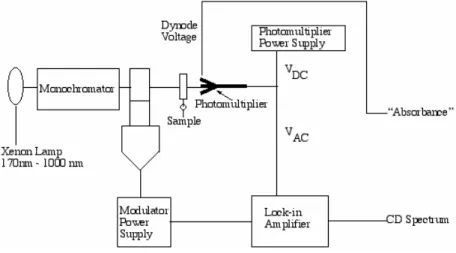
Figura 2.6 Schema interno di uno spettrometro CD.
Nella pratica quando la luce piano polarizzata passa attraverso una celletta che contiene un campione chirale, le due componenti circolari, destra e sinistra, sono assorbite differentemente e la radiazione che si otterrà alla fine non sarà più piano polarizzata ma risulterà ellitticamente polarizzata: il vettore campo elettrico ruoterà in un piano normale alla direzione di propagazione descrivendo un’ellisse. Questo segnale è raccolto da un fotomoltiplicatore ed elaborato da un computer. Sperimentalmente si misura l’ellitticità, indicata con , definita come la differenza tra l’assorbanza della componente destra e quella sinistra.
Gli spettri di dicroismo circolare sono stati registrari con uno strumento MOS-450 Biologic (Claix, France) provvisto di una lampada ad arco di Xenon (Figura 2.6). In tale strumento la temperatura della cella è controllata da un bagno termostatico con la precisione di ± 0.1°C. Sono state utilizzate celle di quarzo con cammino ottico di 1.0 cm nell´intervallo di lunghezze d’onda 200 ÷ 700 nm. Le titolazioni CD sono state effettuate aggiungendo quantità crescenti di colorante al polimero posto in cella e registrando gli spettri dopo ogni aggiunta.
2.2.5 Calorimetria Differenziale a Scansione
La calorimetria differenziale a scansione (DSC) è una tecnica usata per studiare cosa accade a determinati materiali (nel caso di questo lavoro di tesi agli acidi nucleici in soluzione) quando vengono riscaldati. Si utilizza pertanto per studiare le transizioni termiche di opportune sostanze come ad esempio la fusione di un polimero, le transizioni vetrose, la cristallizzazione ed altri cambiamenti di stato. In tutti i casi i processi studiabili con questa tecnica devono essere accompagnati da sviluppo o assorbimento di calore. La tecnica fu inventata E.S. Watson e J. O’Neill nel 1964 (Watson et al., 1964). Il primo calorimetro differenziale per lo studio dei biopolimeri e’ stato sviluppato da Privalov e Monaselidze nel 1964 e il metodo e’ stato usato con successo per studiare il processo di denaturazione della doppia elica del DNA in seguito a riscaldamento (Sturtevant, 1987; Ferriera e Sheardy, 2006).
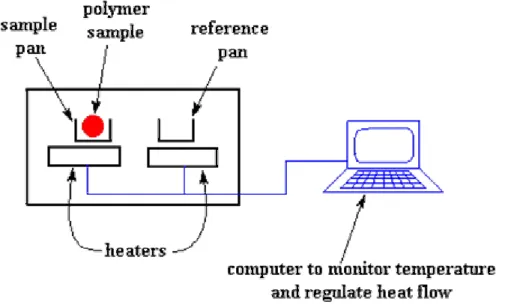
La tecnica DSC consiste nel riscaldare il sistema da studiare secondo le modalità descritte qui di seguito. La Figura 2.7 ne mostra in forma schematica il principio di funzionamento.
Figura 2.7 Rappresentazione schematica della tecnica calorimetrica differenziale
Il sistema calorimetrico contiene due recipienti: in uno, il recipiente “campione” viene posto il materiale da studiare, per esempio un polimero. L'altro, il recipiente di “riferimento” non lo contiene. Ogni recipiente è posto su un riscaldatore. Tramite computer vengono accese delle resistenze che fanno aumentare la temperatura dei riscaldatori. Viene programmato di eseguire il riscaldmento secondo la modalità desiderata, ad esempio 10°C al minuto. Il computer controlla che il flusso di calore rimanga costante per tutta la durata dell’esperimento
e, cosa ancora più importante, controlla che i due recipienti separati, ognuno con la sua resistenza, si scaldino con lo stesso flusso di calore.
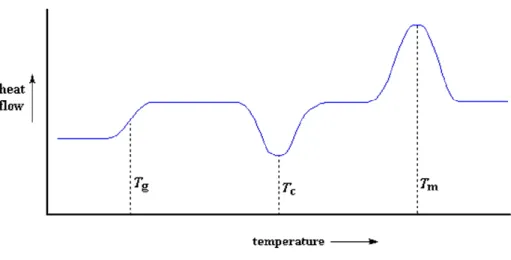
Poichè il “campione” ed il “riferimento” sono diversi (uno contiene il polimero, l'altro no), è diversa la condizione dei recipienti perché uno contiene del materiale in più rispetto all’altro. Avere del materiale in più significa che sarà necessario maggior calore per far sì che il flusso di aumento della temperatura sia uguale nei due recipienti. Nel momento in cui si manifesta la transizione (supponiamo che sia endotermica) la resistenza sotto il recipiente campione deve lavorare di più rispetto a quello sotto il recipiente di riferimento ovvero deve fornire più calore. La quantità di calore in più che deve emanare è il parametro che viene misurato in un esperimento DSC. Il risultato dell’esperimento viene presentato in forma di grafico (Figura 2.8) in cui sull’asse-x viene riportata la temperatura e sull'asse-y la differenza di calore fornito dalle due resistenze in corrispondenza di ogni temperatura. Tale differenza è indicata come flusso di calore (heat flow).
Figura 2.8 Termogramma DSC che mostra le principali caratteristiche delle curve sperimentali a seconda della
transizione osservata: Tg temperatura di transizione vetrosa, Tc temperatura di cristallizzazione, Tm temperatura di fusione.
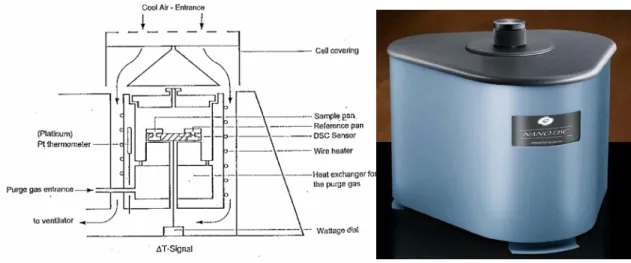
Il calorimetro utilizzato in questo lavoro di tesi è stato un Setaram micro DSC III differential scanning calorimeter (Figura 2.9).
Figura 2.9 Schema e foto di un microcalorimetro differenziale a scansione
Esso consta essenzialmente di un blocco metallico con alta conducibilità termica, che crea un ambiente adiabatico la cui funzione è di comportarsi come un termostato. Al suo interno esistono due celle gemelle, quella del riferimento e del campione, circondate da sensori per misurare il flusso di calore. Un sistema di circolazione di undecano attorno al blocco assicura la sua stabilità termica, essendo la temperatura controllata attraverso un sistema Peltier. Questa tecnica è stata impiegata in un primo momento per determinare i parametri termodinamici del processo di denaturalizzazione del poli(A)∙poli(U) (CP = 2.5×10-4) M sotto
l’influenza dello ione Mg2+ in varie concentrazioni, poi è stata utilizzata con lo stesso scopo anche per il sistema poli(A)∙poli(U)/Coralina a varie concentrazioni di Na+. Da un punto di vista sperimentale è stato utilizzato un volume di 850 L. La velocità di scansione utilizzata per questi esperimenti è di 1°C/min. E’ importante non avere bolle di aria nella cella, per questo tutte le soluzioni vengono degassificate in un degasatore della TA instruments per almeno 15 minuti prima di riempire la cella. I dati sono stati analizzati con il software NanoAnalyze 2.0.