1 Doctoral Programme in
Agrobiodiversity
MAGIC maize population: A new
strategy on dissecting the genetic
basis and heterosis of Fusarium
seedling rot resistance
Author
Popi Septiani
Supervisor
Prof. Mario Enrico Pè
Accademic Year
2015/2016
2
TABLE OF CONTENTS
Page Chapter 1: General Introduction 4
• Footprints of maize domestication: from Teosinte to hybrid maize
5 • The genetic basis underlying heterosis 8 • State of the art of maize cultivation and challenges 10 • Maize diseases and genetic bases of disease resistance in
maize
12 • Strategies to discover QTL and causal genes underlying disease
resistance
15 • An integrated approach based on the MAGIC maize population 19 • Thesis overview and objectives 21
• References 22
Chapter 2: Genetic Architecture of Fusarium Seedling Rot Resistance in Recombinant Inbred Line of MAGIC Maize Population
35 Unravelling the genetic basis of Fusarium seedling rot resistance in the MAGIC maize population: novel targets for breeding (Scientific Manuscript) 37 • Abstract 38 • Introduction 38 • Result 41 • Discussion 46
• Material and Methods 52
• References 57
• Supplementary Materials 69 Chapter 3: Seedling resistance to Fusarium verticillioides and its
heterotic components in the MAGIC maize recombinant intercrosses (RIXs)
78 Seedling resistance to Fusarium verticillioides and its heterotic components in the MAGIC maize recombinant intercrosses (RIXs) (Scientific Manuscript)
80
• Abstract 81
• Introduction 82
• Material and Methods 85
• Result 90
3
• References 103
• Supplementary Materials 118 Chapter 4: General Conclusions 126
4
CHAPTER 1
5
Footprints of maize domestication: from Teosinte to hybrid maize
Maize (Zea mays spp. mays L.) went through a remarkable transformation from a wild grass growing in Mesoamerica into the most productive crop worldwide. Archeological discoveries (Piperno et al., 2009) sided by genetic studies (Matsuoka et al., 2002; van Heerwaarden et al., 2011) have shown that maize was domesticated from its wild ancestor teosinte (Zea mays spp. parviglumis) in the central Balsas river valley, Mexico. From there, maize spread to Northern and Southern America, was traded to Europe (Tenaillon and Charcosset, 2011), and then eventually dispersed worldwide through the global trade network. In this process, maize adapted to a broad range of different environmental conditions beyond the ecological niche in which its progenitor was originally grown (Ruiz Corral et al., 2008). In this sense, maize domestication is one of the greatest achievements of artificial selection and evolution of crops.
Modern maize and teosinte show profound differences in their morphologies as a result of human selection, including modifications in vegetative architecture, ear and kernel morphology (Beadle, 1980). Typically, teosinte features pronounced tillering, i.e. the growth of numerous lateral stalks stemming from the base of the plant, while modern maize, except for pop-corn genotypes, has only one. Their female inflorescences are also strikingly different: teosinte ears possess only about 5 to 12 tightly sealed kernels forming a shell-like fruit cases. At maturity, teosinte ears disarticulate so that the individual units of the fruit case become the dispersal unit. The fruit hardness makes them indigestible when consumed by birds and dispersed by fecal matter (Beadle, 1980; Doebley, 2004). The spread of teosinte is also supported by its pollen which easily dispersed by the wind and furthermore the pollen flow between teosinte and domesticated
6
maize contributed in shaping its genetic diversity (Beadle, 1980; van Heerwaarden et al., 2011).
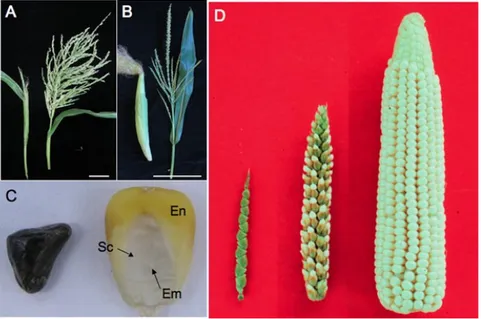
Fig 1. Teosinte compared to maize (A) A teosinte female inflorescence (left), which arises as a secondary branch from tillers, and tassel (right). (B) An ear (left) and tassel (right) of maize. Size bar in A and B is 10 cm. (C) Teosinte kernel (left) and maize kernel (right). The teosinte kernel is hidden by hardened glumes (see Glossary). The maize kernel is exposed and reveals the endosperm (En) and embryo (Em). The embryo is surrounded by the scutellum (Sc), the nutritive tissue of the cotyledon. (D) A comparison of teosinte on the left, maize on the right and the F1 of maize and teosinte in the middle. Image credits: (D) John Doebley, Department of Genetics, University of Wisconsin–Madison; all other images, Sarah Hake (Hake and Ross-Ibarra, 2015).
The most striking morphological differences between modern maize and teosinte may be explained by a relatively small number of mutations with dramatics development effects. Using basic Mendelian ratios from 50,000 maize and teosinte hybrids, George W. Beadle recognized for the first time that as few as five loci could be involved in major ear and plant morphological changes (Beadle, 1972; Beadle, 1980). Ear and plant morphology are characters whose
7
phenotypic variation is continuous and conditioned by allelic variation at multiple loci or often been referred to as quantitative traits (Doebley, 2004). Hence, the individual loci controlling quantitative traits are referred to quantitative trait loci (QTL). Advent of molecular marker or DNA marker on genetic analysis has enabled researchers to map the QTL underlying the quantitative traits (Winter and Kahl, 1995). QTL mapping is based on the principle of detecting an association between phenotype and the molecular markers using statistical method in a segregating mapping population (Jones et al., 1997; Collard et al., 2005).
Decades after Beadle’s findings, thanks to advancement of QTL analyses, Doebley and colleagues validated Beadle’s findings by identifying a limited number of genomic regions in the maize genome with large effect on inflorescence morphology (Doebley, 1990; Doebley and Stec, 1991; Doebley and Stec, 1993). A single major locus, teosinte glume architecture1 (tga1) is responsible for the formation of stone-like fruit cases absent in maize (Dorweiler et al., 1993). The radical phenotypic change from hard glumes to softer glumes was shown to be caused by a single amino acid mutation of tga1, demonstrating how single genetic alterations can induce dramatic changes during domestication and evolution (Wang et al., 2005). Likewise, the switch from long lateral branches terminated by male tassels in teosinte to short lateral branches tipped by female ears in maize is caused by a single locus, teosinte branched1 (tb1) (Doebley et al., 1995; Doebley et al., 1997). Studies on ancient DNA have suggested that selection at this locus may have occurred approximately 4,400 years ago (Jaenicke-Despres et al., 2003). Subsequently, thanks to the development of larger QTL mapping populations and more advanced molecular markers technologies, studies reported a plethora of loci contributing to the morphology of domesticated maize (Briggs et al., 2007).
8
Over time, farmers and plant breeders harnessed maize genetic diversity to adapt to various environments and to amplify the most desirable traits. In the United States, farmers’ selection led to two main types of maize: the Northern Flint, with better adaptability to different environments, and the Southern Dents, with higher yield potential. After thousands of years of sustained selection, during the 20th century maize was ready to a second major step change in its profitability
as a crop. Flint and Dent types became the genetic backbone for modern maize hybrid technology (Troyer, 2009). Already in 1908, Edward M. East and George H. Shull independently revealed the biological basis of hybrid vigor, named by Shull as heterosis in maize (reported in Crow, 1998). Since the introduction of commercial maize hybrids during the 1920s in the United States, it was estimated that the yield increased about 50%, paving the way for maize to become the most successful crop in agricultural industry (Duvick, 2005).
The genetic basis underlying heterosis
The term heterosis or hybrid vigor refers to the phenomenon by which the F1 progeny of inbred varieties is superior to the corresponding parental lines by
means of biomass, speed of development, resistance to disease and yield (Birchler et al., 2010). The genetic study of hybrid vigor has an illustrious forerunner in Charles Darwin, who, in its “The effect of cross and self-fertilization in the vegetable kingdom” from 1876, compared inbred and crossed-pollinated maize. Darwin noted that crossed-pollinated maize (hybrid) plants had greater height, weight and fertility, as compared with their self-pollinated counterparts because of their “greater innate constitutional vigor” (Darwin, 1876). Although heterosis was agronomically exploited for over a century, its molecular basis are only partially understood.
9
Classical quantitative genetics advanced several hypotheses for a genetic model explaining heterosis. Among these, the dominance model is based on complementation, and postulates that the inbred parents carry slightly deleterious alleles at different loci; hence, the superior allele of one parent will complement the inferior allele of the other parent in the resulting hybrid (Xiao et al., 1995; Bruce, 1910; Jones, 1917). The over-dominance model instead considers that the allelic interaction of different alleles provides superior function in heterozygous hybrids than in either of the homozygous parent (East, 1936; Crow, 1948). The pseudo-over-dominance model refers instead to a particular situation in which tightly linked genes with favorable dominant alleles of the parental lines are in repulsion phase (dominant and recessive alleles on opposite homologues for two genes) resulting the characteristic of over-dominance action when combined in the hybrid (Crow, 1948). A fourth interpretation, based on the epistasis model, explains that superior phenotypic expression of a trait in hybrids results from interactions between non-allelic genes from the two parent at two or more loci showing dominance, and/or over-dominance action(Schnell, 1992; Li et al., 2001).
All these genetic hypotheses indicate the combination of a considerable number of genes as responsible for heterosis, and imply that positive and negative effects of loci might compensate each other, making difficult to support one hypothesis over the other (Hochholdinger and Hoecker, 2007). However, with the advancements in functional genomics and transcriptomics related technologies, the riddle of heterosis is being reinvestigated to understand its underlying molecular mechanisms. Changes in gene expression in hybrids are often drastic, deviating from mid-parent values (Song and Messing, 2003). In Arabidopsis, methylation levels are higher in hybrids, and correlate with the
10
transcriptional down-regulation of a number of genes having profound effects heterosis for biomass (Shen et al., 2012). In rice, numerous superior alleles contribute to heterosis associated with higher yield, while only few loci show strong over-dominance effects (X., Huang et al., 2015). In maize, a transcriptomic study identified several genes that were expressed in only one of the two inbred parents (Mo17 x B73) but all of them were expressed in F1 hybrids, suggesting complementation contributes to transcriptome complexity in hybrids relative to their inbred parents (Paschold et al., 2012). One study reported that a high frequency of structural variations (SVs) including copy number variation (CNV) and presence/absence variation (PAV) was observed in two maize inbred lines Mo17 and B73 (Springer et al., 2009). The SVs constitute a portion of dispensable genome, in contrast with the theoretical concept of core genome which defined as minimal genome necessary for the survival (Faster, 2001). On the other hand, it was suggested that SVs substantially contributes to genetic variations and plays important role in shaping the hybrid pattern of gene content and gene expression which in turn may be linked to heterosis (Marroni et al., 2014).
State of the art of maize cultivation and challenges
Today, maize is perhaps the world’s most important crop, providing food, feed, fuel and raw materials for industry. It was estimated that about 1.06 billion metric tons of maize were harvested in 2016 from 188 million hectares cropped worldwide (FAOSTAT, 2018). United states, China and Brazil are the world top three maize producing Countries, contributing to 64% of maize world production (FAOSTAT, 2018). Maize is processed into various industrial products, including starch, sweeteners, oil, beverages, glue, industrial alcohol, and ethanol. An important part of maize production is currently used to generate ethanol fuel,
11
whose increased demand has resulted in increased maize prices and maize acreage (Ranum et al., 2014). However, maize remains a food source of prime importance. Its flour contains approximately 66% carbohydrates, 14% proteins, and 11% fats, with an energy density of 419 kcal/100 g (USDA, n.d.) providing 20% of the human diet calories (Brown et al., 1988). Maize is also important as feed for the livestock in the form of grain and whole plant such as forage and silage (Klopfenstein et al., 2013). As the world population will approach nine billions by 2050, maize is more and more needed to achieve global food security and nutrition (Nuss and Tanumihardjo, 2010). Indeed, maize demand is expected to double by 2050, especially in Developing Countries where needs for food and feed are rising by 1.3 % and 2.9% per annum, respectively (Prasanna, 2009). Increasing demand and production shortage in global maize supplies have worsened market volatility and contributed to surging global maize prices (Ranum et al., 2014; Klopfenstein et al., 2013).
Climate change dynamics, with consequent rise in abiotic and biotic stresses, are an ever-increasing menace to maize cultivation. Global warming is affecting disproportionately food-insecure regions, jeopardizing crop and livestock production. Lobell and colleagues reported that between 1980 and 2008, global maize production was decreased by 3.8% due to a changing climate, countervailing some of the yield gain by breeding efforts and technological advances (Lobell et al., 2011). Increasing temperature may have strong negative effects in grain yield (Bassu et al., 2014) as it shortens the length of growing cycle, decreasing the opportunity to gain more radiation and CO2 assimilation, thus
eventually reducing biomass and yield accumulation (Hatfield, 2016). Climate change can be a driver of emerging infectious disease of plants through gradual changes in climate including increased temperature and changing pattern of
12
climate variability such as precipitation anomalies (Anderson et al., 2004). For example, milder winter and higher nocturnal temperature may determine an increase of the development and survival of plant pathogens, accelerating vector and pathogen life cycle, and increasing sporulation and infectiousness of foliar fungi such as Septoria sp. (Harvell et al., 2002). The combination of extreme weather events such as high temperature, high rain fall and long periods of drought might also favor the growth of the fungus Aspergillus flavus, resulting in an increase of Aflatoxin content in maize kernels, a toxic compound produced by this fungus (Rosenzweig et al., 2001).
Plant disease manifestation is a result of three-way interacting factors combining susceptible host plant, virulence of the pathogen and environment condition suitable for disease development, known as disease triangle (Eastburn et al., 2011). Climatic factors such as high temperature and drought might alter stages and enhance the rate of the development of the pathogen, modify host resistance and change the cellular processes and physiology of the host-pathogen interactions. It also contributes to the alteration on virulence, pathogenicity, aggressiveness and fecundity of the pathogen (Garrett et al., 2006). Developing and deploying climate-resilient maize genotypes in the wake of climate change has become a top priority of modern breeding (Cairns and Prasanna, 2018), especially in relation to disease resistance (Chakraborty and Newton, 2011). To meet this ambitious aim, it is fundamental to enlarge our comprehension of maize-pathogen interaction.
Maize diseases and genetic bases of disease resistance in maize
Global maize yield losses due to fungal pathogens were estimated over 9% in 2001-2003, locally ranging from 4% in Europe to 14 % in Asia and Africa, and
13
with a tendency to increase overtime (Oerke, 2006). The most relevant maize diseases caused by fungal pathogens include foliar disease and stalk and ear rot (The CIMMYT Maize Program, 2004). The main foliar diseases of maize include southern leaf blight, caused by Cochliobolus heterostrophus, northern leaf blight, caused by Setosphaeria turcica, common rust, caused by Puccinia sorghi, gray leaf spot, caused by Cercospora zeae-maydis, and the banded leaf and sheath blight caused by Rhizoctonia solani (Pingali, 2001). Stalk and ear rots are mostly caused by fungi belonging to the genera Aspergillus, Penicillium, and Fusarium. The latter especially are causes of devastating diseases on maize fields worldwide. Several Fusarium species are spread in temperate and semi-tropical areas, including all European maize-growing areas. In Europe, it is estimated that Fusarium spp. causes maize yield losses between 10% and 30% infecting roots, seedling, stalks and ears (Logrieco et al., 2002). Besides reducing yield, Fusarium infections produce mycotoxins in the infected ears, rendering the derived flour unsafe for food and feed. Mycotoxins are becoming increasingly controlled worldwide because their presence was associated with severe diseases in human (Lucic et al., 1999) and livestock (Awad et al., 2008). While the biggest concern is on ear and kernel contamination, the formation of mycotoxins in stalk, infected leaves, and whole plants may also pose to significant risks for forage and silage maize (Alonso et al., 2013). In addition to health risks, mycotoxin contamination in feed creates economic and trade implication in which the products are not marketable if the mycotoxin level is higher than the maximum acceptable limits regulated by local authorities (Pinotti et al., 2016).
There are several ways by which farmers may control diseases in the field, starting from field management: plowing, adjusting sowing dates, and also
14
residues (Munkvold, 2014). When disease occurrence is persistent and imminent, application of fungicides can be used as an alternative. However, it is important to control the excessive use of fungicides to avoid unintended side-effects, such as the development of pathogen resistance (Mundt, 2014). One of the most effective approaches to disease management is rendering the plants genetically resistant to the pathogen. To be effective, plant resistance should be durable and remain efficacious when deployed over wide areas under substantial disease pressure (Johnson, 1984). In this sense, achieving host resistance through plant breeding is a reliable alternative to fungicides (Mundt, 2014) and it may be combined with agronomic practices, creating integrated disease management approaches (Mundt et al., 2002; Nelson et al., 2018).
Plant resistance may be categorized as either qualitative or quantitative (Vanderplank, 1968). Qualitative resistance is the consequence of major genes, called resistance genes (R-genes), which provide complete or near-complete resistance. R-genes typically determine dominant phenotypes, but recessive resistance may also occur because of loss of function in variants of the genes that confer susceptibility to the disease (S-genes). Quantitative disease resistance shows an incomplete or partial phenotype, and is controlled by multiple genes with small effect. Genes providing quantitative resistance are minor genes contributing to QTL (Jamann et al., 2013; Poland et al., 2009).
Studies in Arabidopsis thaliana have provided insights on the mechanisms underlying plant disease resistance (Jones and Dangl, 2006). Plant immune systems may follow two mechanisms: effector-triggered immunity (ETI) and pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) (Thomma et al., 2011; Jones and Dangl, 2006). ETI is often manifested as a hypersensitive response (HR) causing rapid cell death localized at the infection
15
site. ETI is activated when plant resistance proteins, encoded by R-genes, recognize the corresponding pathogenic effector proteins, thus it contributes to qualitative resistance. PTI is instead responsible of broad spectrum resistance that is triggered in response of PAMPs. When pathogens infect the plant, the plant cell surface recognizes the PAMPs via its pattern recognition receptors (PRRs). In contrast to ETI, PTI is the basis of quantitative resistance (Nelson et al., 2018).
Studies looking to qualitative disease resistance may be more straight-forward because resistance can be unambiguously distinguished from susceptibility on a single-plant basis, thanks to the involvement of R-genes (Nelson et al., 2018). However, qualitative resistance is mostly effective for short term due to i) a lack of durability in some plants, particularly when the pathogen has higher evolutionary potential and ii) a lack of availability in other system especially in necrotrophic systems (Poland et al., 2009). On the contrary, quantitative resistance is generally more durable in the field than qualitative resistance (Mcdonald and Linde, 2002), thus it is important from a perspective of sustainable agronomy.
Strategies to discover QTL and the causal genes underlying disease resistance Quantitative disease resistance is the result of multiple QTL that contribute to the continuous distribution of the trait (Nelson et al., 2018). Identifying quantitative resistance QTL is made difficult by the multi-genic interactions and environmental factors that come into play in determining the trait. A substantial number of papers have reported QTL for quantitative resistance in maize, leading to strong candidate for foliar diseases resistance such as northern leaf blight (Hurni et al., 2015; Jamann et al., 2016; Wisser et al., 2011),
16
southern leaf blight and gray leaf spot (Wisser et al., 2011) , but on the contrary, there are no strong candidates resistance to rots caused by Fusarium (Wisser et al., 2006). The genetic architecture of Fusarium resistance appears complex, with minor effect QTL and strongly influenced by environmental conditions (Coan et al., 2018; Robertson-Hoyt et al., 2006). The complexity of Fusarium resistance and the large influence of the environments leads to a moderate heritability, hindering the accurate identification of QTL and the estimation of their effects (Mesterházy et al., 2012; Coan et al., 2018).
An approach to improve and accelerate efforts in mapping Fusarium resistance QTL may rely on three key assets: dense molecular markers, precision phenotyping, and advanced genetic materials (Yang et al., 2017). A genotyping approach with dense molecular marker is necessary to increase the capacity to detect and precisely locate QTL. Thanks to advances in genomics technology that allow high-throughput genotyping capabilities, high-density and genome-wide molecular markers such as single nucleotide polymorphisms (SNPs) may be produced with relative ease. Several genotyping arrays targeting coding SNPs are available on maize, including the Affymetrix Axiom Maize Genotyping Array (Unterseer et al., 2014) and the Illumina MaizeSNP50 BeadChip (Ganal et al., 2011). Genotyping arrays are sided by sequencing based approaches such as the genotyping by sequencing (GBS) technology (Elshire et al., 2011). The rapid development of sequencing technology is supported by the availability of genomic information on maize like the B73 maize genome reference (Schnable et al., 2009). Altogether, these tools enable to achieve high density molecular marker for higher resolution QTL mapping.
The second important factor to increase the capacity to identify the genetic basis of quantitative resistance to Fusarium is an accurate, high precision
17
phenotyping. Phenotyping is indeed a crucial step in QTL mapping. However, a reliable disease evaluation may be hampered by interactions between host, pathogens, and environment (Munkvold and Desjardins, 1997). Indeed, field conditions and pathogen populations are vary across locations, and this reduces the consistency across multiple measurements (Clements et al., 2003). Most studies increase phenotyping reliability using artificial infection of Fusarium inoculation through kernels or silks (Robertson-Hoyt et al., 2006; Munkvold and Desjardins, 1997). Nevertheless, to precisely evaluate the quantitative disease resistance, the phenotyping must be replicated across different environment in different years, making the field phenotyping is time consuming and labor intensive (Robertson-Hoyt et al., 2007; Zila et al., 2014; Maschietto et al., 2017). Bioassays conducted in controlled conditions represent an appealing alternative to field phenotyping, as they are generally more repeatable, and less labor- and resource-intensive. In-vivo bioassays based on rolled towels (Ellis et al., 2011; Stagnati et al., 2019) or petri dishes (Ju et al., 2017) allow the high-throughput screening of hundreds of genotypes with various pathogen and growth conditions in seedlings. These methods permit to evaluate the level of disease infection in seedling and kernels, and were successfully used to map QTL for disease resistance in several species, including soybean (Ellis et al., 2011; Ellis et al., 2014) and maize (Ju et al., 2017; Stagnati et al., 2019) .
A third crucial factor in QTL mapping is the genetic material screened for resistance. Traditionally, QTL mapping in plants used bi-parental crosses combining the genome of two lines with contrasting resistance phenotypes (Robertson-Hoyt et al., 2006). This type of population provides high mapping power, but it has insufficient recombination events and lack of diversity due to the narrow genetic base (Holland, 2015; Huang et al., 2015). An alternative to
bi-18
parental populations are association mapping panels, in which various lines with unknown kinship are selected and measured for a trait of interest. QTL mapping may be conducted on these materials using genome wide association (GWA) approaches, a strategy that led to identification of several Fusarium resistance QTL in maize (Zila et al., 2014; Ju et al., 2017; Coan et al., 2018). Although this method provides high diversity and dense recombinant events, its usefulness is limited by missing pedigree and parental information that lowers the statistical power of QTL identification. The shortcoming of such alternative approaches pushed to the development of multi-parent populations (MPP) encompassing more genetic diversity and resulting in higher mapping power and resolution (Huang et al., 2015), combining the strengths of bi-parental populations and association mapping panels. MPPs are developed by crossing more than two inbred parent lines to produce recombinant inbred lines (RIL) following difference designs. MPPs include Nested Association Mapping (NAM) populations, created by crossing one reference inbred line to a collection of diverse inbred lines to produce F1 seeds. The F1 plants are then reproduced by self-pollination and the
resulting F2 are advanced through single seed descent (SSD) resulting in multiple
interconnected bi-parental RIL families sharing the common parental haplotypes (Yu et al., 2008; McMullen et al., 2009).
The NAM design has been used to study the genetic architecture of morphological (Tian et al., 2011) and disease resistance traits in maize (Poland et al., 2011) and also in wheat (Bajgain et al., 2016). Multi-parent Advance Generation Intercross (MAGIC) populations are instead designed by inter-mating an even number of inbred lines (typically four, eight or sixteen) in a balanced funnel breeding scheme resulting in RILs that are each a mosaic of MAGIC founder diversity (Churchill et al., 2004; Kover et al., 2009; Huang et al., 2012; Dell ’Acqua
19
et al., 2015). The high level of genetic diversity featured by NAM and MAGIC is sufficient to dissect the phenotypic diversity in almost any trait of interest (Ladejobi et al., 2016). Since its conception and after its successful development in mice (Churchill et al., 2004), the MAGIC design was widely employed in plants and especially crops, including Arabidopsis (Kover et al., 2009), wheat (Mackay et al., 2014; Huang et al., 2012), rice (Bandillo et al., 2013), tomato (Pascual et al., 2015), and maize (Dell ’Acqua et al., 2015).
An integrated approach based on the MAGIC maize population
The MAGIC maize (MM) population offers unique properties to facilitate the genetic analysis of complex traits (Dell ’Acqua et al., 2015), including quantitative resistance to pathogens. The MM population derives from eight founder lines, its RILs population has been genotyped with 50K SNP markers (Ganal et al., 2011), and its founder lines were fully sequenced and had their transcriptome characterized. In the MM, the inherited founder haplotypes may be statistically computed in each RIL genomic window and allow accurate mapping of QTL. The genetic properties of the MM population also facilitate GWA study within the QTL intervals, providing the means to conduct high resolution mapping of candidate genes most likely contributing to the observed phenotypes. In addition, the MM population allows to exploit transcriptomic data developed on founder lines in order to further reinforce the identification of candidate genes (Dell ’Acqua et al., 2015).
Each mapping population type has advantages and disadvantages. The use of RILs for a QTL analysis are obviously advantageous because multiple selfing processes can increase the number of recombination events, which results in a finer mapping of QTLs. More importantly, once RILs are established, the
20
genotypes are fixed as homozygotes and can be repeatedly used for investigating QTLs of various phenotypes under different environments (Takuno et al., 2012).
However, the deployment of RILs is limited by the fact that only additive gene action can be measured. As a matter of fact, yield, growth performance and resistance to disease in maize have clearly shown heterotic effects in which not only additive but also dominance gene action may be involved (Lippman and Zamir, 2006). Hence, the use of heterozygous genetic material is important to measure the effects of additive and dominance gene action at specific loci. It is possible to create such heterozygous genetic background by crossing RILs producing many combination of hybrid genotypes known as recombinant intercrosses (RIXs).
Heterozygous genotypes have been used in other crop species to decipher the genetic mechanism underlying heterosis. Genomic analysis in rice reveals that rice hybrids accumulate more superior alleles compare to their parental inbred lines, producing high-yielding hybrids with positive dominance (Huang et al., 2015). A study in Arabidopsis thaliana, identified F1 hybrids manifesting significant heterosis (superiority to both parents) for biotrophic bacterial resistance. The comparison of genome-wide gene expression profiles of A. thaliana hybrids and their parents revealed that salicylic acid biosynthesis is required for heterosis to biotrophic bacterial defense (Yang et al., 2015).
Referring to those studies, we can lay a perspective to utilize the MM RIXs as the heterozygous genetic material to study heterosis for disease resistance and agronomic traits. MM RIXs have several advantages. The RIXs population have multiple parental lines which may contain a lot of dispensable genome segregating in the population. The concept of dispensable portion of genome is
21
referred to the existence of structural variants (SVs) that may provide an important contribution to phenotypic diversity (Marroni et al., 2014). An implication of pervasive effect of SVs is its possible role in shaping the hybrid pattern of gene content and gene expression which may in turn to be linked to heterotic effect on the given phenotype, including disease resistance (Paschold et al., 2012; Stupar and Springer, 2006). Another advantage is that, MM RIXs do not require additional genotyping. Once their parental lines have been genotyped, genotyping can be done in silico, providing cost effective and time saving to conduct the study. Altogether, the unique properties of MM RIXs would create unprecedented tools to reveal new insight on heterosis in maize (Dell ’Acqua et al., 2015).
Thesis overview and objectives
Recent advances in genetics and genomics have provided new opportunities to dissect the genetic basis of quantitative disease resistance in maize. This research is designed to take advantage of genomic tools, advanced phenotyping, and advanced genetic materials to decipher the genetic complexity of resistance to Fusarium verticillioides, a prevalent pathogen in maize. In this research work, the MM population is used in combination with fast-screening phenotyping tools to elucidate the genetic basis underlying resistance to Fusarium seedling rot (FSR), following a two-step approach. In the first step, the study was aimed at characterizing FSR resistance and seedling traits in MM RILs, using this information to map resistance QTL. In a second step, MM RIXs derived from RILs analyzed in step one were used to characterize heterosis for FSR resistance and seedling traits. Altogether, the research output is expected to contribute knowledge on the genetic mechanisms of F. verticillioides resistance
22
at early developmental stages in maize lines and hybrids, an important starting point for further investigations in adult plants and field resistance and for accelerating breeding for resistance.
This thesis consists of four chapters. Chapter 1 focuses on the background of the study, including general review of maize cultivation and challenges following the highlight of the past and current evidences, issues and strategies on dissecting complex trait of disease resistance in maize. Chapter 2 reports experimental work aimed at exploiting the MM RILs to unravel the genetic architecture of FSR resistance. Chapter 3 focuses on the investigation of heterosis for disease resistance in MM RIXs. Chapter 4 closes the thesis, summarizing the most important findings and perspectives of the research undertaken for this PhD.
REFERENCES
Alonso, V.A., Pereyra, C.M., Keller, L.A.M., Dalcero, A.M., Rosa, C.A.R. and Chiacchiera, S.M. (2013) Fungi and mycotoxins in silage : an overview. , 637–643.
Anderson, P.K., Cunningham, A.A., Patel, N.G., Morales, F.J., Epstein, P.R. and Daszak, P. (2004) Emerging infectious diseases of plants: Pathogen
pollution, climate change and agrotechnology drivers. Trends Ecol. Evol., 19, 535–544.
Awad, W.A., Ghareeb, K., Böhm, J., Razzazi, E., Hellweg, P. and Zentek, J. (2008) The Impact of the Fusarium Toxin Deoxynivalenol ( DON ) on Poultry. , 7, 827–842.
Bajgain, P., Rouse, M.N., Tsilo, T.J. and Macharia, G.K. (2016) Nested
Association Mapping of Stem Rust Resistance in Wheat Using Genotyping by Sequencing. PLoS One, 11, 1–22.
Bandillo, N., Raghavan, C., Muyco, P.A., et al. (2013) Multi-parent advanced generation inter-cross ( MAGIC ) populations in rice : progress and potential for genetics research and breeding. , 1–15.
Baranwal, V.K., Zehr, U.B., Tyagi, A.K. and Kapoor, S. (2012) Heterosis : emerging ideas about hybrid vigour. J. Exp. Bot., 63, 6309–6314. Bassu, S., Brisson, N., Durand, J., et al. (2014) How do various maize crop
23
models vary in their responses to climate change factors ? Glob. Chang. Biol., 20, 2301–2320.
Beadle, G.W. (1980) the anchestry of corn. Sci. Am., 242, 112–119.
Beadle, G.W. (1972) The mistery of maize. F. Museum Natl. Hist. Bull, 43, 2–11. Birchler, J.A. (2015) The genetic basis of hybrid vigour. Nat. Publ. Gr., 1, 1–2.
Available at: http://dx.doi.org/10.1038/nplants.2015.20.
Birchler, J.A., Hong, Y., Chudalayandi, S., Vaiman, D. and Veitia, R.A. (2010) Heterosis. Plant Cell, 22, 2105–2112.
Birchler, J.A., Yao, H. and Chudalayandi, S. (2006) Unraveling the genetic basis of hybrid vigor. , 103, 12957–12958.
Bonnafous, F., Fievet, G., Blanchet, N., et al. (2018) Comparison of GWAS models to identify non ‑ additive genetic control of flowering time in sunflower hybrids. Theor. Appl. Genet., 131, 319–332.
Bradbury, P.J., Zhang, Z., Kroon, D.E., Casstevens, T.M., Ramdoss, Y. and Buckler, E.S. (2007) TASSEL : software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635.
Briggs, W.H., Mcmullen, M.D., Gaut, B.S. and Doebley, J. (2007) Linkage Mapping of Domestication Loci in a Large Maize–Teosinte Backcross Resource. Genetics, 1928, 1915–1928.
Brown, W., Bressani, R., Glover, D. and Hallauer, A. (1988) Quality-Protein Maize. In F. Bostid, ed. Report of an Ad Hoc Panel of the Advisory Committee on technology innovation board on science and. National academy press. Available at:
https://scholar.google.com/scholar_lookup?hl=en&volume=Food+Nutr+Bul l&publication_year=1988&pages=%0ABrown%2C+WL%2C+%0ABressani+R %2C+%0AGlover+DV%2C+%0AHallauer+AR%2C+%0AJohnson+VA%2C+%0A
Qualset+CO.+1988.+Quality-protein+maize%3A+report+of+an+ad+hoc+panel [Accessed October 11, 2018].
Bruce, A.B. (1910) The Mendelian theory of heredity and the augmen-tation of vigor. Science (80-. )., 32, 627–628.
Cairns, J.E. and Prasanna, B.M. (2018) Developing and deploying climate-resilient maize varieties in the developing world. Curr. Opin. Plant Biol., 45, 1–5. Available at: https://doi.org/10.1016/j.pbi.2018.05.004.
Chakraborty, S. and Newton, A.C. (2011) Climate change , plant diseases and food security : an overview. Plant Pathol., 60, 2–14.
Chen, J., Shrestha, R., Ding, J., Zheng, H., Mu, C., Wu, J. and Mahuku, G. (2016) Genome-Wide Association Study and QTL Mapping Reveal Genomic Loci Associated with Fusarium Ear Rot Resistance in Tropical Maize Germplasm.
24
G3 (Bethesda)., 6, g3.116.034561. Available at: http://www.ncbi.nlm.nih.gov/pubmed/27742723.
Churchill, G.A., Airey, D.C., Allayee, H., et al. (2004) The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet., 36, 1133–1137. Available at:
http://www.nature.com/doifinder/10.1038/ng1104-1133.
Clements, M.J., Kleinschmidt, C.E., Maragos, C.M., Pataky, J.K. and White, D.G. (2003) Evaluation of inoculation techniques for Fusarium ear rot and fumonisin contamination of corn. Plant Dis., 87, 147–153.
Coan, M.M.D., Senhorinho, H.J.C., Pinto, R.J.B., Scapim, C.A., Tessmann, D.J., Williams, W.P. and Warburton, M.L. (2018) Genome-wide association study of resistance to ear rot by Fusarium verticillioides in a tropical field maize and popcorn core collection. Crop Sci., 58, 564–578.
Collard, B.C.Y., Jahufer, M.Z.Z., Brouwer, J.B. and Pang, E.C.K. (2005) An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica, 142, 169–196.
Crow, J.F. (1998) 90 Years Ago : The Beginning of Hybrid Maize. Genetics, 148, 923–928.
Crow, J.F. (1948) Alternative Hypotheses of Hybrid Vigor. Genetics, 33, 477–487. Darwin, C.R. (1876) The effects of cross and self fertilisation in the vegetable
kingdom, London: John Murray.
Dell ’Acqua, M., Gatti, D.M., Pea, G., et al. (2015) Genetic properties of the MAGIC maize population: a new platform for high definition QTL mapping in Zea mays. genome Biol., 16, 167.
Doebley, J. (1990) Molecular Evidence and the Evolution of Maize. , 44, 6–27. Doebley, J. (2004) The genetics of maize evolution. Annu. Rev. Genet., 38, 37–
59.
Doebley, J. and Stec, A. (1991) Genetic Analysis of the Morphological
Differences Between Maize and Teosinte. Genetics, 129, 285–95. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1682215.
Doebley, J. and Stec, A. (1993) Inheritance of the Morphological Differences Between Maize and Teosinte: Comparison. Genetics, 134, 559–570.
Doebley, J., Stec, A. and Gustus, C. (1995) Teosinte branched1 and the origin of maize: Evidence for epistasis and the evolution of dominance. Genetics, 141, 333–346.
Doebley, J., Stec, A. and Hubbard, L. (1997) The evolution of apical dominane in maize. Nature, 386, 485–488.
25
on Fusarium species pathogenic to cereals. , 755–768.
Dorweiler, J., Stec, A., Kermicle, J. and Doebley, J. (1993) Teosinte glume architecture 1 : A Genetic Locus Controlling a Key Step in Maize Evolution. Science (80-. )., 14, 233–235.
Duvick, D.N. (2001) Biotechnology in the 1930s: The development of hybrid maize. Nat. Rev. Genet., 2, 69–74.
Duvick, D.N. (2005) The contribution of breeding to yield advances in maize (Zea mays L.), Academic Press.
East, E.M. (1936) Heterosis. Genetics, 21, 375–397.
Eastburn, D.M., Mcelrone, A.J. and Bilgin, D.D. (2011) Influence of atmospheric and climatic change on plant – pathogen interactions. , 54–69.
Ellis, M., Broders, K., Paul, P. and Dorrance, A. (2011) Infection of soybean seed by Fusarium graminearum and effect of seed treatments on disease under controlled conditions. Plant Dis., 95, 401–407. Available at:
http://apsjournals.apsnet.org/doi/abs/10.1094/PDIS-05-10-0317.
Ellis, M.L., Jimenez, D.R.C., Leandro, L.F. and Munkvold, G.P. (2014) Genotypic and Phenotypic Characterization of Fungi in the Fusarium oxysporum Species Complex from Soybean Roots. Phytopathology, 104, 1329–1339. Available at: http://apsjournals.apsnet.org/doi/10.1094/PHYTO-02-14-0043-R [Accessed June 22, 2017].
Elshire, R.J., Glaubitz, J.C., Sun, Q., Poland, J.A., Kawamoto, K., Buckler, E.S. and Mitchell, S.E. (2011) A Robust , Simple Genotyping-by-Sequencing ( GBS ) Approach for High Diversity Species. PLoS One, 6, 1–10.
FAOSTAT (2018) FAOSTAT database collections. Food Agric. Organ. United Nations. Available at: http://www.fao.org/faostat/en/?#data/QC/visualize [Accessed July 2, 2018].
Faster, S.N.P. and C.M. (2001) The complexity of simplicity. Geno, 2, comment2002.1–2002.8. Available at:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2447184&tool =pmcentrez&rendertype=abstract%5Cnhttp://onlinelibrary.wiley.com/doi/ 10.1002/cfg.70/full%5Cnhttp://journal.frontiersin.org/Article/10.3389/fmic b.2015.00713/abstract%5Cnhttp://www.scienc.
Frascaroli, E., Cane, M.A., Landi, P., Pea, G., Gianfranceschi, L., Villa, M., Morgante, M. and Pe, M.E. (2007) Classical Genetic and Quantitative Trait Loci Analyses of Heterosis in a Maize Hybrid Between Two Elite Inbred Lines. Genetics, 176, 625–644.
Ganal, M.W., Durstewitz, G., Polley, A., et al. (2011) A large maize (zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS One, 6.
26
Garrett, K.A., Dendy, S.P., Frank, E.E., Rouse, M.N. and Travers, S.E. (2006) Climate Change Effects on Plant Disease : Genomes to Ecosystems. Annu. Rev. Phytopathol., 44, 1–21.
Giraud, H., Bauland, C., Falque, M., et al. (2017) Reciprocal Genetics : Identifying QTL for General and Specific Combining Abilities in Hybrids Between Multiparental Populations from Two Maize. Genetics, 207, 1167– 1180.
Hake, S. and Ross-Ibarra, J. (2015) Genetic, evolutionary and plant breeding insights from the domestication of maize. Elife, 4, 1–8.
Harvell, C.D., Harvell, C.D., Mitchell, C.E., Ward, J.R., Altizer, S., Dobson, A.P., Ostfeld, R.S. and Samuel, M.D. (2002) Climate Warming and Disease Risks for Terrestrial and Marine Biota. Science (80-. )., 296, 2158–2162.
Hatfield, J.L. (2016) Increased Temperatures Have Dramatic Effects on Growth and Grain Yield of Three Maize Hybrids. Agric. Environtmental Lett., 1, 1–5. Heerwaarden, J. van, Doebley, J., Briggs, W.H., Glaubitz, J.C., Goodman, M.M., Jesus Sanchez Gonzalez, J. de and Ross-Ibarra, J. (2011) Genetic signals of origin, spread, and introgression in a large sample of maize landraces. Proc. Natl. Acad. Sci., 108, 1088–1092. Available at:
http://www.pnas.org/cgi/doi/10.1073/pnas.1013011108.
Hochholdinger, F. and Hoecker, N. (2007) Towards the molecular basis of heterosis. Trends Plant Sci., 12, 427–432.
Hoecker, N., Keller, B., Piepho, H.P. and Hochholdinger, F. (2006) Manifestation of heterosis during early maize (Zea mays L.) root development. Theor. Appl. Genet., 112, 421–429.
Holland, J.B. (2015) MAGIC maize: a new resource for plant genetics. Genome Biol., 16, 163. Available at: http://genomebiology.com/2015/16/1/163. Huang, B.E., George, A.W., Forrest, K.L., Kilian, A., Hayden, M.J., Morell, M.K.
and Cavanagh, C.R. (2012) A multiparent advanced generation inter-cross population for genetic analysis in wheat. Plant Biotechnol. J., 10, 826–839. Huang, E.B., Verbyla, K.L., Verbyla, A.P., Raghavan, C., Singh, V.K., Gaur, P.,
Leung, H., Varshney, R.K. and Cavanagh, C.R. (2015) MAGIC populations in crops: current status and future prospects. Theor. Appl. Genet., 128, 999– 1017. Available at: http://dx.doi.org/10.1007/s00122-015-2506-0.
Huang, X., Yang, S., Gong, J., et al. (2015) Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun., 6, 1–9. Available at:
http://dx.doi.org/10.1038/ncomms7258.
Hung, H.Y. and Holland, J.B. (2012) Diallel analysis of resistance to Fusarium ear rot and fumonisin contamination in maize. Crop Sci., 52, 2173–2181.
27
Hurni, S., Scheuermann, D., Krattinger, S.G., et al. (2015) The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc. Natl. Acad. Sci., 112, 8780–8785. Available at: http://www.pnas.org/lookup/doi/10.1073/pnas.1502522112. Jaenicke-Despres, V., Buckler, E.S., Smith, B.D., Gilbert, M.T.P., Cooper, A. and
Doebley, J. (2003) Early Allelic Selection in Maize as Revealed by Ancient DNA. Science (80-. )., 1, 3–6.
Jamann, T., Nelson, R. and Balint-kurti, P. (2013) The genetic basis of disease resistance in maize. In R. K. Varshney and R. Tuberosa, eds. Translational genomics for crop breeding. John Wiley & Sons, Inc., pp. 31–43.
Jamann, T.M., Luo, X., Morales, L., Kolkman, J.M., Chung, C.L. and Nelson, R.J. (2016) A remorin gene is implicated in quantitative disease resistance in maize. Theor. Appl. Genet., 129, 591–602.
Johnson, R. (1984) A critical analysis of durable resistance. Annu. Rev. Phytopathol., 22, 309–330.
Jones, D.F. (1917) Dominance of linked factors as a means of accounting for heterosis. Genetics, 2, 466–479.
Jones, J.D.G. and Dangl, J.L. (2006) The plant immune system. Nature, 444, 323– 329.
Jones, N., OUGHAM, H. and THOMAS, H. (1997) Markers and maping: we are all geneticists now. New Phytol., 165–177.
Ju, C., Zhang, W., Liu, Y., Gao, Y., Wang, X., Yan, J. and Yang, X. (2018) Genetic analysis of seedling root traits reveals the association of root trait with other agronomic traits in maize. BMC Plant Biol., 18, 1–15.
Ju, M., Zhou, Z., Mu, C., Zhang, X., Gao, J. and Liang, Y. (2017) Dissecting the genetic architecture of Fusarium verticillioides seed rot resistance in maize by combining QTL mapping and genome-wide association analysis. Sci. Rep., 7, 46446. Available at: http://dx.doi.org/10.1038/srep46446.
Juroszek, P. and Tiedemann, A. Von (2013) Climatic changes and the potential future importance of maize diseases : a short review. J. Plant Dis. Crop Prot., 120, 49–56.
Kang, H.M., Zaitlen, N.A., Wade, C.M., Kirby, A., Heckerman, D., Daly, M.J. and Eskin, E. (2008) Efficient Control of Population Structure in Model Organism Association Mapping. , 1723, 1709–1723.
Klopfenstein, T.J., Erickson, G.E. and Berger, L.L. (2013) Maize is a critically important source of food, feed, energy and forage in the USA. F. Crop. Res., 153, 5–11. Available at: http://dx.doi.org/10.1016/j.fcr.2012.11.006. Kover, P.X., Valdar, W., Trakalo, J., Scarcelli, N., Ehrenreich, I.M., Michael, D.,
Inter-28
Cross to Fine- Map Quantitative Traits in Arabidopsis thaliana. , 5. Ladejobi, O., Elderfield, J., Gardner, K.A., Gaynor, R.C., Hickey, J., Hibberd,
J.M., Mackay, I.J. and Bentley, A.R. (2016) Maximizing the potential of multi-parental crop populations. Appl. Transl. Genomics, 11, 9–17.
Larièpe, A., Mangin, B., Jasson, S., et al. (2012) The Genetic Basis of Heterosis : Multiparental Quantitative Trait Loci Mapping Reveals Contrasted Level of Apparent Overdominance Among Traits of Agronomical Interest in Maize (Zea myas L.). Genetics, 190, 795–811.
Ledencan, T., ŠIMIĆ, D., BRKIĆ, I., JAMBROVIĆ, A. and ZDUNIĆ, Z. (2003) Resistance of Maize Inbreds and their Hybrids to Fusarium Stalk Rot. Czech J. Genet. Plant Breed, 39, 15–20.
Letunic, I. and Bork, P. (2016) Interactive tree of life ( iTOL ) v3 : an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res., 44, 242–245.
Li, Z., Luo, L.J., Mei, H.W., et al. (2001) Overdominant Epistatic Loci Are the Primary Genetic Basis of Inbreeding Depression and Heterosis in Rice . I . Biomass and Grain Yield. Genetics, 158, 1737–1753.
Lippman, Z.B. and Zamir, D. (2006) Heterosis : revisiting the magic. Trend Genet., 23.
Liu, X., Huang, M., Fan, B., Buckler, E.S. and Zhang, Z. (2016) Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome- Wide Association Studies. PLoS Genet., 12, 1–24.
Lobell, D.B., Schlenker, W. and Costa-Roberts, J. (2011) Climate Trends and Global Crop Production Since 1980. Science (80-. )., 333, 616–621.
Logrieco, A., Mulè, G., Moretti, A. and Bottalico, A. (2002) Toxigenic Fusarium species and mycotoxins associated with maize ear rot in Europe. Eur. J. Plant Pathol., 108, 597–609.
Lucic, A., Pavlovic, M., Peraica, M. and Radic, B. (1999) Toxic effects of mycotoxins in humans. , 77.
Mackay, I.J., Bansept-basler, P., Barber, T., et al. (2014) An Eight-Parent
Multiparent Advanced Generation Inter-Cross Population for Winter-Sown Wheat : Creation , Properties , and Validation. , 4, 1603–1610.
Marasas, W.F.O. (1996) Fumonisins: History, World-Wide Occurrence and Impact. In Springer, Boston, MA, pp. 1–17. Available at:
http://link.springer.com/10.1007/978-1-4899-1379-1_1 [Accessed January 12, 2018].
Marroni, F., Pinosio, S. and Morgante, M. (2014) Structural variation and genome complexity: Is dispensable really dispensable? Curr. Opin. Plant Biol., 18, 31–36. Available at: http://dx.doi.org/10.1016/j.pbi.2014.01.003.
29
Maschietto, V., Colombi, C., Pirona, R., Pea, G., Strozzi, F., Marocco, A., Rossini, L. and Lanubile, A. (2017) QTL mapping and candidate genes for resistance to Fusarium ear rot and fumonisin contamination in maize. BMC Plant Biol., 17, 20. Available at:
http://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-017-0970-1.
Matsuoka, Y., Vigouroux, Y., Goodman, M.M., Sanchez G., J., Buckler, E. and Doebley, J. (2002) A single domestication for maize shown by multilocus microsatellite genotyping. Proc. Natl. Acad. Sci., 99, 6080–6084. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.052125199.
Mcdonald, B.A. and Linde, C. (2002) The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica, 124, 163–180. McMullen, M.D., Kresovich, S., Villeda, H.S., et al. (2009) Genetic Properties of
the Maize Nested Association Mapping Population. Science (80-. )., 325, 737–740. Available at:
http://www.sciencemag.org/cgi/doi/10.1126/science.1174320.
Mesterházy, Á., Lemmens, M. and Reid, L.M. (2012) Breeding for resistance to ear rots caused by Fusarium spp. in maize - A review. Plant Breed., 131, 1– 19.
Meyer, R.C., Witucka-Wall, H., Becher, M., et al. (2012) Heterosis manifestation during early Arabidopsis seedling development is characterized by
intermediate gene expression and enhanced metabolic activity in the hybrids. Plant J., 71, 669–683.
Miedaner, T., Schulthess, A.W., Gowda, M., Reif, J.C. and Longin, C.F.H. (2017) High accuracy of predicting hybrid performance of Fusarium head blight resistance by mid-parent values in wheat. Theor. Appl. Genet., 130, 461– 470.
Mundt, C.C. (2014) Durable resistance: A key to sustainable management of patogen and pests. Infect Genet Evol., 0, 446–455.
Mundt, C.C., Cowger, C. and Garrett, K.A. (2002) Relevance of integrated disease management to resistance durability. Euphytica, 124, 245–252. Munkvold, G. (2014) Crop Management Practices to Minimize the Risk of
Mycotoxins Contamination in Temperate- Zone Maize. Mycotoxin Reduct. Grain Chain., 378.
Munkvold, G.P. (2003) Epidemiology of Fusarium diseases and their mycotoxins in maize ears. Eur. J. Plant Pathol., 109, 705–713.
Munkvold, G.P. and Desjardins, A.E. (1997) Fumonisins in Maize. , 81.
Munkvold, G.P., McGee, D.C. and Carlton, W.M. (1997) Importance of Different Pathways for Maize Kernel Infection by Fusarium moniliforme.
30
Phytopathology, 87, 209–217. Available at:
http://apsjournals.apsnet.org/doi/10.1094/PHYTO.1997.87.2.209 [Accessed August 11, 2017].
Nelson, R., Wiesner-Hanks, T., Wisser, R. and Balint-Kurti, P. (2018) Navigating complexity to breed disease-resistant crops. Nat. Rev. Genet., 19, 21–33. Available at: http://dx.doi.org/10.1038/nrg.2017.82.
Nuss, E.T. and Tanumihardjo, S.A. (2010) Maize: A paramount staple crop in the context of global nutrition. Compr. Rev. Food Sci. Food Saf., 9, 417–436. Oerke, E. (2006) Crop losses to pests. J. Agric. Sci., 144.
Paschold, A., Jia, Y., Marcon, C., et al. (2012) Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res., 22, 2445–2454.
Paschold, A., Marcon, C., Hoecker, N. and Hochholdinger, F. (2010) Molecular dissection of heterosis manifestation during early maize root development. Theor. Appl. Genet., 120, 383–388.
Pascual, L., Desplat, N., Huang, B.E., et al. (2015) Potential of a tomato MAGIC population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol. J., 13, 565–577. Pereira, G.S., Pinho, R.G. V, Pinho, E.V.R. V, et al. (2017) Selection of maize
inbred lines and gene expression for resistance to ear rot. Genet. Mol. Res., 16, gmr16039415. Available at: http://www.geneticsmr.com/sites/default/files/articles/year2017/vol16- 3/pdf/gmr-16-03-gmr.16039415.pdf%0Ahttp://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=ref erence&D=emexa&NEWS=N&AN=617261540%0Ahttp://ovidsp.ovid.com/o vidweb.cgi?T=JS&PAGE=reference&D=med8&NEW.
Peterson, B. (2018) Econometric Tools for Performance and Risk Analysis. Available at: https://github.com/braverock/PerformanceAnalytics.
Pingali, P.L. (2001) CIMMYT 1999–2000 World Maize Facts and Trends. Meeting World Maize Needs: Technological Opportunities and Priorities for the Public Sector, Mexico: CIMMYT.
Pinotti, L., Ottoboni, M., Giromini, C., Dell’Orto, V. and Cheli, F. (2016) Mycotoxin contamination in the EU feed supply chain: A focus on Cereal Byproducts. Toxins (Basel)., 8.
Piperno, D.R., Ranere, A.J., Holst, I., Iriarte, J. and Dickau, R. (2009) Starch grain and phytolith evidence for early ninth millennium B.P. maize from the Central Balsas River Valley, Mexico. Proc. Natl. Acad. Sci., 106, 5019–5024. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.0812525106. Poland, J.A., Balint-Kurti, P.J., Wisser, R.J., Pratt, R.C. and Nelson, R.J. (2009)
31
Shades of gray: the world of quantitative disease resistance. Trends Plant Sci., 14, 21–29.
Poland, J.A., Bradbury, P.J., Buckler, E.S. and Nelson, R.J. (2011) Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc. Natl. Acad. Sci., 108, 6893–6898.
Prasanna, B. (2009) Maize in the Developing World: Trends , Challenges , and Opportunities. In Agriculture and food security under global change: prospects for 2025/2050. IFPRI. pp. 26–38.
R core, T. (2013) R: A language and environment for statistical computing. R Found. Stat. Comput. Vienna, Austria. Available at: https://www.r-project.org/about.html [Accessed October 4, 2017].
Ranum, P., Peña-Rosas, J.P. and Garcia-Casal, M.N. (2014) Global maize production, utilization, and consumption. Ann. N. Y. Acad. Sci., 1312, 105– 112.
Reif, J.C., Gumpert, F.M., Fischer, S. and Melchinger, A.E. (2007) Impact of interpopulation divergence on additive and dominance variance in hybrid populations. Genetics, 176, 1931–1934.
Robertson-Hoyt, L.A., Betrán, J., Payne, G.A., White, D.G., Isakeit, T., Maragos, C.M., Molnár, T.L. and Holland, J.B. (2007) Relationships Among
Resistances to Fusarium and Aspergillus Ear Rots and Contamination by Fumonisin and Aflatoxin in Maize. Am. Phytopathol. Soc., 97, 311–317. Robertson-Hoyt, L.A., Jines, M.P., Balint-Kurti, P.J., Kleinschmidt, C.E., White,
D.G., Payne, G.A., Maragos, C.M., Molnár, T.L. and Holland, J.B. (2006) QTL mapping for fusarium ear rot and fumonisin contamination resistance in two maize populations. Crop Sci., 46, 1734–1743.
Rosenzweig, C., Iglesius, A., Yang, X.B., Epstein, P.R. and Chivian, E. (2001) Climate change and extreme weather events-Implications for food
production, plant diseases, and pests. NASA Publ., 24, 90–104. Available at: http://digitalcommons.unl.edu/nasapub/24 [Accessed October 12, 2018]. Ruiz Corral, J.A., Puga, N.D., Jesús, J. De, González, S. and Parra, J.R. (2008)
Climatic Adaptation and Ecological Descriptors of 42 Mexican Maize Races. Crop Sci., 48, 1502–1512.
Saeki, N., Kawanabe, T., Ying, H., et al. (2016) Molecular and cellular
characteristics of hybrid vigour in a commercial hybrid of Chinese cabbage. BMC Plant Biol., 1–15. Available at: http://dx.doi.org/10.1186/s12870-016-0734-3.
Schnable, P.S., Ware, D., Fulton, R.S., Stein, J.C., Wei, F., Pasternak, S. and Al., E. (2009) The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science (80-. )., 326, 1112–1115. Available at:
32
http://www.sciencemag.org/cgi/doi/10.1126/science.1178534.
Schnell, F.W. (1992) Arbitrary Gene Action in Heterosis. Genetics, 131, 461–469. Septiani, P., Lanubile, A., Stagnati, L., Busconi, M., Nelissen, H., Pè, M.E.,
Dell’Aqcua, M. and Marocco, A. (2019) Unravelling the genetic basis of Fusarium seedling rot resistance in the MAGIC maize population: novel targets for breeding. Sci. Rep.
Shen, H., He, H., Li, J., et al. (2012) Genome-Wide Analysis of DNA Methylation and Gene Expression Changes in Two Arabidopsis Ecotypes and Their Reciprocal Hybrids. Plant Cell, 24, 875–892.
Song, R. and Messing, J. (2003) Gene expression of a gene family in maize based on noncollinear haplotypes. Proc. Natl. Acad. Sci. U. S. A., 100, 9055–60. Available at:
http://www.ncbi.nlm.nih.gov/pubmed/12853580%5Cnhttp://www.pubme dcentral.nih.gov/articlerender.fcgi?artid=PMC166437.
Springer, N.M., Ying, K., Fu, Y., et al. (2009) Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet., 5.
Stagnati, L., Lanubile, A., Samayoa, L.F., Bragalanti, M., Giorni, P., Busconi, M., Holland, J.B. and Marocco, A. (2019) A Genome wide association study reveals markers and Genes Associated with Resistance to Fusarium verticillioides Infection of Seedlings in a Maize Diversity Panel. G3 Genes|Genomes|Genetics, 9, 571–579.
Stupar, R.M. and Springer, N.M. (2006) Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics, 173, 2199–2210.
Takuno, S., Terauchi, R. and Innan, H. (2012) The Power of QTL Mapping with RILs. PLoS One, 7, 1–10.
Tenaillon, M.I. and Charcosset, A. (2011) A European perspective on maize history. Comptes Rendus - Biol., 334, 221–228.
The CIMMYT Maize Program (2004) Maize diseases: a guide for field identification,.
Thomma, B.P.H.J., Nu, T. and Joosten, M.H.A.J. (2011) Of PAMPs and Effectors : The Blurred PTI-ETI Dichotomy. Plant Cell, 23, 4–15.
Tian, F., Bradbury, P.J., Brown, P.J., et al. (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet., 6–11. Available at:
http://dx.doi.org/10.1038/ng.746.
Troyer, A.F. (2009) Development of Hybrid Corn and the Seed Corn Industry. In J. L. Bennetzen and S. Hake, eds. Maize Handbook-Volume II: Genetics and
33
Genomics. Springer Science + Bussuniess Media LLC, pp. 87–114.
Unterseer, S., Bauer, E., Haberer, G., et al. (2014) A powerful tool for genome analysis in maize : development and evaluation of the high density 600 k SNP genotyping array. , 1–15.
USDA Food Composition Databases Show Nutrients List. Available at:
https://ndb.nal.usda.gov/ndb/nutrients/index [Accessed August 14, 2018]. Vanderplank, J.E. (1968) Disease resistance in plants, New York: Academic Press. Vaughan, M., Backhouse, D. and Ponte, E.. Del (2016) Climate change impacts
on the ecology of Fusarium graminearum species complex and
susceptibility of wheat to Fusarium head blight : a review. Worl Mycotoxin J., 9, 685–700.
Wang, H., Nussbaum-wagler, T., Li, B., Zhao, Q., Vigouroux, Y., Faller, M., Bomblies-yant, K., Lukens, L. and Doebley, J. (2005) The origin of the naked grains of maize. Nature, 436, 714–719.
Wang, H., Xu, C., Liu, X., et al. (2017) Development of a multiple-hybrid
population for genome-wide association studies: Theoretical consideration and genetic mapping of flowering traits in maize. Sci. Rep., 7, 1–16.
Available at: http://dx.doi.org/10.1038/srep40239.
Wickham, H. (2016) ggplot2: Elegant Graphics for Data Analysis. Available at: http://ggplot2.org.
Winter, P. and Kahl, G. (1995) Molecular marker technologies for plant improvement. World J. Microbiol. Biotechnol., 11, 438–448.
Wisser, R.J., Balint-Kurti, P.J. and Nelson, R.J. (2006) The genetic architecture of disease resistance in maize: a synthesis of published studies.
Phytopathology, 96, 120–129.
Wisser, R.J., Kolkman, J.M., Patzoldt, M.E., Holland, J.B., Yu, J., Krakowsky, M., Nelson, R.J. and Balint-Kurti, P.J. (2011) Multivariate analysis of maize disease resistances suggests a pleiotropic genetic basis and implicates a GST gene. Proc. Natl. Acad. Sci., 108, 7339–7344. Available at:
http://www.pnas.org/cgi/doi/10.1073/pnas.1011739108.
Wu, F., Bhatnagar, D., Bui-Klimke, T., Hellmich, R.L., Munkvold, G., Paul, P., Payne, G. and Takle, E. (2011) Climate change impacts on mycotoxin risks in US maize. Worl Mycotoxin J., 4, 79–93.
Xiao, J., Li, J., Yuan, L. and Tanksley, S.D. (1995) Dominance Is the Major Genetic Basis of Heterosis in Rice as Revealed by QTL analysis Using Molecular Markers. Genetics, 140, 745–754.
Yang, L., Li, B., Zheng, X., Li, J., Yang, M., Dong, X., He, G., An, C. and Deng, X.W. (2015) Salicylic acid biosynthesis is enhanced and contributes to increased biotrophic pathogen resistance in Arabidopsis hybrids. Nat.
34
Commun., 6, 1–11. Available at: http://dx.doi.org/10.1038/ncomms8309. Yang, Q., Balint-kurti, P. and Xu, M. (2017) Quantitative Disease Resistance :
Dissection and Adoption in Maize. Mol. Plant, 10, 402–413. Available at: http://dx.doi.org/10.1016/j.molp.2017.02.004.
Yin, L., Zhang, H., Zhang, Z., Li, X., Yuan, X., Zhao, S. and Liu, X. (2018) A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool For Genome-Wide Association Study. Available at:
https://github.com/XiaoleiLiuBio/rMVP.
Yu, J., Holland, J.B., Mcmullen, M.D. and Buckler, E.S. (2008) Genetic Design and Statistical Power of Nested Association Mapping in Maize. Genetics, 551, 539–551.
Zila, C.T., Ogut, F., Romay, M.C., Gardner, C.A., Buckler, E.S. and Holland, J.B. (2014) Genome-wide association study of Fusarium ear rot disease in the U . S . A . maize inbred line collection. , 1–15.
35
CHAPTER 2
GENETIC ARCHITECHTURE OF FUSARIUM SEEDLING
ROT RESISTANCE IN RECOMBINANT INBRED LINE OF
36
This chapter is presented in the form of the manuscript entitled “Unravelling the genetic basis of Fusarium seedling rot resistance in the MAGIC maize population: novel targets for breeding”. The manuscript has been accepted for publication in the scientific journal Scientific Report (Accepted: 26 March 2019). In this research, the MAGIC maize population (Dell’Acqua et al., DOI:10.1186/s13059-015-0716-z) has been employed. In the experiment here described we combined advanced genomics and genetic material as key feature of MAGIC maize platform to identified causal variant of Fusarium seedling rot (FSR) combined with a fast-screening phenotyping method at seedling level. We performed high definition QTL mapping and association mapping to identified quantitative trait loci associated to FSR resistance, seedling length and seedling weight. Finally, we leveraged full genome sequencing and RNA sequencing data produced on the founders of the MAGIC population to identify candidate genes. This work may represent a methodological reference to those wanting to perform QTL mapping for resistance in maize and other cereals using a cheap, reliable method.
37
TITLE
Unravelling the genetic basis of Fusarium seedling rot resistance in the MAGIC maize population: novel targets for breeding
AUTHORS
Popi Septiani1, Alessandra Lanubile2, Lorenzo Stagnati2, Matteo Busconi2, Hilde
Nelissen3,4, Mario Enrico Pè1, Matteo Dell’Acqua1, Adriano Marocco2*
1Institute of Life Sciences, Scuola Superiore Sant’Anna, Pisa, 56127, Italy
2Department of Sustainable Crop Production, Università Cattolica del Sacro
Cuore, Piacenza, 29122, Italy
3Department of Plant Biotechnology and Bioinformatics, Ghent University,
Ghent, B-9052, Belgium
4VIB Centre for Plant Systems Biology, Ghent, B-9052, Belgium *Corresponding author: [email protected]
38
ABSTRACT
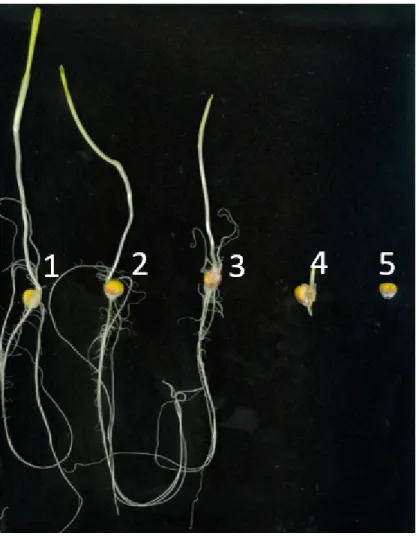
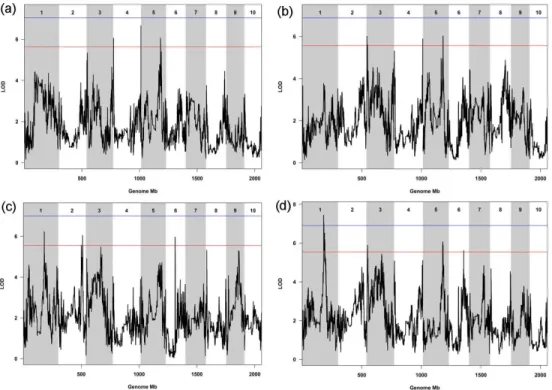
Fungal infection by Fusarium verticillioides is cause of prevalent maize disease leading to substantial reductions in yield and grain quality worldwide. Maize resistance to the fungus may occur at different developmental stages, from seedling to maturity. The breeding of resistant maize genotypes may take advantage of the identification of quantitative trait loci (QTL) responsible for disease resistance already commenced at seedling level. The Multi-parent Advance Generation Intercross (MAGIC) population was used to conduct high-definition QTL mapping for Fusarium seedling rot (FSR) resistance using rolled towel assay. Infection severity level, seedling weight and length were measured on 401 MAGIC maize recombinant inbred lines (RILs). QTL mapping was performed on reconstructed RIL haplotypes. One-fifth of the MAGIC RILs were resistant to FSR and 10 QTL were identified. For FSR, two QTL were detected at 2.8 Mb and 241.8 Mb on chromosome 4, and one QTL at 169.6 Mb on chromosome 5. Transcriptomic and sequencing information generated on the MAGIC founder lines was used to guide the identification of eight candidate genes within the identified FSR QTL. We conclude that the rolled towel assay applied to the MAGIC maize population provides a fast and cost-effective method to identify QTL and candidate genes for early resistance to F. verticillioides in maize. INTRODUCTION
Maize (Zea mays L.) is a key crop for food, feed, and industrial products. It is the cereal species with the highest grain production worldwide, with more than one billion tons harvested each year1. Maize production, however, is menaced by
numerous pathogens that affect both the quantity and the quality of the grain produced2–4. The occurrence of the diseases varies by year and depends on
39
cropping practices5. In recent years, climate change is causing disease outbreaks
even in geographic regions in which they were infrequent in the past6.
Fusarium verticillioides (Sacc.) Nirenberg is a predominant endophyte and pathogen of maize causing substantial yield losses and reduction of grain quality. Maize can be infected by the fungus at all growth stages, from the early vegetative phases to maturity. The fungus can be transmitted through infected kernels and cause systemic infection that eventually contribute to the development of seedling diseases7 including seedling rot8,9, root rot and stalk rot7. Indeed, in
seedling rot, reduction in seed emergence and seedling growth may be observed when seedlings are inoculated with F. verticillioides strains producing fumonisin B110,11. The amount of damage caused by this disease depends on the extent of
rotting: if rot is extensive, the embryo may die and the seed will not germinate. Even for seeds that are not severely rotted, germination in unfavorable environments will be slow or absent due to presence of the internal fungus2. In
many plant-pathogen interactions, the expression of the resistance to pathogens depends on the stage of development at which the plant is infected. Plants are generally more susceptible to diseases in early stages rather than in later stages12.
Plants already resistant to pathogens at early stages may be able to increase their ability to control infection at later stages13. Conversely, genotypes which are
susceptible at early stages may either acquire disease resistance or remain susceptible over time 13,14.
The genetic basis of F. verticillioides resistance in maize is not yet fully understood. Several studies reported genetic sources of resistance to F. verticillioides at maturity stage associated to Fusarium ear rot (FER)15–19.
However, complete resistance to FER has not yet been achieved 20–22. Resistance