Contents lists available atScienceDirect
Vascular Pharmacology
journal homepage:www.elsevier.com/locate/vphReview
Diabetic macroangiopathy: Pathogenetic insights and novel therapeutic
approaches with focus on high glucose-mediated vascular damage
Rosalinda Madonna
a,b, Damiana Pieragostino
c, Carmela Rita Balistreri
d, Claudia Rossi
c,
Yong-Jian Geng
b, Piero Del Boccio
c, Ra
ffaele De Caterina
a,⁎aCenter of Aging Sciences and Translational Medicine - CESI-MeT, Institute of Cardiology, Department of Neurosciences, Imaging and Clinical Sciences,“G. d'Annunzio”
University, Chieti, Italy
bCenter for Cardiovascular Biology and Atherosclerosis Research, Department of Internal Medicine, The University of Texas Health Science Center at Houston, Houston,
TX, United States
cAnalitical Biochemistry and Proteomics Unit Center of Aging Sciences and Translational Medicine - CESI-MeT, "G. d'Annunzio" University, Chieti, Italy dDepartment of Patho-biology and Medical Biotechnologies, University of Palermo, Palermo, Italy
A R T I C L E I N F O
Keywords: Diabetes Hyperglycemia Atherosclerosis Aquaporins Metabolomics ProteomicsA B S T R A C T
Diabetic macroangiopathy– a specific form of accelerated atherosclerosis – is characterized by intra-plaque new vessel formation due to excessive/abnormal neovasculogenesis and angiogenesis, increased vascular perme-ability of the capillary vessels, and tissue edema, resulting in frequent atherosclerotic plaque hemorrhage and plaque rupture. Mechanisms that may explain the premature and rapidly progressive nature of atherosclerosis in diabetes are multiple, and to a large extent still unclear. However, mechanisms related to hyperglycemia cer-tainly play an important role. These include a dysregulated vascular regeneration. In addition, oxidative and hyperosmolar stresses, as well as the activation of inflammatory pathways triggered by a dysregulated activation of membrane channel proteins aquaporins, have been recognized as key events. Here, we review recent knowledge of cellular and molecular pathways of macrovascular disease related to hyperglycemia in diabetes. We also here highlight how new insights into pathogenic mechanisms of vascular damage in diabetes may indicate new targets for prevention and treatment.
1. Introduction
According to the U.S. National Diabetes Statistics Institute, the prevalence of diabetes mellitus (diabetes) in the United States reached 9.3% of the total population in 2014, largely due to an increased pre-valence of type 2 diabetes [1]. This number is particularly worrisome because about 20–30% of diabetic patients have prevalent macro-vascular complications, including ischemic heart disease, cere-brovascular and peripheral arterial disease [1]. While type 1 diabetes may offer a relatively clean model of pure hyperglycemia-related vas-cular damage, type 2 diabetes is characterized by multiple metabolic abnormalities, where the independent effect of high glucose is difficult to separate from other components mostly linked to insulin resistance. Indeed, type 2 diabetic patients are particularly prone to develop atherosclerotic cardiovascular disease due to a combined effect of in-sulin resistance, hyperglycemia and the associated metabolic abnorm-alities.
Hyperglycemia is primarily responsible for the occurrence of mi-crovascular disease, the primary cause of blindness (retinopathy),
end-stage renal failure (nephropathy), peripheral neuropathy and diabetic cardiomyopathy. Hyperglycemia, however, is also known to aggravate macrovascular disease, conferring it a more severe, diffuse, and ac-celerated pattern [2]. Atherosclerosis in diabetes, and especially in type 2 diabetes, is indeed more aggressive than in the non-diabetic popula-tion, being premature, rapidly progressive, and with the involvement of multiple arterial districts at the same time [2]. This is thought to occur in part because the disturbed glucose metabolism in diabetes may modify and increase the impact of other risk factors for atherosclerotic disease. For example, low density lipoproteins (LDL) are susceptible to modification by advanced glycated end products (AGEs), which are modified proteins present in the circulation as well as in the sub-endothelial matrix in diabetes [3]. Increased lipoprotein oxidation, increased uptake of LDL by the LDL receptor, and platelet hyperag-gregability are all perturbed in diabetes. In part, however, disturbed glucose metabolism causes the activation of specific pathways leading to vascular damage.
From a pathological standpoint, diabetic macroangiopathy is indeed characterized by intra-plaque new vessel formation due to excessive or
https://doi.org/10.1016/j.vph.2018.01.009
Received 1 November 2017; Received in revised form 22 December 2017; Accepted 31 January 2018
⁎Corresponding author at: Institute of Cardiology, "G. d'Annunzio" University– Chieti C/o Ospedale SS. Annunziata, Via dei Vestini, Chieti, 66013, Italy.
E-mail address:[email protected](R. De Caterina).
1537-1891/ © 2018 Published by Elsevier Inc.
abnormal neovasculogenesis and angiogenesis, increased vascular per-meability of the capillary vessels, and tissue edema, resulting in more frequent atherosclerotic plaque hemorrhage and plaque rupture, as well as in cardiac microvascular dysfunction [4,5].
Animal models that mimic the interaction of diabetes and athero-sclerosis can virtually help identify factors underlying accelerated vascular disease, as well as the initial vascular abnormalities in dia-betes. However the results are not immediately applicable to humans. In fact, both type 1 and type 2 diabetes are multifactorial diseases, in which a very complex genetic background interacts with environmental factors, contributing to the development of disease. The limited translability of results from in vitro and animal models to humans re-flects the inability of any particular model to reproduce the patho-physiology of the disease in its complexity in humans. Nevertheless, a variety of small and large animal models, as well as in vitro models, have been used, and valuable insights into the molecular mechanisms of diabetic vascular disease have been inferred so far through the use of animal models. They have proven essential for studying the different features or phenotypes of the disease (e.g., hyperglycemia, hyper-insulinemia, insulin resistance, obesity), and their correlation with other factors (mainly environmental, such as diet or physical activity) that can efficiently contribute to the initiation of the disease [6–10]. Such comorbidity models can be used as appropriate tools for in-vestigating the effect of various drugs aimed at preventing and/or at-tenuating vascular complications in diabetes. An important field of research in which animal models have made a significant contribution is that of nutrient-gene interactions. With the help of nutrigenetics/ nutrigenomics, such models have helped to unravel the interactions between obesity and diabetes [7,11–16]. Important data for the early diagnosis of diabetic complications and for a better understanding of the related pathophysiology can now also be obtained by proteomic analyses, performed on body fluids and arterial tissues implicated in macrovascular complications [17].
2. Specific cellular mechanisms of macroangiopathy in diabetes 2.1. Stem/progenitor cells in vascular disease and repair
Pathogenetic mechanisms for vascular dysfunction in diabetes in-clude dysregulated vessel regeneration or impaired function of cells involved in the maintenance of vascular homeostasis and permeability (i.e., endothelial cells, smooth muscle cells, stromal cells, pericytes, inflammatory cells, circulating and tissue-resident vascular stem/pro-genitor cells, reviewed in [18]). Stem/progenitor cells residing in the vessel wall or circulating in the blood have recently attracted much attention as a mechanism to repair vascular damage and to replace exfoliated endothelial cells. Defects in the number of circulating pro-genitor cells have been related to cardiovascular risk factors, and have been associated with a more rapid progression of vascular disease [19,20]. Diabetes in general, and the hyperglycemic and insulin re-sistance components of diabetes specifically [21], have been related to a reduction/loss of function of progenitor cells, as well as to stem cell mobilization defects– a so called bone-marrow “mobilopathy” [22] due to the ineffective egress of stem/progenitor cells from the bone marrow into the peripheral circulation, contributing to the development of vascular disease [19,20]. In particular, hyperglycemia has been shown to negatively affect the growth reserve and repair capacity of the vessel wall [23]. One type of stem cells, mesenchymal stem cells (MSCs), which has been the subject of our own research [24], has been proposed as a potential target of diabetes-related pathogenetic mechanisms [24]. A reduction in the abundance and function of circulating and tissue-resident stem/progenitor cells, including MSCs, have been shown in both type 1 and type 2 diabetes [19,20]. Therefore, macrovascular complications of diabetes may reflect – at least in part – a “stem cell vasculopathy”, whereby the defective stem cell compartment is unable to regenerate dying endothelial or vascular smooth muscle cells, or
where the dysfunctional stem cell compartment itself contributes to the development of macrovascular complications. In type 2 diabetes, the reduction of vascular stem/progenitor cells is directly related to the degree of glycemic control [25,26], although some reduction is already apparent in subjects with impaired glucose regulation (pre-diabetes) [27–29]. An inverse correlation between the number and function of vascular stem/progenitor cells and blood glucose (fasting and 2 h after an oral glucose tolerance test) has here been shown [27,29]. Whether the reduction and/or dysfunction of stem/progenitor cells is, however, the direct consequence of glucose levels derangements, and to what extent this putative mechanism can be attributed to the hyperosmolar component of hyperglycemia is difficult to unravel. In patients with type 1 diabetes, a reduction of vascular stem/progenitor cells propor-tional to hemoglobin A1c (HbA1c) levels has been reported (Fig. 1). 2.2. Vascular calcification and macrophage polarization
Calcification may occur either within atherosclerotic plaques or in the tunica media of large and medium-size arteries with several me-chanisms. In atherosclerosis, intimal calcified nodules contribute to destabilize the plaque [30,31], while in the peripheral vasculature medial calcification leads to arterial stiffening and raises systolic blood pressure [32,33]. There is a subset of circulating cells positive for the bone protein osteocalcin (OC) and bone alkaline phosphatase (BAP) that have been termed osteoprogenitor cells (OPCs) [34,35]. These cells descend from the myeloid lineage, retaining monocyte/macrophage markers, may recirculate from peripheral tissues, and may differentiate from peripheral blood monocytes, contributing to ectopic calcifications in vivo [35]. In addition to having plaques with a larger necrotic core and significantly greater inflammation, diabetic patients also have more extensive calcifications in the coronary and carotid arteries. In type 2 diabetic patients, coronary artery calcium score (CACs), assessed by computed tomography, had the best prognostic value and offered sig-nificantly better accuracy in predicting atherosclerotic events compared to non-invasive markers of atherosclerosis, including brachial artery endothelial function, carotid artery atheroma burden, ankle-brachial index, arterial stiffness [36]. Oxidative stress, changes in the metabo-lism of minerals, and increased mobilization of OPCs from the marrow to the blood and to the arterial wall are key players of coronary and
3. OPCs
Calcifications
Neointimal
formation
Angiogenesis
1. ECs
EPCs
2. SMCs
Atherosclerotic
artery
LumenECs
Fig. 1. Cellular mechanisms associated with diabetic macroangiopathy. Thefigure de-picts the main cellular mechanisms underlying arterial calcification, neointimal formation and angiogenesis in the atherosclelrotic aorta. Abbbreviations: ECs, endothelial cells; SMCs, smooth muscle cells; OPCs, osteoprogenitor cells; EPCs, endothelial progenitor cells.
carotid atherosclerotic calcification in diabetes [37]. Vascular calcifi-cation is also a consequence of aging [38]. In people with an average age of 80, several parameters, such as male sex, the white race, cardi-ovascular disease, high triglyceride levels, smoking and chronic ob-structive pulmonary disease, have been independently associated with CACs, which is relevant in the stratification of cardiovascular risk and in allowing the early assessment of preclinical atherosclerosis [39]. However, the altered expression of some recently identified longevity-related genes might be associated with premature calcification. The reduced expression of mammalian sirtuin 1 (SIRT1), a histone deace-tylase, is associated with hyperphosphoremia and with coronary artery disease [40]. Furthermore, some SIRT1 polymorphisms are associated with coronary artery calcification in hemodialysis patients [41,42]. Insulin resistance and the metabolic syndrome are associated with SIRT1 gene and protein expression in peripheral blood mononuclear cells (PBMC) [43]. Importantly, SIRT1 gene expression negatively correlates with carotid intima-media thickness (IMT) [43,44]. Type 2 diabetes is also associated with chronic sterile low-grade inflammation, usually caused by hyperglycemia and its associated biochemical ab-normalities, as well as by excess body weight/obesity. One key element of this type of inflammation is the anti-inflammatory polarization of circulating monocytes and tissue macrophages. Diabetes indeed causes an imbalance of this polarization, favoring the proinflammatory (M1) macrophages at the expenses of antiinflammatory (M2) monocytes [45,46]. Cells belonging to the monocyte/macrophage lineage are of great importance in diabetes pathophysiology, as they are involved in atherosclerosis and adipose tissue biology, both of which determine diabetes outcomes. It is now recognized that M1/M2 polarization relies on the expression of chemokines/cytokines and their respective re-ceptors [46,47]. Compared with nondiabetic controls, diabetic patients feature an imbalance in M1/M2 polarization, in favor of M1 cells. Hyperglycemia per se may indeed affect M1/M2 polarization [48] (Fig. 1).
3. Molecular mechanisms of macroangiopathy in diabetes 3.1. The AGEs/RAGEs system and the new molecular mediator, CTRP1: linking inflammation and altered metabolic processes to endothelial/ vascular dysfunction
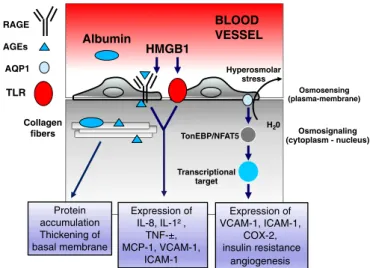
Advanced glycation endproducts (AGEs) are a heterogeneous group of complex structures, which form non-enzymatically when reducing sugars react with free amino groups in proteins, lipids, or nucleic acids. AGEs formation and accumulation are processes associated with phy-siological aging driven by high glycemic levels. In diabetic patients, AGE related processes occur at an accelerated rate in diabetic patients, and represent one of the most important mechanisms involved in the complex pathophysiology of macrovascular complications of diabetes [49] (Fig. 2). AGEs alter cellular mechanisms via their receptor, RAGE, a member of the immunoglobulin superfamily of cell surface molecules. RAGE is a multi-ligand receptor expressed in multiple cell types in-cluding vascular cells, immune cells such as monocytes/macrophages, dendritic cells and lymphocytes, cardiomyocytes, neurons, kidney po-docytes epidermal cells, that play a key role in inflammatory processes [50,51] Unlike other receptors that are downregulated by increased levels of their ligands, the RAGE-ligand interaction leads to a positive feedback activation, which further increases receptor expression [50–52]. Among all the biochemical mechanisms involved in diabetic vascular damage, the AGE-RAGE pathway appears to be the most im-portant in the pathogenesis and progression of vascular complications. There is increasing evidence that, in diabetic patients, activation of the AGEs-RAGE pathway triggers a proatherogenic cascade of events. Firstly, it influences the expression and activity of different cytoplasmic and nuclear transcription factors, thus modulating gene transcription [53,54]. In particular, among the activation of multiple cellular sig-naling pathways, the AGEs/RAGEs interaction leads to the activation of
nuclear factor (NF)-κB. As a result, increased expression of cytokines and adhesion molecules, induction of oxidative stress, and augmented formation of cytosolic reactive oxygen species (ROS) occur [55]. Fur-thermore, AGEs, independent of RAGE, also lead to the formation of stable cross-links with collagen, which causes chemical and biophysical changes of collagen structure and consequent functional changes, such as a thickening of the basement membrane and increased resistance to proteolytic digestion [49].
In diabetic patients, other ligands interact with the upregulated RAGE, primarily including members of the S100 calgranulin family and high mobility group box 1 (HMGB1) [56]. The interaction of these li-gands with RAGE can trigger interaction with the innate immune system signaling molecules, such as Toll-Like Receptors (TLR), parti-cularly TLR-4 [57]. In particular, it has been demonstrated that HMGB1, being able to interact with both the two receptors, RAGE and TLR4 (as well as TLR2) (Fig. 2) [57], determines the activation of NF-κB, which induces the expression of genes encoding for adhesion mo-lecules, proinflammatory cytokines, and proangiogenic molecules [58] (Fig. 2).
The total pool of sRAGEs in the plasma is involved in a wide range of macrovascular damage, even in non-diabetic subjects [59,60]. In addition to the full length RAGE, several variants of sRAGEs, i.e., two distinct forms of“soluble RAGE (sRAGEs)”, including the endogenous soluble RAGEs (esRAGEs) and circulating truncated variants of the RAGE isoform, have recently added its contribution to this complex scenario [61]. The two soluble RAGE forms may derive: a) from cell surface cleavage mechanisms induced by matrix metalloproteinases (MMPs) and A-distintegrin and Metalloprotease (ADAM)-10, as in the case offirst soluble RAGE form; b) from pre-messenger (m)RNA alter-native splicing, as in the case of the second soluble RAGE form [61]. These isoforms are capable of neutralizing the AGE-mediated damage by competing with cell-surface RAGEs for ligand binding, and perhaps with other cell surface receptors [61,62]. For example, HMGB1 and certain S100 s have been shown to bind to both RAGE and TLR re-ceptors, as described above [51,57,61]. Consequently, treatments with soluble RAGE molecules would reduce the ability of these ligands to bind these receptors, thereby attenuating inflammation and cellular stress [61]. Recently, Santilli and co-workers [63] have evaluated, in individuals affected or not by non-alcoholic fatty liver disease (NAFLD),
Collagen fibers AGEs HMGB1 Protein accumulation Thickening of basal membrane Expression of IL-8, IL-1² , TNF-±, MCP-1, VCAM-1, ICAM-1 AQP1 Osmosensing (plasma-membrane) Hyperosmolar stress H20 Osmosignaling (cytoplasm - nucleus) TonEBP/NFAT5 Expression of VCAM-1, ICAM-1, COX-2, insulin resistance angiogenesis BLOOD VESSEL Albumin Transcriptional target RAGE TLR
Fig. 2. Molecular pathways associated with diabetic macroangiopathy. Thefigure depicts some of the key points discussed in the text on the role of AGE products and hyperosmolar stress in diabetic macroangiopathy. Abbreviations: RAGE, receptor for advanced glyca-tion endproducts; AGEs, advanced glycaglyca-tion endproducts; AQP1, aquaporin isoform 1; TLR, Toll-Like Receptor; HMGB1, high mobility group box 1; TonEBP/NFAT5, Tonicity-responsive enhancer binding protein/nuclear factor of activated T-cells 5; IL, interleukin; TNF-α, tumor necrosis factor alpha; MCP-1, Monocyte chemoattractant protein-1; VCAM-1 vascular cell adhesion molecule VCAM-1; ICAM-VCAM-1, intercellular adhesion molecule VCAM-1; COX-2 cycloxygenase 2.
plasma levels of several inflammatory mediators, including soluble RAGE molecules. They have observed significantly lower levels of so-luble RAGE molecules in NAFLD patients, and consequently concluded that lower levels of soluble RAGE were associated with higher degrees of inflammation, oxidative stress and prothrombotic potential and with NAFLD itself [63]. Additional studies have examined their role in other disease states, including types 1 and 2 diabetes, with and without complications, neurodegenerative disorders, such as Alzheimer's dis-ease and amyotrophic lateral sclerosis, autoimmune/inflammatory disorders, and chronic obstructive pulmonary diseases [61]. Here si-milar data have been obtained, although further confirmatory studies are here encouraged [61]. EsRAGEs are a reflection of the AGEs and of several intra- and extracellular targets of AGEs, so that they can be seen as markers of intracellular and extracellular diabetic damage. More-over, whatever the level of hyperglycemia, AGE-related intracellular glycation of mitochondrial respiratory chain proteins has been found to induce a more abundant production of reactive oxygen species (ROS) [59], which further promotes AGE formation. Excessive AGE formation leads to a thickening of the large vessels, hypertension, endothelial dysfunction, loss of pericytes, the induction of growth factors with consequent increased angiogenesis and neovascularization [64], as well as decreased platelet survival and increased platelet aggregation, with the consequent promotion of a prothrombotic state [65], all favoring atherosclerosis, its complications, and the ensuing vascular occlusion. The exacerbation of inflammation and proangiogenic effects in diabetic patients also seem to be mediated by additional molecules, some of which able directly to interact with TLR pathways and to up-regulate the activation of NF-κB, as recently discovered [66]. They are members of the C1q/TNF-related protein (CTRP) family, and are considered as new molecular mediators connecting inflammation and metabolic dis-eases to vascular complications [66]. Of relevance in this context, is CTRP1, a secreted glycoprotein predominantly produced by the stromal-vascular cells of adipose tissue, with insulin sensitizing effects and associated with coronary artery disease [67]. In 2016, Shen's group [68] showed that CTRP1 upregulates the expression of adhesion mo-lecules through the p38 MAPK/NF-κB pathway, promoting athero-sclerosis. Accordingly, previous studies also found that activation of the p38 MAPK/NF-κB pathway (via TLR or other innate/immune in-flammatory pathways) causes angiotensin II-induced endothelial dys-function, and that arginase inhibition increases endothelial function in patients with coronary artery disease and type 2 diabetes [69,70]. These data have been recently confirmed [71], evidencing that CTRP1 is a mechano-sensitive proinflammatory factor that mediates disturbed flow-induced vascular barrier dysfunction. As a consequence, inhibition of CTRP1 might revert the pathogenesis of atherosclerosis at early stages.
Interestingly, the same authors [72] have recently demonstrated, in newly diagnosed subjects with type 2 diabetes, the presence of sig-nificant circulating levels of CTRP1, which appears sigsig-nificantly asso-ciated with age, diastolic blood pressure, and glucose levels, consistent with the current literature [73–75]. Precisely, these authors have de-monstrated for the first time that age, glucose and CTRP12 predict serum CTRP1 concentrations [72]. Thus, the authors propose that pharmacological agents targeting CTRP1 and CTRP12 might represent a new treatment for type 2 diabetes. Thus CTRP-1 is now proposed as both a marker and a mediator, and may be a target for pharmacological interference [68].
3.2. The TLR-2 and -4 signaling pathways
Inflammation is a key event characterizing and promoting the early steps of atherogenesis [76] in general, and also of diabetic macro-angiopathy [77]. Here, the activation of NF-κB caused by high glucose determines an enhanced expression of inflammatory molecules in var-ious cell types, including endothelial cells (ECs), monocyte/macro-phages, and smooth muscle cells. In addition, TLR-2 and -4 signaling
pathways appear to be main triggers of these inflammatory responses. Recent in vitro studies have shown that high concentrations of glucose significantly increase TLR-2 and TLR-4 mRNA and protein expression in human ECs, as well as the activation of the p65 NF-κB component, with the consequent expression of inflammatory mediators and markers, such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, monocyte chemoattractant protein (MCP)-1, vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, and IL-8 [78]. All these events are reverted by TLR-4 or TLR-2 inhibition, or by the dual pathway inhibition by a TLR4/2 inhibitory peptide. In addition, anti-oxidant treatments reduce TLR-2 and TLR-4 expression and down-stream inflammatory events. Collectively, these data suggest that hy-perglycemia induces TLR-2 and TLR-4 activation and its downstream inflammatory signaling, possibly through increased generation of ROS [78]. It has been also shown that endogenous ligands related to hy-perglycemic conditions (i.e., endogenous damage-associated molecular patterns—DAMPs, such as saturated fatty acids and necrotic cell pro-ducts) can, in particular, activate TLR4 [79], expressed on pancreatic beta-cells and resident macrophages), can induce insulin resistance, pancreatic beta-cell dysfunction, and changes in glucose homeostasis. TLR4 activation seems also to be exacerbated by low-grade en-dotoxemia (due to circulating lipopolysaccharide (LPS)) correlated with an altered gut microbiome, which characterizes subjects with metabolic diseases, such as type 2 diabetes [80]. A cooperation between RAGE and TLR2/4 has been recently suggested to coordinate and regulate immune and inflammatory responses in diabetic patients [80,81]. RAGE co-operation with TLR-2 and -4 results in the amplification of inflammatory responses, with increasing evidence supporting their potential synergism. RAGE and TLRs indeed share several common li-gands, including HMGB1, as described above (Fig. 2). RAGE also ap-pears to interact with TIRAP and MyD88, intracellular adaptor proteins used by TLRs to activate their downstream signaling pathway. These strong interactions among HMGB1-TLR-RAGE constitute a tripod trig-gering NF-κB activation [81–83]. This suggests that the blockade or the downregulation of HMGB1 and/or a dampening of such inflammatory tripod may be a promising therapeutic approach for the treatment of diabetic macroangiopathy and its complications [81–83].
In addition, alterations of gut microbiota and the consequent onset of intestinal dysbiosis, along with increased intestinal permeability and high circulating levels of lipopolysaccharides, a condition known as endotoxemia, characterize diabetic patients [84]. In turn, endotoxemia contributes to exacerbate inflammation and the onset of diabetic complications, including macroangiopathy, by further activating RAGE and TLR4 receptors [81]. The key role of TLR-4 has also been confirmed by data showing that deletions or mutations resulting in decreased or gained function in the TLR-4 gene may either protect against or ex-acerbate fatty acid-induced insulin resistance and obesity induced by diet [84,85]. Of note, the +896 A > G single nucleotide poly-morphism (SNP) that causes the substitution of Asp299 with Gly, modifying the normal structure of the extracellular region of TLR-4, is associated with decreased ligand recognition or protein interaction, and altered responsiveness to lipopolysaccharide. As a consequence, type 2 diabetic patients carrying the AG or GG genotypes feature an increased risk of developing macroangiopathy compared with patients carrying the AA genotype [85,86].
3.3. Endothelial nitric oxide synthase expression and nitric oxide bioavailability
ECs in the aorta play an important role in the control of vascular smooth muscle ell contractility and the autoregulation of vascular homeostasis through the release of a variety of vasconstricting or va-sodilating autacoids, including thromboxane, prostacyclin, and nitric oxide (NO). In 1980, Furchgott and Zwadaki reported that the relaxa-tion of isolated rabbit aorta and other arteries induced by acetylcholine and other agonists depends on the presence of ECs in the preparation
[87]. In the diabetic rat aorta, endothelium-dependent vasodilatation is impaired, largely due to the production of vasoconstrictive prostanoids and/or oxygen-derived free radicals [88]. An abnormal release of NO, or altered responses to NO have been proposed to contribute to vascular and endothelial dysfunction in the diabetic state [88]. It has been re-ported that eNOS mRNA and protein levels are increased in the aortic wall of diabetic rats [89]. Nevertheless, NO levels are actually sig-nificantly reduced in the aorta of diabetic mice [89]. One hypothesis exploring this apparent paradox refers to inadeguate levels and of availability of the eNOS cofactor tetrahydrobiopterin (BH4), a critical determinant of eNOS activity. When BH4 levels are inadequate, the enzymatic reduction of molecular oxygen by eNOS is no longer coupled to L-arginine oxidation, resulting in superoxide, rather than NO, pro-duction, thereby inducing vascular oxidative stress and endothelial dysfunction [90]. Thus, diabetes may increase eNOS expression and reduce NO bioavailability in the vascular wall. One possible explana-tion for inadequate levels of BH4 in diabetes is given by the inhibiexplana-tion and/or increased proteasome-dependent degradation of guanosine tri-phosphate (GTP) cyclohydrolase 1 (GTPCH1), a homodecameric pro-tein that catalyzes the rearrangement of GTP to dihydroneopterin tri-phosphate, subsequently converted to BH4 through the sequential action of 6-pyruvoyl-tetrahydrobiopterin synthase and sepiapterin re-ductase [91]. GTPCH1 is the rate-limiting enzyme responsible for the intracellular BH4 content, and several in vitro studies have shown that GTPCH1 gene transfer reverses BH4 deficiency, increases NO synthesis, and restores eNOS function in both endothelial cells and vessels isolated from diabetic rats [92]. Furthermore, in vivo study in diabetic spon-taneously hypertensive (SHR) rat showed that dietary supplementation with green tea was able to restore BH4 levels and NOS coupling, through a normalization of GTPCH1 activity, which is reduced by high glucose [93].
3.4. High glucose-induced hyperomolar stress: the role of membrane-channel proteins aquaporins
The accelerated atherosclerosis in diabetes is usually thought to be promoted by a widespread endothelial damage caused by hypergly-cemia and promoted by oxidative stress, leading to an overactivity of classical metabolic pathways, such as the protein kinase C (PKC), the AGEs, the polyol and the hexosamine pathways (for a complete review of glucose metabolic pathways see [18]). One puzzling aspect of dia-betic endotheliopathy is the relative contribution of the hyperosmotic component of high glucose. In overt type 1 and type 2 diabetes, hy-perglycemia causes an increase in plasma osmolarity, which determines an osmotic efflux of water that reduces intracellular volume (cell shrinking) and promotes adaptive responses, including the activation of hypertonicity-responsive genes, which may be themselves involved in glucotoxicity and vascular injury [94]. Aquaporin-1 (AQP1) [95] is an example of proteins induced by the hyperosmotic stress. Aquaporins are a family of 13 different water-specific, membrane-channel proteins expressed in diverse tissues [96]. Aquaporins have been shown to play an important role in controlling the interplay between vascular per-meability and angiogenesis [96,97]. AQP1 is specifically expressed in most microvascular endothelial cells and plays an important role in increasing cell membrane water permeability and promoting water transport, driven by osmotic pressure, across cells. AQP1 is also ex-pressed in atherosclerotic lesions following balloon injury, especially in neointimal vascular smooth muscle cells [98]. We have shown that concentrations of glucose attainable in conditions of hyperglycemia increase the expression of early proinflammatory genes such as ICAM-1, VCAM-1 and cycloxygenase (COX)-2, and decrease NO bioavailability in human ECs. All these events occur, at least in part, through the ac-tivation of the water channel protein AQP1, suggesting that hyper-osmotic stress may contribute, through this mechanism, to hypergly-cemia-induced endothelial inflammation and the early development of atherosclerosis [99,100]. We have also shown that, in the retina of
diabetic Ins2Akita mice and in micro- and macrovascular endothelial cells exposed to high glucose or its hyperosmolar control mannitol, COX-2 and AQP1 are upregulated in conjunction with increased an-giogenesis and tubulization [101]. Finally, interruption of the AQP1 axis by siRNA attenuated the in vivo angiogenesis occurring in response to high glucose [101]. These results showed that hyperosmolar stress is a biophysical mechanism through which excessive angiogenesis can occur in micro- and macrovascular endothelium in conditions of dia-betes (Fig. 2).
4. Proteomics and metabolomics as tools for characterizing diabetic macroangiopathy
Proteomics today is considered as a promising tool for detecting signaling pathways as well as for the identification and quantification of proteins specifically expressed in target tissues of diabetes. Despite the innumerable applications of proteomics, few data are available on tissue samples from diabetic patients. Using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS) technology, serum from individuals with normal glucose tolerance and patients with type 2 diabetes was profiled [102]. Mass spectra from subjects with normal glucose tolerance and type 2 diabetic patients identified four differentially expressed proteins, such as apolipoprotein C3 (9.4 kDa), transthyretin (13.9 kDa), albumin (66 kDa) and trans-ferrin (79 kDa). While apolipoprotein C3 and transthyretin were up-regulated, albumin and transferrin were down-regulated in type 2 diabetes [102]. Using the same technology, Cho and coworkers [103] examined aortic lysate and sera of 102 streptozotocin (STZ)-induced diabetic and 85 control male Sprague-Dawley rats at the 4th, 8th and 12th week after STZ injection. The authors showed 7 single potential biomarkers in the descending aorta, as well as 7 single potential bio-markers in the sera differentially expressed in the two groups of rats [103]. Among biomarkers more expressed in the diabetic group, the authors identified islet amyloid polypeptide (IAPP, also known as amylin), a protein produced by the pancreatic beta-cell that selectively inhibits insulin-stimulated glucose utilization and glycogen deposition in the muscle, while not affecting adipocyte glucose metabolism [104]) and resistin (a hormone produced by adipocytes [105]). A more recent study by Preil et al. [106] compared non-atherosclerotic tissue from internal mammary arteries from patients with type 2 diabetes and tissue from non-diabetic patients, focusing on matrix proteins. This study showed an increase in basement membrane protein content in arteries from diabetic patients and lower levels among metformin users [106]. Larger amount of data are available in animal models of diabetes. Using an integrative proteomic and bioinformatics analysis, a recent study characterized aortic tissues in the low-dose SZT-induced diabetic mouse model [107]. The authors observed a significant dysregulation of pro-teins involved in myogenesis, vascularization, hypertension, hyper-trophy (associated with aortic wall thickening), and a substantial re-duction in fatty acid storage. Moreover, the authors found a rere-duction in glycogen synthase kinase-3β (Gsk3β) and an upregulation of proteins linked to the tricarboxylic acid cycle. In addition, dysregulation of pathways involving primary alcohols and the breakdown of amino acids, which potentially lead to the production of ketones, were shown [108,109]. Finally using a label-free proteomics analysis, two in-dependent research groups recently showed a dysregulation of AQPs in diabetic kidney tissues, particularly a decrease in AQP2 protein in the inner medullary collecting ducts from diabetic rats [109]. Proteomic analyses are therefore welcome for further characterizing mechanisms in vascular dysfunction leading to diabetic macroangiopathy. While few proteomics studies are available [110], a greater number of studies was conducted with a metabolomics approach. Characterization of meta-bolic alterations may have a key role in the early detection, treatment and dissection of molecular mechanisms of diabetes [111]. In fact, specific metabolites have been described as “metabolic signature” of diabetes [111,112]. In particular, the accumulation of long- and
medium-chain acylcarnitines have long been implicated in the devel-opment of type 2 diabetes [113], and changes of amino acids such as branched-chain amino acids were correlated with increasing risk of developing type 2 diabetes [114]. The possibility of integrating meta-bolomics and proteomics approaches may thus give new hope for a better understanding of the molecular circuitries responsible for dia-betic mascrovascular disease [115,116].
5. Conclusions
Causes of increased cardiovascular risk in diabetes are many, and still incompletely understood. The identification of novel pathogenic pathways in principle promises to devise new therapeutic strategies. Because of the explosion of diabetic epidemics worldwide and of the problems associated with the administration of insulin or insulin se-cretagogues as a way to control hyperglycemia (including weight gain and the potential proliferative, proatherogenic and prothrombotic ef-fects of high insulin concentrations in the setting of insulin resistance [117–120]), there is a strong need for devising new strategies to control macrovascular complications in diabetes. A significant number of novel cellular and molecular targets have been shown in preclinical models of diabetic macroangiopathy. A better understanding of the translational impact of these preclinical observations will however help unraveling novel biological/pharmacological targets for hyperglycemia-related macrovascular complications. To this aim, and given the metabolic nature of atherosclerosis, large scale, unbiased, global metabolomic and proteomic approaches, capable of identifying multiple signaling net-works activated in atherosclerotic vascular disease might lead to novel diagnostic or therapeutic targets.
Conflicts of interest declaration
The authors declare no conflicts of interest as to the content of this review paper.
References
[1] Centers for Disease Control and Prevention, National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014, U.S. Department of Health and Human Services, Atlanta, GA, 2014.
[2] G.S. Getz, Report on the workshop on diabetes and mechanisms of atherogenesis. September 17th and 18th, 1992, Bethesda, Maryland, Arterioscler. Thromb. 13 (1993) 459–464.
[3] M. Brownlee, A. Cerami, H. Vlassara, Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications, N. Engl. J. Med. 318 (1988) 1315–1321.
[4] Y.C. Chen, A.V. Bui, J. Diesch, et al., A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling, Circ. Res. 113 (2013) 252–265. [5] Y.C. Chen, J. Rivera, K. Peter, Tandem stenosis to induce atherosclerotic plaque
instability in the mouse, Methods Mol. Biol. 1339 (2015) 333–338. [6] C. Renard, E. Van Obberghen, Role of diabetes in atherosclerotic pathogenesis.
What have we learned from animal models? Diabetes Metab. 32 (2006) 15–29. [7] P.A. Wood, Genetically modified mouse models for disorders of fatty acid
meta-bolism: pursuing the nutrigenomics of insulin resistance and type 2 diabetes, Nutrition 20 (2004) 121–126.
[8] L. Plum, F.T. Wunderlich, S. Baudler, W. Krone, J.C. Bruning, Transgenic and knockout mice in diabetes research: novel insights into pathophysiology, limita-tions, and perspectives, Physiology 20 (2005) 152–161.
[9] D. LeRoith, O. Gavrilova, Mouse models created to study the pathophysiology of type 2 diabetes, Int. J. Biochem. Cell Biol. 38 (2006) 904–912.
[10] A. Nitenberg, Macrovascular disease in type 2 diabetes: we do need animal models for in vivo studies, Cardiovasc. Res. 73 (2007) 450–452.
[11] W.T. Cefalu, Animal models of type 2 diabetes: clinical presentation and patho-physiological relevance to the human condition, ILAR J. 47 (2006) 186–198. [12] D. Chen, M.W. Wang, Development and application of rodent models for type 2
diabetes, Diabetes Obes. Metab. 7 (2005) 307–317.
[13] K. Srinivasan, P. Ramarao, Animal models in type 2 diabetes research: an over-view, Indian J. Med. Res. 125 (2007) 451–472.
[14] E. Shafrir, E. Ziv, R. Kalman, Nutritionally induced diabetes in desert rodents as models of type 2 diabetes: Acomys cahirinus (spiny mice) and Psammomys obesus (desert gerbil), ILAR J. 47 (2006) 212–224.
[15] P. Masiello, Animal models of type 2 diabetes with reduced pancreatic beta-cell mass, Int. J. Biochem. Cell Biol. 38 (2006) 873–893.
[16] E.H. Leiter, P.C. Reifsnyder, Differential levels of diabetogenic stress in two new mouse models of obesity and type 2 diabetes, Diabetes 53 (Suppl. 1) (2004) S4–11. [17] M. Maris, L. Overbergh, W. D'Hertog, C. Mathieu, Proteomics as a tool to discover biomarkers for the prediction of diabetic complications, Expert Opin. Med. Diagn. 2 (2008) 277–287.
[18] R. Madonna, R. De Caterina, Cellular and molecular mechanisms of vascular injury in diabetes—part I: pathways of vascular disease in diabetes, Vasc. Pharmacol. 54 (2011) 68–74.
[19] G.P. Fadini, A reappraisal of the role of circulating (progenitor) cells in the pa-thobiology of diabetic complications, Diabetologia 57 (2014) 4–15.
[20] J.H. Moon, M.K. Chae, K.J. Kim, et al., Decreased endothelial progenitor cells and increased serum glycated albumin are independently correlated with plaque-forming carotid artery atherosclerosis in type 2 diabetes patients without docu-mented ischemic disease, Circ. J. 76 (2012) 2273–2279.
[21] M.B. Kahn, N.Y. Yuldasheva, R.M. Cubbon, et al., Insulin resistance impairs cir-culating angiogenic progenitor cell function and delays endothelial regeneration, Diabetes 60 (2011) 1295–1303.
[22] DiPersio JF, Diabetic stem-cell“mobilopathy”, N. Engl. J. Med. 365 (2011) 2536–2538.
[23] G.P. Fadini, C. Agostini, S. Sartore, A. Avogaro, Endothelial progenitor cells in the natural history of atherosclerosis, Atherosclerosis 194 (2007) 46–54.
[24] R. Madonna, Y.J. Geng, R. De Caterina, Adipose tissue-derived stem cells: char-acterization and potential for cardiovascular repair, Arterioscler. Thromb. Vasc. Biol. 29 (2009) 1723–1729.
[25] M.R. De Pascale, G. Bruzzese, E. Crimi, et al., Severe type 2 diabetes induces re-versible modifications of endothelial progenitor cells which are ameliorate by glycemic control, Int. J. Stem Cells 9 (2016) 137–144.
[26] W.S. Yue, K.K. Lau, C.W. Siu, et al., Impact of glycemic control on circulating endothelial progenitor cells and arterial stiffness in patients with type 2 diabetes mellitus, Cardiovasc. Diabetol. 10 (2011) 113.
[27] G.P. Fadini, S. Sartore, C. Agostini, A. Avogaro, Significance of endothelial pro-genitor cells in subjects with diabetes, Diabetes Care 30 (2007) 1305–1313. [28] A. Angelidi, A. Melidonis, I. Protopsaltis, et al., Endothelial progenitor cells as a
cardiometabolic risk factor marker in prediabetes, Hormones 13 (2014) 244–251. [29] G.P. Fadini, P. Mancuso, F. Bertolini, S. de Kreutzenberg, A. Avogaro,
Amelioration of glucose control mobilizes circulating pericyte progenitor cells in type 2 diabetic patients with microangiopathy, Exp. Diabetes Res. 2012 (2012) (274363).
[30] M. Sudo, Y. Li, T. Hiro, et al., Inhibition of plaque progression and promotion of plaque stability by glucagon-like peptide-1 receptor agonist: serial in vivofindings from iMap-IVUS in Watanabe heritable hyperlipidemic rabbits, Atherosclerosis 265 (2017) 283–291.
[31] R. Lin, S. Chen, G. Liu, Y. Xue, X. Zhao, Association between carotid athero-sclerotic plaque calcification and Intraplaque hemorrhage: a magnetic resonance imaging study, Arterioscler. Thromb. Vasc. Biol. 37 (2017) 1228–1233. [32] A. Laszlo, G. Reusz, J. Nemcsik, Ambulatory arterial stiffness in chronic kidney
disease: a methodological review, Hypertens. Res. 39 (2016) 192–198. [33] J.N. Stabley, D.A. Towler, Arterial calcification in diabetes mellitus: preclinical
models and translational implications, Arterioscler. Thromb. Vasc. Biol. 37 (2017) 205–217.
[34] M. Pirro, G. Schillaci, M.R. Mannarino, et al., Circulating immature osteopro-genitor cells and arterial stiffening in postmenopausal osteoporosis, Nutr. Metab. Cardiovasc. Dis. 21 (2011) 636–642.
[35] G.Z. Eghbali-Fatourechi, J. Lamsam, D. Fraser, D. Nagel, B.L. Riggs, S. Khosla, Circulating osteoblast-lineage cells in humans, N. Engl. J. Med. 352 (2005) 1959–1966.
[36] K.K. Lau, Y.K. Wong, Y.H. Chan, et al., Prognostic implications of surrogate markers of atherosclerosis in low to intermediate risk patients with type 2 dia-betes, Cardiovasc. Diabetol. 11 (2012) 101.
[37] K. Yahagi, F.D. Kolodgie, C. Lutter, et al., Pathology of human coronary and carotid artery atherosclerosis and vascular calcification in diabetes mellitus, Arterioscler. Thromb. Vasc. Biol. 37 (2017) 191–204.
[38] M. Tesauro, A. Mauriello, V. Rovella, et al., Arterial ageing: from endothelial dysfunction to vascular calcification, J. Intern. Med. 281 (2017) 471–482. [39] R. Madonna, S. Selvaggio, G. Selvaggio, M. Coronelli, N. Cocco, "State-of-art"
paper of the Italian working group on atherosclerosis: preclinical assessment of early coronary atherosclerosis, Int. J. Cardiol. 214 (2016) 442–447. [40] A. Takemura, K. Iijima, H. Ota, et al., Sirtuin 1 retards
hyperphosphatemia-in-duced calcification of vascular smooth muscle cells, Arterioscler. Thromb. Vasc. Biol. 31 (2011) 2054–2062.
[41] P. Ramkaran, A. Phulukdaree, S. Khan, D. Moodley, A.A. Chuturgoon, Sirtuin 1 rs1467568 and rs7895833 in South African Indians with early-onset coronary artery disease, Cardiovasc. J. Afr. 27 (2016) 213–217.
[42] Y. Shimoyama, Y. Mitsuda, Y. Tsuruta, K. Suzuki, N. Hamajima, T. Niwa, SIRTUIN 1 gene polymorphisms are associated with cholesterol metabolism and coronary artery calcification in Japanese hemodialysis patients, J. Renal Nutr. 22 (2012) 114–119.
[43] S.V. de Kreutzenberg, G. Ceolotto, I. Papparella, et al., Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syn-drome: potential biochemical mechanisms, Diabetes 59 (2010) 1006–1015. [44] D. Bunout, G. Barrera, M.P. de la Maza, L. Leiva, S. Hirsch, Effect of weight
maintenance or gain in a 10 years period over telomere length, sirtuin 1 and 6 expression and carotid intima media thickness, J. Hum. Nutr. Diet. 28 (2015) 155–164.
[45] N. Wang, H. Liang, K. Zen, Molecular mechanisms that influence the macrophage m1-m2 polarization balance, Front. Immunol. 5 (2014) 614.
[46] G. Chinetti-Gbaguidi, B. Staels, Macrophage polarization in metabolic disorders: functions and regulation, Curr. Opin. Lipidol. 22 (2011) 365–372.
[47] S. Fujisaka, I. Usui, A. Bukhari, et al., Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice, Diabetes 58 (2009) 2574–2582.
[48] A. Castoldi, C. Naffah de Souza, N.O. Camara, P.M. Moraes-Vieira, The macro-phage switch in obesity development, Front. Immunol. 6 (2015) 637. [49] R. Lopez-Diez, A. Shekhtman, R. Ramasamy, A.M. Schmidt, Cellular mechanisms
and consequences of glycation in atherosclerosis and obesity, Biochim. Biophys. Acta 1862 (2016) 2244–2252.
[50] A.M. Schmidt, S.D. Yan, S.F. Yan, D.M. Stern, The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses, J. Clin. Invest. 108 (2001) 949–955.
[51] R. Ramasamy, S.F. Yan, A.M. Schmidt, The diverse ligand repertoire of the re-ceptor for advanced glycation endproducts and pathways to the complications of diabetes, Vasc. Pharmacol. 57 (2012) 160–167.
[52] G. Basta, A.M. Schmidt, R. De Caterina, Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes, Cardiovasc. Res. 63 (2004) 582–592.
[53] M.A. Reddy, S.L. Li, S. Sahar, et al., Key role of Src kinase in S100B-induced ac-tivation of the receptor for advanced glycation end products in vascular smooth muscle cells, J. Biol. Chem. 281 (2006) 13685–13693.
[54] P. Lewis, N. Stefanovic, J. Pete, et al., Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice, Circulation 115 (2007) 2178–2187.
[55] G. Basta, G. Lazzerini, S. Del Turco, G.M. Ratto, A.M. Schmidt, R. De Caterina, At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products, Arterioscler. Thromb. Vasc. Biol. 25 (7) (2005) 1401.
[56] L.J. Wang, L. Lu, F.R. Zhang, Q.J. Chen, R. De Caterina, W.F. Shen, Increased serum high-mobility group box-1 and cleaved receptor for advanced glycation endproducts levels and decreased endogenous secretory receptor for advanced glycation endproducts levels in diabetic and non-diabetic patients with heart failure, Eur. J. Heart Fail. 13 (2011) 440–449.
[57] J.S. Park, D. Svetkauskaite, Q. He, et al., Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein, J. Biol. Chem. 279 (2004) 7370–7377.
[58] J.R. van Beijnum, W.A. Buurman, A.W. Griffioen, Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1), Angiogenesis 11 (2008) 91–99.
[59] E. McNair, M. Qureshi, K. Prasad, C. Pearce, Atherosclerosis and the hypercho-lesterolemic AGE-RAGE axis, Int. J. Angiol. 25 (2016) 110–116.
[60] A.M. Marsh, A.H. Nguyen, T.M. Parker, D.K. Agrawal, Clinical use of high mobility group box 1 and the receptor for advanced glycation end products in the prognosis and risk stratification of heart failure: a literature review, Can. J. Physiol. Pharmacol. 95 (2017) 253–259.
[61] A.M. Schmidt, Soluble RAGEs - prospects for treating & tracking metabolic and inflammatory disease, Vasc. Pharmacol. 72 (2015) 1–8.
[62] W.Q. Ma, Q.R. Qu, Y. Zhao, N.F. Liu, Association of RAGE gene Gly82Ser poly-morphism with coronary artery disease and ischemic stroke: a systematic review and meta-analysis, Medicine 95 (2016) e5593.
[63] F. Santilli, P. Blardi, C. Scapellato, et al., Decreased plasma endogenous soluble RAGE, and enhanced adipokine secretion, oxidative stress and platelet/coagula-tive activation identify non-alcoholic fatty liver disease among patients with fa-milial combined hyperlipidemia and/or metabolic syndrome, Vasc. Pharmacol. 72 (2015) 16–24.
[64] G. Eelen, P. de Zeeuw, M. Simons, P. Carmeliet, Endothelial cell metabolism in normal and diseased vasculature, Circ. Res. 116 (2015) 1231–1244.
[65] D.A. Rubenstein, Z. Maria, W. Yin, Glycated albumin modulates endothelial cell thrombogenic and inflammatory responses, J. Diabetes Sci. Technol. 5 (2011) 703–713.
[66] A. Schaffler, C. Buechler, CTRP family: linking immunity to metabolism, Trends Endocrinol Metab 23 (2012) 194–204.
[67] J.M. Peterson, S. Aja, Z. Wei, G.W. Wong, CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition, J. Biol. Chem. 287 (2012) 1576–1587. [68] L. Lu, R.Y. Zhang, X.Q. Wang, et al., C1q/TNF-related protein-1: an adipokine
marking and promoting atherosclerosis, Eur. Heart J. 37 (2016) 1762–1771. [69] A. Rahman, K.N. Anwar, M. Minhajuddin, et al., cAMP targeting of p38 MAP
ki-nase inhibits thrombin-induced NF-kappaB activation and ICAM-1 expression in endothelial cells, Am. J. Physiol. Lung Cell. Mol. Physiol. 287 (2004) L1017–24. [70] A. Shatanawi, M.J. Romero, J.A. Iddings, et al., Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway, Am. J. Physiol. Cell Physiol. 300 (2011) C1181–C1192. [71] Z.H. Liu, C. Li, J.W. Chen, et al., C1q/TNF-related protein 1 promotes endothelial barrier dysfunction under disturbedflow, Biochem. Biophys. Res. Commun. 490 (2017) 580–586.
[72] B. Bai, B. Ban, Z. Liu, M.M. Zhang, B.K. Tan, J. Chen, Circulating C1q complement/ TNF-related protein (CTRP) 1, CTRP9, CTRP12 and CTRP13 concentrations in type 2 diabetes mellitus: in vivo regulation by glucose, PLoS One 12 (2017) e0172271. [73] S. Han, J.D. Kim, S. Lee, et al., Circulating CTRP1 levels in type 2 diabetes and
their association with FGF21, Int. J. Endocrinol. 2016 (2016) (5479627). [74] X. Pan, T. Lu, F. Wu, et al., Circulating complement-C1q TNF-related protein 1
levels are increased in patients with type 2 diabetes and are associated with insulin sensitivity in Chinese subjects, PLoS One 9 (2014) e94478.
[75] S. Rodriguez, X. Lei, P.S. Petersen, S.Y. Tan, H.C. Little, G.W. Wong, Loss of CTRP1 disrupts glucose and lipid homeostasis, Am. J. Phys. Endocrinol. Metab. 311 (2016) E678–E697.
[76] J. Frostegard, Immunity, atherosclerosis and cardiovascular disease, BMC Med. 11 (2013) 117.
[77] G. Grandl, C. Wolfrum, Hemostasis, endothelial stress, inflammation, and the metabolic syndrome, Semin. Immunopathol. (2017),http://dx.doi.org/10.1007/ s00281-017-0666-5(Epub ahead of print).
[78] R. Pahwa, P. Nallasamy, I. Jialal, Toll-like receptors 2 and 4 mediate hypergly-cemia induced macrovascular aortic endothelial cell inflammation and perturba-tion of the endothelial glycocalyx, J. Diabetes Complicat. 30 (2016) 563–572. [79] S. Sirisinha, Insight into the mechanisms regulating immune homeostasis in health
and disease, Asian Pac. J. Allergy Immunol. 29 (2011) 1–14.
[80] C.R. Balistreri, C. Caruso, G. Candore, The role of adipose tissue and adipokines in obesity-related inflammatory diseases, Mediat. Inflamm. 2010 (2010) 802078. [81] Z.A. Ibrahim, C.L. Armour, S. Phipps, M.B. Sukkar, RAGE and TLRs: relatives,
friends or neighbours? Mol. Immunol. 56 (2013) 739–744.
[82] J.A. Nogueira-Machado, C.M. Volpe, C.A. Veloso, M.M. Chaves, HMGB1, TLR and RAGE: a functional tripod that leads to diabetic inflammation, Expert Opin. Ther. Targets 15 (2011) 1023–1035.
[83] Y. Wang, J. Zhong, X. Zhang, et al., The role of HMGB1 in the pathogenesis of type 2 diabetes, J. Diabetes Res. 2016 (2016) 2543268.
[84] A. Sabatino, G. Regolisti, C. Cosola, L. Gesualdo, E. Fiaccadori, Intestinal micro-biota in type 2 diabetes and chronic kidney disease, Curr. Diabetes Rep. 17 (2017) 16.
[85] M.J. Hommels, A.A. Kroon, M.G. Netea, et al., The Asp299Gly toll-like receptor 4 polymorphism in advanced aortic atherosclerosis, Neth. J. Med. 65 (2007) 203–207.
[86] C.R. Balistreri, A.R. Bonfigli, M. Boemi, et al., Evidences of +896 A/G TLR4 polymorphism as an indicative of prevalence of complications in T2DM patients, Mediat. Inflamm. 2014 (2014) (973139).
[87] R.F. Furchgott, J.V. Zawadzki, The obligatory role of endothelial cells in the re-laxation of arterial smooth muscle by acetylcholine, Nature 288 (1980) 373–376. [88] M.J. Durand, D.D. Gutterman, Diversity in mechanisms of endothelium-dependent
vasodilation in health and disease, Microcirculation 20 (2013) 239–247. [89] S. Cai, J. Khoo, S. Mussa, N.J. Alp, K.M. Channon, Endothelial nitric oxide
syn-thase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation, Diabetologia 48 (2005) 1933–1940.
[90] Y. Hattori, S. Hattori, X. Wang, H. Satoh, N. Nakanishi, K. Kasai, Oral adminis-tration of tetrahydrobiopterin slows the progression of atherosclerosis in apoli-poprotein E-knockout mice, Arterioscler. Thromb. Vasc. Biol. 27 (2007) 865–870. [91] M.A. Kolinsky, S.S. Gross, The mechanism of potent GTP cyclohydrolase I
in-hibition by 2,4-diamino-6-hydroxypyrimidine: requirement of the GTP cyclohy-drolase I feedback regulatory protein, J. Biol. Chem. 279 (2004) 40677–40682. [92] C.J. Meininger, S. Cai, J.L. Parker, et al., GTP cyclohydrolase I gene transfer re-verses tetrahydrobiopterin deficiency and increases nitric oxide synthesis in en-dothelial cells and isolated vessels from diabetic rats, FASEB J. 18 (2004) 1900–1902.
[93] A.M. Faria, A. Papadimitriou, K.C. Silva, J.M. Lopes de Faria, J.B. Lopes de Faria, Uncoupling endothelial nitric oxide synthase is ameliorated by green tea in ex-perimental diabetes by re-establishing tetrahydrobiopterin levels, Diabetes 61 (2012) 1838–1847.
[94] A. Schaffler, H. Arndt, J. Scholmerich, K.D. Palitzsch, Amelioration of hypergly-cemic and hyperosmotic induced vascular dysfunction by in vivo inhibition of protein kinase C and p38 MAP kinase pathway in the rat mesenteric micro-circulation, Eur. J. Clin. Investig. 30 (2000) 586–593.
[95] F. Umenishi, R.W. Schrier, Hypertonicity-induced aquaporin-1 (AQP1) expression is mediated by the activation of MAPK pathways and hypertonicity-responsive element in the AQP1 gene, J. Biol. Chem. 278 (2003) 15765–15770. [96] D. Brown, The discovery of water channels (Aquaporins), Ann. Nutr. Metab. 70
(Suppl. 1) (2017) 37–42.
[97] A.S. Verkman, Mammalian aquaporins: diverse physiological roles and potential clinical significance, Expert Rev. Mol. Med. 10 (2008) e13.
[98] C.M. Shanahan, D.L. Connolly, K.L. Tyson, et al., Aquaporin-1 is expressed by vascular smooth muscle cells and mediates rapid water transport across vascular cell membranes, J. Vasc. Res. 36 (1999) 353–362.
[99] R. Madonna, V. Doria, R. De Caterina, High glucose and hyperosmolar stress in-crease the expression of COX2 and AQP1 in aorta of type 1 diabetic mice devel-oping atherosclerosis: implication for unstable vascular disease in diabetes, Eur. Heart J. (2017) (supplement).
[100] R. Madonna, E. Montebello, G. Lazzerini, M. Zurro, R. De Caterina, NA+/H+ exchanger 1- and aquaporin-1-dependent hyperosmolarity changes decrease nitric oxide production and induce VCAM-1 expression in endothelial cells exposed to high glucose, Int. J. Immunopathol. Pharmacol. 23 (2010) 755–765.
[101] R. Madonna, G. Giovannelli, P. Confalone, F.V. Renna, Y.J. Geng, R. De Caterina, High glucose-induced hyperosmolarity contributes to COX-2 expression and an-giogenesis: implications for diabetic retinopathy, Cardiovasc. Diabetol. 15 (2016) 18.
[102] T. Sundsten, M. Eberhardson, M. Goransson, P. Bergsten, The use of proteomics in identifying differentially expressed serum proteins in humans with type 2 diabetes, Proteome Sci. 4 (2006) 22.
[103] W.C. Cho, T.T. Yip, W.S. Chung, A.W. Leung, C.H. Cheng, K.K. Yue, Differential expression of proteins in kidney, eye, aorta, and serum of diabetic and non-dia-betic rats, J. Cell. Biochem. 99 (2006) 256–268.
[104] M. Nishi, S.J. Chan, S. Nagamatsu, G.I. Bell, D.F. Steiner, Conservation of the sequence of islet amyloid polypeptide infive mammals is consistent with its
putative role as an islet hormone, Proc. Natl. Acad. Sci. U. S. A. 86 (1989) 5738–5742.
[105] C.M. Steppan, E.J. Brown, C.M. Wright, et al., A family of tissue-specific resistin-like molecules, Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 502–506.
[106] S.A. Preil, L.P. Kristensen, H.C. Beck, et al., Quantitative proteome analysis reveals increased content of basement membrane proteins in arteries from patients with type 2 diabetes mellitus and lower levels among metformin users, Circ. Cardiovasc. Genet. 8 (2015) 727–735.
[107] H. Husi, T. Van Agtmael, W. Mullen, et al., Proteome-based systems biology analysis of the diabetic mouse aorta reveals major changes in fatty acid bio-synthesis as potential hallmark in diabetes mellitus-associated vascular disease, Circ. Cardiovasc. Genet. 7 (2014) 161–170.
[108] J. Nielsen, J.D. Hoffert, M.A. Knepper, P. Agre, S. Nielsen, R.A. Fenton, Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: mechanisms for aquaporin 2 down-regulation and cellular proliferation, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 3634–3639.
[109] S. Khositseth, P. Uawithya, P. Somparn, et al., Autophagic degradation of aqua-porin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus, Sci. Rep. 5 (2015) 18311.
[110] T.M. Jensen, D.R. Witte, D. Pieragostino, et al., Association between protein sig-nals and type 2 diabetes incidence, Acta Diabetol. 50 (2013) 697–704. [111] T.R. Koves, J.R. Ussher, R.C. Noland, et al., Mitochondrial overload and
in-complete fatty acid oxidation contribute to skeletal muscle insulin resistance, Cell Metab. 7 (2008) 45–56.
[112] T.J. Wang, M.G. Larson, R.S. Vasan, et al., Metabolite profiles and the risk of developing diabetes, Nat. Med. 17 (2011) 448–453.
[113] J.Z. Villarreal-Perez, J.Z. Villarreal-Martinez, F.J. Lavalle-Gonzalez, et al., Plasma and urine metabolic profiles are reflective of altered beta-oxidation in non-diabetic obese subjects and patients with type 2 diabetes mellitus, Diabetol. Metab. Syndr. 6 (2014) 129.
[114] C.B. Newgard, J. An, J.R. Bain, et al., A branched-chain amino acid-related me-tabolic signature that differentiates obese and lean humans and contributes to insulin resistance, Cell Metab. 9 (2009) 311–326.
[115] P. Del Boccio, C. Rossi, M. di Ioia, I. Cicalini, P. Sacchetta, D. Pieragostino, Integration of metabolomics and proteomics in multiple sclerosis: from biomarkers discovery to personalized medicine, Proteomics Clin. Appl. 10 (2016) 470–484. [116] C. Rossi, V. Marzano, A. Consalvo, et al., Proteomic and metabolomic
character-ization of streptozotocin-induced diabetic nephropathy in TIMP3-deficient mice, Acta Diabetol. (2017),http://dx.doi.org/10.1007/s00592-017-1074-y(Epub ahead of print).
[117] R. Madonna, R. De Caterina, Atherogenesis and diabetes: focus on insulin re-sistance and hyperinsulinemia, Revista espanola de cardiologia 65 (2012) 309–313.
[118] R. De Caterina, R. Madonna, H. Sourij, T. Wascher, Glycaemic control in acute coronary syndromes: prognostic value and therapeutic options, Eur. Heart J. 31 (2010) 1557–1564.
[119] R. Madonna, R. De Caterina, Prolonged exposure to high insulin impairs the en-dothelial PI3-kinase/Akt/nitric oxide signalling, Thromb. Haemost. 101 (2009) 345–350.
[120] G. Patti, M. Lucerna, I. Cavallari, et al., Insulin-requiring versus noninsulin-re-quiring diabetes and thromboembolic risk in patients with atrialfibrillation: PREFER in AF, J. Am. Coll. Cardiol. 69 (2017) 409–419.