Stabulazione di Xenopus laevis e cura degli embrioni
Gli esemplari di X.laevis sono stabulati in un ambiente climatizzato, in vasche di acqua a pH, salinità e temperatura controllata.
Gli embrioni utilizzati per gli studi in vivo sono ottenuti tramite fecondazione in vitro secondo i protocolli descritti da Newport e Kirshner. Il maschio viene anestetizzato immergendolo per circa 30’ in una soluzione allo 0,1% di MSS-222 (metansulfonato dell’estere etilico dell’acido 3-aminobenzoico) prima venire sottoposto a chirurgia per la rimozione dei testicoli. I testicoli vengono poi conservati a 4°C (per un periodo massimo di due settimane) in una soluzione di MMR 1X + gentamicina 20 μg/ml fino al momento dell’uso.
La femmina viene prestimolata da 4 a 11 giorni prima della deposizione con 100 UI di “Folligon Intervet” per uso veterinario (ormoni estrogeni), e in seguito stimolata 12-15 h prima della deposizione con 800/1000 UI di gonadotropina corionica “Profasi HP 5000 Serono”. Gli ormoni vengono somministrati per iniezione parenterale.
Le uova vengono ottenute “spremendo” con delicatezza l’animale e vengono raccolte in piastre petri contenenti MMR 0,1X. Sulle uova viene poi sparso un omogenato di testicolo (per una fecondazione è sufficiente una piccola porzione dell’organo). Dopo circa 30’-1h il fenomeno della rotazione corticale evidenza l’avvenuta fecondazione.
Le uova di X.laevis sono, alla deposizione, immerse in una soluzione gelatinosa che in natura ha la funzione di proteggerle e mantenerle ancorate a
Stadio Tempo (22°C) Stadio Tempo (22°C) 1 (1 cell) 0 24 26h 15’ 2 (2 cell) 1h 15’ 25 27h 30’ 3 (4 cell) 2h 26 29h 30’ 4 (8 cell) 2h 15’ 27 31h 15’ 5 (16 cell) 2h 45’ 28 32h 30’ 6 (32 cell) 3h 29/30 35h 7 (64 cell) 3h 30’ 31 37h 30’ 8 4h 32 1g 16h 9 5h 33/34 1g 21h 10 7h 35/36 2g 2h 11 11h 45’ 37/38 2g 6h 12 13h 15’ 39 2g 8h 13 14h 45’ 40 2g 18h 14 16h 15’ 41 3g 4h 15 17h 30’ 42 3g 8h 16 18h 15’ 17 18h 45’ 18 19h 45’ 19 20h 45’ 20 21h 45’ 21 22h 30’ 22 24h 23 24h 45’
Eventi salienti dello sviluppo:
St.8: Mid blastula transitino St.10: Inizio della gastrulazione St. 13: Inizio della neurulazione St. 18: Chiusura del tubo neurale
St. 28: Rottura del sacco vitellino, embrione natante
St. 40: Apertura della bocca
Variazioni del ritmo di crescita in base alla temperatura:
(secondo Rob Grainger)
22 °C: 1X 20 °C: 0,75X 16 °C: 0,5X 14 °C: 0,33X
Table 1: Andamento temporale dello sviluppo di Xenopus laevis secondo Nieuwkoop e Faber ed
supporti (piante e rocce) subacquei o superficiali. Tale sostanza è di disturbo per le procedure di manipolazione degli embrioni, e viene pertanto rimossa immergendo gli zigoti per 5’-10’ in una soluzione degelificante contenente DTT (ditiotreitolo). Dopo la degelificazione, gli embrioni vengono nuovamente immersi in MMR 0,1X, in cui vengono mantenuti durante lo sviluppo in piastre petri di 10 cm di diametro.
Durante lo sviluppo l'embrione viene mantenuta a temperature comprese tra 14°C e 22°C. La temperatura influenza la velocità di sviluppo senza avere particolari effetti negativi su di esso. E’ possibile pertanto sfruttare una certa plasticità nei tempi e nelle temperature di crescita, che consente di pianificare gli esperimenti in anticipo. Uno strumento indispensabile è lo schema temporale dello sviluppo elaborato da Nieuwkoop e Faber nel 1967 (vedi tab. 1), modificato da Rob Grainger tenendo conto della temperatura. Lo stadio degli embrioni viene identificato osservandone la morfologia esterna (esempi dei diversi stadi sono indicati in fig. 9)
Il principale rischio per gli embrioni in crescita è dovuto allo shock derivante dalla microchirurgia, che può provocare gravi anomalie di sviluppo quali mancata gastrulazione o gastrulazione esterna, mancata neurulazione ed errata specificazione degli assi corporei o semplicemente morte. Un secondo grave problema è dato dall’infezione batterica, che si manifesta tramite l’opacizzazione del mezzo di crescita e la comparsa di ammassi di microrganismi sopra gli embrioni. Per evitarla questi vengono travasati 1-2 volte al giorno in una nuova piastra contenente MMR 0,1X pulito utilizzando una pipetta sterile. Nei casi più gravi è possibile aggiungere gentamicina in
Fig. 9: Morfologia esterna di embrioni di Xenopus Laevis a vari stadi
concentrazione 10 mg/ml diluita 1:1000 in MMR 0,1X, anche se l’utilità di quest’ultima contromisura non è più garantita, in seguito all’insorgenza di ceppi batterici resistenti.
La contaminazione batterica è solitamente ingente al di fuori del sacco vitellino, mentre l’embrione all’interno è generalmente esente da contaminazione. Per evitare la sua contaminazione al momento della rottura del sacco è possibile rimuovere chirurgicamente quest’ultimo con una coppia di pinzette immediatamente prima della rottura spontanea., ripulendo poi l’embrione e trasferendolo in una piastra pulita.
Un terzo fattore di rischio è lo shock termico, frequente nel caso in cui gli embrioni vengano mantenuti in un ambiente sottoposto a variazioni repentine di temperatura o vengano spostati frequentemente. Per evitare questo problema si è preferito, quando possibile, lasciare che l'intero sviluppo si svolgesse nelle medesime condizioni di temperatura.
Il trattamento farmacologico con ciclopamina o idrossiurea/afidicolina ha effetti notevoli sullo sviluppo. Gli embrioni sottoposti a questi trattamenti presentano di frequente gravi malformazioni alla parte anteriore, alla testa e al tubo neurale. Nel caso della ciclopamina gli occhi si presentano più vicini del normale e possono arrivare a fondersi (fenomeno noto come oloprosencefalia, o
ciclopismo), oltre a essere di dimensioni ridotte, mentre nel caso dell'HUA le dimensioni oculari
sono ridottissime, tanto che talvolta non è possibile individuare un occhio vero e proprio.
La manipolazione degli embrioni, quando necessario, viene eseguita con anse a capello (ottenute da un capello umano sterilizzato e da una pipetta pasteur tagliata) o con puntali sterili.
Dissezione di tessuto oculare da embrioni di Xenopus laevis
Il tessuto oculare necessario per l'estrazione dell'RNA totale utilizzato negli studi successivi viene estratto per dissezione da embrioni a st. 33 / st. 42 di X.Laevis. Gli embrioni vengono posti in una piastra petri contenente un sottile velo di MMR 0,1X. In seguito, utilizzando un micro-bisturi e un paio di pinzette da dissezione (serie “Dumont biologie” n.5), vengono decapitati. Si esegue poi un taglio sagittale mediano nella testa, esponendo il lato interno dei globi oculari, inclusi in una matrice extracellulare relativamente poco compatta. Evitando manovre brusche con il bisturi (per non danneggiare il tessuto) i globi oculari vengono a questo punto, con il piatto della lama, rimossi e in seguito puliti, sempre con l'aiuto del bisturi, dal tessuto estraneo (tipicamente l'epitelio esterno). Il fatto che il globo oculare sia notevolmente più compatto della matrice in cui è incluso facilita di molto queste operazioni.
Gli occhi dissezionati vengono a questo punto prelevati (badando di aspirare la minore quantità di liquido possibile) con una micropipetta da 10 μl e posti in una provetta da 1,5 ml contenente 500 μl di reagente “Trizol” posta in ghiaccio. Si procede poi immediatamente con l'estrazione dell'RNA.
Estrazione di RNA da tessuto di Xenopus / linee cellulari
Per ottenere l'RNA necessario agli esperimenti di ibridazione su microarray o di RT-PCR è stato utilizzata la tecnica di purificazione per precipitazione in fenolo/cloroformio utilizzando il reagente commerciale “Trizol” (fornito dalla ditta “Invitrogen”). Si tratta di una miscela di fenolo e guanidina isocianato miscelati a una concentrazione ideale per la purificazione di RNA.
L'estrazione è stata effettuata a partire da 60-100 occhi per gli embrioni di X.laevis (utilizzando 500 μl di “Trizol”) o da 1x106 cellule per linee HEK293 e HeLa (1 ml).
I tessuti sono stati dapprima omogenati nel reagente, secondo il protocollo standard fornito dalla Invitrogen. In seguito sono stati aggiunti 0,2 volumi di cloroformio, dopodichè la miscela è stata agitata per unire le due fasi e centrifugata per 15' a ~15000 RCF a 4°C. E' stata poi recuperata la fase acquosa (contenente gli acidi nucleici) prestando la massima attenzione a non prelevare parti della fase inferiore per non contaminare l'estratto.
Si è poi proceduto alla precipitazione alcoolica aggiungendo 1 volume di isopropanolo e centrifugando per ulteriori 15' a 15000 RCF a 4°C. Il pellet risultante è stato lavato con etanolo, lasciato seccare e risospeso in H2O di purezza mQ sterile. Nonostante si trattasse di campioni di
RNA, quindi molto soggetti a degradazione, non è stata utilizzato l'inibitore delle nucleasi
dietilpirocarbonato (DEPC) in quanto avrebbe rischiato di diminuire l'efficienza degli enzimi
utilizzati negli esperimenti successivi.
Dopo l'estrazione, i campioni sono stati conservati a -20°C fino al momento dell'utilizzo.
Controllo di qualità degli RNA
Affinchè l'ibridazione su microarray o la retro-trascrizione riescano nel migliore dei modi, gli RNA estratti devono trovarsi nelle migliori condizioni possibili. Questo include sia la purezza chimica (non devono essere presenti tracce di contaminanti derivanti dal processo di estrazione,
come l'isopropanolo o il fenolo, che inibirebbero le reazioni enzimatiche necessarie) che l'integrità (gli RNA non devono essere degradati, ed in particolare i miRNA devono essere integri.
Nonostante sia estremamente complesso e poco pratico verificare direttamente la presenza di un pool di miRNA valido, le tecniche tradizionali per il controllo della qualità di soluzioni di acidi nucleici si sono dimostrate adeguate ai nostri scopi. In particolar modo abbiamo effettuato sui nostri campioni una analisi di assorbanza tramite spettrofotometria e una analisi elettroforetica.
La purezza chimica è stata calcolata misurando il rapporto tra i valori di assorbanza del campione se attraversato da una luce di lunghezza d'onda pari a 260 e 280 nm. Sono stati utilizzati nelle analisi successive solo campioni con rapporto superiore a 1.70.
L'integrità è stata valutata tramite corsa elettroforetica su gel di agarosio all' 1% p/V condotta per 30' a ~150 V. Assieme ai campioni sono stati caricate miscele di tRNA a concentrazione nota, utilizzati per costruire una curva di calibrazione da cui è stata in seguito stimata la loro concentrazione approssimativa. Dopo l'acquisizione dell'immagine della corsa, è stato valutato il rapporto tra lo spessore / luminosità della banda relativa al rRNA 28S e quella relativa al rRNA 18S, che sono le due principalmente visibili in un estratto totale di RNA. Tale rapporto deve essere pari a circa 2 perchè i campioni possano essere considerati integri.
Ibridazione e analisi di microarray per miRNA
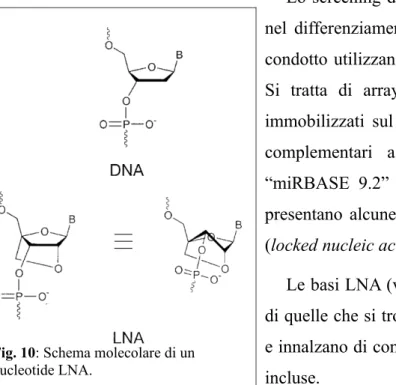
Lo screening di massa per la ricerca di miRNA implicati nel differenziamento delle cellule bipolari retiniche è stato condotto utilizzando microarray “Exiqon miRCURY LNA”. Si tratta di array specifici per miRNA in cui i probes immobilizzati sul vetrino sono costituiti da oligonucleotidi complementari a tutti i miRNA inclusi nel database “miRBASE 9.2” del Sanger Institute. Tali oligonucleotidi presentano alcune basi sostituite con acidi nucleici bloccati (locked nucleic acid, LNA).
Le basi LNA (vedi fig. 10) sono estremamente più stabili di quelle che si trovano comunemente nel DNA e nell'RNA, e innalzano di conseguenza la Tm della molecola in cui sono
incluse.
Fig. 10: Schema molecolare di un
Negli arrays tradizionali, le Tm dei probes possono venire uniformate modificandone la
lunghezza, ma questo non è possibile per i miRNA, dato che questi sono molto corti e presentano molta omologia nelle prime basi, specie all'interno dei cosiddetti cluster (quindi per ottenere una sufficiente specificità è necessario usare l'intera sequenza). L'uso degli LNA consente, sostituendo un numero variabile di basi, di ridurre moltissimo la variabilità delle Tm, anche in questo caso, e di
condurre quindi l'ibridazione dell'interno vetrino ad una stessa temperatura, peraltro abbastanza elevata (cosa che contribuisce a ridurre il disturbo da legami aspecifici).
Su ogni vetrino sono stati ibridati due diversi campioni di RNA, marcati con due differenti fluorocromi (hy3/cy3 e hy5/cy5). I miRNA marcati presenti in ciascuno dei due campioni competono per il legame al probe specifico. Il rapporto hy3/hy5 osservato per ogni probe costituisce, quindi, un indice della differente abbondanza del relativo miRNA nei campioni (un rapporto 1:1 indica che non ci sono variazioni).
La marcatura dei campioni di RNA totale è stata effettuata come da protocollo “Exiqon miRCURY array power labeling kit”. In seguito i due campioni sono stati miscelati e, dopo l'aggiunta di 1 volume di “soluzione di ibridazione” (presente nel kit commerciale) la miscela è stata posta sull'array, poi coperto da un vetrino coprioggetto 24x60 mm e posto in una camera di ibridazione, identica a quelle di uso standard per ISH su sezioni, inumidita con la stessa soluzione di ibridazione.
Gli array sono stati ibridati per un tempo compreso tra le 12 e le 16 ore in incubatore termico umidificato mediante bacinelle conteneti acqua deionizzata, a una temperatura di 56°-60°C. In seguito ai lavaggi, effettuati come da protocollo (escluso l'ultimo passaggio in H2O) abbiamo
proceduto all'analisi.
Il segnale di ibridazione di ciascun vetrino è stato acquisito con uno scanner “Axon Genepix” con risoluzione minima pari a 5 μm utilizzando il software “Axon Genepix pro 6”. Il valore dei fotomoltiplicatori relativi ai canali dei fluorocromi hy3 e hy5 è stato tarato empiricamente con l'obiettivo di portare il più vicino possibile a 1 il valore del rapporto di intensità per i probes di controllo. Si tratta di probes disegnati per legarsi specificamente a RNA (in questo caso gli RNA del complesso di splicing U6) considerati “housekeeping” e quindi presumibilmente invarianti nelle varie tesi sperimentali.
Dopo l'acquisizione, utilizzando i file di descrizione della geometria dell'array forniti dalla Exiqon (files GAL), è stata disegnata una griglia comprendente tutti i probes ibridati, poi allineata all'immagine. Le regioni del vetrino male ibridate, o in qualche modo danneggiate, sono state
escluse dalle analisi successive. A questo punto sono stati rilevati i valori di intensità di ogni singolo probe e dello sfondo (disturbo) su ciascun canale.
Su ogni vetrino sono presenti 4 ripetizioni di ogni probe. I dati relativi a ciascuna di esse sono stati mediati e utilizzati per calcolare l'espressione relativa di ogni miRNA (definita come il rapporto tra l'intensità di fluorescenza del canale hy3 sul canale hy5, generalmente utilizzato per marcare l'RNA scelto come linea di base dell'esperimento), poi convertita in scala logaritmica. Oltre a questo valore sono stati presi in considerazione nell'analisi anche il rapporto segnale-rumore (definito come il rapporto tra la fluorescenza all'interno dello spot e quella media dello sfondo) e l'intensità totale (definita come la somma dei 4 replicati delle mediane dell'intensità di fluorescenza dei due canali).
I dati ottenuti sono poi stati analizzati utilizzando il software “Axon Acuity” e posti in forma grafica sotto forma di mappa cromatica (heatmap) utilizzando il software open source “JcolorGrid”.
Reazione di retro-trascrizione per l'analisi di miRNA
Il primo passo dell'analisi per qRT-PCR dell'espressione di miRNA è la reazione di retro-trascrizione. Al fine di poter esaminare molecole di RNA di così piccole dimensioni, questa deve essere effettuata in modo particolare.
Le reazioni tradizionali di RT effettuate su estratti di RNA totale o poli-A, infatti, non produrrebbero alcun cDNA relativo a miRNA. In queste reazioni solitamente si fornisce come primer di innesco della trascrittasi inversa un oligonucleotide poli-dT o una miscela di esameri a sequenza casuale. I miRNA però non vengono poli-adenilati, ed essendo molto brevi vi è una elevata probabilità che alcuni di essi non presentino regioni di complementarietà con nessuno degli esameri presenti nella miscela.
Per ovviare a questo problema vi sono diverse alternative. Quella da noi utilizzata è presente nei kit commerciali “miScript reverse transcription kit” prodotto dalla Qiagen e “QuantiMIR” prodotto dalla Systems Biosciences, entrambi utilizzati in questo studio (i dati presentati nella sezione
risultati si riferiscono al primo).
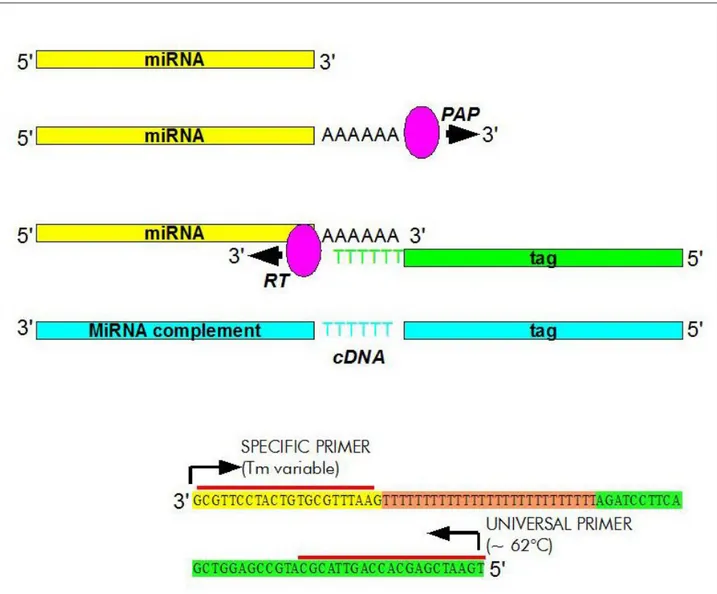
Essenzialmente si tratta di aggiungere ai passaggi tradizionali della reazione di RT una reazione iniziale di poli-adenilazione. I campioni di RNA vengono dapprima incubati in presenza di un enzima dotato di attività poli-adenil-polimerasica, che aggiunge alle estremità 3' di tutte le
molecole, inclusi miRNA e in genere piccoli RNA nucleari, una sequenza di adenine. In seguito viene eseguita la retro-trascrizione, utilizzando come primer di innesco un oligonucleotide, detto
universal RT primer, dotato di una sequenza poli-dT al 3' e di una sequenza nota (tag) al 5'.
La ragione di quest'ultima sequenza sta ancora nelle piccole dimensioni dei miRNA. A causa di esse sarebbe impossibile, infatti, nella fase di PCR vera e propria, utilizzare una coppia di primer entrambi specifici (dovrebbero essere troppo brevi). L'uso di una sequenza universale al 3' del primer di innesco consente invece di “allungare” l'amplicone, utilizzando come primer reverse della PCR un oligonucleotide complementare al tag. Questo permette una analisi efficace anche se non esente da problemi (vedi risultati).
L'intera reazione è stata svolta come da protocollo incluso nel kit. (E' stato utilizzato ~1 μg di RNA per ogni campione). Al termine ogni cDNA è stato diluito in TE 1X (10mM tris-HCl pH 8,0; 2mM EDTA) fino a portarlo ad una concentrazione pari a ~ 0,4 ng / μl e aliquotato in modo da poter utilizzare campioni identici nei diversi esperimenti di PCR.
Uno schema dell'intera reazione di retro-trascrizione e della successiva PCR è presente in fig.11
PCR quantitativa real-time per la quantificazione di miRNA
Le reazioni di PCR real-time sono state svolte dapprima con un termociclatore “Roche LightCycler 480/96” in specifiche piastre multiwell da 96 pozzetti, e in seguito in provette individuali da 0,2 ml con l'ausilio di un roto-termociclatore “Corbett Rotor-Gene 6000”, seguendo la procedura descritta nei rispettivi manuali di utilizzo. Il secondo macchinario si è dimostrato di maggiore praticità e precisione, specie per quanto riguarda l'uniformità termica e di lettura ottica (e quindi la precisione dei replicati interni), ed è per questo stato utilizzato per ottenere i dati mostrati in risultati.
Per la rilevazione dell'amplificazione è stato utilizzato l'intercalante fluorescente SYBR green, che si lega in modo specifico al solo DNA a doppio filamento e fluoresce solo in questa forma. L'intensità di fluorescenza è dunque un indice della quantità di amplificato sintentizzata. Dapprima abbiamo fatto uso della miscela “Qiagen miScript PCR kit” ma, in seguito ai risultati non ottimali ottenuti con l'analisi di alcuni miRNA, siamo passati ad una miscela contenente una maggiore quantità di intercalante (“Corbett MESA Green”). Le reazioni sono state allestite come di seguito indicato.
Come primer forward è stato utilizzato un oligonucleotide con sequenza identica al miRNA da analizzare, e quindi complementare al cDNA. Le piccole dimensioni dei miRNA fanno sì che, di fatto, questo primer copra completamente la sequenza da amplificare. Come già detto, non è possibile “puntare” due diversi primer su uno stesso miRNA.
Come primer reverse, come già detto, è stato utilizzato un oligonucleotide complementare alla parte finale della sequenza tag inclusa nel primer di innesco della retro-trascrizione. Sono stati utilizzati diversi primer reverse, di diversa lunghezza, in modo da adattarsi alle temperature di melting dei vari primer forward specifici.
Ogni reazione è stata condotta in triplicato. Dopo una fase di attivazione iniziale dell'enzima (tutte le miscele utilizzate contenevano una taq ad attivazione termica, o hot start) di 5' a 95°C, sono stati effettuati 40 cicli con i seguenti passaggi. La temperatura di annealing è stata ottimizzata per ogni reazione tenendo conto della Tm del primer specifico. (vedi risultati)
1. Denaturazione 95°C 15''
2. Annealing temperatura variabile (fig. 11) 30''
3. Estensione 72°C 30''
La lettura dell'intensità di fluorescenza è stata effettuata al termine del terzo passaggio. Immediatamente prima della prima acquisizione il fotomoltiplicatore del sistema è stato tarato in modo che il valore di fluorescenza iniziale fosse compreso tra 1 e 3 in una scala che va da 1 a 100. Questo ci ha permesso di essere certi di includere l'intera curva di amplificazione nel campo dinamico dello strumento.
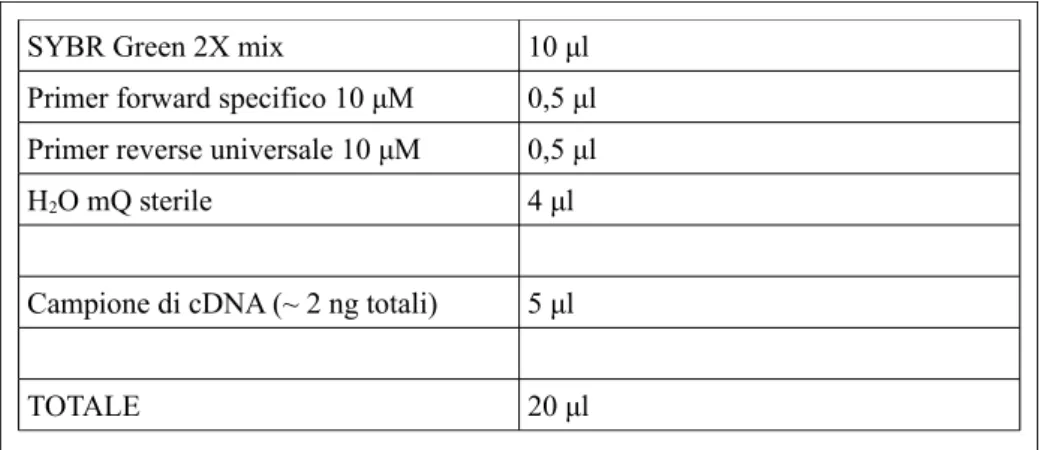
SYBR Green 2X mix 10 μl
Primer forward specifico 10 μM 0,5 μl Primer reverse universale 10 μM 0,5 μl
H2O mQ sterile 4 μl
Campione di cDNA (~ 2 ng totali) 5 μl
TOTALE 20 μl
Al termine dell'esperimento i campioni sono stati sottoposti a un aumento graduale della temperatura da 40° C a 96°C, durante il quale l'intensità di fluorescenza è stata letta in modo continuo. La curva temperatura/fluorescenza ottenuta presenta dei flessi corrispondenti alle temperature di melting delle specie presenti nell'amplificato (idealmente solo una). Tali flessi compaiono come picchi in un grafico temperatura/derivata seconda(fluorescenza), o curva di
melting, e consentono di verificare se l'amplificazione è stata effettuata correttamente o se, al
contrario, sono presenti ampliconi derivanti da legami aspecifici dei primer.
Fig. 11: Schema della reazione di retro-trascrizione e PCR per miRNA. E' qui indicato il metodo utilizzato dai kit
Analisi dei dati di qRT-PCR
Una volta verificata la specificità dell'amplificazione, abbiamo eseguito l'analisi secondo due diversi metodi, utilizzati rispettivamente con i dati ricavati dal sistema Roche e dal sistema Corbett.
Per quanto riguarda il primo, abbiamo dapprima interpolato i dati del grafico tempo/fluorescenza che descrive l'andamento dell'amplificazione, ottenendo una curva che è possibile dividere idealmente in 3 segmenti:
1. Plateau In questa fase la fluorescenza dell'agente intercalante incorporato nel DNA non è sufficiente per essere rilevata dallo strumento. La curva rimane piatta sul valore iniziale
2. Fase esponenziale In questa fase la reazione procede esattamente come previsto dalla teoria, vale a dire in modo esponenziale. La quantità di DNA, e quindi di fluorescenza, aumenta ad ogni ciclo con una curva di tipo y=Ex, dove x corrisponde al numero di cicli
ed E all'efficienza di amplificazione (teoricamente pari a 2)
3. Fase logaritmica La reazione inizia a rallentare via via che si esauriscono i pool di primers e di dNTP, e tende alla saturazione, giungendo fino a un secondo plateau corrispondente al valore finale della fluorescenza.
A questo punto il grafico è stato convertito in forma logaritmica, in modo che la fase esponenziale comparisse come una retta con pendenza proporzionale all'efficienza dell'amplificazione. Questa è stata calcolata attraverso l'algoritmo contenuto nel software open-source “LinRegPCR” (Ramakers et al., 2003).
La quantificazione degli amplificati è stata invece effettuata tracciando sul grafico una retta corrispondente ad un valore di fluorescenza in cui tutte le curve di amplificazione si trovassero nella fase esponenziale. L'intercetta di ciascuna delle curve con tale retta, o punto di take-off è stata utilizzata come indicatore della quantità iniziale della specie amplificata. A questo punto abbiamo eseguito una quantificazione relativa della quantità dei vari miRNA, utilizzando uno dei campioni come “linea di base”.
eff mediaamplificato da quantificaretakeoff medio BASE −TRATTATO
eff mediaamplificato di riferimentotakeoff medio BASE −TRATTATO (metodo di quantificazione relativa di Pfaffl, Tichopad et al.,
Dove con “amplificato di riferimento” si intende l'RNA U6, quantificato per ogni diverso cDNA ed utilizzato per normalizzare eventuali differenze nella quantità di RNA iniziale e nell'efficienza della retro-trascrizione.
Il risultato del calcolo ha fornito il cosiddetto fold ratio, vale a dire il rapporto tra la quantità di amplificato presente nel trattato e quella presente nel campione di riferimento, normalizzato per differenze “globali” nella concentrazione di cDNA. Tali rapporti sono poi stati espressi in forma logaritmica nei grafici mostrati in risultati.
Nel caso del sistema “Corbett Rotor Gene”, invece, il grafico tempo/intensità di fluorescenza è stato dapprima modificato sostituendo all'intensità di fluorescenza la propria derivata seconda. In questa forma, il grafico si presenta come una sorta di sinusoide.
In primo luogo viene individuato il valore di x (numero di cicli) corrispondente al massimo della curva derivata della fluorescenza (Xmax), in seguito il valore corrispondente al 20% di tale massimo,
che viene assunto come take off della curva (Xto). L'efficienza viene invece ricavata sulla base della
pendenza del segmento di retta incluso tra Xto e Xmax, che si assume essere lineare. Anche in questo
caso, chiaramente, l'efficienza massima teorica è pari a due.
La quantificazione relativa viene a questo punto calcolata con il metodo di Pfaffl già descritto.
Fissazione, inclusione e crioprotezione di embrioni di Xenopus laevis
Prima dell’osservazione, gli embrioni da esaminare necessitano di una preparazione adeguata che comprenda un passaggio di fissazione, affinché sia conservata la morfologia cellulare, ed uno di inclusione, che renda possibile la realizzazione di sezioni abbastanza sottili da essere osservate o sottoposte ad ulteriori trattamenti.
Il metodo comunemente utilizzato per gli embrioni di X.Laevis a stadio di sviluppo 30 (st. 30) o superiore consiste nella loro inclusione in un blocco di resina acquosa a basso punto di congelamento (detta O.C.T o ‘Tissue-Tek’ , composta per oltre il 98% da sostanze non reattive e per la restante parte di Alcol Polivinilico e “carbowax”, prodotto commerciale) e nel loro taglio tramite un criostato. Questo rende necessario l’inserimento, nel protocollo di fissazione, di un passaggio di crio-conservazione che permetta la conservazione della morfologia cellulare nonostante il congelamento (diversamente i cristalli di ghiaccio distruggerebbero i tessuti).
Gli embrioni sono stati innanzi tutto prelevati dalle piastre di crescita e immersi in una soluzione al 4% P/V di paraformaldeide (PFA) in PBS 1X per un tempo variabile tra 1h e 1h30’ in agitazione orizzontale oppure over night (O.N.) senza agitazione a 4°C (a seconda delle dimensioni). Questo passaggio ne provoca la morte, e fissa le strutture morfologiche dato che la PFA forma legami crociati tra le proteine.
Quindi gli embrioni sono stati travasati in una soluzione di saccarosio in PBS 1X con concentrazione pari al 20% P/V, e incubati per 1-3h in agitazione a RT oppure O.N a 4°C. In questo intervallo di tempo il saccarosio permea completamente i tessuti agendo come crioprotettivo, impedendo cioè la formazione di micro-cristalli di ghiaccio durante il successivo congelamento.
E’ essenziale che la paraformaldeide e il saccarosio siano diluiti in PBS. Questa è una soluzione fisiologica iso-osmotica rispetto alle cellule, contenente sali e tamponata a pH 7,4. Se per le diluizioni venisse utilizzata acqua, ipo-osmotica rispetto al citoplasma, le cellule esploderebbero per osmolisi con conseguente perdita sia della morfologia tissutale sia del contenuto in acidi nucleici e proteine da rilevare in seguito.
A questo punto gli embrioni sono stati posti in una vaschetta di plastica, coperti con la resina di inclusione (O.C.T.) e allineati con il dorso in alto e con le teste rivolte tutte nella stessa direzione, lungo un margine della vaschetta. Le vaschette sono state poi immerse in un bagno di etanolo a -80°C e conservate alla stessa temperatura fino al momento dell’uso.
Il passaggio cruciale di questo protocollo è il congelamento. Infatti, perché la morfologia tissutale sia conservata, è necessario che questo sia molto rapido e che non ci siano differenze di temperatura tra i vari punti del blocco di resina (che, a causa dei movimenti di dilatazione termica, provocherebbero strappi nei tessuti). Per questo si utilizza un bagno di etanolo a bassissima temperatura (generalmente -80°C), che è in grado di portare in breve tempo tutta la vaschetta al punto di congelamento.
Taglio al criostato
Per produrre le sezioni necessarie agli esperimenti successivi è stato utilizzato un criostato (Leica). Abbiamo scelto questo approccio in luogo della classica inclusione in paraffina per la sua rapidità, essendo il grado di conservazione della morfologia tissutale fornito da questa tecnica (comunque inferiore a quello della paraffina) sufficiente per i nostri scopi.
I blocchetti contenenti gli embrioni sono stati fatti aderire al portablocco del criostato con un velo di O.C.T e allineati alla lama. In seguito all'eliminazione di tutta la resina in eccesso il blocco è stato ritagliato fino ad ottenere un profilo rettangolare in più possibile preciso comprendente una fila ordinata di embrioni sezionati trasversalmente. A questo punto è stato posizionato il vetrino stendifetta e sono state effettuate sezioni con spessore di 12 μm, recuperate in seguito avvicinando ad esse, mentre sono ancora stese sulla lama, un vetrino la cui superficie è stata precedentemente coperta con una matrice di poli-lisina per favorire l'adesione (“Menzel-Glaser POLYSINE slides”)
Il taglio è una operazione critica per il processo di preparazione degli embrioni, e necessita di una certa manualità. E’ necessario prestare attenzione a molte variabili, che possono tutte danneggiare il risultato finale. In particolare:
1. Temperatura: tutti i componenti devono avere temperatura omogenea, diversamente i movimenti di dilatazione termica peggiorerebbero la qualità delle sezioni
2. Posizione del vetrino stendifetta e degli altri componenti dell’apparecchiatura (lama,supporto): da questo deriva la corretta stesura delle sezioni
3. Forma del blocco da sezionare: una forma non geometricamente precisa produce fette che tendono a rovinarsi o ad incurvarsi
4. Presenza di intaccature sulla lama/sul vetrino: le intaccature possono strappare le sezioni 5. Recupero delle sezioni: il vetrino su cui si recuperano le sezioni deve essere mantenuto il
più possibile immobile e parallelo alla lama durante l’adesione, che avviene, anche in assenza di contatto diretto, per attrazione elettrostatica. Movimenti anche impercettibili possono strappare gli embrioni
Generalmente su un comune vetrino 25x75 mm è possibile recuperare dalle 16 alle 20 sezioni di spessore pari a 12 μm. Dopo il recupero, i vetrini devono essere lasciati per 30’ – 1h all’aria a seccare, per consentire una migliore adesione delle sezioni, e utilizzati il prima possibile (eventualmente conservandoli a -80°C per preservare il contenuto di RNA interno alle sezioni)
Ibridazione in-situ su sezione ottimizzata per miRNA
Per l'ibridazione in situ delle sezioni istologiche di X.laevis abbiamo utilizzato un protocollo da noi perfezionato a partire da quello descritto da S.Banfi (Karali et al., 2007).
Il probe è costituito da un oligonucleotide modificato LNA complementare alla sequenza del miRNA sotto studio, recante all'estremità 3' una molecola di digossigenina (DIG) che ne consente la rilevazione tramite un apposito anticorpo monoclonale. I probes sono stati forniti dalla ditta Exiqon come nel caso di tutti gli altri prodotti contenenti LNA descritti in questo lavoro di tesi.
L'uso degli LNA consente di migliorare la specificità di legame ed è al momento l'unica tecnica affidabile per la detezione di miRNA su preparati istologici (Kloosterman et al., 2006). In questo caso il vantaggio fondamentale derivante dall'utilizzo di molecole di acido nucleico modificato non è tanto l'uniformità tra le Tm di diversi probe, dato che ciascun miRNA viene analizzato a sé, quanto
la possibilità di utilizzare temperature, in assoluto, più elevate. Per ottenere una ibridazione efficiente di molecole così brevi e poco abbondanti, infatti, in assenza delle modificazioni sarebbe necessario utilizzare temperature di ibridazione estremamente basse. In altre parole, in una sezione istologica, il legame tra probe e bersaglio sarebbe tanto instabile da venire rotto spesso già a temperatura ambiente, con conseguente insuccesso dell'esperimento.
L'uso degli LNA, al contrario, permette di aumentare la stabilità e di ottenere un sufficiente segnale specifico, anche se le temperature di ibridazione restano comunque relativamente basse (vedi tab.3) e i lavaggi devono essere effettuati in modo poco stringente.
I vetrini contenenti le sezioni vengono dapprima immersi per circa 5' in una soluzione di PBT 1X (PBS 1X addizionato con lo 0,1% di detergente tween 20) allo scopo di rimuovere la resina utilizzata per l'inclusione.
Su ogni vetrino viene in seguito gocciolato ~1 ml di una soluzione contenente 0,25 ng/μl di proteinasi K in PBT 1X, che viene mantenuto sulle sezioni per 5' esatti. Questo passaggio fessure le membrane cellulari e nucleari, permettendo un migliore accesso dei probes alle molecole di miRNA.
La proteinasi K viene in seguito neutralizzata lavando i vetrini due volte per 10' in una soluzione di glicina 2mg/ml in PBT 1X. La glicina satura l'enzima, bloccandone l'azione. Le sezioni vengono poi post-fissate per 15' in una soluzione di paraformaldeide al 4% p/V in PBT 1X, per fissare la morfologia cellulare parzialmente danneggiata dal passaggio in proteinasi.
Dopo tre ulteriori lavaggi di 3' ciascuno in PBT 1X sui vetrini viene gocciolato ~1 ml di buffer di ibridazione, basato sul buffer SSC 5X e contenente il 50% V/V di formammide, lo 0,1% di tween 20 ed eparina e RNA totale di torula (per saturare i siti di legame aspecifici) in concentrazione pari rispettivamente a 50 μg/ml e 500 μg/ml. La soluzione viene portata a pH 6 (dato che l'RNA è più stabile in condizioni acide) con acido citrico 1M
In questo primo passaggio al buffer non viene aggiunto il probe. Questa pre-ibridazione, che viene effettuata per 1h alla stessa temperatura dell'ibridazione vera e propria, ha lo scopo di saturare i siti aspecifici di legame, facendo sì che venga rilevato un minor segnale di fondo.
Come ultimo passaggio su ogni vetrino vengono posti circa 150 μl di buffer di ibridazione, contenente stavolta circa 150 ng (1 ng/ml) di probe LNA 3'DIG, coperti in seguito con un vetrino copri-oggetto 24x60 facendo attenzione a non lasciare bolle d'aria.
Il tutto viene poi posto in una camera umida (ricavata da una piastra petri di 15 cm di diametro sul cui fondo, coperto da un doppio strato di carta 3 MM imbevuta di buffer di ibridazione, sono state incollate sezioni di pipette da 10 ml come sostegno) e incubato per 14h-16h a una temperatura dipendente dal probe (vedi tab. 3)
Dopo l'incubazione, i copri-oggetto vengono rimossi immergendo per un attimo i vetrini in una soluzione di SSC (sodio-sodio citrato) 2X alla temperatura di ibridazione. I vetrini vengono di seguito lavati per due volte 10' in SSC 2X e per una volta 10' in SSC 0,2X (lavaggi a stringenza decrescente). Dopo l'ultimo lavaggio in SSC se ne effettuano ancora 5 di 3' ciascuno in PBT 1X allo scopo di allontanare i sali dell'SSC.
A questo punto si passa alla rilevazione dei gruppi DIG tramite un anticorpo policlonale conigato a fosfatasi alcalina (Anti-DIG Fab-fragments Roche1:2000). I vetrini vengono dapprima trattati per 30' / 1h con una soluzione di pre-immunomarcatura contenente l' 1% di “Blocking solution Roche”, lo 0,1% di BSA (albumina serica bovina) e lo 0,1% di Tween 20 in buffer MAB 1X, allo scopo di
xtr-mir-129 LNA 3'DIG 42° C xtr-mir-155 LNA 3'DIG 37° C xtr-mir-222 LNA 3'DIG 42 °C xtr-mir-214 LNA 3'DIG 42 °C xtr-mir-124 LNA 3'DIG 42 °C xtr-mir-scrambled LNA 3'DIG 42 °C
bloccare i siti aspecifici (questo passaggio è analogo a quello di pre-ibridazione). L'immunomarcatura vera e propria viene effettuata per 12h-14h a 4°C, con l'anticorpo diluito nella stessa soluzione .
L'ultimo passaggio del protocollo è la rilevazione degli anticorpi anti-DIG attraverso la reazione dell'enzima fosfatasi alcalina loro coniugato con un substrato fluorescente (“Fast RED”). A questo scopo i vetrini rimossi dall'incubazione con l'anticorpo, dopo aver subito 3 lavaggi in PBT 1X, vengono immersi per 3 volte 10' in una soluzione contenente NaCL 1M, CaCl2 0,1M, Tris pH 9,5
1M, Tween 20 0,1% e “Tetramisole” in concentrazione di 500 ng/ml (un inibitore delle fosfatasi endogene, che reagirebbero con il substrato fluorescente dando segnale di fondo). I lavaggi in questa soluzione (AP buffer) creano sulle sezioni le condizioni ideali per l'azione dell'enzima sul substrato.
Il substrato è fornito sotto forma di tavolette solubili che vengono disciolte in 2 ml di tampone Tris pH 8.2 0,1 M. La soluzione risultante viene gocciolata sui vetrini poi coperti con vetrini coprioggetto. La reazione di colorazioni viene fatta preseguire per un tempo variabile tra i 10' e i 180' a temperatura ambiente, dopodichè i vetrini vengono lavati un'ultima volta in PBT 1X, colorati con hoechst per marcare i nuclei cellulari (come da protocollo standard) e montati con una resina a base acquosa (“Aqua POLY-mount”)
Acquisizione delle immagini
I vetrini ibridati sono stati osservati utilizzando un microscopio ad epifluorescenza “Zeiss Axioplan” dotato di obiettivi 4X, 10X, 20X e 40X. Le osservazioni in fluorescenza sono state effettuate utilizzando set ottici con filtri predisposti per gli spettri di eccitazione dei tre tipi di fluorocromo maggiormente utilizzati nella pratica di laboratorio: rodamina (luce rossa, utilizzato anche per la RFP), fluoresceina (luce verde, utilizzato anche per la GFP) e DAPI (luce blu, utilizzato per la rivelazione della contro-colorazione nucleare con hoechst). In alcuni casi sono state utilizzate gelatine colorate poste direttamente sopra il filtro di emissione per ridurre ulteriormente la banda di frequenze osservate.
Le foto sono state acquisite utilizzado una fotocamera digitale monocromatica con sensore raffreddato (per minimizzare il disturbo dovuto alle resistenze elettriche interne) da 2 megapixel “Zeiss AxioCam ” e il software “Zeiss AxioVision LE”.
Le foto sono state poi ottimizzate (manipolando esclusivamente i parametri di luminosità,