UNIVERSITA’ DI PISA
SCUOLA DI SPECIALIZZAZIONE IN FARMACOLOGIA
TESI DI SPECIALIZZAZIONE
FARMACI AGONISTI DEI RECETTORI ATTIVATI DAL
PROLIFERATORE DEL PEROSSISOMA (PPAR) NEL
TRATTAMENTO DEL DIABETE DI TIPO II: DALLO SVILUPPO
CLINICO AL RITIRO DI ROSIGLITAZIONE PER TOSSICITÁ
CARDIOVASCOLARE
Relatore
Chiar.mo Prof. Corrado Blandizzi
Candidato
Dott.ssa Pamela Giambastiani
Anno Accademico 2010/2011
INDICE
1. Introduzione
32. Obiettivo
43. Metodi
44. Fisiopatologia e decorso naturale del diabete di tipo II
55. Terapia del diabete di tipo II e razionale per lo sviluppo dei glitazoni
75.1 Recettori PPAR: ruolo fisiologico 7
5.2 Secrezione e sensibilità all’insulina 12
5.3 Aumento della sensibilità all’insulina 12
5.4 Effetti indiretti nel tessuto adiposo 14
6. Efficacia clinica dei glitazoni
176.1 Effetti in pazienti con diabete di tipo II 17
6.2 Effetti ipoglicemizzanti 17
6.3 Effetti sui lipidi 18
6.4 Effetti in condizioni di insulino resistenza 19
6.5 Steatosi nonalcolica 20
6.6 Sindrome dell’ovaio policistico 20
6.7 Lipodistrofie 21
6.8 Effetti sui fattori di rischio cardiovascolare 22
6.9 Aumento ponderale e pressione arteriosa 22
6.10 Plasma e fattori urinari 23
6.11 Funzione vascolare 24
7. Sicurezza e tollerabilità dei glitazoni
257.1 Epatotossicità 25
7.1.1 Studi sulla tossicità epatica di troglitazone 25
7.1.2 Studi sulla tossicità epatica di pioglitazone e rosiglitazone 28
7.2 Ritenzione di fluidi, edema e rischio di insufficienza cardiaca 30
7.2.1 Ritenzione idrica 30
7.2.2 Edema 31
7.2.3 Rischio di insufficienza cardiaca congestizia 32
7.3 Aumento ponderale 35
7.4 Effetti procarcinogenici dei PPARγ 36
7.5 Teratogenesi 38
8. Tossicità cardiovascolare di Rosiglitazone: evidenza clinica
399. Tossicità cardiovascolare di Rosiglitazone: azioni regolatorie
429.1 European Medicines Agency 42
9.2 Food and Drug Administration 43
10. Conclusioni
461.
INTRODUZIONEIl diabete mellito è un disturbo metabolico di eziologia variabile, caratterizzato da iperglicemia cronica e da diverse complicanze microvascolari e macrovascolari che possono aggravare la patologia e portare a mortalità. Esistono due tipi di diabete: di tipo 1 (insulino-dipendente) e di tipo 2 (non-insulino dipendente) che differiscono per eziologia, decorso e trattamento [Alberti, 1998]. Negli ultimi decenni, l’incidenza e la prevalenza del diabete di tipo 2 sono aumentate a livello mondiale, e si prevede una ulteriore diffusione della malattia nell’immediato futuro in considerazione dei dati epidemiologici dell’obesità, patologia “gemella” del diabete nel contesto della sindrome metabolica [Mokdad, 2001; Amos, 1997]. Viceversa, l’incidenza del diabete di tipo 1 è stabile da diversi anni e non sembra destinata ad aumentare [Amos, 1997]. Il diabete è uno dei fattori più importanti che favoriscono la malattia cardiovascolare e la prima causa di malattia terminale renale e di cecità nei paesi occidentali [American Diabetes Association, 2007]. Per questo motivo la prevenzione e il trattamento del diabete di tipo 2 rappresentano uno dei problemi più pressanti per la salute pubblica.
L’approccio terapeutico nel diabete di tipo 2 si basa sull’adozione di misure dietetiche corrette e sull’acquisizione di un adeguato stile di vita, caratterizzato ad esempio da una corretta attività fisica [N Engl J Med, 2002]. Quando questa strategia non è sufficiente deve essere considerato l’impiego di farmaci in grado di correggere l’insulino-resistenza. Gli strumenti terapeutici attualmente disponibili sono insoddisfacenti nel migliorare alcune conseguenze della resistenza all’insulina, quali iperglicemia, dislipidemia diabetica, coagulazione anormale, fibrinolisi e ipertensione [Yki-Jarvinen, 2003], ognuno dei quali necessita dell’uso di terapie ulteriori con farmaci specifici. Pertanto è importante pianificare lo sviluppo di farmaci in grado di migliorare tutti gli aspetti patologici secondari all’insulino-resistenza. I glitazoni, farmaci agonisti selettivi per il recettore nucleare attivato dal proliferatore del perossisoma (PPAR), costituiscono agenti terapeutici promettenti per il trattamento dell’insulino-resistenza (e delle patologie ad essa correlate) in
pazienti con diabete di tipo 2 e in pazienti con insulino-resistenza di tipo non diabetico [Lehmann, 1995].
2. OBIETTIVO
Questa tesi si pone l’obiettivo di effettuare una revisione delle informazioni disponibili in letteratura relative alla classe terapeutica dei glitazoni con particolare riferimento al loro profilo di tollerabilità.
3. METODI
Per mezzo di ricerche bibliografiche in PubMed, nel database di Medline, sono stati
identificati studi che descrivono i trattamenti disponibili per il diabete mellito di tipo II,
con particolare riferimento alla classe dei glitazoni, utilizzando i seguenti termini:
“Diabetes”, “PPAR”, “thiazolidinediones”, “pioglitazone”, “rosiglitazone”, “troglitazone”,
“safety”, “tolerability” e “adverse drug reactions”. Sono state valutate anche le bibliografie
degli articoli richiamati per identificare altri studi di interesse.
4. FISIOPATOLOGIA E DECORSO NATURALE DEL DIABETE DI TIPO 2
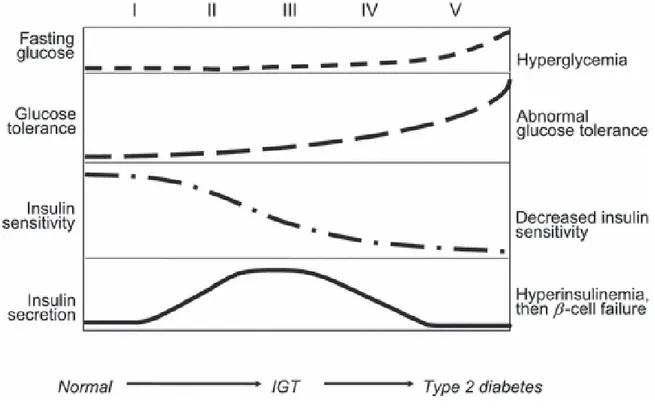
Il diabete mellito di tipo 2 è una malattia a patogenesi complessa e poco conosciuta. E’ caratterizzata da due principali alterazioni: secrezione inadeguata dell’insulina dal pancreas e resistenza periferica all’azione dell’insulina in particolar modo nel muscolo e nel fegato [Alberti, 1998; De Fronzo, 1992]. Queste anomalie sono generalmente riscontrabili al momento della diagnosi. Dalla gravità di queste alterazioni, legate all’insulina, dipendono l’insorgenza di iperglicemia e lo sviluppo di molteplici sindromi correlate al diabete di tipo 2 [Alberti, 1998], associati alla tossicità del glucosio [De Fronzo, 1992; Tack, 2006]. Nei paesi occidentali il diabete di tipo 2 si associa molto frequentemente ad obesità. L’aumento dei casi di obesità è considerato un fattore in gran parte responsabile del corrispondente incremento dell’incidenza e della prevalenza del diabete di tipo 2 [Mokdad, 2003; Amos, 1997]. Inoltre, anche molti pazienti con diabete di tipo 2 non obesi presentano anomalie nella percentuale di massa grassa localizzata nella regione addominale (obesità addominale o centrale) [Alberti, 1998]. Nei pazienti non obesi affetti da diabete di tipo 2, l’alterazione della secrezione di insulina è il difetto predominante, mentre l’insulino resistenza tende ad essere meno grave [Edelman, 2003]. In ogni caso, in soggetti diabetici di tipo 2 obesi o con obesità addominale, che rappresentano la grande maggioranza dei diabetici di tipo 2, il principale disturbo è rappresentato dall’insulino-resistenza [Edelman, 2003]. L’obesità, relativa all’aumento dell’insulino-resistenza, può precedere l’insorgenza del diabete conclamato di diversi anni o addirittura decenni [Sarafidis, 2007] e può associarsi anche ad altre complicanze della sindrome metabolica quali ipertrigliceridemia o ipertensione [Sarafidis, 2006]. Secondo quanto detto sopra, il difetto più importante, nella maggior parte dei pazienti con diabete di tipo 2, è l’insulino-resistenza [De Fronzo, 1992; Turner, 1998; Ramlo-Haslsted, 1999]. Come illustrato in figura 1, in soggetti con normale funzionalità delle cellule -pancreatiche, un aumento della secrezione di insulina (iperinsulinemia compensatoria) può inizialmente controllare l’aumento fisiologico delle concentrazioni di glucosio.
L’aggravamento dell’obesità e il protrarsi dell’insulino-resistenza costringe le cellule -pancreatiche a secernere un elevato quantitativo di insulina e gradualmente il paziente passa da uno stato di alterata tolleranza al glucosio (IGT) caratterizzata da insulino-resistenza, a iperinsulinemia compensatoria ed a iperglicemia postprandiale lieve. Dopo un periodo variabile di tempo, le cellule non saranno più in grado di compensare con quantità adeguate di insulina e, con un deficit relativamente lieve di insulina, il paziente comincerà a manifestare iperglicemia a digiuno, che da un punto di vista clinico rappresenta l’insorgenza del diabete di tipo 2. Quando i livelli di insulina si abbassano ulteriormente, l’effetto inibitore dell’insulina sulla produzione di glucosio epatico diminuisce con conseguente aggravamento dell’iperglicemia a digiuno. Poichè la diagnosi di diabete di tipo 2 normalmente viene effettuata a questo stadio avanzato della malattia, non sorprende il fatto che in soggetti con recente diagnosi di diabete di tipo 2 la funzionalità delle cellule -pancreatiche sia ridotta del 50% [Tack, 2006; Edelman, 2003].
L’ampia diffusione del diabete di tipo 2, diagnosticato attraverso valori glicemici specifici, necessita principalmente del trattamento dell’iperglicemia che può migliorare lo stato morboso della malattia [Nathan, 2006]
5. TERAPIA DEL DIABETE DI TIPO II E RAZIONALE PER LO SVILUPPO DEI
GLITAZONI
La terapia del diabete di tipo 2 si basa sull’adozione di misure dietetiche corrette e sull’acquisizione di un adeguato stile di vita, caratterizzato ad esempio da una corretta attività fisica [N Engl J Med, 2002]. Quando questa strategia non è sufficiente deve essere considerato l’impiego di farmaci in grado di correggere l’insulino-resistenza. Gli strumenti terapeutici attualmente disponibili sono insoddisfacenti nel migliorare gli effetti della resistenza all’insulina quali iperglicemia, dislipidemia diabetica, anomalie della coagulazione, fibrinolisi e ipertensione [Yki-Jarvinen, 2003], ognuno dei quali necessita dell’uso di terapie con farmaci specifici. Pertanto è importante studiare lo sviluppo di farmaci specifici in grado di migliorare tutti gli aspetti patologici secondari all’insulino-resistenza. I glitazoni, farmaci agonisti selettivi per il recettore nucleare attivato dal proliferatore del perossisoma (PPAR), costituiscono agenti terapeutici promettenti per il trattamento dell’insulino-resistenza (e delle patologie ad essa correlate) in pazienti con diabete di tipo 2 e in pazienti con insulino-resistenza di tipo non diabetico [Lehmann, 1995].
5. 1 Recettori PPAR: ruolo fisiologico
I recettori attivati dal proliferatore del perossisoma (PPAR) fanno parte di una superfamiglia
costituita da 48 recettori nucleari [Chawla, 2001] cheregolano l’espressione genica in risposta ad
interazioni molecolari [Berger J, 2002; Barbier, 2002]. I ligandi endogeni per i PPAR sono rappresentati generalmente da acidi grassi, mentre per alcuni recettori appartenenti a questa superfamiglia (recettore X farnesoide) il ligando endogeno è rappresentato da acidi biliari e per altri (recettori X epatici) da ossisteroli [Chawla, 2001].
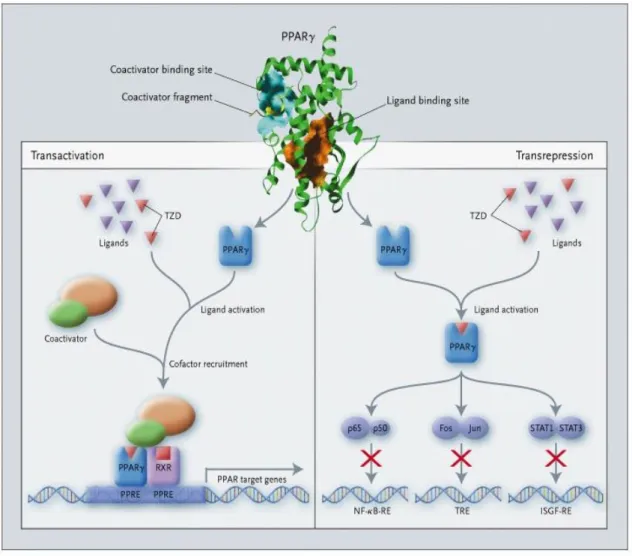
La famiglia dei PPAR comprende attualmente 3 sottotipi di recettori, denominati PPARα, PPARδ (detto anche PPARβ ) e PPARγ, i quali presentano una differente distribuzione tissutale. I PPAR, in seguito all’interazione con i ligandi endogeni, subiscono specifiche alterazioni conformazionali che favoriscono il richiamo di uno o più coattivatori proteici [Willson, 2001] determinando risposte biologiche diverse. [Berger J, 2002; Barbier, 2002; Chinetti, 2000; Berger JP, 2003; Picard, 2002]. I PPAR regolano la trascrizione genica attraverso due meccanismi (Figura 2): il primo, la transattivazione, è un processo DNA-dipendente, e prevede il legame di fattori proteici specifici attivati dai PPAR con sequenze geniche specifiche, denominate PPAR Response Elements (PPRE), e la successiva eterodimerizzazione con il recettore X retinoide [Willson, 2001]; il secondo, la transrepressione, è invece un processo DNA-indipendente, e spiega l’attività antinfiammatoria dei PPAR, attraverso il coinvolgimento di altri fattori [Chinetti, 2000].
FIGURA 2: Il recettore attivato dal proliferatore del perossisoma (PPAR) è un fattore di trascrizione attivato dai glitazoni (TZD). Nella transattivazione, DNA-dipendente, PPAR forma un eterodimero con il recettore X retinoide (RXR) e riconosce regioni specifiche del DNA denominate
PPAR Response Elements (PPRE) nella regione promoter di geni specifici. Questo meccanismo
provoca la trascrizione di geni specifici sensibili ai PPAR. Dopo il legame con i ligandi, i PPARs subiscono alterazioni conformazionali che favoriscono il richiamo di coattivatori proteici. I coattivatori interagiscono con i recettori nucleari in maniera ligando-dipendente e influenzano una serie di geni trascritti. Nella transrepressione, PPARs reprimono la trascrizione genica interferendo negativamente con processi di trasduzione del segnale, come nuclear factor-kB, in maniera DNA-indipendente. STAT indica i trasduttori del segnale ed attivatori della trascrizione, ISGF-RE gli elementi di risposta interferon-stimulated factor e TRE TPA elementi di risposta dove TPA è un estere.
PPARα è localizzato in prevalenza a livello di fegato, cuore, muscolo e parete vascolare [Barbier, 2002]. I fibrati, quali fenofibrato, bezafibrato, ciprofibrato e gemfibrozil, agiscono come agonisti completi o parziali dei PPAR. L’attivazione di PPAR promuove l’ossidazione degli acidi grassi liberi, modula l’espressione di molti geni che regolano le concentrazioni di lipoproteine e determina effetti antinfiammatori (Figura 3). Gli agonisti dei PPAR prevengono o ritardano l’aterosclerosi nel topo e nell’uomo [Duez, 2002; Rubins, 1999; The Diabetes Atherosclerosis Intervention Study, 2001].
PPARδ si trova in molti tessuti, soprattutto nella cute, nel cervello e nei tessuti adiposi. In topi mutati con ablazione di PPAR [Michalik, 2003], i tessuti mostrano alterazioni specifiche quali ritardo della rimarginazione delle ferite e ridotta mielinizzazione.
PPARγ è presente abbondantemente nel tessuto adiposo ma anche nelle cellule β-pancreatiche, nell’endotelio vascolare e nei macrofagi [Willson, 2001; Dubois, 2000], mentre la sua espressione è ridotta nei tessuti con elevata densità di PPARα. La scoperta dei PPAR come specifici bersagli per i glitazoni è stata promossa da studi preclinici e clinici su varie molecole [Aronoff, 2000; Scherbaum, 2002; Rosenblatt, 2001; Einhorn, 2000; Kipnes, 2001; Rosenstock, 2002; Lebovitz, 2002; Fonseca, 2000; Gomez-Perez, 2002; Vongthavaravat, 2002; Raskin, 2001]. Nel gennaio 1997 fu approvato per l’uso clinico negli Stati Uniti il primo glitazone, troglitazone, come terapia ipoglicemizzante nei pazienti con diabete di tipo 2. Tuttavia, nel marzo 2000, troglitazone è stato ritirato dal commercio in seguito alla segnalazione di casi di tossicità epatica. Gli agonisti dei PPAR attualmente disponibili, rosiglitazone e pioglitazone, sono stati approvati negli Stati Uniti nel 1999.
5.2 Secrezione e sensibilità all’insulina
Studi clinici indicano che i glitazoni riducono le concentrazioni postprandiali e a digiuno di glucosio, di acidi grassi liberi e di insulina [Nolan, 1994; Suter, 1992; Miyazaki, 2001; Bajaj, 2004]. La capacità di alterare questi parametri suggerisce che i glitazoni agiscano come sensibilizzanti della risposta tissutale all’insulina, effetto che è stato dimostrato da misurazioni dirette in studi in vivo nell’uomo. Per esempio, il trattamento da tre a sei mesi con troglitazone, rosiglitazone o pioglitazone in soggetti non diabetici o in soggetti con diabete di tipo 2 aumenta la sensibilità all’insulina nei tessuti periferici [Nolan, 1994; Miyazaki, 2001-2002; Tack, 1998]. In studi simili i glitazoni hanno dimostrato di poter aumentare la sensibilità all’insulina del fegato (misurata attraverso la capacità dell’insulina di annullare la produzione endogena di glucosio) e nel tessuto adiposo (misurata attraverso la capacità di minimizzare le concentrazioni di acidi grassi liberi) [Miyazaki, 2001]. Inoltre, i glitazoni aumentano la secrezione di insulina in soggetti con Figura 3: PPAR è localizzato prevalentemente a livello di fegato, muscolo scheletrico, cuore (non mostrato), cellule endoteliali e cellule del muscolo liscio della parete vascolare. PPAR è localizzato principalmente nel tessuto adiposo dove regola geni necessari per la differenziazione degli adipociti, per la captazione e l’accumulo di acidi grassi liberi e per la captazione del glucosio. Inoltre stimola anche la lipolisi intravascolare. “CPT”: carnitina palmitolo transferasi, “” regolazione dei geni da parte di agonisti PPAR, “” regolazione dei geni da parte di agonisti PPAR, “” regolazione dei geni da parte di agonisti PPAR (PPAR), “GLUT4” trasportatore del glucosio insulino-sensibile, “PDK-4” piruvato deidrogenasi chinasi 4, “HSD1” idrossisteroide deidrogenasi di tipo 1, “LPL” lipoproteina lipasi, “IRS” substrato insulino-recettoriale, “CAP”-Cbl-proteina-associata, “GyK” glicerolo chinasi, “ABCA1” ATP-binding cassette A1, “SR” recettore idrofilo, “iNOS” ossido nitrico sintetasi inducibile, “TNF-” fattore di necrosi tumorale , “MMP-9” matrice metalloproteinai 9, “MCP-1” proteina chemotattica per i monociti 1 e “TZD” glitazoni.
ridotta tolleranza glucidica [Cavaghan, 1997] e in soggetti con diabete di tipo 2 [Miyazaki, 2002]. Paradossalmente questi miglioramenti sono associati all’aumento di peso e all’aumento della massa del tessuto adiposo sottocutaneo [Miyazaki, 2001-2002; Carey, 2002; Adams, 1997].
5.3 Aumento della sensibilità insulinica
Il recettore PPAR è essenziale per la normale differenziazione e proliferazione degli adipociti e per la captazione e l’accumulo di acidi grassi liberi. Studi su modelli animali indicano che i glitazoni aumentano il numero degli adipociti di piccole dimensioni e la massa del tessuto adiposo sottocutaneo [Picard, 2002; Miyazaki, 2002; Okuno, 1998]. Queste osservazioni, unite all’espressione elevata di PPAR nel tessuto adiposo, permettono di ipotizzare che i glitazoni agiscono come sensibilizzanti all’insulina insulina sia direttamente (ipotesi del “sequestro di acidi grassi”) che indirettamente, alterando la liberazione di adipochine in grado di modulare la sensibilità all’insulina fuori dal tessuto adiposo. Secondo l’ipotesi del “sequestro degli acidi grassi”, i glitazoni promuovono la captazione e l’accumulo di acidi grassi liberi nel tessuto adiposo aumentando la massa del tessuto adiposo sottocutaneo e risparmiando altri tessuti insulino-sensibili, quali muscolo scheletrico, fegato, e cellule beta pancreatiche, dagli effetti metabolici dannosi associati ad alte concentrazioni di acidi grassi liberi. In accordo con questa ipotesi, i glitazoni riducono le concentrazioni di acidi grassi liberi circolanti, i livelli di trigliceridi nel fegato, ma non nel muscolo scheletrico e la lipodistrofia [Sutinen, 2003] in pazienti con diabete di tipo 2 [Bajaj, 2004; Carey, 2002; Mayerson, 2002; Bajaj, 2003; Tiikkainen, 2004]. La metformina aumenta la sensibilità all’insulina nel fegato senza alterare il suo contenuto di grassi in pazienti con diabete di tipo 2 [Tiikkainen, 2004], mentre i glitazoni possono ridurre i livelli di insulina a digiuno senza aumentare la massa grassa sottocutanea in pazienti con lipodistrofia [Sutinen, 2003]. Nel topo, la specifica delezione del recettore PPAR nel tessuto adiposo non provoca insulino-resistenza nel muscolo [He, 2003], mentre la delezione muscolo-specifica del PPAR promuove questa resistenza [Hevener, 2003]. In questi animali, l’insulino-resistenza nel muscolo non risponde all’azione dei glitazoni, e questa osservazione suggerisce che i glitazoni sensibilizzano il tessuto
muscolare in maniera diretta stimolando i recettori PPAR muscolari [Hevener, 2003]. Nel topo con delezione di PPAR nel tessuto adiposo, l’insulino-resistenza epatica risponde ai glitazoni [Henever, 2003]. Questi dati suggeriscono che gli effetti insulino-sensibilizzanti dei glitazoni nel fegato e nel muscolo del topo non sono mediati dai recettori PPAR presenti nel tessuto adiposo nei casi in cui tale tessuto non è in grado di rispondere normalmente a questi farmaci. Tuttavia, la lipoatrofia associata alla delezione tessuto-specifica dei PPAR-, può rendere l’azione di agonisti PPAR dipendente in modo anomalo dall’espressione dei PPAR in altri tessuti. Per esempio il rosiglitazone è in grado di inibire la trigliceridemia, l’iperglicemia e l’iperinsulinemia nel topo normale mentre risulta inefficace nel topo lipoatrofico [Gavrilova, 2003]. Presi nell’insieme, questi dati ottenuti da modelli di topo knockout sostengono l’ipotesi che il tessuto adiposo è il più importante sito d’azione per i glitazoni.
5.4 Effetti indiretti nel tessuto adiposo
Nonostante i glitazoni possano aumentare la sensibilità all’insulina favorendo l’afflusso di grassi al tessuto adiposo, essi possono indurre anche effetti indiretti. Studi sull’espressione genica condotti per mezzo di microarrays in adipociti differenziati 3T3-L1 mostrano che rosiglitazone e pioglitazone regolano l’espressione di più di 100 geni e che questi geni non sono identici, nonostante siano raggruppati in un cluster [Berjer, 2003]. Alcuni geni, identificati come bersaglio dei PPAR e che sembrano regolati anche nel tessuto adiposo umano in vitro, sono mostrati in Figura 2. [Guan, 2002; Rieusset, 1999; Smith, 2001]. Nei roditori, gli agonisti dei PPAR regolano l’espressione di varie adipochine, come l’adiponectina [Maeda, 2002; Matsuda, 2002], il tumor necrosis factor (TNF) [Peraldi, 1998], la resistina [Steppan, 2001], e l’11-idrossisteroide-deidrogenasi-1 (enzima deputato alla produzione di cortisolo nel tessuto adiposo) [Picard, 2002; Berjer J, 2001]. Tra questi, l’adiponectina aumenta la sensibilità all’insulina mentre il TNF, la resistina e l’11-idrossisteroide-deidrogenasi-1 [Masuzaki, 2001] inducono insulino-resistenza nei roditori.
L’adiponectina, una adipocitochina prodotta dal tessuto adiposo, ha proprietà sia insulino-sensibilizzanti che antiaterogene nel topo [Maeda, 2002; Matsuda, 2002]. Gli agonisti dei PPAR aumentano l’espressione dell’adiponectina nel tessuto adiposo in vitro [Maeda N, 2001]. I livelli di adiponectina sono bassi nei pazienti obesi, con diabete di tipo 2 [Hotta, 2000; Arita, 1999; Hirose, 2002; Phillips, 2003; Yu, 2002; Weyer, 2001] e anche con lipodistrofia [Haque, 2002; Mynarcik, 2002; Addy, 2003; Sutinen, 2003]. Il trattamento con i glitazoni in vivo [Bajaj, 2004; Maeda, 2001; Hirose, 2002; Phillips, 2003; Yu, 2002; Yang, 2002; Combs, 2002] aumenta in maniera significativa le concentrazioni di adiponectina circolanti [Maeda, 1996]. Non è chiaro se l’adiponectina aumenti l’insulino-sensibilità epatica nell’uomo come avviene nel topo, nonostante le concentrazioni di adiponectina nel plasma siano correlate con il contenuto di lipidi nel fegato sia prima che dopo il trattamento con i glitazoni in pazienti con diabete di tipo 2 [Bajaj, 2004; Tiikkainen, 2004]. Nel fegato e nel tessuto adiposo, l’11-idrossisteroide-deidrogenasi-1 catalizza la reazione di conversione del cortisone in cortisolo [Walker, 2000].
Nel topo con iper-espressione dell’enzima 11-idrossisteroide-deidrogenasi-1 nel tessuto adiposo si sviluppa una grave sindrome metabolica caratterizzata da obesità e accumulo di grasso viscerale e da un aumento dei livelli di cortisolo nella vena porta ma non del cortisolo sistemico [Masukazi, 2001]. I glitazoni determinano down-regulation dell’espressione dell’11-idrossisteroide-deidrogenasi-1 nel tessuto adiposo [Berjer J, 2001] e possono alleviare alcune alterazioni della sindrome metabolica. Tuttavia, non sono disponibili dati sugli effetti dei glitazoni sull’attività o sull’espressione dell’ enzima 11-idrossisteroide-deidrogenasi-1 nell’uomo.
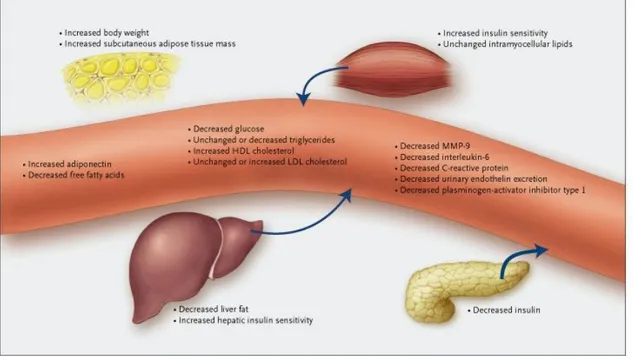
I molteplici effetti dei glitazoni nei diversi tessuti non permettono di definire esattamente i meccanismi che sostengono la loro azione insulino-sensibilizzante in vivo nell’ uomo. I dati disponibili suggeriscono che probabilmente sono coinvolti diversi meccanismi (Figura 4). Un meccanismo prevede la stimolazione dell’immagazzinamento degli acidi grassi liberi nel tessuto adiposo risparmiando altri tessuti, quali fegato, muscolo scheletrico e possibilmente le cellule beta del pancreas, dalla tossicità legata all’accumulo di lipidi [Unger, 2003]. Questi farmaci svolgono
anche effetti insulino-sensibilizzanti indiretti, soprattutto nel fegato, attraverso la secrezione di adiponectina dal tessuto adiposo.
Figura 4: I glitazoni sono in grado di aumentare la lipogenesi nel tessuto adiposo abbassando le concentrazioni di acidi grassi nel sangue e aumentando la massa di tessuto adiposo sottocutaneo e il peso corporeo. Inoltre aumentano le concentrazioni sieriche e l’espressione di adiponectina nel tessuto adiposo, che, insieme alla riduzione dei livelli di acidi grassi liberi nel sangue, può contribuire ad aumentare la sensibilità epatica all’insulina, la riduzione del contenuto di grassi nel fegato e l’inibizione della produzione del glucosio epatico. Inoltre riducono la concentrazione di glucosio nel plasma. La riduzione della concentrazione circolante di insulina è una conseguenza dell’aumento della liberazione e sensibilità all’insulina. I glitazoni sono in grado di ridurre i livelli di fattori di rischio cardiovascolare e infiammazione vascolare circolatori o urinari quali l’inibitore di tipo 1 dell’attivatore del plasminogeno, proteina C reattiva, matrice metalloproteinasi 9 (MMP-9) ed escrezione urinaria di endotelina. HDL, lipoproteine ad alta densità; LDL, lipoproteine a bassa densità.
6. EFFICACIA CLINICA DEI GLITAZONI
6.1 Effetti in pazienti con diabete di tipo 2
In molti Paesi, rosiglitazone e pioglitazone sono stati approvati per il trattamento dell’ iperglicemia in pazienti con diabete di tipo 2 sia in monoterapia che in associazione con sulfoniluree o metformina. Negli Stati Uniti invece, entrambi i farmaci sono stati approvati anche per l’uso in associazione con insulina, purchè vengano seguite specifiche precauzioni.
6.2 Effetti ipoglicemizzanti
Studi controllati con placebo suggeriscono che pioglitazone e rosiglitazone sono moderatamente efficaci nel controllo della glicemia (Tab. 1). Alle massime dosi, entrambi i farmaci diminuiscono i livelli di emoglobina glicosilata in media dall’1 all’1,5 %, quindi, in un paziente tipico affetto da diabete di tipo 2 ci si aspetterebbe che il trattamento con questi farmaci possa ridurre il livello di emoglobina glicosilata partendo da valori compresi tra il 7 e l’8,5% (il livello normale si colloca infatti tra il 4 e il 6%). Pioglitazone e rosiglitazone riducono le concentrazioni di emoglobina glicosilata maggiormente rispetto ad altri farmaci ipoglicemizzanti (ad es. nateglinide e inibitori dell’ -glucossidasi) ma in misura inferiore rispetto a dosi piene di glimepiride (da 4 a 6 mg), glibenclamide (da 10 a 15 mg) o metformina (da 2 a 2,5 g) [Gale, 2001; Nathan, 2002; De Fronzo, 1995]. L’uso dei glitazoni in monoterapia o in combinazione con altre terapie non sembra modificare la loro efficacia ipoglicemizzante. Non sono disponibili dati sulla possibilità di predire una buona risposta dei pazienti al trattamento con rosiglitazone o pioglitazone. Dati recenti riguardo il mantenimento del controllo glicemico a lungo termine in confronto ad altre terapie in uso sono stati forniti dallo studio ADOPT (A Diabetes Outcome Progression Trial), effettuato in pazienti con diabete di tipo 2 non precedentemente trattati e randomizzati al trattamento in monoterapia con rosiglitazone, gliburide o metformina. Questo studio ha evidenziato alcune differenze sul piano dell’efficacia e della tollerabilità tra i tre trattamenti che devono essere considerati al momento della scelta della terapia. [Viberti, 2002; Khan, 2006].
6.3 Effetti sui lipidi
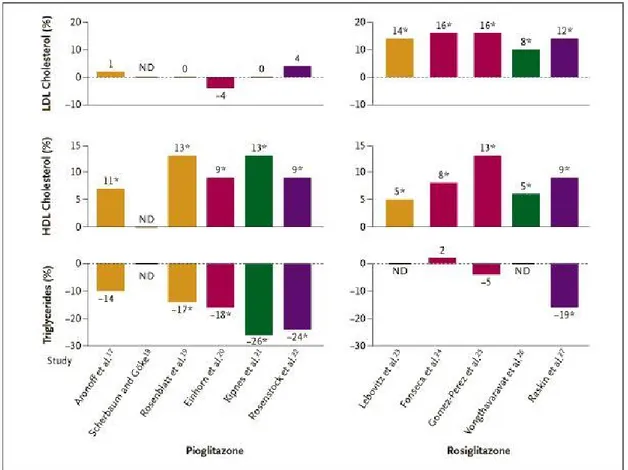
Non ci sono studi di confronto diretto in doppio cieco che documentino gli effetti di pioglitazone e rosiglitazone sui lipidi sierici e sulle lipoproteine. Tuttavia, i livelli di colesterolo-lipoproteine a bassa densità (c-LDL) sono rimasti immutati nel trattamento con pioglitazone sia in monoterapia che in associazione con sulfaniluree, metformina o insulina, mentre questi valori sono aumentati dall’ 8 al 16 % negli studi con rosiglitazone. I livelli di colesterolo-lipoproteine ad alta densità (c-HDL) sono aumentati approssimativamente del 10% negli studi su entrambi i farmaci (Fig. 5).
Figura 5: Effetti comparativi alle dosi massime di rosiglitazone (8 mg) e pioglitazone (da 30 a 45 mg) sui lipidi e sulle lipoproteine nel siero in studi randomizzati e controllati. Tutti le variazioni sono mostrate come variazione percentuale in relazione alla variazione osservata nel gruppo placebo (ad esempio, la variazione percentuale rispetto al basale con pioglitazione o rosiglitazone alla quale si sottrae la variazione percentuale rispetto al basale osservata nel gruppo placebo). Gli asterischi indicano P0.05. LDL, lipoproteine a bassa densità, HDL lipoproteine ad alta densità, ND, nessun dato. Gli istogrammi gialli indicano monoterapia, gli istogrammi rossi terapia in associazione con metformina, gli istogrammi verdi terapia in associazione con sulfanilurea e quelli viola terapia in associazione con insulina.
Gli effetti dei glitazoni sui trigliceridi appaiono più variabili. Una riduzione dei livelli di trigliceridi è stata riscontrata più frequentemente con pioglitazione piuttosto che con rosiglitazone (Fig. 5). L’unico confronto diretto tra rosiglitazone e pioglitazone è stato condotto in uno studio in aperto nel quale furono arruolati 127 pazienti precedentemente trattati con troglitazone. Lo studio ha mostrato che rosiglitazone e pioglitazone svolgono effetti simili su glicemia e peso corporeo [Khan, 2002]. Lo stesso studio ha mostrato che pioglitazone è più efficace di rosiglitazone nel controllare i livelli delle c-LDL e dei trigliceridi nel siero. La differenza tra gli effetti dei due farmaci non può essere attribuita alle differenze dei loro effetti sulle concentazioni di acidi grassi nel siero, che si riducono in modo simile, approssimativamente del 20-30% [Miyazaki, 2001]. E’ possibile che pioglitazone agisca anche come agonista parziale di PPAR in vitro, mentre rosiglitazone sia un agonista puro di PPAR [Sakamoto, 2000].
Non sono disponibili dati attendibili sui meccanismi che sottendono l’azione dei glitazoni sui lipidi nell’uomo. Ad esempio mancano dati circa gli effetti dei glitazoni sulla produzione e sulla rimozione di particelle lipoproteiche contenenti apolipoproteina A-I o apoliproteina B. Pertanto la causa dell’aumento dei livelli di c-HDL e c-LDL durante il trattamento con rosiglitazone rimane sconosciuta. Gli effetti di rosiglitazone o di pioglitazone sulla dimensione delle particelle LDL non sono stati ricercati in uno studio in doppio-cieco e controllato con placebo. Modelli di topo e ratto non sono ideali per lo studio delle lipoproteine nell’uomo, poiché in questi modelli una alterazione della clearance è il principale difetto responsabile dell’ipertrigliceridemia, rispetto all’eccessiva produzione di lipoproteine a densità molto bassa come accade nell’uomo [Malmstrom, 1997].
6.4 Effetti in condizioni di insulino-resistenza
Attualmente, la terapia con i glitazoni è approvata solo per il trattamento del diabete di tipo 2. Tuttavia, terapie sperimentali con i glitazoni hanno evidenziato buoni risultati in altre condizioni di insulino-resistenza quali steatosi non-alcolica [Clark, 2002], sindrome dell’ovaio policistico [Franks, 1999] e lipodistrofie [Reitman, 2000].
6.5 Steatosi non-alcolica
Il diabete di tipo 2 è strettamente correlato alla steatosi non-alcolica, una patologia caratterizzata da danno epatico di gravità variabile dalla steatosi benigna alla cirrosi potenzialmente fatale [Clark, 2002]. Secondo la terza National Health and Nutrition Examition Survey, negli USA questa patologia interessa 6,4 milioni di adulti [Clark, 2003]. La steatosi epatica non-alcolica rappresenta la causa principale di incremento dei livelli di enzimi del fegato [Clark, 2003], e i livelli elevati di alanina aminotransferasi rappresentano un fattore predittivo di diabete indipendente dall’obesità [Vozarova, 2002]. La steatosi epatica è associata ad un aumento dell’insulino-resistenza nell’uomo [Seppala-Lindroos, 2002] ed è correlata al fabbisogno di insulina in pazienti con diabete di tipo 2 [Ryysy, 2000].
In studi recenti, i glitazoni hanno dimostrato di ridurre l’accumulo di grasso nel fegato in pazienti con diabete di tipo 2 [Carey, 2002; Mayerson, 2002; Bajaj, 2003; Tiikkanen, 2004] e in pazienti con lipodistrofia associata al trattamento con antiretrovirali [Sutinen, 2003]. Coerentemente con queste osservazioni, i livelli di enzimi epatici, monitorati strettamente in questi pazienti per prevenire l’epatotossicità, sembrano diminuire piuttosto che aumentare durante il trattamento con pioglitazone e rosiglitazone [Mayerson, 2002; Sutinen, 2003].
La steatosi epatica rappresenta uno stadio avanzato nello spettro delle malattie degenerative grasse non-alcoliche del fegato ed è definita istologicamente dalla presenza di steatosi unita ad aree di necrosi e di infiammazione. Studi recenti hanno suggerito che i glitazoni non solo sono in grado di ridurre il contenuto di grassi nel fegato ma inducono anche miglioramenti istologici nel tessuto epatico [Neuschwander-Tetri, 2003; Promrat, 2004].
6.6 Sindrome dell’ovaio policistico
La sindrome dell’ovaio policistico è una patologia ad eziologia sconosciuta che interessa approssimativamente il 4% delle donne in età riproduttiva [Knochenhauer, 1998]. Pazienti con questa sindrome sono spesso insulino-resistenti e a rischio per lo sviluppo di diabete di tipo 2 [Solomon, 1999]. E’ opinione comune che l’iperinsulinemia associata all’insulino-resistenza possa contribuire allo sviluppo di iperandrogenismo nelle pazienti affette da sindrome dell’ovaio
policistico [Franks, 1999; Nestler, 1991]. Trattamenti in grado di determinare riduzione dei livelli di insulina, quali la perdita di peso e la terapia farmacologica (metformina, diazoxide o analoghi della somatostatina), riducono l’iperandrogenismo e l’insulino-resistenza [Iuorno, 2001]. Uno studio controllato con placebo e condotto su 410 donne ha mostrato che il trattamento con troglitazone è associato a miglioramento significativo della funzione ovulatoria, irsutismo, iperandrogenemia e resistenza all’insulina [Azziz, 2001]. Risultati simili sono stati riscontrati in un piccolo studio controllato con placebo in donne randomizzate al trattamento con rosiglitazone più placebo o rosiglitazone più clomifene. In questo studio, il 56% delle pazienti con storia di resistenza a clomifene ha mostrato ovulazione normale (33% nel gruppo rosiglitazone più placebo e 77% nel gruppo rosiglitazone più clomifene) [Ghazeeri, 2003]. Nonostante la terapia con metformina sia considerata sicura per donne in gravidanza, rosiglitazone e pioglitazone sono classificati come farmaci di categoria C per la gravidanza poiché studi sperimentali su modelli animali suggeriscono che questi farmaci provocano ritardo nello sviluppo fetale nella fase intermedia e tardiva della gestazione. I farmaci di categoria C hanno indotto effetti tossici in studi su modelli animali ma i risultati in studi sull’uomo sono insufficienti. Questi farmaci dovrebbero quindi essere usati in gravidanza soltanto se il beneficio potenziale supera il rischio per il feto. La terapia con i glitazoni non è attualmente approvata per la sindrome dell’ovaio policistico.
6.7 Lipodistrofie
La forma più comune di lipodistrofia è quella associata all’uso della terapia antiretrovirale nei pazienti affetti dal virus di immunodeficienza umana (HIV). Generalmente, almeno un sintomo relativo alla lipodistrofia si sviluppa tra i 12 e 18 mesi in circa la metà dei pazienti trattati con terapia antiretrovirale [Carr, 2000]. La lipodistrofia, in particolare la lipoatrofia facciale, può avere conseguenze estetiche invalidanti (volto sfigurato). Non è disponibile una terapia farmacologica per la lipoatrofia, una patologia che si associa sempre a condizioni di forte insulino-resistenza. La terapia con i glitazoni potrebbe rappresentare il trattamento ideale per contrastare l’insulino-resistenza e la lipoatrofia causate dalla terapia antiretrovirale, poichè i farmaci aumentano sia la sensibilità all’insulina che la massa grassa sottocutanea. E’ stato condotto uno studio controllato
con placebo nel quale pazienti con lipodistrofia associata a terapia antiretrovirale sono stati sottoposti a trattamento con rosiglitazone (8 mg al giorno, per 6 mesi). Questo studio non ha mostrato nessun miglioramento nella distribuzione del tessuto adiposo o variazione del peso corporeo dei pazienti [Sutinen, 2003], al contrario di quanto osservato in studi su pazienti con diabete di tipo 2 [Miyazaki, 2001; Miyazaki, 2002]. In rare forme di lipodistrofia umana, il trattamento con troglitazone per 6 mesi è stato associato a riduzione dei valori di emoglobina glicosilata e trigliceridi, con leggero aumento (2,4 %) del grasso sottocutaneo [Arioglu, 2000].
6.8 Effetti sui fattori di rischio cardiovascolare
La malattia cardiovascolare è la prima causa di morte nel mondo e la principale complicanza del diabete di tipo 2 [Haffner, 1998]. Al contrario di insulina, sulfoniluree e metformina, che si sono dimostrati efficaci o almeno sicuri [UK Prospective Diabetes Study, 1999], nel United Kingdom Prospective Diabetes Study, per i glitazoni sono attualmente disponibili soltanto dati su marcatori di rischio cardiovascolare, mentre mancano dati di mortalità o per end point specifici quali infarto del miocardio e ictus.
6.9 Aumento ponderale e pressione arteriosa
La terapia con i glitazoni aumenta il peso corporeo di 2-3 kg per ogni punto percentuale di riduzione dei valori di emoglobina glicosilata (Tabella. 1). L’entità di questo aumento si presenta in maniera simile sia in monoterapia che in combinazione con insulina [Yki-Jarvinen, 2001] o metformina [Einhorn, 2000; Fonseca, 2000; Gomez-Perez, 2002] in pazienti con diabete di tipo 2. L’incremento di peso potrebbe essere attribuito all’espansione dei depositi adiposi sottocutanei, e in alcuni pazienti all’edema, quando la massa grassa sottocutanea risulti inalterata [Carey, 2002] o diminuita [Miyazaki, 2002]. La rilevanza clinica attribuibile a questi cambiamenti in pazienti con patologia cardiovascolare deve essere stabilita. In letteratura non sono stati riscontrati dati che documentino gli effetti terapeutici dei glitazoni sulla pressione arteriosa [Czoski-Murray, 2004].
6.10 Plasma e fattori urinari
Pioglitazone e rosiglitazone sono in grado di ridurre il rischio cardiovascolare aumentando i livelli di colesterolo HDL e riducendo le concentrazioni di glucosio, insulina e acidi grassi liberi. Il significato clinico dell’incremento dei livelli di colesterolo LDL osservato durante trattamento con rosiglitazone non è chiaro, poiché non sono disponibili dati sugli effetti del farmaco sulla dimensione delle particelle LDL. Livelli ridotti di adiponectina si osservano anche in pazienti con malattia coronarica [Hotta, 2000; Kumada, 2003].
Esistono pochi dati sugli effetti dei glitazoni su altri marcatori di rischio cardiovascolare. Uno studio in doppio-cieco e controllato con placebo in pazienti con diabete di tipo 2 ha mostrato che la monoterapia con rosiglitazone si associa a riduzione del rapporto albumina urinaria-creatinina [Lebovitz, 2001]. Un altro studio controllato con placebo ha suggerito che il trattamento con rosiglitazone riduce i livelli plasmatici dell’ inibitore di tipo 1 dell’attivatore del plasminogeno in pazienti HIV-positivi con lipodistrofia associata all’impiego di terapia antiretrovirale
[Yki-Jarvinen, 2003]. Inoltre, altri dati suggeriscono che rosiglitazone riduce i livelli sierici di MMP-9 (matrix metallopreinase 9; enzima coinvolto nella rottura della matrice extracellulare sia in condizioni fisiologiche quali sviluppo embrionale, riproduzione e rimodellamento tissutale, che patologiche, quali artrite e metastasi), proteina C-reattiva ed interleuchina 6 (Fig. 3) [Haffner, 2002; Marx, 2003]
6.11 Funzione vascolare
Due studi in doppio-cieco e controllati con placebo hanno analizzato gli effetti di troglitazone sulla vasodilatazione dipendente e indipendente dell’endotelio vascolare nell’uomo: il primo indica che la terapia con troglitazone per 8 settimane non influisce sulla funzionalità vascolare nei pazienti obesi [Tack, 1998], mentre il secondo mostra miglioramenti nella vasodilatazione flusso-mediata in un sottogruppo di pazienti con diabete di tipo 2 [Caballero, 2003]. Un altro studio placebo-controllato ha evidenziato che rosiglitazone riduce la progressione dell’ispessimento della tonaca intima e della tonaca media dell’arteria carotide comune nei pazienti con diabete di tipo 2 [Sidhu, 2004].
7. SICUREZZA E TOLLERABILITA’ DEI GLITAZONI
A fronte di una certa efficacia nel trattamento dell’iperglicemia in pazienti con diabete di tipo 2, l’uso dei glitazoni si associa allo sviluppo di alcuni eventi avversi. In alcuni casi, questi eventi avversi riguardano il singolo glitazone, mentre altri sono considerati effetti di classe. L’evento avverso dei glitazoni per il quale esiste maggiore documentazione e che ha causato il ritiro dal mercato di troglitazone, capostipite della classe, è rappresentato dall’epatotossicità [Watkins, 1998]. Studi successivi sembrano suggerire tuttavia che l’epatotossicità non è un effetto comune a tutti i glitazoni [Lebovitz, 2002]. Altre possibili reazioni avverse sono rappresentate da aumento del peso corporeo, emodiluizione (riduzione dell’ematocrito e dell’emoglobina), edema periferico e anemia lieve. Nel 2007 un nuovo segnale di allarme è stato lanciato per il possibile rischio di insufficienza cardiaca congestizia associato all’uso dei glitazoni [Lago, 2007].
7.1 Epatotossicità
Come già accennato, dati clinici hanno dimostrato che il trattamento con troglitazone si associava ad epatotossicità [Gitlin, 1998; Terrine, 1999; Neuschwander-Tetri, 1998] che spesso insorgeva con un aumento dell’alanina aminotransferasi (ALT) nel siero [Watkins, 1998]. Troglitazone sembra caratterizzato da una tossicità epatica particolarmente marcata, in confronto a pioglitazone e rosiglitazone [Watkins, 1998]. Il meccanismo di tossicità epatica indotta dai glitazoni è poco conosciuto e potrebbe essere PPAR-indipendente, soprattutto se si considera che in condizioni normali PPAR non è espresso a livelli funzionalmente rilevanti nel fegato.
7.1.1 Studi sulla tossicità epatica di troglitazone
Troglitazone è stato il primo glitazone introdotto nella pratica clinica. Gli studi pre-marketing avevano suggerito che l’1,9% dei pazienti trattati con troglitazone sviluppava livelli di alanina amino transferasi 3 volte superiori rispetto al limite di riferimento, causando epatotossicità lieve o moderata [Watkins, 1998]. In seguito all’immissione del farmaco nella pratica clinica, sono stati riscontrati casi rari ma rilevanti di danno epatico che hanno determinato il ritiro di troglitazone dal
mercato [Gale, 2001; Graham, 2003]. Le caratteristiche cliniche dell’epatotossicità da troglitazone suggeriscono che il mecanismo di questa reazione avversa sia classificabile tra le idiosincrasie metaboliche [Bissell, 2001] e, nonostante questo tipo di epatotossicità non sia stata riscontrata in esperimenti su animali, sono stati condotti studi su larga scala, compresi esperimenti in vitro con epatociti, per comprendere i meccanismi che sostengono l’epatotossicità [Smith, 2003; Chojkier, 2005]. Troglitazone, analogamente ad altri farmaci epatotossici, viene metabolizzato in un metabolita reattivo che si lega covalentemente alle macromolecole cellulari, nonostante la sua attività epatotossica rimanga discutibile.
La citotossicità del farmaco e dei suoi metaboliti è stata esaminata in vari sistemi cellulari, comprese cellule HepG2 ed epatociti, in esperimenti su linee cellulari animali e umane. Nella maggior parte degli studi in vitro, la citotossicità è stata riscontrata quando le cellule venivano esposte a concentrazioni di troglitazone di circa 50 M, mentre sono state riscontrate concentrazioni non superiori a 3,6 e 6,3 M nel plasma umano, dopo trattamento con dosi terapeutiche pari a 400 e 600 mg al giorno, rispettivamente [Loi, 1999]. Inoltre, è stato evidenziato che l’assenza dell’albumina nel mezzo di coltura aumentava la biodisponibilità di troglitazone [Chojkier, 2005] e la tossicità di troglitazone si riduceva notevolmente quando l’albumina veniva aggiunta alla cultura [Toyoda, 2001]. In generale, le cellule epatiche sono state mantenute in coltura con siero di feto bovino, mentre l’assenza di siero o la sua bassa concentrazione potenziano le risposte cellulari [Haskins, 2001;Yamamoto, 2001]. Pertanto, i dati provenienti da studi in vitro sulla citotossicità dovrebbero essere valutati con attenzione, poiché potrebbero sopravvalutare l’epatotossicità di questi farmaci.
Come già accennato, troglitazone viene metabolizzato in un metabolita reattivo che si lega covalentemente alle macromolecole cellulari. Tuttavia il ruolo dell’adduzione covalente nella citotossicità indotta da troglitazone rimanga discutibile. Studi su epatociti umani, conservati criogenicamente e dotate di varie attività enzimatiche per CYP P450, UGT e fenolsulfotransferasi (PST), hanno evidenziato che non esiste correlazione tra la citotossicità indotta da troglitazone e le specifiche attività enzimatiche, nonostante sia stata stabilita una correlazione negativa tra la citotossicità indotta da troglitazone negli epatociti e la somma delle attività del CYP3A4 e
dell’UGT e dell’attività PST, suggerendo che troglitazone solfato aggiunto a troglitazone può risultare tossico [Hewitt, 2002]. Gli inibitori degli enzimi responsabili del metabolismo del troglitazone non sono stati efficaci nel prevenire la citotossicità indotta da troglitazone. L’inibizione della solfatazione di troglitazone provocata dal 2,4-dicloro-4-nitrofenolo e dal pentaclorofenolo ha promosso notevolmente la tossicità di troglitazone sugli epatociti umani [Kostrubsky, 2000]. SKF-525A, un inibitore degli enzimi P450, e ketoconazolo, un potente inibitore del CYP3A4, sono risultati inefficaci nell’attenuare la citotossicità di troglitazone [Yamamoto, 2001; Tirmenstein, 2002]. In uno studio recente, gli epatociti G2 esposti a microsomi contenenti CYP3A4 espressi sul DNA o epatociti G2 transfettati con CYP3A4, sono stati in grado di metabolizzare i composti testati (SKF-525A e ketoconazolo) aumentando la tossicità [Vignati, 2005]. La co-incubazione del ketoconazolo, inibitore del CYP3A4, proteggeva le cellule dalla tossicità, confermando l’ipotesi che l’aumento della tossicità è mediato dalla formazione di metaboliti reattivi dipendenti dal CYP3A4 [Vignati, 2005].
Il metabolita chinonico di troglitazone, un presunto precursore di un metabolita reattivo, è risultato meno citotossico rispetto a troglitazone sia negli epatociti del ratto che negli epatocitiG2 [Tettey, 2001; Yamamoto, 2002; Haskins, 2001; Yamamoto, 2001]. Risultati simili sono stati ottenuti con le cellule hepatoma (N1S1) del ratto [Narayanan, 2003]. Non è semplice valutare la tossicità del metabolita chinonico prodotta da troglitazone negli epatociti servendosi di dati in vitro, anche se lo studio sopra descritto, che la tossicità originava da metaboliti extracellulari [Vignati, 2005]. Ciò suggerisce che alcuni metaboliti citotossici, che non possono derivare da troglitazone chinone, potrebbero essere prodotti dal CYP3A4, ma il loro contributo alla tossicità è relativamente basso non solo negli epatocitiG2 ma anche negli epatociti umani normali poichè questi metaboliti non potrebbero formarsi e accumularsi nelle cellule tanto da esercitare i loro effetti tossici.
7.1.2 Tossicità epatica di pioglitazone e rosiglitazone
La tossicità epatica osservata per troglitazone non sembra rappresentare un effetto di classe. In 13 studi in doppio-cieco, l’1,91% di 2510 pazienti, lo 0,26% di 1526 pazienti e lo 0.17% di 3503 pazienti trattati rispettivamente con troglitazone, pioglitazone e rosiglitazone hanno mostrato livelli
di alanina aminotransferasi 3 volte maggiori rispetto al limite di riferimento [Lebovitz, 2002]. Inoltre sono stati osservati, nello 0.68% dei pazienti sottoposti a trattamento con troglitazone, livelli di alanina aminotransferasi 10 volte al di sopra rispetto al limite di riferimento al contrario di rosiglitazone e pioglitazone per i quali non è stata riscontrata nessuna alterazione di entità simile dei livelli di alanina aminotransferasi.
Studi su modelli in vitro hanno confrontato la tossicità di rosiglitazone e pioglitazone con troglitazone. In epatociti isolati di ratto, rosiglitazone non ha indotto alcuna perdita enzimatica dagli epatociti in presenza delle condizioni in cui troglitazone induceva una perdita significativa [Haskins, 2001]. Inoltre, è stato riscontrato che troglitazone, al contrario di rosiglitazone e pioglitazone, induceva apoptosi negli epatociti del ratto [Toyoda, 2001]. La citotossicità di troglitazone e rosiglitazone, valutata in epatociti umani raccolti da molteplici donatori e criopreservati, ha indicato che rosiglitazone è meno tossico [Lloyd, 2002]. In uno studio condotto su un singolo donatore, troglitazone ha mostrato maggiore tossicità rispetto a rosiglitazone (ad esempio contenuto di ATP e deplezione di glutatione). [Lloyd, 2003]. Non è stata evidenziata tossicità di rosiglitazone o pioglitazone negli epatociti umani normali immortalizzati con SV40 e transfetti al CYP3A4, mentre troglitazone in questo sistema risulta tossico [Gao, 2004].
Come già accennato, sembra che la citotossicità di troglitazone non sia mediata da un metabolita reattivo, a meno che che l’enzima attivante sia sovraespresso. La produzione di metaboliti reattivi è dovuta a differenze nella struttura molecolare e, all’interno della classe dei glitazoni, soltanto troglitazone possiede un anello cromanico che può convertirsi in un chinone metide reattivo [Smith, 2003; Park, 2005]. Al contrario, tutti i glitazoni hanno struttura ad anello e la sua apertura genera intermedi reattivi che non giustificano l’alta tossicità di troglitazone. Infatti, addotti glutationici sono stati rilevati dopo incubazione di microsomi epatici umani con rosiglitazone e pioglitazone in presenza di NADPH e glutatione o estere etilico glutationico [Soglia, 2004]. Gli addotti glutationici di pioglitazone ottenuti nel ratto e nei microsomi epatici umani e in epatociti del ratto isolati di recente ma non in epatociti umani, furono identificati quali prodotti di apertura dell’anello tiazolidinedionico [Baughman, 2005].
Troglitazone, al contrario di rosiglitazone o pioglitazone, ha indotto citotossicità dose-dipendente alterando la vitalità delle cellule, favorendo l’apoptosi e la perdita di LDH negli epatociti G2 [Yamamoto, 2001; Bae, 2003]. Risultati simili, unitamente all’arresto del ciclo cellulare ottenuto in fase G1 osservato con troglitazone ma non con rosiglitazone, sono stati ottenuti in cellule di epatoma di ratto e in epatociti G2 [Bae, 2003]. L’acido fenossiacetico tiazolidinedionico, un composto dotato dall’anello cromanico come rosiglitazone, non ha indotto citotossicità negli epatociti di ratto nelle stesse condizioni in cui troglitazone induceva tossicità [Narayanan, 2003]. Poichè i dati sulla tossicità dei glitazoni in vitro erano simili ai dati ottenuti in studi clinici, questi risultati sono in grado di fornire informazioni significative sull’epatotossicità a livello clinico. Oltre all’attivazione metabolica, non sono noti altri meccanismi di tossicità. Un confronto sugli effetti dei glitazoni sull’espressione genica in epatociti in coltura per mezzo di microarryas, ha evidenziato che troglitazone agisce sull’espressione genica in misura maggiore rispetto a rosiglitazone e pioglitazone [Kier, 2004]. Inoltre, uno studio di proteomica eseguito per mezzo di elettroforesi bidimensionale ha mostrato che troglitazone, e non rosiglitazone, è in grado di indurre chaperonine (proteine fondamentali per il corretto “ripiegamento” delle strutture cellulari e quindi per il raggiungimento della conformazione attiva delle proteine cellulari) nelle cellule HepG2 [Maniratanachote, 2005]. Studi in corso stanno verificando gli effetti dei glitazoni sui mitocondri come conseguenza della loro attività epatotossica.
7.2 Ritenzione di fluidi, edema e rischio di insufficienza cardiaca
Il trattamento con i glitazoni si associa ad aumento moderato ma significativo del peso corporeo dovuto all’incremento del tessuto adiposo e alla ritenzione idrica. La ritenzione idrica è associata ad emodiluizione ed edema periferico, aumentando il rischio di insufficienza cardiaca. I glitazoni sono quindi controindicati in pazienti a rischio di insufficienza cardiaca [Nesto, 2004].
7.2.1 Ritenzione idrica
Nella pratica clinica, sia i glitazoni che gli agonisti di PPAR- riducono ematocrito, globuli rossi ed emoglobina [Werner, 2001; Hollenberg, 2003; Fagerberg, 2005; Henry, 1997]. A dosi
terapeutiche, questi effetti sono associati generalmente ad emodiluizione che si presenta con ritenzione idrica. Nonostante studi recenti si siano interessati principalmente del ruolo di PPAR
nella modulazione del trasporto di Na nell’urotelio, è probabile che la ritenzione idrica riconosca
meccanismi multifattoriali. Recentemente, in modelli di roditori, due gruppi indipendenti hanno suggerito che l’incremento dell’attività o dell’espressione dei canali al sodio epiteliali (ENaC) nel dotto collettore renale rappresenta la causa principale responsabile del riassorbimento di sodio e liquidi. Nel topo con soppressione genica di PPAR a livello del dotto collettore, l’aumento del peso corporeo e l’espansione del volume del plasma non è maggiore rispetto al topo normale, dimostrando il ruolo diretto di PPAR nel rene [Zhang, 2005; Guan, 2005]. Secondo questi autori, la ritenzione idrica potrebbe essere associata in parte all’espressione dei ENaC indotta dai glitazoni [Guan, 2005], tuttavia Chen et al. hanno dimostrato che l’agonista specifico PPAR (farglitazar) aumentava il riassorbimento di sodio nel nefrone distale del ratto [Chen, 2005], ma non provocava nessuna alterazione nell’espressione delle subunità di trasporto di EnaC [Chen, 2005] o alterazioni della filtrazione renale [Yang, 2003]. Pertanto Chen et al. hanno ipotizzato che farglitazar aumentava l’assorbimento di sodio nel neurone distale, probabilmente attraverso stimolazione del sistema NaKATPasi. Inoltre, gli stessi autori sostengono che l’inibitore diretto di ENaC, amiloride, non è in grado di prevenire l’espansione del volume del plasma suggerendo che potrebbero essere coinvolti altri meccanismi ENaC-indipendenti.
Gli studi su animali che hanno stabilito un effetto dei PPAR sul trasporto di sodio come meccanismo alla base della ritenzione idrica associata all’uso dei glitazoni, sono stati confermati da osservazioni cliniche. Nell’uomo, i glitazoni potrebbero agire anche a livello dei tubuli prossimali del rene aumentando il riassorbimento di sodio [Zanchi, 2004]. Alcuni studiosi hanno dimostrato che i modulatori dei PPAR potrebbero aumentare la translocazione della subunità ENaC del trasportatore del sodio verso la superficie cellulare, indirettamente attraverso induzione trascrizionale della SGK1, in colture cellulari umane [Hong, 2003]. La modulazione dell’attività del trasporto di Na nell’urotelio potrebbe essere coinvolta nel meccanismo di ritenzione di liquidi e
di sodio indotta da glitazoni attraverso un meccanismo PPAR-dipendente sia nell’uomo che negli animali. Tuttavia potrebbero essere coinvolti altri meccanismi, anche PPAR-indipendenti.
7.2.2 Edema
Dati clinici dimostrano che i glitazoni provocano edema nel 10-15% dei pazienti [Fonseca, 1998; Lebovitz, 2001; Aranoff, 2000], una percentuale che aumenta nel trattamento di associazione con insulina [Rosenstock, 2002; Buse, 1998], determinando talvolta la sospensione della terapia [49, 50]. I glitazoni non aumentano la pressione arteriosa, ma l’espansione del volume del plasma provocata dal riassorbimento dei liquidi a livello del rene può fornire un innalzamento della pressione del lume dei microvasi aumentando il gradiente pressorio attraverso la parete dei microvasi e un innalzamento netto del flusso attraverso il compartimento interstiziale. Inoltre, i glitazoni, attraverso il blocco dei canali al calcio di tipo L [Zhang, 1994; Bauchanan, 1995], causano probabilmente edema periferico per una riduzione della resistenza arteriolare che provoca aumento della pressione idrostatica nella circolazione precapillare. L’innalzamento del gradiente di pressione idrostatica attraverso la parete vascolare potrebbe costituire la causa principale dello stravaso dei fluidi verso il compartimento interstiziale, con sviluppo di edema periferico. E’ probabile che i glitazoni alterino la permeabilità intrinseca dei fluidi attraverso la barriera endoteliale. Diversi possibili meccanismi potrebbero essere coinvolti in questo effetto, ma non sono stati pubblicati studi su misurazioni dirette dei flussi dei fluidi attraverso l’endotelio. I glitazoni hanno dimostrato di indurre, probabilmente attraverso attivazione dei PPAR [Marx, 2004], l’espressione del fattore di crescita dell’endotelio vascolare (VEGF) [Yamakawa, 2000; Baba, 2001; Inoue, 2001], che ha dimostrato di agire come un fattore di permeabilità vascolare. E’ stato dimostrato inoltre che l’insulina contribuisce al rischio di edema periferico [Chelliah, 2004]. Il miglioramento di sensibilità all’insulina indotto dal trattamento con glitazoni può causare edema, con un meccanismo che favorisce la vasodilatazione mediata dall’insulina [Walker, 1998] e/o la permeabilità endoteliale determinata dall’insulina [Idris, 2003]. Tuttavia, Rennings et al. recentemente hanno affermato che la ritenzione idrica associata ai glitazoni non è provocata da
modificazioni delle funzioni vascolari in risposta all’insulina, ma suggerirono alcune relazioni tra gli effetti di questi farmaci sull’assorbimento di glucosio e la quantità del liquido interstiziale [Rennings, 2006]. Infine, numerosi studi hanno dimostrato uno stretto legame tra l’ attivazione dei PPAR, il sistema renina-angiotensina, e la liberazione di endotelina-1 e di ossido nitrico nei vasi, che potrebbero essere causati dalla vasodilatazione favorita dai glitazoni con possibile aumento della permeabilità vascolare [Hinder, 1997].
Pertanto, nonostante l’incremento pressorio del lume dei microvasi sia associato allo sviluppo di edema periferico, sono stati proposti altri meccanismi che suggeriscono azioni dirette dei glitazoni sull’endotelio e sul conseguente aumento della permeabilità vascolare.
7.2.3 Rischio di insufficienza cardiaca congestizia
Lo studio PROactive, che ha arruolato più di 5000 pazienti affetti da diabete di tipo 2, ha cercato di valutare l’influenza dei glitazoni sul rischio cardiovascolare e sulla mortalità. Nonostante questo studio non abbia verificato l’end point primario, né una combinazione di end points correlati alla patologia (mortalità, infarto miocardio non fatale, stroke e sindrome coronarica acuta) e terapeutici (rivascolarizzazione coronarica e della gamba e amputazione della gamba), lo studio PROactive ha verificato il suo obiettivo secondario, dimostrando che rosiglitazone riduceva il rischio di infarto, stroke e morte del 16% dei pazienti ad alto rischio con diabete di tipo 2 [Dormandy, 2005].
Lo studio DREAM ha dimostrato successivamente che rosiglitazone previene lo sviluppo del diabete di tipo 2 in pazienti non diabetici in condizioni di insulino-resistenza e sindrome metabolica [Lancet, 2006].
Tuttavia, in entrambi gli studi, l’incidenza di diagnosi di insufficienza cardiaca recente è risultata maggiore nel gruppo trattato con glitazone rispetto a quello trattato con placebo e questo è dovuto al fatto che i glitazoni favoriscono l’edema periferico che può aggravarsi fino all’insufficienza cardiaca congestizia dovuta ad aumento del carico cardiaco conseguente all’espansione del volume plasmatico [Patel, 2005]. Per questo motivo, nonostante l’edema associato ai glitazoni si presenti principalmente a livello periferico senza danneggiare direttamente la funzionalità del ventricolo sinistro, soggetti con insufficienza cardiaca di 3° o 4° grado (valutata secondo i criteri di New York
Heart Association - NYHA) sono stati esclusi dagli studi clinici sulla sicurezza ed efficacia di rosiglitazone e pioglitazone [Nesto, 2004].
Sebbene alcune evidenze suggeriscano che soggetti sensibili possono sviluppare segni e sintomi di insufficienza cardiaca congestizia durante trattamento con glitazoni, questi farmaci non sembrano esercitare tossicità diretta sul cuore. Al contrario, studi clinici in soggetti con diabete di tipo 2 dimostrano che i glitazoni non inducono effetti avversi sull’attività cardiaca e che la terapia a lungo termine con questi farmaci è associata a miglioramento della funzionalità cardiaca [Guazzi, 1997; Sutton, 2002]. Analogamente, in uno studio eseguito in pazienti diabetici con malattia cardiaca, Tang et al. non hanno osservato alcuna associazione tra ritenzione idrica e il livello basale del danno cardiaco [Tang, 2003]. Inoltre, il tasso di incidenza di insufficienza cardiaca e di ricovero sono risultati minori in pazienti esposti a glitazoni rispetto a pazienti trattati con insulina, in soggetti non affetti da insufficienza cardiaca basale [Rajagopalan, 2004].
Nonostante non siano disponibili dati relativi alla tossicità cardiaca diretta indotta da glitazoni, modelli animali suggeriscono che l’azione diretta degli agonisti dei PPAR sul cuore è in grado di produrre effetti terapeutici. In effetti, gli agonisti dei PPAR migliorano la contrattilità e l’attività sistolica [Guazzi, 1997; Shimoyama, 1999; Shimabukuro, 1996], aumentano l’attività diastolica [Shimoyama, 1999; Shimabukuro, 1996; Yao, 2001] e riducono l’ipertrofia cardiaca indipendente da condizioni di carico [Asakawa, 2002;Yamamoto, 2001; Tsuji, 2001;]. Altri studi su animali dimostrano che i glitazoni producono effetti terapeutici positivi nel rimodellamento del ventricolo sinistro e miglioramento della sua funzionalità in seguito a lesione ischemica [Yue, 2001; Shiomi, 2002].
In una meta-analisi condotta da Nissen e Wolski è stato rilevato il rischio di eventi avversi cardiovascolari associati all’uso di glitazoni. Gli autori hanno osservato che l’uso di rosiglitazone è associato ad un aumento significativo del rischio di infarto del miocardio e di decessi causati da eventi avversi cardiovascolari. I risultati ottenuti dalla meta-analisi non sono stati esaustivi a causa della mancanza di disponibilità di dati sui singoli pazienti, e degli studi che non sono stati condotti con l’intento di valutare gli eventi cardiovascolari. Nissen e Wolski affermano che i rischi cardiovascolari associati all’uso di rosiglitazone nel trattamento del diabete dovrebbero essere
valutati maggiormente sia dai medici che dai loro pazienti. L’FDA ha deciso di inserire un avvertimento nel foglietto illustrativo del farmaco sul possibile rischio di eventi avversi cardiovascolari nei pazienti affetti da insufficienza cardiaca congestizia. Sono state condotte altre analisi sui glitazoni nel NEJM, e due review su JAMA, una che riguardava il rischio di eventi cardiovascolari associati a pioglitazone, l’unico altro glitazone disponibile (Actos®, Takeda), e l’altra sul rischio cardiovascolare di rosiglitazone a lungo termine. Entrambi i farmaci presentavano un aumento del rischio di insufficienza cardiaca ma non di mortalità. Lago e coll. hanno osservato che i pazienti che assumono glitazoni sembrano sviluppare un maggior rischio di insufficienza cardiaca congestizia, ma non maggior rischio di mortalità per cause cardiovascolari. Gli studi di meta-analisi condotti sui glitazoni comprendono marker surrogati; questi studi non sono stati condotti per valutare gli obiettivi cardiovascolari ma per migliorare il controllo glicemico. L’attuale importanza clinica sul controllo del glucosio è associata a complicanze microvascolari e macrovascolari, alla qualità della vita e alla sopravvivenza della maggior parte dei pazienti. In futuro dovranno essere condotti studi clinici basati sui risultati ottenuti dagli studi finora esaminati sui glitazoni. Le aziende produttrici dovranno effettuare studi postmarketing per valutare i limiti di sicurezza a lungo termine dei loro farmaci e le agenzie regolatorie dovranno ispezionare le ‘case produttrici’ affinché queste misure vengano adottate correttamente e istituire delle linee guida per la prescrizione di questi farmaci. Le agenzie regolatorie come FDA in USA dovrebbero impedire ciò che potenzialmente potrebbe risultare dannoso per la popolazione. I glitazoni potrebbero diventare semplicemente l’ultimo di una serie di disastri farmacologici evitabili, se non verranno affrontati i problemi di comprensione, analisi e comunicazione dei dati sulla sicurezza del farmaco.
7.3 Aumento ponderale
L’aumento ponderale è stato osservato per tutti i glitazoni e in tutte le specie animali [Chilcott, 2001; Khan, 2002]. Poiché l’indice di massa corporea è correlato ad insulino-resistenza, l’aumento di peso potrebbe essere considerato come un effetto avverso dannoso del trattamento con i glitazoni in pazienti con diabete di tipo 2, molti dei quali sono già sovrappeso o obesi. Nonostante l’aumento