1
1. Introduzione
1.1. Cenni sulla fisiologia e sulla funzione del mitocondrio
I mitocondri sono organelli cellulari presenti nel citoplasma di tutte le cellule animali a metabolismo aerobico e nelle cellule eucariote vegetali.
Questi organelli sono considerati la centrale energetica della cellula, poiché in grado di convertire l’energia in forme utili a promuovere le reazioni cellulari. La loro funzione primaria è, infatti, quella di estrarre energia chimica dai substrati energetici e di immagazzinarla in una molecola facilmente utilizzabile: l’adenosina trifosfato (ATP).
Tuttavia questi organelli sono strettamente coinvolti in una serie di processi essenziali per la cellula, quali il metabolismo degli steroidi, la biosintesi degli amminoacidi, l’ossidazione degli acidi grassi e l’apoptosi (Morán et al., 2012). La loro forma può variare da granulare a bastoncellare in relazione ai differenti momenti funzionali della cellula.
Anche le dimensioni sono molto variabili, ma in genere la lunghezza è compresa tra 1 e 6 µm mentre il diametro varia tra 0.5 e 1 µm.
Questi possono variare , infatti, considerevolmente come forma, numero e localizzazione a seconda della funzione della cellula o del tessuto, ma anche all’interno della stessa cellula in relazione al suo sviluppo, al ciclo cellulare ed in relazione a stimoli esterni (Lehninger et al., 2006).
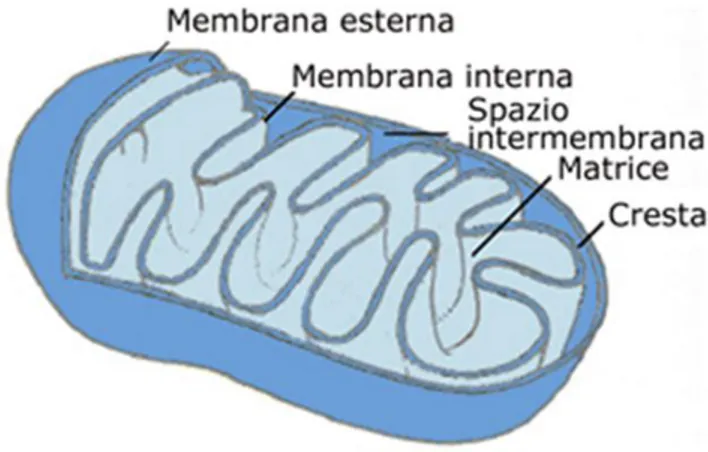
I mitocondri presentano una membrana interna ed una esterna, che circoscrivono due spazi ben differenziati (Fig.1).
La membrana esterna, che circonda completamente l’organello, è liscia ed è impermeabile alle molecole più grandi di 5000 Da e delimita lo spazio intermembrana, una regione che gioca un ruolo importante nella produzione mitocondriale di ATP (Silverthon, Johnson., 2005).
2
Essa è caratterizzata dalla presenza di molte copie di una proteina di trasporto chiamata porina, la quale forma grossi canali acquosi attraverso il doppio strato lipidico. Tale membrana, quindi, risulta permeabile a tutte le molecole di dimensioni inferiori a 5 kDa, comprese le piccole proteine. Queste molecole possono giungere allo spazio intermembrana, ma la maggior parte di esse non possono attraversare la membrana interna impermeabile. Così mentre lo spazio intermembrana risulta chimicamente equivalente al citosol, la matrice invece contiene una serie selezionata di piccole molecole.
La membrana interna, invece, è altamente specializzata, in quanto contiene un’alta proporzione del fosfolipide cardiolipina, il quale presenta quattro acidi grassi e contribuisce a rendere la membrana particolarmente impermeabile agli ioni.
Essa contiene anche una varietà di proteine di trasporto che permettono il passaggio selettivo di piccole molecole che sono metabolizzate o richieste dagli enzimi mitocondriali presenti nello spazio interno.
La membrana interna, inoltre, forma numerose introflessioni chiamate creste mitocondriali, che aumentano notevolmente la sua area superficiale. Su queste creste sono ancorate le proteine che costituiscono la catena di trasporto degli elettroni, l’apparato di fosforilazione ed i trasportatori di membrana.
Lo spazio intermembrana, localizzato tra le due membrane mitocondriali, contiene un distinto gruppo di proteine, che includono il trasportatore mobile di elettroni, ossia il citocromo c (Lesnefsky e Hoppel, 2006).
Il compartimento delimitato dalla sola membrana interna è invece chiamato matrice mitocondriale ed ha consistenza gelatinosa a causa della elevata concentrazione di enzimi e di intermedi chimici coinvolti nel metabolismo energetico della cellula. Questa contiene, inoltre, numerosi ribosomi e molecole di DNA circolare a doppio filamento.
I mitocondri, infatti, possiedono un proprio materiale genetico che viene trascritto e tradotto all’interno del mitocondrio stesso, nella matrice. Le proteine mitocondriali
3
vengono codificate in parte da geni nucleari e in parte da geni del DNA mitocondriale.
Fig. 1 Rappresentazione schematica della struttura di un mitocondrio, in cui sono evidenziate la membrana esterna
e la membrana interna ripiegata in creste.
1.1.1. La respirazione cellulare
Il ruolo principale del mitocondrio è quello di produrre energia sfruttabile dalla cellula partendo dai prodotti della glicolisi: piruvato e NADH.
In presenza di ossigeno, la produzione cellulare di energia si articola in tre fasi principali. Nella prima fase, le molecole delle sostanze nutrienti organiche (glucosio, ac. grassi ed alcuni amminoacidi) sono ossidate fino a formare frammenti a due atomi di carbonio, e cioè il gruppo acetilico dell’acetil-coenzima A (acetil-CoA). Nella seconda fase, questi gruppi acetilici sono immessi nel ciclo degli acidi tricarbossilici o ciclo di Krebs, in cui vengono ossidati enzimaticamente a CO2. L’energia rilasciata dall’ossidazione viene conservata riducendo contemporaneamente i trasportatori di elettroni NAD+ e FAD rispettivamente a NADH e FADH2. Nella terza fase, la respirazione, i cofattori ridotti sono a loro volta ossidati liberando protoni ed elettroni. Gli elettroni sono trasferiti lungo una catena di molecole di trasportatori di elettroni, nota come catena respiratoria,
4
all’ossigeno, che si riduce formando acqua. Durante questo processo di trasferimento degli elettroni viene rilasciata molta energia, che è poi convertita in ATP in un processo chiamato fosforilazione ossidativa (Lesnefsky e Hoppel, 2006) (Fig. 2).
Fig.2 Rappresentazione schematica della respirazione cellulare.
1.1.1.1 Il flusso elettronico mitocondriale
I trasportatori di elettroni della catena respiratoria sono organizzati in complessi sovramolecolari. I complessi I e II trasferiscono elettroni all’ubichinone (detto anche coenzima Q) da due donatori diversi: il NADH (complesso I) ed il succinato (complesso II). Il complesso III trasporta gli elettroni dall’ubichinone al citocromo c ed il complesso IV completa la sequenza trasferendo elettroni dal citocromo c all’ossigeno (Fig. 3).
5
Fig.3 Rappresentazione schematica della catena di trasporto degli elettroni e dell’ATP-sintetasi. Il flusso degli elettroni è accompagnato da una traslocazione di protoni attraverso la membrana mitocondriale, producendo un gradiente chimico ed elettrico. La forza motrice protonica, che spinge i protoni verso la matrice, fornisce l’energia per la sintesi di ATP, catalizzata dal complesso ATP-sintetasi.
Complesso I: detto anche NADH-ubichinone ossido-reduttasi, è un enzima
contenente più di 42 catene polipeptidiche, inclusa la flavoproteina contenente il cofattore flavinico FMN e sei centri ferro-zolfo.
Tale complesso catalizza due processi accoppiati e simultanei:
1- Il trasferimento esoergonico all’ubichinone di uno ione idruro dal NADH e di un protone dal solvente acquoso della matrice
NADH + H+ + Q → NAD+ + QH2
2- Il trasferimento endoergonico di quattro protoni dalla matrice (m) allo spazio intermembrana (si).
Il complesso I è quindi una pompa protonica, guidata dall’energia derivante dal trasferimento degli elettroni. L’intera reazione può essere scritta:
6
L’ubichinolo, la forma completamente ridotta QH2, diffonde all’interno della
membrana mitocondriale interna dal Complesso I al Complesso III, dove viene ossidato a Q, in un processo che coinvolge anche lo spostamento verso l’esterno di protoni H+.
Complesso II: detto anche succinato-deidrogenasi, contiene al suo interno due tipi di
gruppi prostetici ed almeno quattro proteine diverse. Una proteina ha legato covalentemente un FAD ed un centro Fe-S con quattro atomi di ferro; è presente anche una seconda proteina Fe-S. Si pensa che gli elettroni passino dal succinato al FAD e poi attraverso i centri Fe-S all’ubichinone (Fig.4)
Fig.4 Struttura del Complesso II.
L’ubichinone ridotto viene poi riossidato dal Complesso III, successivo componente della catena mitocondriale di trasporto degli elettroni.
Complesso III: detto anche ubichinone:citocromo c ossidoreduttasi, accoppia il
trasferimento degli elettroni dall’ubichinolo (QH2) al citocromo c con il trasporto
vettoriale protonico dalla matrice allo spazio intermembrana. L’equazione netta per le reazioni redox di questo ciclo dell’ubichinone è:
QH2 + 2Cit c1(ossidato) + 2H+m → Q + 2Cit c1(ridotto) + 4H+si
Il citocromo c è una proteina solubile dello spazio intermembrana. Quando il suo gruppo eme accetta un elettrone dal Complesso III, il citocromo c si sposta verso il
7
Complesso IV per donare l’elettrone ad un centro rameico binucleare di questo enzima.
Complesso IV: detto anche citocromo ossidasi, nella tappa finale della catena
respiratoria, trasporta gli elettroni dal citocromo c all’ossigeno molecolare, riducendolo ad H2O. Ogni volta che quattro elettroni attraversano questo complesso, l’enzima utilizza quattro ioni H+
dalla matrice per convertire l’O2 in due molecole di
H2O. Il complesso utilizza anche l’energia di questa reazione redox per pompare un protone nello spazio intermembrana, per ogni protone che lo attraversa, aumentando ulteriormente il potenziale elettrochimico prodotto dal trasporto protonico attraverso il Complesso I ed il Complesso III.
L’intera reazione catalizzata dal Complesso IV è:
4Cit c(ridotto) + 8H+m + O2 → 4Cit c(ossidato) + 4H+si + 2H2O
1.1.1.2. La sintesi di ATP
Il flusso di elettroni attraverso i complessi I, III e IV è accompagnato da una traslocazione di protoni dalla matrice allo spazio intermembrana, come descritto dalla teoria chemiosmotica di Mitchell, 1961. In accordo con tale modello, durante il trasporto degli elettroni ad alta energia dagli idrogeni del NADH e del FADH2 lungo la catena respiratoria fino alla membrana mitocondriale interna, l’energia rilasciata nel passaggio da un trasportatore al successivo è usata per pompare protoni (H+) attraverso la membrana interna dalla matrice mitocondriale allo spazio intermembrana.
Il trasferimento di elettroni comporta, infatti, un cambiamento conformazionale delle proteine trasportatrici che consentono il trasferimento di protoni dalla matrice allo spazio intermembranale contro gradiente di concentrazione, generando un potenziale elettrochimico, negativo all’interno. I protoni in seguito rientrano nella matrice per diffusione facilitata attraverso un canale protonico associato all’enzima ATP-sintetasi, o Complesso V, il quale sfrutta l’energia così ottenuta per
8
aggiungere un gruppo fosfato ad una molecola di ADP (adenosina difosfato), al fine di ottenere ATP (Voet et al., 2007).
ADP + Pi + nH+si → ATP + H2O + nH+m
L’ATP-sintetasi si comporterebbe , dunque, come una permeasi protonica che tende a riequilibrare il gradiente elettrochimico dei protoni fra i due lati della membrana, trasducendo così l’energia del gradiente in energia conformazionale e, quindi, in energia di legame chimico dell’ATP. L’ATP prodotto dalla fosforilazione all’interno dei mitocondri esce dal mitocondrio grazie all’enzima traslocasi dei nucleotidi adeninici (ANT) inserito nella membrana mitocondriale interna, che catalizza lo scambio con un ADP citosolico entrante nel mitocondrio (Kim et al.,
1988).
Il trasferimento dei protoni, attraverso la membrana interna, dallo spazio della matrice allo spazio intermembrana ha come conseguenza la formazione di due tipi di gradienti:
1. un gradiente di pH (∆pH) attraverso la membrana mitocondriale interna, con il pH della matrice più alto (basico) di quello dello spazio intermembrana;
2. un gradiente di voltaggio (∆V) (o potenziale di membrana, ∆Ψm) derivante
principalmente dal flusso netto di ioni positivi verso l’esterno, per cui la membrana interna risulterà carica positivamente all’esterno e negativamente all’interno.
Queste due grandezze (∆pH e ∆Ѱm) determinano la forza motrice dei protoni, il
cosiddetto gradiente protonico elettrochimico (∆µH+). Tale gradiente elettrochimico
di H+ rappresenta quindi la driving-force necessaria per la sintesi di ATP. La misurazione in maniera continuativa del ∆Ψm risulta essere, quindi, un parametro di
fondamentale importanza nella valutazione della funzionalità mitocondriale.
Nel processo di fosforilazione della catena respiratoria, l’ADP e il fosfato sono dei reagenti necessari per il trasporto degli elettroni dal NADH all’ossigeno. Infatti, il trasporto degli elettroni procede alla massima velocità nei mitocondri isolati soltanto quando fosfato e ADP sono presenti nel mezzo in cui sono sospesi in
9
quantità saturanti. In assenza di ADP, la velocità della respirazione è molto bassa e non avviene alcuna fosforilazione poiché non vi è accettore per il fosfato, e naturalmente, non vi è sintesi di ATP. Questa condizione, nota come Fase 2 della respirazione, rappresenta lo stato di riposo della respirazione stessa. Se ad una sospensione di mitocondri nella Fase 2, in presenza degli opportuni substrati (succinato o glutammato + piruvato), si aggiunge una quantità saturante di ADP, il consumo di ossigeno aumenta bruscamente e contemporaneamente l’ADP aggiunto viene fosforilato ad ATP. Questa è chiamata Fase 3 o respirazione attiva. Quando tutto l’ADP aggiunto è stato fosforilato, la velocità di consumo dell’ossigeno ritorna allo stato di riposo della respirazione, denominato Fase 4.
Questo fenomeno, in cui la velocità di trasporto degli elettroni è controllata dalla concentrazione di ADP, è chiamato controllo da parte dell’accettore o controllo respiratorio. I mitocondri, infatti, hanno un’affinità molto elevata per l’ADP e continuano a fosforilarlo, purchè siano presenti tutti gli altri componenti necessari, finchè la concentrazione di ADP non diventa molto bassa.
1.1.1.3. Conduttanza protonica mitocondriale
La sintesi di ATP e il trasporto degli elettroni lungo la catena respiratoria appaiono, dunque, due processi strettamente accoppiati. Tuttavia, l’accoppiamento tra l’ossidazione dei substrati e la sintesi dell’ATP non è pari al 100%. Infatti, una parte dell’energia è persa sotto forma di calore mediante delle reazioni che consentono di dissipare il gradiente protonico senza portare alla consueta sintesi di ATP. Si verifica, quindi, un vero e proprio disaccoppiamento della fosforilazione ossidativa. Tra i fattori che influenzano il grado di accoppiamento termodinamico, la permeabilità della membrana mitocondriale interna a ioni H+ svolge un ruolo molto importante.
Infatti, il disaccoppiamento basale è dovuto ad una via di perdita protonica passiva, determinato dalle caratteristiche intrinseche della membrana mitocondriale interna, definito anche conduttanza protonica basale (Nicholls, 1974).
10
Questa non risulta essere un artefatto dell’isolamento dei mitocondri, poiché è stato dimostrato che essa è presente anche in mitocondri di cellule ed organi intatti, come il fegato, i timociti, i linfociti, il muscolo scheletrico ed il cuore (Rolfe e Brand,
1996; Challoner, 1968).
1.1.1.4. Conduttanza protonica inducibile
Vi sono, inoltre, alcune condizioni o reagenti in grado di disaccoppiare il trasporto degli elettroni lungo la catena respiratoria dalla sintesi di ATP (disaccoppiamento inducibile). Quando i mitocondri intatti vengono rotti con un trattamento con detergenti o con sistemi meccanici, i frammenti di membrana sono ancora capaci di trasferire gli elettroni dal NADH o dal succinato all’ossigeno, ma la sintesi di ATP non è più accoppiata a questa respirazione.
Alcuni composti chimici sono in grado di determinare un disaccoppiamento anche senza la distruzione della struttura del mitocondrio. Tra i disaccoppianti chimici sono compresi il 2,4-dinitrofenolo (DNP) ed il carbonilcianuro-3-clorofenilidrazone (CCCP), che agiscono da protonofori. Questi disaccoppianti sono acidi con proprietà idrofobiche , caratteristica che permette loro di diffondere spontaneamente attraverso la membrana mitocondriale. Dopo essere entrati nella matrice in forma protonata, possono rilasciare un protone, dissipando così il gradiente protonico
(Lehninger, 2006).
1.1.1.5. Produzione di ROS
Le ROS (specie reattive dell’ossigeno) sono generate in molti processi all’interno della cellula. Proteine di membrana, enzimi citosolici (come le ciclossigenasi), metabolismo dei lipidi nei perossisomi coinvolgono tutti una piccola produzione di specie radicaliche , ma la grande maggioranza di ROS (stimata intorno al 90%) è prodotta dai mitocondri durante la fosforilazione ossidativa (Miquel et al., 1980;
11
Fig.5 Schema riassuntivo della fosforilazione ossidativa e della produzione di ROS.
Durante questo processo il NADH o il FADH2 vengono utilizzati per generare un
gradiente protonico tra la matrice e lo spazio intermembrana. Normalmente gli elettroni generati dal NADH passano attraverso la catena respiratoria e vengono utilizzati per produrre acqua e per trasferire protoni nello spazio intermembrana generando un potenziale di membrana. La produzione di ROS avviene quando nel mitocondrio sussiste un eccesso di elettroni, che causa la riduzione permanente dei trasportatori; questi elettroni possono, quindi, reagire con una molecola di ossigeno per dare l’anione superossido, O2
•-. Ciò accade , ad esempio, quando si assumono troppe calorie oppure si riduce l’attività fisica.
Le osservazioni che, nei mitocondri isolati, gli elettroni possano fuoriuscire dalla catena respiratoria sotto forma di radicali superossidi (O2•- ) ha portato un gruppo di ricercatori a sostenere che la formazione di O2•- possa essere un sottoprodotto
obbligatorio della respirazione (Staniek et Nohl, 2000).
Per far fronte a questi squilibri, il mitocondrio è dotato di sistemi di difesa enzimatici. L’ O2•- può essere convertito in perossido di idrogeno (H2O2)
dall’enzima superossido dismutasi manganese-dipendente (localizzato nella matrice) o dall’enzima superossido dismutasi rame/zolfo-dipendente (localizzato nel citoplasma e nello spazio intermembrana). H2O2 è più stabile di O2•- e può
12
diffondere attraverso le membrane al di fuori del mitocondrio, fino al nucleo, o può essere inattivata dall’enzima glutatione perossidasi o dall’enzima catalasi. Tuttavia H2O2, in presenza di metalli di transizione ridotti, può anche essere convertita nella specie altamente reattiva radicale idrossilico (•OH).
Dei quattro complessi enzimatici che compongono la catena di trasporto degli elettroni, i complessi I e III sono attualmente considerati come i principali responsabili della produzione di superossido nei mitocondri delle cellule normali
(Selivanov VA et al., 2011).
Essendo, quindi, i mitocondri la principale fonte di ROS nella cellula, risultano essere anche i target maggiormente colpiti dallo stress ossidativo cellulare. I deficit di energia cellulare causati da declini nella funzione mitocondriale possono indebolire le normali attività cellulari e compromettere l’abilità delle cellule ad adattarsi a vari stress fisiologici.
1.1.2. I canali del potassio mitocondriali
Canali del potassio simili a quelli presenti nella membrana plasmatica di varie cellule sono stati trovati anche nella membrana interna mitocondriale (Fig.6)(Szewczyk et al., 2009) e sono:
- canali del potassio ATP-dipendenti (mitoKATP) (Inoue et al., 1991);
- canali del potassio Ca2+-dipendenti a larga conduttanza (mitoBKCa) (Siemen
et al., 1999);
- canali del potassio Ca2+-dipendenti a conduttanza intermedia (mitoIKCa) (De Marchi et al., 2009);
- canali del potassio voltaggio-dipendenti (mitoKv1.3) (Szabo et al., 2005); - canali del potassio twin-pore (mitoTASK-3) (Rusznak et al., 2008).
13
Fig.6 Canali del potassio mitocondriali
Questi canali sono coinvolti nella regolazione del volume mitocondriale (Halestrap,
1994; Bednarkzyk et al., 2008a), del potenziale di membrana (Debska et al., 2001) ,
del pH(Bednarczyk et al., 2008b) e dell’apoptosi (Szabo et al., 2008).
1.1.2.1 Canali mitoK
ATPI canali del potassio sensibili all’ATP (KATP) furono descritti per la prima volta da
Noma nel 1983 in esperimenti effettuati su cellule cardiache di cavia (Noma, 1983). In seguito questi canali furono trovati anche in altri tessuti, tra i quali le cellule β del pancreas, alcuni tipi di neuroni, cellule dei tubuli renali e nei muscoli (Bajgar et al.,
2001; Brochiero et al., 2002; Mironova et al., 1997).
Sempre nel 1983 venne evidenziato, sulla membrana interna del mitocondrio, la presenza del canale del potassio dipendente da ATP, chiamato in seguito mitoKATP (Noma , 1983).
Più recentemente, studi biochimici ed elettrofisiologici hanno dimostrato la presenza di questo canale a livello della membrana mitocondriale interna di vari tipi di cellule. Il canale mitoKATP è stato descritto, infatti, nei mitocondri di fegato
(Inoue et al., 1991), cuore (Paucek et al., 1992), cervello (Bajgar et al., 2001; Debska et al., 2001; Kulawiak e Bednarczyk, 2005), rene (Cancherini et al., 2003) ,
14
Nonostante i tentativi di vari gruppi di ricerca, l’identità molecolare di questo canale non è ancora ben chiara. Diverse osservazioni riguardanti il suo profilo farmacologico suggeriscono che il canale mitoKATP appartiene alla famiglia dei canali del potassio inward rectifier, Kir6.x (Suzuki et al., 1997; Zhou et al., 1999). Infatti, recenti studi suggeriscono che il canale mitoKATP cardiaco possa essere costituito dalle subunità inward rectifying Kir6.1 e Kir6.2 insieme con la subunità regolatoria recettore per la sulfonilurea SUR2 , mentre la subunità SUR1 sembra essere esclusa (Von Cuong D. et al., 2005). Il canale cardiaco sarcKATP , invece, presente nella membrana sarcolemmale, è un complesso ottamerico composto da quattro subunità Kir6.2 e quattro subunità SUR2A (Yokoshiki H. et al., 1998).
Un’ipotesi alternativa sulla natura molecolare di questi canali immagina un supercomplesso comprendente la succinato deidrogenasi, il complesso V (ATP-sintasi), il translocatore nucleotide adenina (ANT), il carrier del fosfato(piC) e la proteina 1 ATP-binding cassette (ABC1)(Ardehali et al., 2004). L’evidenza farmacologica supporta l’idea che il complesso II della catena respiratoria (succinato deidrogenasi) possa essere una componente regolatoria dei canali mitoKATP e ANT è stato proposto come mediatore del flusso del potassio, indotto dall’apertura dei canali mitoKATP , alla matrice mitocondriale (Kopustinskiene et al., 2003).
Il canale KATP miocardico risponde altamente alle condizioni metaboliche: viene inibito dall’ATP intracellulare e attivato dai nucleosidi difosfato intracellulari. In condizioni normossiche, i canali KATP sono preferenzialmente in uno stato inattivato, mentre in condizioni di stress cellulare metabolico (quali l’ischemia), vengono attivati dalla riduzione della concentrazione di ATP intracellulare e/o dall’accumulo di metaboliti ischemici (O’Rourke et al., 1994; Fujita e Kurachi ,
2000).
L’attivazione del canale sarcKATP , dovuta all’ipossia, ischemia e ad agenti
farmacologici, accelera la ripolarizzazione della membrana dei miocardiociti, riduce la durata del potenziale d’azione (APD) e previene l’inversione dello scambiatore Na+/K+ , con la conseguente inibizione dell’entrata di Ca2+ nella cellula e suo
15
sovraccarico (Noma , 1983). Questi eventi possono spiegare l’aumentata resistenza contro il danno ischemico a causa dell’attivazione dei canali sarcKATP; comunque,
numerosi e recenti studi attribuiscono all’attivazione del canale mitoKATP un ruolo
preminente nella protezione cardiaca anti-ischemica (Mubagwa e Flameng , 2001;
Sato et al., 2000a; Sato et al., 2000b). Questa ipotesi è rafforzata dall’osservazione
che alcuni attivatori KATP , quali bimakalim e cromakalim, sono abili nel produrre gli effetti cardioprotettivi anche a dosi prive di alcuna influenza sull’ADP, cioè senza interessare il potenziale di membrana sarcolemmale. Questa evidenza suggerisce indirettamente l’esistenza di un sito intracellulare , indipendente dai canali sarcolemmali, responsabile dell’azione cardioprotettiva (Yao e Gross , 1994;
Grover et al., 1995a; Grover et al., 1996; Grover et al., 1995b).
L’osservazione che, nei mitocondri di cuore bovino, l’attivatore KATP diazossido
induceva l’apertura del canale mitoKATP a concentrazioni più basse di quelle necessarie per l’apertura del canale sarcKATP , rappresentava il primo diretto
suggerimento per quanto riguarda il ruolo dei canali mitoKATP.
Nei cuori isolati di ratto, diazossido e cromakalim, a basse concentrazioni che non influenzano i canali sarcKATP , producevano un effetto cardioprotettivo . Questi effetti del diazossido e cromakalim venivano aboliti dal bloccante selettivo dei canali mitoKATP , il 5-idrossidecanoato(5-HD), dimostrando così il ruolo chiave di questo tipo di canale (Garild et al., 1997). Al contrario, P-1075, un pinacidil analogo che non aumentava l’autoflorescenza della flavoproteina (un indice dell’apertura dei mitoKATP) ma che provocava una corrente sarcKATP inibita dal
bloccante sarcKATP-selettivo HMR 1098, non produceva alcun effetto
cardioprotettivo (Sato et al., 2000b).
Altri induttori dell’attività dei mitoKATP sono le ROS (Fornazari et al., 2008; Grigoriev et al., 1999), GTP e GDP (Mironova et al., 1999; Paucek et al., 1996),
UDP (Mironova et al., 2004), il pH alcalino (Bednarczyk et al., 2008) e il derivato benzopiranico spirociclico denominato temporaneamente composto A (Calderone
et al., 2010). Anche il Levosimedan apre i canali mitoKATP (Kopustinskiene et al., 2001). D’altra parte, l’NO (Sasaki et al., 2000), il chinino (Bednarczyk et al.,
16
2004), l’ acido 5-idrossidecanoico (5-HD)(Jabůrek et al., 1998), la glibenclamide (Inoue et al., 1991) e l’acilCoA a lunga catena (Paucek et al., 1996) inibiscono i
mitoKATP.
Inoltre, l’attivazione del canale mitoKATP è stato proposto essere il target finale del
“signalosoma cardioprotettivo”, identificato da Casey e colleghi (Quinlan et al.,
2009).
Tecniche del doppio strato lipidico planare e di patch-clamp sono state applicate con successo per lo studio delle proprietà del singolo canale mitoKATP (Zhang et al., 2001; Nakae et al., 2003; Bednarczyk et al., 2004). Gli studi di patch-clamp hanno
mostrato che il canale mitoKATP umano è modulato dal calcio e dall’ossido nitrico
(Dahlem et al., 2004). Gli studi sul doppio strato lipidico planare hanno mostrato
che lo stress ossidativo provoca l’attivazione del canale mitoKATP , inibito dall’acido
5-idrossidecanoico (5-HD) o da un composto sulfidril alchilante, l’N-etilmaleimide
(Zhang et al., 2001). Inoltre, l’anestetico isoflurano è stato visto attivare
direttamente il canale mitoKATP (Nakae et al., 2003).
Recentemente è stata descritta la regolazione dei mitoKATP cardiaci attraverso la chinina, gli ioni magnesio, i protoni e i nucleotidi adeninici (Tab.1)(Bednarczyk et
al., 2004; 2005; 2008). Inoltre, il canale mitoKATP è regolato probabilmente da
17
1.1.2.2. Canali mitoBK
Cae mitoIK
CaUna delle prime relazioni sui canali del potassio calcio-dipendenti proviene da studi effettuati su neuroni di molluschi attraverso l’utilizzo di un microelettrodo (Meech,
1974;Meech e Standen, 1974).
I canali BKCa sono ampiamente distribuiti nella membrana plasmatica di cellule eccitabili e non-eccitabili. Questi canali sono localizzati nel muscolo liscio
(Giangiacomo et al., 1995), nel cervello ( Knaus et al., 1996) e nelle cellule
cromaffini (Lingle et al., 1996).
Il canale BKCa funzionale è un tetramero di subunità α che formano il poro del
canale ionico (Fig.7) (Butler et al., 1993). La subunità α è codificata da un singolo gene, Kcnma1 o Slo1, che subisce un esteso splicing pre-mRNA (Butler et al.,
18
uno o due domini transmembrana (Fig.7) che modificano la cinetica del canale e la sensibilità al voltaggio e al Ca2+.
Fig.7 Topologia dei canali BKCa e subunità modulatorie.
Le subunità β1-β4 hanno due domini transmembrana e influenzano la farmacologia del canale e la sua risposta ai lipidi (Knaus et al., 1994; Wallner et al., 1999; Xia et
al., 1999; Brenner et al., 2000; Meera et al., 2000; Uebele et al., 2000; Vaithianathan et al., 2008), mentre le proteine LRRC (leuchine-rich
repeat-containing proteins) 26, LRRC38, LRRC52 e LRRC55 sono proteine di membrana di singolo passaggio con LRRC26 che risulta essere il più potente attivatore producente uno shift negativo di 140 mV dell’attivazione voltaggio-dipendente
(Yan e Aldrich, 2010, 2012).
Più recentemente, i canali BKCa sono stati trovati anche nei mitocondri del cervello (Kulawiak e Bednarzyk, 2005) e nella linea cellulare LN229 di glioma umano (Siemen et al., 1999).
19
In linea con la visione secondo la quale i canali BKCa sono localizzati negli
organelli cellulari per una specifica funzione, nei cardiomiociti neonatali si ritiene che questi canali, localizzati nei mitocondri (mitoBKCa), possano essere utili per proteggere il cuore dall’insulto ischemico(Redel et al., 2008).
Un canale con proprietà simili al canale BKCa della membrana plasmatica è stato osservato nei mitoplasti delle cellule ventricolari di cavia. Il canale è stato stimolato con NS1619 e bloccato da caribdotossina (ChTx), iberiotossina (IbTx) e paxillina.
Immunoblots di mitocondri cardiaci con anticorpi diretti contro la regione C-terminale del canale hanno identificato una proteina di 55kDa come presunto
canale, che può contribuire all’effetto cardioprotettivo (Xu et al., 2002). Inoltre, è stato riportato che l’attivazione dei mitoBKCa cardiaci da parte di NS1619 è
amplificata dal monofosfato 8-bromoadenosina-3’,5’-ciclico e dalla forskolina ma non dal forbol estere PKC-attivante (PMA). Questi risultati suggeriscono che l’apertura dei mitoBKCa è modulata da una proteina chinasi cAMP-dipendente (Sato et al., 2005).
Un recente studio ha mostrato che copie della subunità β1 dei canali BKCa sono
espresse nei mitocondri cardiaci di mammifero (Ohya et al., 2005).
Il canale è attivato da concentrazioni micromolari di Ca2+ e dall’acido 12,14-diclorodeidroabietico (diCl-DHAA)(Samavati et al., 2002), da NS1619 e NS11021
(Aon et al., 2010; Bentzen et al., 2009), da CGS7181 e CGS7184 (Lawson, 2000);
è invece bloccato da specifici inibitori quali caribdotossina (Gu et al., 2007;
Skalska et al., 2009), iberiotossina (Cheng et al., 2008; Cheng et al.,2011) e
paxillina (Heinen et al., 2007a; Heinen et al., 2007b).
Recentemente, è stato trovato che i canali ionici mitocondriali sono sensibili a bassi livelli di ossigeno. Usando la tecnica del patch-clamp, è stato riportato che l’ipossia inibisce il “permeability transition pore” ma aumenta sostanzialmente l’attività dei canali mitoBKCa nei mitocondri di fegato di ratto e degli astrociti. Le osservazioni del gruppo Siemen indicano un possibile ruolo dei canali mitoBKCa durante l’ipossia, che potrebbe essere interpretato come un meccanismo che inibisce l’apoptosi (Cheng et al., 2008) .
20
La risposta mitocondriale ai cambiamenti della concentrazione citosolica di calcio ha un forte impatto sul metabolismo e la vitalità delle cellule neuronali. E’ stato osservato che l’aggiunta di Ca2+ ai mitocondri isolati di cervello di ratto induceva
dissipazione del potenziale di membrana mitocondriale e di conseguenza aumentava la respirazione mitocondriale. Gli effetti del calcio venivano bloccati da IbTx o ChTx, inibitori del canale BKCa. Inoltre, NS1619, che apre il canale BKCa, induceva un flusso del potassio simile a quello indotto dal calcio. Questi risultati suggeriscono la presenza di un canale del potassio Ca2+-regolato a larga conduttanza (mitoBKCa )nei mitocondri di cervello di ratto (Skalska et al., 2009).
I canali mitoIKCa (mitoKCa3.1; MIMIK) sono stati recentemente osservati
attraverso il patch-clamp di mitoplasti isolati da cellule di carcinoma del colon umano (De Marchi et al., 2009). Questo canale è stato rilevato anche in altri tipi di cellule, cioè in quelle HeLa dell’adenocarcinoma della cervice umana e nei fibroblasti embrionali del topo (Sassi et al., 2010). Le caratteristiche biofisiche sono simili a quelle del canale del potassio Ca2+-dipendente presente nella membrana plasmatica (canale KCa3.1). Il canale è potassio-selettivo, con una conduttanza che va da 10 a 90 pS (Tab. 1). Anche le proprietà farmacologiche dei mitoIKCa sono simili a quelle dei canali ionici presenti nella membrana plasmatica.
L’attività è completamente abolita a bassi livelli di calcio, e gli inibitori del canale KCa3.1 come il clotrimazolo e TRAM-34 inibiscono la sua attività a concentrazioni
nanomolari (De Marchi et al., 2009). Inoltre, la localizzazione di questo canale è stata confermata dal Western blotting e da strumenti di fluorescenza. Fino ad ora, un canale simile al mitoIKCa non è stato rilevato nel tessuto di cervello (Bednarczyk P., 2009).
Il canale mitoIKCa può avere un ruolo protettivo simile a quello proposto per i canali mitoBKCa e mitoKATP; il suo ruolo fisiologico, comunque, non è stato ancora
studiato in dettaglio. Per verificare il ruolo dei mitoIKCa nella morte cellulare, è stato utilizzato il permeante di membrana TRAM-34, ma l’applicazione di questo inibitore da solo non ha indotto la morte cellulare (Sassi et al., 2010).
21
1.1.2.3. Canali mitoK
V1.3
I canali del potassio voltaggio-dipendenti (canali KV) appartengono alla famiglia dei canali ionici regolati da cambiamenti del potenziale di membrana (Gutman et al.,
2005). Essi giocano un ruolo cruciale durante i potenziali d’azione attraverso il
ritorno della cellula depolarizzata allo stato di riposo.
Nel 2005, un canale voltaggio dipendente, KV 1.3, sensibile alla margatossina, è stato identificato nei mitocondri dei linfociti T (Tab. 1)(Szabo et al., 2005).
Nei linfociti T, è stato dimostrato che questi canali giocano un ruolo cruciale nella regolazione della proliferazione e del volume (Chandy et al., 2004). Di conseguenza, una disfunzione dei canali KV causa vari disturbi neuronali, immunitari e cardiaci (Shieh et al., 2000).
Dati biofisici, biochimici, farmacologici e genetici hanno confermato l’espressione funzionale dei KV1.3 nei mitocondri dei linfociti:
- studi con microscopio elettronico su linfociti di sangue periferico mostravano la presenza del canale nella membrana mitocondriale interna;
- uno studio di patch-clamp mostrava la localizzazione del canale mitoKV1.3 nella membrana mitocondriale interna, con proprietà simili a questo tipo di canale presente nella membrana plasmatica.
E’ stato dimostrato che il canale KV1.3 è presente nelle membrane plasmatica e
mitocondriale, nonostante la mancanza della sequenza target N-terminale mitocondriale.
Questo tipo di canale è anche presente nel rene (Yao et al., 1996), nell’epitelio
(Grunnet et al., 2003) e nel sistema nervoso centrale (Mourre et al., 1999).
E’ possibile che il canale KV1.3 mitocondriale rappresenti un importante fattore
nella trasduzione del segnale apoptotico. E’ stato dimostrato, infatti, che la proteina Bax (proteina pro-apoptotica della famiglia Bcl-2) media il rilascio del citocromo c
22
e la depolarizzazione mitocondriale, almeno in parte, attraverso la sua interazione con il canale mito KV1.3 (Szabo et al., 2008).
Quindi, mentre l’attivazione dei canali KV1.3 presenti nella membrana plasmatica è associata alla proliferazione cellulare (Cahalan e Chandy , 2009), la loro controparte mitocondriale è coinvolta nella morte cellulare (Gulbins et al., 2010;
Szabò et al., 2008; Szabò et al., 2011; Szabò et al., 2010). Infatti, l’espressione dei
mitoKV1.3 era sufficiente a sensibilizzare la linea cellulare KV1.3-less CTTL-2 dei linfociti T , resistente all’apoptosi, ad una varietà di stimoli di morte; mentre, la soppressione dell’espressione dei KV1.3 nei linfociti umani del sangue periferico
indeboliva l’apoptosi in queste cellule (Szabò et al., 2008).
Quanto al meccanismo, il mitoKV1.3 è stato identificato come nuovo target della famiglia di proteine pro-apoptotiche Bcl-2, tra cui Bax, ed è stato mostrato il verificarsi di una interazione fisica tra le due proteine, ma solo nelle cellule apoptotiche.
La proteina Bax ricombinante inibiva l’attività del canale KV1.3 con una efficienza
simile a quella osservata per le specifiche tossine inibitrici margatossina ed ShK (Stichodactyla toxin). Infatti, l’incubazione di mitocondri isolati, che presentavano il canale mitoKV1.3, con la proteina Bax o concentrazioni nanomolari di
margatossina o Shk innescava gli eventi apoptotici; mentre i mitocondri in cui erano assenti i mitoKV1.3 si mostravano resistenti. I risultati indicano che durante
l’apoptosi, l’inibizione dei mitoKV1.3 localizzati nella membrana mitocondriale
interna attraverso la Bax inserita nella membrana mitocondriale esterna porta all’iperpolarizzazione, che risulta nella riduzione dei componenti della catena respiratoria e nell’aumentata produzione di ROS (Szabò et al., 2008). Le ROS emergono come giocatori chiave nel promuovere il rilascio del citocromo c dai mitocondri (Kaul et al., 2005; Petrosillo et al., 2001). Il citocromo c è normalmente legato alla membrana mitocondriale interna, nello spazio intermembrana, attraverso l’associazione con la cardiolipina (Petrosillo et al.,
2003). La perossidazione della cardiolipina da parte delle ROS può portare alla
23
mitocondriale esterna nel citosol attraverso un meccanismo ancora dibattuto. Le ROS sono capaci di ossidare i gruppi tiolici e attivare così l’mPTP (mitochondrial permeability transition pore) la cui apertura porta alla depolarizzazione (Costantini
et al., 1996; Giorgio et al., 2005; Zoratti e Szabò , 1995) ed è stata correlata al
rilascio del citocromo c (Kumarswamy e Chandna , 2009; Scorrano et al., 2002). La proteina Bax ricombinante e le tossine inibitrici dei canali KV1.3 presenti nella
membrana plasmatica, applicate ai mitocondri isolati inducevano così un aumento della produzione di ROS seguito da una depolarizzazione e rilascio del citocromo c. Tutti questi eventi, che hanno luogo nei mitocondri di vari modelli apoptotici, dipendono dalla presenza dei mitoKV1.3 e non si verificano nei mitocondri delle
cellule CTLL-2/pJK che non presentano suddetti canali (fig. 8).
Fig.8 Vie di segnale che portano al rilascio del citocromo c subito dopo l’inibizione dei mitoKV1.3 e mitoBKCa
mediata dalla Bax pro-apoptotica.
1.1.2.4. Canali mitoTASK-3
Recentemente, il TASK-3 (TWIK-related acid-sensitive K+ channel-3), un canale twin-pore del potassio che risiede nella membrana plasmatica, è stato identificato nei mitocondri delle cellule di melanoma e cheratinociti attraverso metodi immunochimici e di biologia molecolare (Rusznák et al., 2008). Nella membrana
24
plasmatica, TASK-3 ha un ruolo nell’apoptosi e nella cancrogenesi (Patel e
Lazdunski , 2004). Una ricerca sulle proprietà funzionali del canale TASK-3 nella
membrana interna mitocondriale attraverso metodi elettrofisiologici e/o spettroscopici non è ancora stata fatta. In particolare, una linea cellulare di melanoma TASK-3 knockdown mostra una funzione mitocondriale compromessa, suggerendo che i canali TASK-3 sono funzionalmente presenti nei mitocondri delle cellule di melanoma, e la loro funzione è essenziale per la sopravvivenza di queste cellule (Kosztka et al., 2011).
1.1.3. Pre-condizionamento ischemico (IPC)
E’ attualmente ben riconosciuto che brevi episodi ischemici miocardici innescano un fenomeno cardioprotettivo, risultante in una significativa attenuazione del danno determinato da una successiva e più prolungata ischemia, come pure una riduzione delle aritmie (Shiki e Hearse , 1987) e del metabolismo energetico nelle prime fasi dell’ischemia (Murry et al., 1990), un aumentato recupero post-ischemico (Cave e
Hearse , 1992) e la protezione dell’endotelio coronarico (Richard et al., 1994).
Questo meccanismo di “self-defence” è conosciuto come “pre-condizionamento ischemico” (IPC) ed è stato osservato in diverse specie mammifere ed anche nell’uomo (Murry et al., 1986; Li et al., 1990;Liu e Downey , 1992;Liu et al.,
1991;Vahlhaus et al., 1998; Ikonomidis et al., 1994;Deutsch et al.,1990; Cribier et al, 1992;Yellon et al., 1993; Schott et al., 1990). L’IPC presenta due fasi
distinte: una iniziale (conosciuta come “classic” IPC) che dura da 1 a 3 ore dopo lo stimolo precondizionante, ed una ritardata (“second window” dell’IPC) che dura dalle 24 alle 96 ore.
Diversi agenti endogeni, quali adenosina, bradichinina, norepinefrina e oppioidi, sono coinvolti in questo meccanismo cardioprotettivo e molti recettori, che sono il bersaglio di questi agenti cardioprotettivi, sono accoppiati alla stimolazione intracellulare finale della proteina chinasi C (PKC), che gioca un ruolo chiave nell’IPC (Liu et al., 1991; Thornton et al., 1993; Simkhovich et al., 1998; Goto et
25
1994; Dempsey et al., 2000). Recentemente, un’ interessante teoria ha ipotizzato
l’esistenza di un presunto “signalosoma cardioprotettivo” (cioè l’assemblaggio e organizzazione di rilevanti proteine in vescicole intracellulari), responsabile della trasmissione di questi segnali cardioprotettivi dai recettori di superficie alle membrane mitocondriali (Quinlan et al., 2009).
I livelli di adenosina cardiaca sono aumentati da brevi periodi di ischemia nei ratti, conigli, cani e suini (Kuzmin et al., 2000; Lasley et al., 1995; Miura 1996; Mei et
al., 1998; Shulz et al., 1998) e migliorano l’equilibrio domanda-offerta di ossigeno
attraverso la dilatazione dei vasi di resistenza e la riduzione dell’inotropismo cardiaco. Questi effetti sono ampiamente mediati dai recettori A1 e A3, mentre il
sottotipo recettoriale A2 sembra essere escluso nell’IPC (Liu et al., 1991; McCully
et al., 2001).
I recettori per gli oppiodi sono espressi nei miociti cardiaci di ratto e la loro attivazione è coinvolta nella riduzione delle dimensioni dell’infarto (Zhang et al.,
1996; Weil et al. 1998). Diversi dati sperimentali dimostrano che il recettore di tipo
δ per gli oppioidi è coinvolto nell’IPC (Schulz et al., 1995; Schwartz et al., 1997) e, in particolare, che una stimolazione oppioide-selettiva di δ1 può indurre una cardioprotezione ritardata (Fryer et al., 1999). Ad oggi, è controverso un possibile ruolo del recettore κ per gli oppioidi nell’IPC (Wang et al., 2001a).
Anche la bradichinina gioca un ruolo importante, durante una breve IPC nel suino
(Schulz et al., 1998) o un singolo ciclo di ischemia-riperfusione nei conigli, ed è
stato suggerito che l’adenosina e la bradichinina possono agire sinergicamente
(Goto et al., 1995).
Un potenziale coinvolgimento di prostaglandine, noradrenalina e angiotensina è attualmente dibattuto (Bugee et al., 1995; Moolman et al., 1996; Tanno et al.,
2000; Liu et al., 1995; Wang et al., 1996a).
Per quanto concerne i meccanismi recettore-indipendenti, l’ossido nitrico (NO), i cui livelli cardiaci aumentano durante l’ischemia, è stato indicato come un importante fattore endogeno coinvolto nella cardioprotezione. L’inibizione della
26
NO-sintetasi (NOS) riduce gli effetti cardioprotettivi dell’IPC nei ratti, mentre la somministrazione di NO-donors esogeni porta ad una significativa riduzione del danno miocardico nei cuori di diverse specie animali danneggiati dall’ischemia
(Lochner et al., 2000; Jones e Bolli , 2006). Attualmente, sebbene sia ben
riconosciuto un maggior ruolo dell’NO endogeno nella fase ritardata dell’IPC, il suo contributo alla fase iniziale non è ancora chiaro (Bolli , 2001).
La PKC è stata indicata come una delle molecole segnale cruciali dell’IPC. Questo ruolo è stato chiaramente dimostrato con la staurosporina o la polimixina, inibitori della PKC che prevengono l’IPC, mentre l’attivazione della PKC con il forbol estere promuove la cardioprotezione nei cuori umani e di coniglio (Armstrong e
Ganote , 1994; Ytrehus et al., 1994; Ikonomidis et al., 1997). L’effetto
cardioprotettivo dovuto alla PKC coinvolge il processo di fosforilazione di diverse proteine, e fra loro, i canali del potassio ATP-sensibili (KATP).
1.1.4. Post-condizionamento ischemico (I-Post)
Zhao et al. (2003) hanno evidenziato come brevi periodi di riperfusione, intervallati da brevi periodi di occlusione coronarica, durante la prima fase della riperfusione, conferiscono un effetto cardioprotettivo contro il danno da ischemia/riperfusione. Questo meccanismo prende il nome di “post-condizionamento ischemico” (I-Post)
(Zhao et al., 2003).
Il post-condizionamento diminuisce la lesione a livello di molti organi, come ad esempio cuore, midollo spinale, cervello, reni, fegato, muscoli, polmoni, intestino
(Zhao et al., 2010).
Il meccanismo di protezione cardiaca dipendente da questo processo non è ancora del tutto chiaro; si presume che sia dovuto all’accoppiamento di alcuni composti con la proteina G (Zhao e Vinten-Johansen 2006; Hausenloy 2009).
Sono state proposte varie strade di protezione cardiaca mediate da I-Post. Quest’ultimo sembra che provochi una riduzione di ROS, una diminuzione del
27
sovraccarico di Ca2+ e di infiammazione (Zhao et al., 2003; Sun et al., 2005); riduce inoltre l’apoptosi, le aritmie causate dalla riperfusione, il danno dell’endotelio, l’adesione dei neutrofili alle arterie coronarie post-ischemiche dell’endotelio vascolare e l’accumulo di neutrofili nel miocardio (Galagudza et al., 2004; Zhao et
al., 2005; Kloner et al., 2006; Kin et al., 2007; Granfeldt et al., 2009).
Inoltre, attiva le proteine chinasi come la PI3K e la Akt/proteina chinasi B, insieme alla RISK (Reperfusion injurysalvage kinase), la quale garantisce cardioprotezione,
(Downey e Cohen 2005; Hausenloy et al., 2005) fosforilando la proteina GSK-3β,
che inibisce l’apertura del poro di transizione di permeabilità mitocondriale (mPTP)
(Tsang et al., 2004; Burley e Baxter 2009).
Anche la proteina chinasi C (PKC) e la proteina chinasi G (PKG) hanno un ruolo di rilievo nella protezione cardiaca mediata da post-condizionamento (Hausenloy e
Yellon 2006; Burley et al., 2007).
Infine, nell’I-Post sono coinvolti i canali KATP, la sintesi di NO e il suo successivo rilascio (Yang et al., 2004; Hausenloy 2009; Kaur et al., 2009; Cai et al., 2011). Da quanto è stato detto fino ad ora, possiamo aggiungere che esistono dei composti farmacologici che mimano l’effetto cardioprotettivo di I-Post.
Il post-condizionamento farmacologico (P-Post), è caratterizzato dalla somministrazione di agenti farmacologici nel momento della riperfusione, dopo un periodo prolungato di ischemia, per conferire cardioprotezione (Zhao et al., 2003). Esistono molti farmaci con questa funzione. Questi includono gli attivatori del recettore B2 della bradichinina (Penna et al., 2010) e quelli della PKC-adenosina A2B (Philipp et al., 2006).
Considerando che il pre-condizionamento è limitato dal fatto che risulta impossibile innescarlo quando siamo già in presenza di infarto, e quindi quando l’arteria coronarica del paziente al momento del ricovero è già occlusa, si capisce come il ruolo del post-condizionamento non risulti affatto marginale.
28
Il meccanismo di pre- e post-condizionamento hanno molte vie in comune, ma quella che ci interessa maggiormente è legata all’attivazione dei canali KATP , e in
particolare dei mitoKATP , nonché di quelli mitoBKCa, che sappiamo essere coinvolti entrambi nella cardioprotezione.
1.1.5. Ruolo dei canali mitoK
ATPnell’IPC
Il potenziale della membrana interna mitocondriale controlla il flusso degli ioni K+ nella matrice mitocondriale, e la loro fuoriuscita è controbilanciata attraverso un antiporter K+/H+ . L’attivazione dei canali mitoKATP, presenti nella membrana interna mitocondriale, permette l’entrata degli ioni K+ nella matrice. Questo flusso produce diversi effetti nel cuore, dipendenti dagli stati bioenergetici. In condizioni fisiologiche, l’entrata del K+ è molto bassa, quindi l’influenza sul potenziale
mitocondriale di membrana è quasi trascurabile, sebbene questo piccolo aumento dell’influsso di K+
causi un significativo aumento del volume della matrice. Per contro, in condizioni di stress o ischemia, un’ attivazione più sostenuta dei canali mitoKATP porta ad un significativo influsso di ioni K+ , con diverse conseguenze
(Garild e Paucek, 2003). L’apertura dei mitoKATP assicura l’omeostasi del volume
della matrice, preservando la vicinanza tra la membrana interna e quella esterna, e una corretta regolazione della permeabilità ai nucleotidi (ADP e ATP). Infatti, sotto condizioni normossiche, la permeabilità della membrana per ATP e ADP è abbastanza modesta, e i trasferimenti di energia tra i mitocondri e il citosol sono controllati dalla creatinina e creatinina-fosfato; in condizioni ipossiche, la conduttanza della membrana esterna aumenta, con successiva contrazione della matrice e conseguente espansione dello spazio intermembrana. Questo cambiamento causa la dissociazione della creatinina chinasi dal canale, e un aumento della permeabilità della membrana esterna (Rostovtseva e Colombini ,
1997; Schlattner et al., 2001). In queste condizioni, l’apertura dei mitoKATP induce un significativo influsso di K+, accompagnato dalla diffusione di acqua e l’uptake di anioni (principalmente fosfati, ma anche Cl-), risultante così nello swelling della matrice. Questo effetto garantisce la conservazione di una bassa permeabilità della
29
membrana esterna per i nucleotidi, riduce l’idrolisi di ATP, crea un gradiente favorevole per la sintesi di ATP, e induce un effettivo trasferimento di energia tra i mitocondri e l’ATP-asi cellulare. Pertanto, il recupero funzionale al momento della riperfusione risulta migliorato (Garild , 2000).
La depolarizzazione della membrana interna collegata all’apertura dei mitoKATP è
anche un importante fattore che contrasta il sovraccarico di Ca2+ intra-mitocondriale, a causa di una ridotta driving-force per l’uptake del Ca2+
(Holmuhamedov et al., 1998; Holmuhamedov et al., 1999). Quando l’energia del
metabolismo cellulare è compromessa, come nell’ischemia, i livelli di Ca2+
intracellulare aumentano (a causa dell’entrata del Ca2+
extracellulare) e questa crescita della concentrazione citosolica di ioni liberi del Ca2+ è parzialmente compensata dall’uptake del Ca2+
nei mitocondri, principalmente assicurata da un canale ionico altamente selettivo conosciuto come uniporter mitocondriale del Ca2+
(Zhang et al., 2006), e in altri compartimenti di accumulo. Questi alti livelli di Ca2+
mitocondriale, insieme con altri fattori presenti durante la riperfusione (ma non durante l’ischemia), sono causalmente collegati alla formazione dell’mPTP, che durante la riperfusione, produce rilascio di Ca2+ dalla matrice (Halestrap et al.,
2004). L’apertura dell’mPTP rappresenta una causa cruciale del danno irreversibile
riperfusione-indotto dei miocardiociti, dato che porta al rilascio di proteine pro-apoptotiche , quali il citocromo c, dallo spazio intermembrana mitocondriale al citosol. La relazione tra l’attivazione dei canali mitoKATP e lo stato dell’mPTP è
ancora poco chiara. Da una parte, l’attivazione farmacologica dei canali mitoKATP promuove la fuoriuscita di Ca2+ dal mitocondrio pre-caricato, attraverso l’apertura del canale mPTP, secondariamente alla depolarizzazione mitoKATP-indotta e lo swelling dei mitocondri. Dall’altra parte, il trattamento farmacologico con un attivatore mitoKATP riduce il sovraccarico di Ca2+ nei mitocondri isolati esposti ad
alte concentrazioni di calcio attraverso un ridotto gradiente elettrochimico per questo ione, a causa della depolarizzazione della membrana mitocondriale
(Holmuhamedov et al., 1998; Holmuhamedov et al., 1999). Pertanto, durante la
fase ischemica, l’attivazione dei canali mitoKATP , con la conseguente
30
attivazione dell’mPTP), previene il sovraccarico di Ca2+
nella matrice mitocondriale. Questo abbassamento dell’entrata di calcio nella matrice mitocondriale inibisce una successiva apertura sostenuta dell’mPTP nella fase di riperfusione. Quindi, questa inibizione (forse indiretta) dell’mPTP è probabile che sia il meccanismo prevalente, considerando l’effetto cardioprotettivo degli attivatori dei canali mitoKATP (Halestrap et al., 2004) .
Inoltre, è stato suggerito che l’apertura dei mitoKATP può proteggere il cuore
attraverso un ulteriore percorso: il disaccoppiamento della fosforilazione ossidativa. Questo è considerato un fenomeno protettivo, perché è collegato alla riduzione del rilascio delle specie reattive dell’ossigeno (ROS)(Brand et al., 2004; Korshunov et
al., 1997;Miwa e Brand , 2003; Ferranti et al., 2003; Forbes et al., 2001; Krenz et al., 2002). Più recentemente, è stato proposto che i canali mitoKATP potrebbero
agire come sensori del bilancio redox mitocondriale, regolando questo stato sotto condizioni fisiologiche, e prevenendo efficacemente il rilascio di ROS mitocondriali e lo stress ossidativo, sotto condizioni patologiche, quali ischemia e riperfusione post-ischemica (Facundo et al., 2007). Tuttavia, come teoria alternativa e opposta, è stato proposto che l’attivazione dei mitoKATP produce una alcalinizzazione della
matrice e un aumento nella produzione mitocondriale di ROS. Questi, a loro volta, attivano le chinasi, che portano ad una sostenuta attivazione dei mitoKATP (Garild et
al., 2003).
1.1.6. Ruolo dei canali mitoBK
Canell’IPC
Recentemente, è stato dimostrato che anche il canale mitoBKCa, presente a livello della membrana interna mitocondriale, media la cardioprotezione contro il danno da ischemia/riperfusione (Xu et al., 2002), allo stesso modo del canale mitoKATP (Garild et al., 1997).
L’apertura di entrambi i canali aumenta l’uptake di potassio nel mitocondrio, richiesto per il funzionamento ottimale della fosforilazione ossidativa (Kowaltowski
31
cardioprotezione mediata da IPC, Cao et al. (2005) hanno ipotizzato che lo stesso discorso potesse valere anche per l’attivazione dei mitoBKCa .
In accordo con un precedente studio (Wang et al., 2004), Cao et al. hanno visto che l’attivazione del canale con NS1619 conferisce cardioprotezione, proprio come nel caso dell’IPC o dell’attivazione dei canali mitoKATP con diazossido. Queste
osservazioni forniscono una inequivocabile evidenza che il canale mitoBKCa è
coinvolto nella cardioprotezione mediata dal precondizionamento con insulti ischemici. E’ importante notare, sempre in questo studio, che quando il canale mitoBKCa viene bloccato con paxillina 30 minuti prima dell’ischemia, l’effetto
dell’IPC nel cuore isolato e perfuso viene abolito. Dall’altra parte, invece, quando la paxillina viene aggiunta durante l’ischemia e la riperfusione, l’effetto dell’IPC si conserva. Queste ricerche indicano che questo canale è un innesco, piuttosto che un mediatore, della cardioprotezione da precondizionamento.
Un’altra importante scoperta è la relazione causale tra il canale mitoBKCa e l’mPTP,
un passo fondamentale nella cardioprotezione da precondizionamento (Hausenloy et
al., 2002, 2004; Javadov et al., 2003). Cao et al. (2005) hanno visto che l’apertura
dell’mPTP con atractyloside, che abolisce l’effetto benefico dell’IPC sul cuore in termini di protezione contro il danno e funzione contrattile, abolisce anche gli effetti benefici dell’attivazione dei mitoBKCa con NS1619. Questa è la prima
dimostrazione che l’mPTP gioca un ruolo permissivo per la cardioprotezione dovuta all’attivazione dei mitoBKCa.
Inoltre, sempre in questo studio, è stato visto che la cardioprotezione conferita dall’attivazione dei mitoKATP con diazossido non viene alterata dal blocco dei
mitoBKCa , nè viceversa, indicando che questi canali agiscono indipendentemente.
Questo è confermato anche da un ulteriore studio, in cui la paxillina bloccava gli effetti del NS1619 ma non del diazossido, e il 5-idrossidecanoato bloccava gli effetti del diazossido ma non dell’ NS1619 (Sato et al., 2005).
Per concludere, Cao et al. (2005) hanno osservato che il blocco del canale mitoBKCa abolisce gli effetti protettivi dell’IPC accompagnati da un indebolimento
32
della contrazione, mentre l’attivazione di tale canale conferisce cardioprotezione accompaganta da una migliore funzione contrattile.
1.1.7. mPTP (mitochondrial permeability transition pore)
Fig.9 Componenti molecolari dell’mPTP.
Sotto normali condizioni fisiologiche, la membrana interna mitocondriale è impermeabile a tutti i metaboliti e ioni: questo è essenziale per mantenere il potenziale di membrana e il gradiente di pH che insieme guidano la sintesi di ATP attraverso la fosforilazione ossidativa. Inoltre, in condizioni di elevati livelli di Ca2+ nella matrice, specialmente quando questo è accompagnato dallo stress ossidativo, un alto livello di fosfato e una bassa concentrazione di nucleotide adenina, nella membrana mitocondriale interna si apre un poro non-specifico conosciuto come mPTP (mitochondrial permeability transition pore). L’mPTP è un megacanale mitocondriale con un’ampia conduttanza ( 1nS) che permette il passaggio non selettivo di soluti con peso molecolare fino a 1.5kDa.
33
Per prima cosa, permette un libero movimento di protoni attraverso la membrana interna mitocondriale, portando la fosforilazione ossidativa ad essere disaccoppiata. Non solo previene la sintesi di ATP ma consente anche di invertire la direzione della translocazione di protoni dell’ATPasi e così idrolizza attivamente ATP piuttosto che sintetizzarlo. Sotto queste condizioni, le concentrazioni intracellulari di ATP decadono rapidamente, portando alla rottura dell’omeostasi ionica e metabolica e alla attivazione degli enzimi degradativi quali le fosfolipasi, le nucleasi e le proteasi (Halestrap et al., 2004). Questi cambiamenti causano danno irreversibile alla cellula.
Inoltre, l’apertura di solamente un singolo poro in un mitocondrio è probabile che causi l’immediata depolarizzazione del ΔΨm. Questo poi attiverà l’ulteriore apertura di un altro poro nello stesso mitocondrio, dato che l’apertura dell’mPTP dei mitocondri ricchi di calcio è attivata dalla depolarizzazione del ΔΨm (Scorrano
et al., 1997). Così i mitocondri sono o pienamente aperti o pienamente chiusi, ed è
lo stato pienamente aperto che porta allo swelling dei mitocondri (Bernardi , 1999). L’apertura dell’mPTP è promossa da un alto livello di Ca2+ nella matrice, dal
fosfato inorganico, dalle ROS, dagli acidi grassi e dalla depolarizzazione del ΔΨm, che si verificano durante l’ischemia/riperfusione; Mg2+
, nucleotidi adenina, basso pH e ciclosporina A, invece, inibiscono l’apertura dell’mPTP.
Basandosi sugli studi biochimici e farmacologici, è stato proposto che il poro consiste di un canale anionico voltaggio-dipendente (VDAC) nella membrana esterna, della adenina nucleotide translocasi (ANT) nella membrana interna e della CypD (cyclophilin D) che media la sensibilità al Ca2+ di questo canale nella matrice
(Halestrap et al., 2004) (fig.9). Esiste una considerevole evidenza che l’mPTP si
apre durante la riperfusione, e questo è stato collegato al rilascio del citocromo c e all’innesco dell’apoptosi (Crompton , 1999). Griffiths et al. hanno riportato che l’mPTP non si apre durante l’ischemia ma è attivato al momento della riperfusione
(Griffiths e Halestrap , 1995). E’ stato postulato, inoltre, che l’attivazione del canale
del potassio mitocondriale è coinvolta nella cardioprotezione e nel meccanismo di inibizione dell’apoptosi attraverso l’inibizione dell’mPTP (Nishida et al., 2010).
34
1.1.8. Ruolo dei canali ionici mitocondriali nella cardioprotezione
Nel sistema cardiovascolare i mitocondri sono gli organelli bersaglio degli insulti ischemici ma anche gli effettori per la cardioprotezione. Come indicato da Jennings e Ganote oltre 30 anni fa, il cuore è strettamente aerobico e quindi vulnerabile ad una ridotta fornitura di ossigeno (Jennings e Ganote , 1976). Di conseguenza, un’ ischemia miocardica causa squilibri mitocondriali profondi e immediati. Questi includono la cessazione della sintesi di ATP, l’inibizione della respirazione, e la depolarizzazione della membrana mitocondriale (ΔΨm). Tutto questo è accompagnato da un cambiamento cellulare, specialmente un aumento di calcio e fosfato durante l’ischemia, e un grosso aumento delle ROS che originano dalla catena respiratoria durante la riperfusione (Turrens , 2003). Nell’ottica di una prospettiva terapeutica, è importante notare che sono specialmente i canali ionici mitocondriali a giocare un ruolo chiave. Un aumento del flusso di K+ mitocondriale, per esempio, nei cuori trattati con composti che aprono i canali del K+ , attivando i canali mitoKATP o quelli mitoKCa, è stato visto ridurre significativamente la dimensione dell’infarto e migliorare la ripresa funzionale dopo l’ischemia/riperfusione. Dall’altra parte, l’inibizione dell’mPTP può prevenire la perdita della funzione mitocondriale, causa della morte cellulare necrotica o apoptotica. Dato che la cardioprotezione coinvolge l’attivazione dei canali mitoKATP
o mitoKCa ed una riduzione dell’apertura dell’mPTP, è ragionevole ipotizzare che questi due fenomeni facciano parte della stessa via di segnalazione(Fig.10). Infatti, questa connessione è stata già dimostrata in un precedente lavoro (Costa et al.,
35
Fig.10 Rappresentazione schematica del meccanismo di protezione mediato dai canali ionici mitocondriali.
Circa 25 anni fa Lamping et al. hanno mostrato che gli agenti farmacologici capaci di aprire i canali del K+ avevano un effetto cardioprotettivo contro il danno da ischemia/riperfusione (Lamping et al., 1984). I primi studi naturalmente supponevano che il target di questi composti era il canale del potassio sarcolemmale ATP-sensibile (sarcKATP). Tuttavia, fu aperta una nuova porta nel 1991, con la
scoperta dei canali mitoKATP nella membrana interna mitocondriale del fegato (Inoue et al., 1991). Queste scoperte furono rafforzate dall’evidenza che una varietà
di attivatori e inibitori del canale del potassio influenzavano la funzione mitocondriale (Garild et al., 1996; Paucek et al., 1992; Szewczyk et al., 1993;
Garild et al., 1997), stabilendo così un collegamento tra il canale mitoKATP e la cardioprotezione contro il danno da ischemia/riperfusione nei cuori intatti (Garild
et al., 1997) e nei miociti isolati (Liu et al., 1998).
I canali mitoKATP sono considerati gli effettori finali della cardioprotezione, responsabili dell’attenuazione del danno cellulare (Wang et al., 2001b). Queste conclusioni si basano sul fondamento logico che i mitocondri agiscono come organelli di vita o di morte. Questi canali, essendo abbondanti nei mitocondri, riducono l’accumulo di Ca2+
al loro interno, riducono l’apoptosi e aumentano la sintesi di ATP, preservando così la struttura e la funzione dei mitocondri. Tutte
36
queste caratteristiche contribuiscono alla protezione cardiaca contro il danno ischemico.
Fig.11 Meccanismi di attivazione precondizionamento-indotta dei canali sarcKATP e mitoKATP nella cardioprotezione.
Una delle maggiori caratteristiche fisiologiche dei mitocondri è la generazione di un grosso potenziale transmembrana attraverso la membrana interna mitocondriale. Questa è una diretta conseguenza delle reazioni biochimiche che costituiscono la catena respiratoria. Così, i substrati forniti ai mitocondri quali il piruvato, prodotti di β-ossidazione degli acidi grassi, e qualche amminoacido partecipano al ciclo TCA e mantengono lo stato ridotto delle coppie NADH/NAD+ e FADH2/FAD. Queste sostanze forniscono elettroni alla catena respiratoria, i quali eventualmente vengono trasferiti all’ossigeno. Il processo trasferisce anche protoni attraverso la membrana interna mitocondriale, generando un gradiente protonico, una forza motrice protonica che è espressa come ΔΨm, stimato usualmente a -180mV.