Role of the exosomes on the
epigenetic modulation of myocardial
plasticity in a rodent model of
myocardial infarction
Phd Course
Translational medicine
Accademic Year
2016/2017
Role of the exosomes on the epigenetic modulation of
myocardial plasticity in a rodent model of myocardial
infarction
Author
Gaia Papini
Supervisor
Prof. Vincenzo Lionetti
Tutor
CONTENTS ABBREVIATIONS 4 ABSTRACT 12 INTRODUCTION 15 CARDIOVASCULAR DISEASE 16 MYOCARDIAL INFARCTION 17 CARDIAC REMODELING 21 CARDIAC HYPERTROPHY 23
ANGIOTENSIN II IN CARDIOVASCULAR DISEASE 34
EPIGENETICS IN CARDIOVASCULAR DISEASE 36
DNA METHYLATION 37
HISTONE MODIFICATIONS 38
CHROMATIN REMODELING 39
EPIGENETICS IN THE HEART 40
MASPIN, AN INHIBITOR OF HISTONE DEACETYLASE 41
SULFORAPHANE 44
SODIUM BUTYRATE 48
EXOSOMES 48
BIOGENESIS OF EXOSOMES 49
EXOSOMES UPTAKE MECHANISMS 53
EXOSOMES IN THE HEART 55
AIM OF THE STUDY 58
MATERIALS AND METHODS 61
IN VITRO STUDY 62
CELL CULTURES 63
PRIMARY CARDIAC FIBROBLASTS FROM ADULT MICE AND CHARACTERIZATION 63
NIH-3T3,HL-1 AND MCEC 63
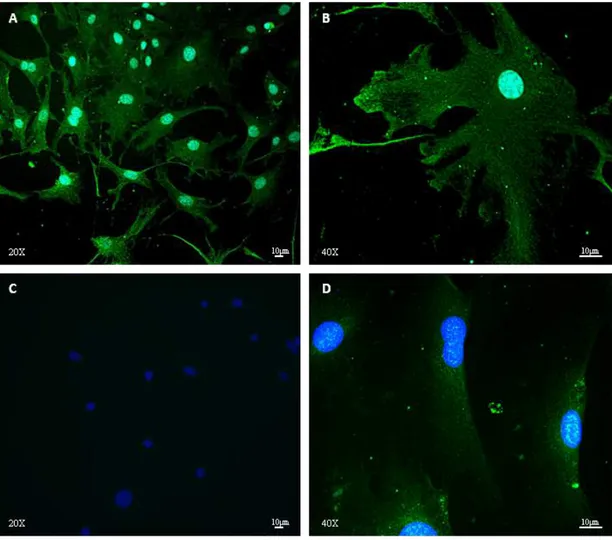
PHENOTYPE CHARACTERIZATION OF FIBROBLASTS BY IMMUNOFLUORESCENCE 64
SULFORAPHANE PREPARATION 64
MTT ASSAY ON NIH-3T3, HL-1 AND MCEC 66
ENDOGENOUS EXOSOMES DETECTION IN PRIMARY CARDIAC FIBROBLASTS AND
NIH-3T3 66
SULFORAPHANE TREATMENT AND EXOSOMES ISOLATION 67
PROTEIN EXTRACTION 67
WESTERN BLOTTING 68
UPTAKE STUDY ON HL-1 AND MCEC 68
HYPERTROPHY STUDY ON HL-1 69
MATRIGEL-BASED ANGIOGENESIS ASSAY ON MCEC 70
EXOSOMES QUANTIFICATION FOR INTRAMYOCARDIAL INJECTION 74 LEFT ANTERIOR DESCENDING (LAD) CORONARY ARTERY LIGATION 74
TRANSTHORACIC ECHOCARDIOGRAPHY 75
HISTOLOGICAL ANALYSIS 75
INFARCT SCAR SIZE 76
CAPILLARY DENSITY 76
MYOCARDIAL HYPERTROPHY 76
RNA EXTRACTION FROM HEART TISSUE 76
REVERSE TRANSCRIPTION AND REAL TIME PCR 77
STATISTICAL ANALYSIS 78
RESULTS 79
IN VITRO 80
PRIMARY CARDIAC FIBROBLASTS ISOLATION AND CHARACTERIZATION 81
SULFORAPHANE TOXICITY ON CARDIAC CELL LINES 81
BIOGENESIS OF CD63+ EXOSOMES IN PRIMARY CARDIAC FIBROBLASTS AND
NIH-3T3 83
LOWER DOSE OF SULFORAPHANE DOES NOT AFFECT H4 HISTONE ACETYLATION
LEVELS, CD63+ EXOSOMES LEVELS AND NRF2 EXPRESSION 84
SULFORAPHANE INCREASES THE LEVELS OF MASPIN INTO CD63+ EXOSOMES 86
MASPIN CLONING AND TRANSFECTIONS 86
SULFORAPHANE DOES NOT AFFECT THE LEVELS OF HDAC1 AND AP3 88 ENRICHMENT OF MASPIN IN EXOSOMES AS ZN-BINDING PROTEIN 89 DOSE-DEPENDENT TOXICITY OF FIBROBLAST-DERIVED EXOSOMES ON HL-1 AND
MCEC UNDER REST AND STRESS CONDITIONS 90
F-EXO-SFN ARE HIGHLY TAKEN UP BY HL-1 CARDIOMYOCYTES 91 F-EXO AND F-EXO-SFN DO NOT AFFECT ANGIOGENIC POTENTIAL IN MCEC 92
F-EXO-SFN DO NOT AFFECT AMPK ACTIVATION 96
F-EXO-SFN INCREASE ACETYLATION OF H4 HISTONE IN HL-1 97
IN VIVO 100
EFFECTS OF INTRAMYOCARDIAL DELIVERY OF EXOSOMES ON CARDIAC FUNCTION OF
INFARCTED RAT HEARTS 101
EFFECTS OF INTRAMYOCARDIAL DELIVERY OF EXOSOMES ON INFARCT SCAR SIZE 103 EFFECTS OF INTRAMYOCARDIAL DELIVERY OF EXOSOMES ON CARDIOMYOCYTE
HYPERTROPHY IN INFARCTED HEART 105
EFFECTS OF INTRAMYOCARDIAL DELIVERY OF EXOSOMES ON CAPILLARY DENSITY IN
INFARCTED HEARTS 108
DISCUSSION 109
BIBLIOGRAPHY 128
PUBLICATIONS AND ABSTRACTS 150
ACKNOWLEDGMENTS 153
4E-BP1: eukaryotic translation initiation factor 4E-binding protein 1; ACE: Angiotensin II converting enzyme;
ADP: adenosine diphosphate; Akt1: protein kinase B (PKB), α -SMA: α-smooth muscle actin; ALT: alanine transaminase;
AMPK: AMP-activated protein kinase; AngII: angiotensin II;
ANP: atrial natriuretic peptide;
APAF-1: apoptotic protease activating factor -1; AST: aspartate aminotransferase;
AT-R: angiotensin II receptor; ATP: adenosine triphosphate; AMP: adenosine monophosphate; Bcl-2: B cell lymphoma 2;
BNP: B-type natriuretic peptide;
CAMK: Ca2+/calmodulin-dependent protein kinase; cAMP: cyclic adenosine monophosphate;
CBP: creb binding protein; CD36: fatty acid translocase;
CM: chylomicron; Cr: creatine;
CVD: cardiovascular disease; DAG: diacylglycerol;
DMEM: dulbecco's modified eagle medium; DMSO: dimethyl sulfoxide;
DNA: deoxyribonucleic acid; DNMT: DNA methyl transferase; ECM: extracellular matrix;
EEF: eukaryotic translation elongation factor; eIF4e: eukaryotic translation initiation factor 4e; eNOS: endothelial NOS;
ERK: extracellular signal-related kinase; ERM: ezrin, radixin and moesin;
ESCRT: endosomal sorting complexes required for transport; ET-1: endothelin-1;
F-EXO-SFN: exosomes isolated from fibroblasts NIH-3T3 after treatment with Sulforaphane 3µM;
F-EXO: exosomes isolated from fibroblasts NIH-3T3; FasL: Fas ligand;
FBS: fetal bovine serum; FcR: Fc receptor;
GLUT1/4: glucose transporter 1/4; GMP: guanosine monophosphate; gp130: glycoprotein 130;
GPCR: G protein coupled receptor; GSK3: glycogen synthase kinase 3; HAT: histone acetyl transferase; HDACi: histone deacetylase inhibitor; HDAC: histone deacetylases;
HDM: histone demethylase; HF: heart failure;
HIF-1α: hypoxia-inducible factor 1α; HL-1: cardiac mouse cell line; HMGB1: high motility group box 1; HMT: histone methyl transferase; HO-1: heme oxigenase-1;
HRE: hormone response element;
HSF-1: heat shock transcription factor-1; HSP: heat shock protein;
ICAM-1: intercellular adhesion molecule-1; IGF-1: insulin-like growth factor 1;
IGF1R: insulin-like growth factor 1 receptor; IL-6: interleukin-6;
IL-10: interleukin-10; IL-6: interleukin-6;
ILV: intraluminal vesicles; IP3: inositol trisphosphate;
IPS: induced pluripotent stem cells; JAK: janus activated kinase;
JNK: jun amino-terminal kinase;
Keap1: kelch-like ECH-associated protein 1; Nrf2: nuclear factor-erythroid 2-related factor 2 LDH: lactate dehydrogenase;
LDL: low density lipoprotein; lncRNA: long non coding RNA; LTCC: L-type calcium channel; LV: left ventricle;
LVDP: left ventricular developed pressure; LVEDP: left ventricular end diastolic pressure; M-CSF: macrophage-colony stimulating factor; MBD: methyl binding domain;
MCEC: Murine coronary endothelial cells; MEF2: myocyte enhancer facor-2;
MFGE8: milk fat globule EGF factor 8 protein; MI: myocardial infarction;
MnSOD: manganese superoxide dismutase; mPTP: mitochondrial permeability transition pore; mRNA: messanger RNA;
mTOR: mammalian target of rapamycin;
MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MVB: multivesicular body;
MVP: major vault protein;
MYBPC3: myosin binding protein C;
NADPH: nicotinamide adenine dinucleotide phosphate; NCX: Na2+/Ca2+ exchanger;
NFAT: nuclear factor of activated T cells; NFkβ: nuclear factor k β
NIH-3T3: Mouse embryo fibroblasts NO: nitric oxide;
NOS: nitric oxide synthase;
NPRA: atrial natriuretic peptide receptor; NuRD: nucleosome remodeling deacetylase; OMM: outer mithocondrial membrane; PBS: phosphate buffered saline;
PCAF: p300/CPB associated factor; PCr: phosphocreatine kinase; PDE: phosphodiesterase;
PGC1α: PPARγ coactivator 1; PI3K: phosphoinositide 3-kinase; PKA: protein kinase A;
PKC: protein kinase C; PKG: protein kinase G; PLC: phospholipase C; PLN: phospholamban;
PPARα: peroxisome proliferator-activated receptor α; PPARβ: peroxisome proliferator-activated receptor β; PRA: plasma renin activity;
PRAS40: proline-rich AKT substrate; PSGL-1: P-Selectin glycoprotein ligand-1; PtDSer: phosphatidylserine;
RAGE: receptor for advanced glycation end-products; RAP1B: RAS related protein 1B;
RAS: renin-angiotensin system; RCL: Reactive site loop; RNA: Ribonucleic acid; ROS: reactive oxygen species; RYR2: Ryanodine receptor 2;
SERCA2a sarco/endoplasmic reticulum Ca2+-ATPase; Serpin: serine protease inhibitors;
SORBS2: sorbin and SH3 domain containing 2 SR: sarcoplasmic reticulum;
STAT: signal transducer and activator of transcription; TAC: transverse aortic constriction;
TAG: triacylglycerol;
TGF-β: transforming growth factor-β;
TIMPS: tissue inhibitors of metalloproteinases; TIP60: Tat-interactive protein, 60 kDa;
TLR: toll-like receptor;
TNF-α: tumor necrosis factor-α; TnT: troponin T;
TRAIL: TNF-related apoptosis-inducing ligand; TSA: trichostatin A;
TSG101: tumor susceptibility gene 101 TSP1/2: thrombospondin 1/2;
TSS: transcription start site;
VCAM-1: vascular cell adhesion molecule-1; VLDL: very low density lipoprotein;
WGA: wheat germ agglutinin; α-MHC: α-myosin heavy chain; β-MHC: β-myosin heavy chain.
Introduction: Myocyte hypertrophy is a hallmark of remodeling of the left ventricle
following myocardial infarction and it is mainly induced by the exposure to angiotensin-II (Angangiotensin-II). The communication between fibroblasts and cardiomyocytes underlies the hypertrophic response to AngII, which contributes to the onset of heart failure. Recently, fibroblast-derived exosomes (F-EXO) have been implicated in mediating the cardiomyocyte hypertrophy under AngII. However, it is still unknown whether fibroblasts are able to release anti-hypertrophic exosomes and their biogenesis is promoted by chemical compounds. Sulforaphane (SFN), a naturally occurring isothiocyanate extracted from cruciferous vegetables, attenuates AngII-induced cardiomyocytes hypertrophy. We tested the effects of SFN on the release of anti-hypertrophic F-EXO (F-EXO-SFN) in
vitro and we assessed the protective effects of F-EXO-SFN in a rat model of
non-reperfused myocardial infarction (MI).
Methods: Murine embryo fibroblasts were long-term treated with non-toxic dose of SFN
(3µM/7 days). Cell viability was assessed by MTT assay. Intact exosomes were isolated from cell culture media of SFN treated cells (F-EXO-SFN) or not treated cells (F-EXO, control) by differential centrifugation. Levels of released F-EXO were quantified by Western blot detection of tetraspanin CD63, an established exosomal protein marker. Exosomal content of maspin, a serine protease inhibitor B5 with function of inhibitor of class I histone deacetylase, was assessed by Western blot. In vitro, the uptake by HL-1 and MCEC of DiA-labeled exosomes was measured at rest or during exposure to AngII. In order to investigate the functional effects of F-EXO and F-EXO-SFN on hypertrophic HL-1 (AngII 100 nM/12h), the cell surface area, an indicator of cell hypertrophy, was measured after 3 or 24 h co-incubation of vehicle, 30 µg F-EXO or F-EXO-SFN with AngII. The effects of F-EXO or F-EXO-SFN on cardiomyocytes apoptosis was assessed by detection of Caspase-3 activation, by Western Blotting. In each experimental condition, ROS production in resting and stressed HL-1 was detected by dihydroethidium staining. Finally, the angiogenic potential of F-EXO or F-EXO-SFN treated cardiac endothelial cells (MCEC) was investigated by Matrigel-based tube formation assay.
In vivo, the anti-remodeling effects of F-EXO or F-EXO-SFN were evaluated in a rat
model of non-reperfused MI. The left anterior coronary artery (LAD) of male Wistar rats was surgically ligated and PBS, F-EXO or F-EXO-SFN were intramyocardially injected in the left ventricular infarct border zone. At 48 hours and 28 days, transthoracic
hearts were explanted for histological analysis to measure infarct scar size, cardiomyocytes size and capillary density. Hypertrophy related genes expression was investigated by real time PCR.
Results: In vitro, fibroblast-derived exosomes significantly reduced HL-1 viability at rest,
but not during exposure to AngII. Conversely, the viability of MCEC was not affected by exosomes. SFN induced the release of fibroblast-derived exosomes containing high levels of maspin, which are significantly taken up by HL-1 cardiomyocytes at rest and under stress, but not by MCEC. Higher intake of F-EXO-SFN was related with higher levels of acetylated H4 histone (P<0.05). The cell surface area of stressed HL-1 cardiomyocytes treated for 24h with F-EXO-SFN was similar to resting cells. The levels of cleaved Caspase-3 in stressed HL-1 treated with F-EXO-SFN were significantly lower than other experimental conditions under stress. Finally, stressed HL-1 treated with F-EXO-SFN have shown a significant reduction of anion superoxide generation, a major trigger of proteasome activation. F-EXO-SFN did not affect the pro-angiogenic ability of MCEC at rest and under stress.
In vivo, the infarct scar size was significantly reduced in MI + F-EXO-SFN group,
compared to MI + PBS and MI + F-EXO groups. The decay of LV ejection fraction (EF) from 48 hours to 4 weeks after coronary ligation was attenuated in F-EXO-SFN group. However, we did not detect significant change of LV fractional shortening between F-EXO and F-F-EXO-SFN group. Similarly, the capillary density at LV border zone was unaffected by each treatment. The size of cardiomyocytes at both LV infarct border and remote zone of MI + F-EXO-SFN rats was smaller than those of MI + PBS hearts. However, mRNA levels of α-MHC, ß-MHC, ANP and BNP were similar in both groups. Interestingly, the levels of SERCA2a mRNA were significantly higher in the LV remote zone of MI + F-EXO-SFN group.
Summary/conclusion: I have demonstrated, for the first time, that long-term treatment
of fibroblasts with SFN induces the release of exosomes containing high levels of maspin, which leads to simultaneous anti-hypertrophic and anti-apoptotic effects. The intramyocardial delivery of single dose of F-EXO-SFN prevents the enlargement of the cardiomyocyte size and the infarct scar size. My data open new avenue in the non-invasive use of natural chemical compounds modulating the release of endogenous anti-remodeling exosomes.
Introduction
Cardiovascular disease
Up to date cardiovascular disease (CVD) constitute one of the main death causes worldwide. In 2010 almost 52 million deaths have been recorded.1 FDA declares that just in United States, 2,200 people everyday die for cardiovascular disease. 1 of 3 deaths is driven by cardiovascular disease.
Cardiovascular disease include ischemic heart disease or coronary artery disease, cerebrovascular disease (e.g. stroke), disease of aorta and arteries including hypertension and peripheral vascular disease, but also congenital heart disease, rheumatic heart disease, cardiomyopathies and cardiac arrhythmias.
Coronary artery disease (heart attack) and cerebrovascular disease are mainly driven by atherosclerosis. Atherosclerosis is a complex process characterized by the accumulation of fatty deposit and cholesterol in the arteries. This deposition, which is called plaque, causes obstruction of arteries, making blood flow irregular. In some cases, plaques can rupture causing the formation of blood clots. Depending on where blood clots develop, it can cause heart attack or stoke. 2 The main risk factors for atherosclerosis include tobacco and alcohol use, physical inactivity, unhealthy diet, hypertension, hypercholesterolemia, diabetes, obesity.3
Rheumatic heart disease results from an abnormal autoimmune response to group A streptococcal infection. Acute rheumatic fever can affect different organs and cause damages to valves leading to heart failure.4
Congenital heart disease, also known as congenital heart defects, consist of malformations of the heart structure at birth. Examples of congenital heart disease include holes in the septum of the heart, abnormal valves and abnormalities in heart chambers 5. Other CVDs such as disorders of the heart muscle (e.g. cardiomyopathy), disorders of the electrical conduction system of the heart (e.g. cardiac arrhythmias) and heart valve diseases are less common than heart attacks and strokes.
Among all the CVD, in this thesis we will mainly discuss myocardial infarction and cardiac remodeling focusing on cardiac hypertrophy, and the currently used therapies for these pathologies.
Myocardial infarction
Myocardial infarction (MI) is the most common and clinically significant form of acute cardiac injury and results in loss of a large number of cardiomyocytes. Other processes such as chronic pressure or volume overload and chronic cardiomyopathy conditions may cause more sporadic loss of cardiac cells and activate hypertrophic and pro-fibrotic responses. 6 Majority of myocardial infarctions results from coronary atherosclerosis,
complicated by rupture of a vulnerable plaque and formation of thrombus. This causes cardiomyocytes loss in the specific zone that was fed by that artery, as show in Fig.1.7
Figure 1. Occlusion of coronary artery with subsequent myocardial infarction.8
Cell loss stimulates inflammatory cascade with recruitment of immune cells in order to eliminate dead cells. Dead cells are replaced by fibrotic tissue, leading to a massive reorganization of heart architecture and structure, which is called ‘Ventricular
remodeling’. 9,10 Ventricular remodeling affects both infarcted and non infarcted zones of the heart leading to chamber dilation, cardiac hypertrophy and increased sphericity of the left ventricle which worsen cardiac function.11 Remodeling is associated with bad clinical
prognosis following myocardial infarction; it is an important predictor of mortality and adverse cardiac events, including heart failure development and ventricular arrhythmias.12–15 The extent of ventricular remodeling depends on the size of myocardial infarction but also on the healing process. Defects in healing process may result in lethal complications, such as cardiac rupture and ventricular aneurysm formation. The cardiac rupture syndromes involve tearing of infarcted tissue and result from mechanical weakening that occurs in the necrotic and inflamed myocardium. Rupture of the ventricular free wall results in cardiac tamponade and it is usually fatal; whereas rupture of the ventricular septum leads to a left-to-right shunt, while the left papillary muscle
results in atrial mitral regurgitation. Formation of ventricular aneurysm, on the other hand, is a late complication associated with a large transmural infarction that heals into a large region of thin scar tissue that paradoxically bulges during systole. 15
Physiopathology of myocardial infarction
From a pathological point of view, MI is defined as cardiomyocyte death caused by an ischemic insult. 16 Loss of cardiomyocytes is used to diagnose MI. Instead, the detection of cardiac troponins allows the clinician to detect myocardial damage. At this regard, it should be considered that an increase in circulating troponins not always reflects myocardial damage, but it can also reveal cell wall permeability, release of proteolytic troponin degradation products, or slow normal cardiomyocytes turnover. 17,18
Myocardial ischemia is a consequence of imbalance of oxygen demand and supply. Atherosclerotic plaque has a gradual growth and since it does not reach more then 75% luminal narrowing it does not cause blood flow reduction at rest. Conversely, in case of physical activity, the flow reduction can lead to development of ischemia or angina pectoris. In most of the cases, MI results from coronary atherosclerotic disease complicated by thrombosis. 19 Plaque rupture is the most common cause of thrombosis. In mammalian heart, cardiomyocytes contain high energy phosphate reserves which is sufficient to maintain contractility for a few minutes of total ischemia. If blood flow is restored after 4-5 minutes, damage is reversible. However, longer blood flow interruption, also for 10-20 minutes, so called ‘myocardial stunning’ does not cause cardiomyocytes death, but cardiac cells are not completely functional for 24 hours. Ultimately, cardiac function will come back to normal.20
From an electrophysiological point of view, ischemia is associated to an increase of interstitial K+. Increased extracellular K+ results in a reduction of action potential. K+ increases more when cardiomyocytes start to die inducing inexcitability and conduction block. In general K+ increase induces acidosis, leading to inhibition of ionic channels, fall in resting potential, prolongation of resting potential and early afterpolarizations. 21–24
Cell death in ischemic heart
Cardiomyocytes loss due to ischemia, may be due to both apoptosis or necrosis. Most of cardiac cells die in 24 hours after ischemic insult, after pro-inflammatory response may cause cell death even if in a lesser extent.25,26
Apoptosis in ischemic hearts is activated by both intrinsic and extrinsic pathways. Outer mitochondrial membrane (OMM) permeabilization is a key process which involves interaction of B cell lymphoma 2 (Bcl-2) family members. Members of Bcl-2 family are classified in 3 classes: anti-apoptotic, pro-apoptotic and BH-3-only pro-apoptotic proteins. OMM permeabilization induces release of cytochrome C in the cytoplasm. Cytochrome C binds to Apaf-1 leading to activation of caspase-9, which in turn activates caspase-3 and caspase-7. This pathway may also be activated in cardiac hypertrophy.27,28 The role of Bcl-2 members in infarcted heart is well documented; it was shown that over-expression of Bcl-2 reduces cardiomyocytes apoptosis and improves cardiac function. Also, deficiency of pro-apoptotic members of Bcl-2 family (e.g. Bax) attenuates post-ischemic dysfunction. 29,30
During necrosis, mitochondrial permeability transition pore (mPTP) opens leading to cessation of ATP synthesis and accumulation of water in mitochondria causing mitochondria swelling. 31
Also, autophagy has been identified in ischemic hearts. It is induced by activation of AMP activated protein kinase (AMPK), but its role is still under investigation, since in literature both protective and non-protective roles of autophagy have been reported.32,33
Myocardium is a heterogeneous tissue made of different cell types, not only cardiomyocytes but also cardiac fibroblasts, endothelial cells, pericytes, macrophages and other cell types. The fate of these cells after ischemic insult is not well understood yet since the cardiac scenario is complicated by inflammatory response. In general, it is conceivable those cells are more resistant to ischemic stress compared to cardiomyocytes.16
Cardiac repair after myocardial infarction
In humans, MI results in loss of about one billion cardiomyocytes, which are replaced by non-contractile cells in order to maintain the ventricle structure. After ischemic insult, inflammatory response is activated, and it can be divided in 3 steps, as shown in Fig. 2.
Figure 2. Inflammatory response after myocardial infarction. It can be divided in three steps. I) Inflammatory phase with leukocytes recruitment leading to clearance of dead cells; II) Proliferative phase with production of matrix proteins by recruited mesenchymal cells; III) Maturation phase with cross-linking of matrix proteins in order to maintain the ventricle structure.16
These three steps are simultaneous. The first phase, inflammatory phase is characterized by recruitment and infiltration of leukocytes leading to clearance of dead cells. In the second phase, proliferative phase, pro-inflammatory signals are shut down and mesenchymal cells are recruited in order to produce matrix proteins. Finally, in the maturation phase, reparative cells turn to a quiescent state and matrix proteins are cross-linked. 34–36
Myocardial infarction therapeutic strategies
Up to date possible therapeutic strategies for acute MI are mainly constituted by ß-blockers, ACE inhibitors and statins. Reperfusion for the ischemic myocardium is also the hallmark of treatment for patients with STEMI (ST Segment elevation of MI). Anticoagulants and anti-platelets are used for reducing new thrombotic events.
ß-blockers have been shown to have arrhythmogenic, apoptotic, anti-inflammatory and anti-fibrotic and also reduce adverse ventricular remodeling.37–40 It was reported that ACE inhibitors decrease mortality and prevent development of heart failure in patients with acute MI. Also, ACE inhibitors reverse ventricular remodeling. 41 ACE inhibition mechanism may be associate to decreased circulating Angiotensin II and decreased degradation of bradykinin. In AT1 KO mouse model, ACE inhibition decreased dilative remodeling, suggesting that ACE inhibition is not only mediated by AT1 receptor.42
Other possible therapies frequently used in the clinic are the statins (3-Hydroxy-3-methylglutaryl-coenzyme A reductase). Statins are known to reduce lipids and to have
anti-inflammatory effects, by modulation of the coagulation and fibrinolytic systems, to protect cardiomyocytes and endothelial cells. 43 Also anti-fibrotic effect and ability of statins to suppress MMP expression has been reported. 44,45
Of course, an important issue would be to develop a therapy which is able to protect cardiomyocytes after ischemic insult. Experimental studies have suggested promising therapeutic approaches such as ischemic post conditioning, inhibition of oxidative stress, attenuation of calcium overload, cyclosporine, correction of pH, suppression of pro-apoptotic pathways and activation of pro-survival pathways.46–48 Briefly, post conditiong consists of cycles of ischemia/reperfusion after the ischemic event which have been related to reduced infarct size.49 Cyclosporine protect ischemic myocardium through
mPTP inhibition. 50
The stimulation of intrinsic regenerative ability of myocardium would be a very promising strategy for ischemic heart. It has been found that also human myocardium possesses regenerative properties, even if it counts for just 1% of cardiomyocytes renewed every year at the age of 20, as published by Bergmann et al. 17 Therapeutic augmentation of this rare regenerative events is a major goal of research in the cardiovascular field.
Cardiac remodeling
Cardiac remodeling was initially described as the pathological changes occurring after myocardial infarction 9,51. Then, cardiac remodeling has been defined as a group of molecular, cellular and interstitial changes associate to modifications in cell shape, size and function of the heart resulting from cardiac injury. 11 These processes may occur in response to different stimuli, such as pressure overload (aortic valve stenosis or hypertension), inflammatory disease (myocarditis), idiopathic dilates cardiomyopathy, and volume overload (valve regurgitation). Two types of cardiac remodeling have been described, physiological (adaptive) remodeling and pathological remodeling. The clinical diagnosis of remodeling is related to detection of morphological changes, such as cavity diameter, mass (hypertrophy and atrophy), geometry of the heart with changes in the wall thickness and shape, areas of scar tissue after MI and fibrosis and inflammation. 11 Cardiac hypertrophy, as extensively discussed later in this thesis, is a common type of cardiac remodeling. It can result from pressure or volume overload, or after MI.
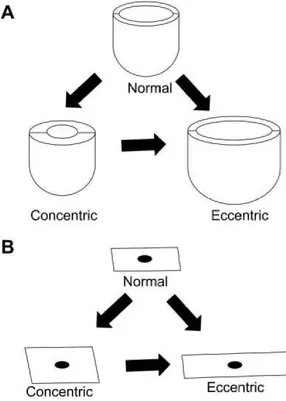
shape with thinner walls. 52 In volume overload hypertrophy, the internal radius of the ventricle increases resulting in eccentric hypertrophy. In contrast, pressure overload stress usually leads to increased wall thickness, while ventricular radius is not modified, in a process called concentric hypertrophy 53 (Fig. 3A)
At the cardiomyocyte level, as shown in Fig 3B, in the concentric hypertrophy, contractile-protein units are assembled in parallel, resulting in width increase of individual cardiomyocytes; on the other hand, contractile-protein units are organized in series in the eccentric hypertrophy, causing cardiomyocytes length increase.
Fig. 3. Macroscopic and microscopic changes during cardiac hypertrophy 54
In the clinic, the most used diagnostic techniques for cardiac remodeling are echocardiography, ventriculography and magnetic resonance imaging (MRI). The detection of remodeling biomarkers is also frequently used. Instead, cardiac remodeling is associated to changes in gene expression such as decrease in α-MHC and increase in ß isoform of MHC, increased expression of GLUT-1, α-actin, natriuretic peptide, galectin, caveolin, neuronal nitric oxide synthase, angiotensin-converting enzyme, a decrease in GLUT-4, SERCA2a and a switch from glucose to fatty acid oxidation. 55–58
Cardiac hypertrophy
Cardiac hypertrophy is usually considered as an adaptive response to different stimuli, such as mechanical and neurohumoral stress. It leads to changes in cardiomyocytes gene expression, protein synthesis, sarcomere structure and cell metabolism. 59–61
Mammalian cardiomyocytes possess reduced ability to proliferate during adulthood 62, but heart is able to respond to different environmental stimuli by cell growth, shrink or loss.63
Cardiac hypertrophy can be classified as physiological or pathological. Physiological hypertrophy is usually associated to growth of the body and is also related to increased blood capillary network which is able to properly feed cardiac cells; on the other hand, pathological hypertrophy occurs when neurohumoral mediators are produced and it is linked to cell injury, cell loss and remodeling. The first is related to normal growth of children and pregnancy and it is usually associated to normal cardiac function, while the second is related to cardiac dysfunction following MI or other cardiac pathologies.64
It is usually induced by different common disease stimuli, such as, long-lasting hypertension, MI, ischemia associated with coronary artery disease, valvular insufficiency and stenosis, myocarditis induced by infectious agents, congenital malformations, familial hypertrophic and dilated cardiomyopathies and diabetic cardiomyopathy. 54,65
Cardiac hypertrophy is characterized by adaptive phase in which cardiac function is preserved. Prolonged pressure overload leads to maladaptive phase, associated to reduced ejection fraction and left ventricular dilatation, causing heart failure.
Figure 4. Processes and main signaling involved in cardiac remodeling and hypertrophy. Schematic view of morphological and molecular changes associated to the transition from compensated hypertrophy to decompensated hypertrophy and heart failure.66
In response to pressure overload or other insults the heart increases its size and mass to normalize the wall stress and to allow normal cardiovascular function at rest; this is usually known as pathological cardiac hypertrophy, as a consequence of MI, the heart undergoes regional heart failure. (Fig. 4)
Different mechanisms are known to be involved in pathological cardiac hypertrophy. Both physiological and pathological hypertrophy are associated to increased intracellular Ca2+. Cyclic Ca2+ changes regulate heart contraction. During contraction, a high action potential causes Ca2+ to enter cardiomyocytes through L-type Ca2+ channels (LTCC) located within T-tubules. Binding of Ca2+ to ryanodine receptor (RyR2) in sarcoplasmic reticulum (SR) membrane leads to accumulation of Ca2+ into cardiomyocytes which in turn binds to Troponin C within sarcomeres. This causes the contraction. 67 Relaxation
occurs when Ca2+ is pumped back into the SR by Ca2+-ATPase (SERCA2a) or outside the
cell by Na+/ Ca2+ exchanger (NCX). 68 SERCA2a activity is regulated by phospholamban (PLN) a protein which is able to inhibit SERCA2a when it is dephosphorylated. When PKA or Ca2+ / calmodulin – dependent protein kinase II (CaMKII) phosphorylate PLN, PLN stops its inhibitory activity on SERCA2a, which can play its function. Impaired activity of SERCA2a or reduced phosphorylation of PLN lead to Ca2+ accumulation in cardiomyocytes, which have been observed in several experimental models of heart failure. 69–71
signaling. Catecholamines and Angiotensin II bind G protein coupled receptors (GqPCR)
which activate phospholipase C, inducing the synthesis of inositol triphosphate leading to accumulation of Ca2+ into cardiomyocytes. Ca2+ activates Calcineurin/NFAT signaling. Calcineurin is a serine/threonine phosphatase which dephosphorylates NFAT; NFAT translocates into the nucleus causing transcription of pro-hypertrophic genes. This signaling by itself is able to induce cardiac hypetrophy.72 Also CaMKII is activated by increased intracellular Ca2+. CaMKII phosphorylates class II histone deacetylases (HDAC), mainly HDAC4 and HDAC5, leading to repression of MEF and to cardiac hypertrophy.73,74
Another possible mechanism associated to cardiac hypertrophy is cGMP/protein kinase G (PKG). Activation of cGMP and PKG has anti-hypertrophic effect. 75 cGMP and PKG
have been found reduced in heart failure patients. 76 Inhibition of cGMP by
phosphodiesterases which cleaved cGMP, is related to anti-hypertrophic effect.77 Also, it was shown that the α-adrenergic receptors coupled with Gq, and Gq-protein kinase C (PKC) signaling and mitogen-activated protein kinase (MAPK) signaling promote cardiac hypertrophy in mice, while suppression of PKC conversely inhibits GPCR-mediated cardiac hypertrophy.78,79
Metabolism in normal heart
During the day normal heart uses 15-20 times its weight in ATP, which is produced by mitochondria. 80 In cardiomyocytes, mitochondria occupy 30% of cell volume. The heart produces the required energy mainly from fatty acids (FAs), but also from glucose and lactate. 81 Circulating fatty acids may come from two sources, as shown in Fig. 5. The first is as a component of triacylglycerol (TAGs) contained in circulating chylomicrons from the liver or very low density lipoprotein (VLDL) from the gut; second possible source is as free fatty acids bound to plasma albumin. Chylomicrons and VLDL-TAGs are cleaved to release fatty acids that can enter cardiomyocytes through fatty acids translocate CD36 or ‘flip-flop’.82 Once in the cytoplasm, fatty acids undergo ß-oxidation in the mitochondria for ATP production, or may be esterified to be stored in lipid droplets.
Figure 5. Energy metabolism in normal cardiomyocytes. Circulating fatty acids, glucose and lactate are uptaken by cardiomyocytes through active or passive transportation. Fatty acids enter the mitochondria through carnitine palmitoytransferase 1 (CPT I) conversion of acyl-CoA to acylcarnitine. Acylcarnitine is then shuttled into the mitochondria by carnitine:acylcarnitine translocase (CT) where CPT II then reverts acylcarnitine to acyl-CoA. Acyl-CoA then undergoes β-oxidation and oxidative phosphorylation to generate ATP. ATP generated is transported via the creatine kinase (CK) shuttle to be utilized in calcium cycling processes and contraction of myofilaments. Additional fatty acids that are unused undergo enzymatic conversion into TAG lipid droplets for storage till further use. ATP can be generated from glucose via glycolysis, or similar to lactate, can be converted to pyruvate before converging with the fatty acid oxidation pathways. 66
Metabolism in pathological cardiac hypertrophy
Pathological cardiac hypertrophy is characterized by:
1. Reduced fatty acids oxidation and use of glucose as main energy source 84;
2. Changes in transcriptional expression of proteins involved in glycolysis and fatty acids oxidation such as PPAR α, PPAR γ and HIF1-α 85–87;
3. Increased glucose uptake, glycolysis and decreased fatty acids oxidation 88–90. Hypertrophy is characterized by an increase in energy demand and as a consequence stored ATP is depleted with reduced phosphocreatine (PCr)/ATP ratio. As shown in Fig. 5, PCr is a small protein which is part of the CK energy shuttle; it transfers ATP from mitochondria to myofibrils. Mitochondrial CK transfers a phosphate from ATP to creatine to form PCr and ADP. PCr diffuses from the mitochondria into the myofibrils where the myofibrils isoform of CK makes ATP from PCr. Free creatine which is created from the
hypertrophy, the increased need for energy leads to decreased PCr production to maintain ATP levels at the cost of elevated levels of ADP which causes impairment of many intracellular processes. This results in the progression to heart failure with impaired contractility and loss of ATP reserves.91,92
Cardiac fibrosis
Fibrosis is the deposition of extracellular matrix (ECM) proteins such as collagens, fibronectin, MMPs and tissue inhibitors of metalloproteinases (TIMPS), which is a common feature of pathological heart conditions. In the normal heart, cardiac fibroblasts are mainly responsible for the deposition of organized ECM where cardiomyocytes and other cardiac cells are embedded. Cardiac fibroblasts have a key role in maintaining balance between degradation of old ECM and replacement with new ECM. 93
Cardiomyocytes death, such as after MI, stimulates cardiac fibrosis, through different signals such as TNF-α, TGF-ß, and endothelin 1 (ET-1) that can be secreted not only by cardiac fibroblasts, but also by macrophages, mast cells, lymphocytes, cardiomyocytes, endothelial cells. Myofibroblasts may also constitute an important source of ECM. 94
Oxidative stress
Oxidative stress is the imbalance between the reactive oxygen species (ROS) and the ability to detoxify these reactive intermediates through the activity of antioxidant systems such as superoxide dismutase (SOD), catalase and glutathione peroxidase. 95
Excessive ROS production has been associate to pathological cardiac hypertrophy and heart failure in experimental models and in patients. 96–98
There are three major ROS sources in the heart: a) the membrane-bound enzyme complex nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase, b) mitochondrial respiratory chain c) uncoupled endothelial nitric oxide synthase eNOS.
Also it was shown that pro-hypertrophic stimuli such as Angiotensin II, ET-1, catecholamines and others may induce ROS production in cardiomyocytes. 99,100 This can lead to the activation of pro-hypertrophic signaling such as ERK1/2 and NF-kß.101 ROS production by NADPH oxidase is associated with the development of pathological hypertrophy, fibrosis, reduced contractility and apoptosis. 102
Cell death in the heart (apoptosis, necrosis and autophagy)
It was shown that transition from compensated heart growth and decompensated heart growth and heart failure is strongly regulated from cell death. Of course, it is very well known that apoptosis, necrosis and autophagy have different features and cells undergo one or another mechanism depending on cell type, type of stimulus and time of insult. Mainly apoptosis leads to the fragmentation of the cell into apoptotic bodies without causing any inflammatory response103. In normal heart, regeneration is low, so apoptosis is not very common, but under pathological conditions, apoptosis rate increases and contributes to decompensated hypertrophy and heart failure 104–106. Conversely, necrosis leads to an inflammatory response since it is characterized by loss of cell membrane, swelling of organelles and cells 107. Autophagy is defined as a cellular process which is characterized by degradation and recycle of aged proteins and clear damaged organelles via a lysosomal-mediated pathway 108. Under stress conditions, autophagy seems to be up regulated with an increase in cardiomyocytes size and sarcomere remodeling 109. Autophagy is reported to protect cardiomyocytes helping degradation of ubiquitinated proteins that would otherwise accumulate in the proteasome 110. It was also shown that autophagy can protect from pathological remodeling and contractile dysfunction but also, excessive autophagy may lead to cellular dysfunction and cell death 111,112.
Typical cardiac gene expression in cardiac hypertrophy
Cardiac hypertrophy is associated to re-expression of fetal genes not usually expressed in the adult heart. Studies in animal models, but also in patients have shown an up regulation of atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), α skeletal actin mRNA 113–115. Cardiac hypertrophy has also been associated with down regulation of SERCA2a and a shift in the expression of α myosin heavy chain (α-MHC, fast contracting isoform) to ß-myosin heavy chain (ß-MHC, slow MHC isoform) 108.
Cardiac hypertrophy is a very complex process from molecular point of view. It may involve many different molecular signalings at the same time. Also it has been reported that different pathways are activated in the case of compensated or decompensated heart growth, as reported in Fig. 6.
Figure 6. A schematic view of the major signaling pathways involved in maladaptive and adaptive cardiac hypertrophy. Dashed lines indicate translocation to a different intracellular compartment. Proteins in green have been shown to be critical for adaptive cardiac hypertrophy.
Among pathways that have been reported in the compensated heart growth and beneficial effect, the most studied are:
IFG1-PI3K-Akt pathway: there is evidence that this pathway is activated in adaptive
physiological heart growth such as in postnatal heart growth and exercise-induced growth. It is also suggested that this pathway is activated under pathological conditions. It was shown that IFG1, IFG1R, PI3K and/or Akt preserve the heart function under stress conditions by activating different processes such as adaptive cardiomyocytes growth, cardiomyocytes survival, angiogenesis, attenuating fibrosis and cell death, and protecting against oxidative stress and ROS accumulation. In these processes, Akt is not always essential, but some signalings may also involve GSK3116.
CEBP/ß: it is a transcription factor involved in cardiomyocytes proliferation. PI3K-Akt
pathway attenuates expression of CEBP/ß, which is able to interact with CBP/p300 and other genes involved in cardiomyocytes proliferation such as Tbx5, Gata and Nkx2.5 117.
physiological heart growth. It was shown that PRAS40 overexpression in transgenic mice, is associated to attenuated pressure overload induced by hypertrophy and to prevent cardiac function 118.
ERK1/2: Mitogen-activated protein kinases (MAPK) are typically divided in three
subfamilies: extracellular signal-regulated kinases (ERKs), c-Jun amino-terminal kinases (JNK) and p38. Activation of ERK1/2 have been associated to both adaptive and maladaptive hypertrophic signaling. Bueno et al., showed that transgenic constitutive expression of MEK1 (upstream of ERK1/2) in mice induced an adaptive response with improved cardiac function without fibrosis 119. Another study suggests that loss of ERK1 and ERK2 from cardiomyocytes did not reduced cardiac enlargement after transverse aortic constriction (TAC)120. In seems that involvement of ERK1/2 in adaptive or
maladaptive response may be associated to activation through phosphorylation at different sites121. The other two subfamilies, JNK and p38 are usually activated in setting of stress and may contribute to pathology and transition to heart failure. 108
PKC: is a family of serine/threonine kinases and it is downstream of GPCR. There are
many PKC isoforms but just four have been reported to have a role in cardiac hypertrophy (PKCα, PKCß, PKCγ, PKCε). PKCε overexpression induced mild cardiac hypertrophy with preserved cardiac function. PKCε down regulation developed severe hypertrophy associated to cardiac dysfunction after TAC78,122. PKCα and PKCß have been reported to play a role in maladaptive hypertrophy.
Hsps and HSF1: Hsps are induced by heat shock or other stresses 123. HSF1 regulates Hsps expression and seems to have a specific role in the adaptive response and not in maladaptive. Indeed, constitutive activation of HSF1 in a mouse model developed less hypertrophy, apoptosis, fibrosis and cardiac function was preserved124. Other Hsps which have been reported to have a role in adaptive cardiac growth are Hsp70, Hsp27, HspB6/Hsp20125–127.
Gp130/JAK/STAT: this signaling may be activated by IL-6, cardiotrophin-1, leukemia
inhibitory factor, produced by cardiomyocytes in response to stress128. Role of JAK/STAT has been assumed as a key process in the early stages of protection in adaptive heart growth, by inducing anti-apoptotic genes, ROS scavengers, and promoting angiogenesis129–131. Conversely, constitutive pathway activation may lead to oxidative stress and inflammation and progress to heart failure128.
AMPK: Adenosine monophosphate-activated protein kinase (AMPK) is a key regulator
of cell metabolism in the heart. AMPK may be activated by increased AMP, ROS production, and it is phosphorylated by upstream kinases such as LKB1-STRAD-MOD25 complex and CaMKK2β132,133. Once phosphorylated, AMPK activates many downstream targets such as PGC-1α, FoxO proteins, PPAR-γ, GLUT4 to regulate cardiac energetic homeostasis. In animal models, MAPK activation was associated to mTOR inhibition and pressure overload-induced hypertrophy attenuation. MAPK down regulation in mice, resulted in severe LV hypertrophy and cardiac dysfunction.
The most investigated pathways associated with pathological hypertrophy and progression to heart failure are listed below:
GPCRs: are a family of transmembrane proteins; interaction of GPCRs with
heterodimeric G proteins leads to signaling activation. G proteins are made of three subunits: Gα (Gαq, Gαs, Gαi), Gβ and Gγ. Gαq have been reported to be involved in cardiac
hypertrophy. Instead, AngII, ET-1, α adrenergic agonists bind AT1, Endothelin receptors or α1 adrenergic receptors respectively, resulting in the activation of downstream targets,
such as PKC, phospholipase C (PLC), MAPK108. It was demonstrated that over activation of Gαq leads to heart failure development and early death, while Gαq down regulation is
associated to attenuated hypertrophy134,135.
PI3K signaling: PI3Kγ (not PI3Kα which is associated to adaptive hypertrophy) is
activated by GPCR. Its activation reduces cardiomyocytes contractility by modulating different mediators such as phosphodiesterases (PDEs) and cAMP. The role of PI3K signaling in cardiac hypertrophy is controversial: PI3K silencing in a mouse experimental model improved contractility but also increased susceptibility to myocardial injury. These animals were also protected from heart failure induced by isoproterenol, suggesting a key role of PI3K in cardiac hypertrophy and progression to heart failure.
PKC: two PKC isoforms have been associated to maladaptive hypertrophy, PKCα and
PKCβ. Braz et al., reported that PKCα contributes to myocardial dysfunction and its silencing leads to improved cardiac contractility 136,137. PKCβ have been shown to induce pathological heart growth with cardiac dysfunction, fibrosis and early death 138.
Calcineurin and CaMK: Calcineurin and CaMK are two calcium dependent proteins.
regulated in cardiac hypertrophy and heart failure patients. CaMKII overexpression is associated to cardiac hypertrophy, while its down regulation protects from pressure overload-induced hypertrophy and heart failure 140,141.
HDACs: It has been reported that mainly class I HDAC are pro-hypertrophy while class
II HDAC are negative regulators of cardiac hypertrophy. HDAC 5 or HDAC9 deletion is associated to increased hypertrophic response 142,143. HDACs will be extensively discussed later on.
Non coding RNAs in pathological hypertrophy
Protein-coding sequences constitute <2% of the human genome. The majority of sequences are mainly non coding RNA, such as miRNAs and long non coding RNAs (LncRNA).
As already established, miRNAs are 22 nucleotides which are able to repress gene expression by binding to target mRNAs. As a result, mRNA may be cleaved or lead to translation inhibition. The role of miRNAs in the myocardium is still under investigation but it is clear that miRNAs play a key role since inhibition of Dicer (an enzyme involved in the miRNA processing) leads to heart failure and premature death in a mouse experimental model 144. The first miRNA to be studied in the cardiac hypertrophy was mir-208, which was found to regulate LV cardiac hypertrophy 145. Later, is was seen that miRNAs in the heart may also play differential roles in the left and right ventricles. Other miRNAs which have been studied in the cardiac hypertrophy are expression of miR-24, miR-21, miR199a, that are up regulated in the LV of human failing hearts 146–148.
LncRNAs are defined as transcripts with 200 nucleotides or more with no evidence of protein-coding function. Unlike miRNAs, lncRNAs may lead to gene expression repression but also enhancement, as well as chromatin remodeling 149. LncRNAs, may act in cis (locally, regulating expression of close genes), but also in trans (to influence gene expression on multiple chromosomes). LncRNAs may interact with proteins (to form scaffolds) and miRNAs for an additional level of transcriptional regulation. It was shown, for example, that cardiac hypertrophy related factor (CHRF) binds and inhibits miR-489, repressing the miR-489 ability to inhibit its target mRNA, Myd88. Myd88 down regulation has been associated to cardiac hypertrophy. Yang et al., reported that approximately 500-700 lncRNAs were differentially expressed in heart failure patients and were normalized after assisted implantation, suggesting a key role of lncRNAs in the
pathogenesis of heart failure 150.
Of course it may be worth to mention that, both miRNAs and lncRNAs which are differentially expressed in cardiac hypertrophy and heart failure patients compared to healthy subjects, may constitute possible biomarkers. In table 1, lncRNAs which have been studied in cardiac hypertrophy and heart failure are listed.
Table 1. List of the LncRNAs which have been studied on cardiovascular disease.66
Current cardiac hypertrophy and heart failure therapies
The main goal of therapies for cardiac hypertrophy is to reduce premature death and patient’s hospitalization and to increase patient’s survival. Promising results have been obtained with LV assist devices and cardiac transplantation. Of course the best choice is related to drug therapy because it is more widely available and has lower costs. The most used therapeutic approaches used in the clinic are ACE inhibitors, β-Blockers and MRAs. Briefly ACE inhibitors prevent the formation of AngII and reduce pathological signaling through the AT-1 receptor. This causes relaxation of blood vessels, facilitation of salt and water excretion, and reduction in blood pressure151. ACE inhibitors improve symptoms of heart failure patients, improve heart function and reduce the risk of MI. ACE inhibitors and ARBs (which are use in ACE inhibitor intolerant patients) may have a direct effect on heart growth via inhibition of AT-1 receptors. Therefore, ACE inhibitors and ARBs are important components of standard HF therapy in patients with HF.
β-Blockers are administered to control HF symptoms (such as shortness of breath or weakness), which occur due to the release of catecholamines. β-Blockers may work by
slowing heart rate, allowing the chambers of the heart to fill more effectively and improve function of the heart, and also by decreasing blood pressure by dilating blood vessels152. Also β-Blockers and ACE inhibitors may be used together with good results in HF patients.
Mineralocorticoid receptor activation in the heart drives cardiac fibrosis and inflammation, which leads then to HF 153,154. Blocking the mineralocorticoid may represent an attractive therapeutic option. Mineralocorticoid receptor antagonists (MRAs) are used together with ACE inhibitors, ARBs and β-blockers.
Even if these therapies reduced mortality and hospitalization, mortality remains still high. Moreover, some patients may experience side effects related to these drugs. ACE inhibitors may be associated to low blood pressure, β-blockers to fluid retention, bradycardia, fatigue and worsening HF during initiation of treatment 152.
Thus, new therapeutic approaches are needed; new molecules may be designed to interfere or stimulate the complicated cascade signaling which is driving cardiac hypertrophy and progression to HF.
Angiotensin II in cardiovascular disease
Renin-Angiotensin system (RAS) has a key role in the development of CVD. Its primary effector, AngII is a key hormone which affect different organs such as heart, kidney, vasculature and brain. Acute AngII stimulation regulates salt/water homeostasis, vasoconstriction and blood pressure. Chronic AngII stimulation may lead to cardiac hypertrophy and remodeling. Basically, as shown in Fig. 7, angiotensinogen (produced in the liver) is sequentially cleaved in Angiotensin I and then in AngII. Renin, which is produced in the kidney, (PRA) and Angiotensin converting enzyme (ACE) are responsible of these two cleavages, respectively.
Figure 7. Angiotensin II synthesis. Angiotensin II is produced from Angiotensinogen through sequential cleavage by Plasma Renin Activity (PRA) and Converting enzyme (CE) enzymes. Once Angiotensin II is produced, it can play its role by binding to Angiotensin type 1 receptor (AT-1). AT-1 activation stimulates aldosterone release and promotes vasoconstriction and structural remodeling, which may also be promoted by aldosterone. AT-1 is a GPCR distributed in all organs, including heart, vasculature, kidney and brain. AT-2 also has been identified; it is a GPCR as well, and it was reported to counteract AT-1 activity since, it activates anti-proliferative and pro-apoptotic changes in endothelial cells. 155
After binding to AT-1 receptor AngII activates different pathways in distinct myocardial cell types, mainly in cardiac fibroblasts and cardiomyocytes156. In cardiac fibroblasts, AngII- AT-1interaction leads to fibroblasts proliferation, ECM proteins synthesis and deposition (such as collagen, fibronectin and laminin) 157,158. Conversely, in cardiomyocytes, AngII causes cell growth, through extracellular-signal regulated kinase (Erk) in neonatal cells, and through other stimuli e.g. α-adrenoreceptors in adult myocytes
159,160.
On endothelial cells, AngII induces several effects such as ROS production, apoptosis, and thrombosis. Importantly, in endothelial cells, AngII regulates nitric oxide (NO) production by Nitric oxide synthase (NOS). Increased AngII results in NO production leading to oxidative stress, which results in impaired endothelial relaxation and endothelial dysfunction. ROS trigger transcription of NF-kB and degradation of its inhibitor IB, leading to increase of VCAM-1, a key factor for endothelial cells adhesion. Also, it was shown that AngII stimulates expression of TNF-α, which play an important role in inflammation. Moreover, AngII inhibits fibrinolysis. This, together with over expression of adhesion molecules such as VCAM-1 and ICAM-1 (through NF-kB activation) leads to first steps for atherosclerosis development. In endothelial cells, AngII induces LDL receptor which is critical in atherosclerosis lesion formation. Thus, AngII plays a key role in the modulation of endothelial function, impaired AngII activation may
contribute to endothelial dysfunction and inflammation.
It is clear that altered AngII-mediated signaling is associated to severe consequences such as ROS production, inflammation, hypertrophy, HF, fibrosis, chronic kidney disease, etc.161
As shown in Fig. 7, the RAS system may be counteracted at different levels. The two major classes of drugs are ACE inhibitors (ACEI) and the AT-1 receptor blockers (ARBs). The ACE inhibitors block the conversion from Angiotensin I to AngII, inhibiting both AT-1 and AT-2activation. The ARBs act by antagonizing the binding of AngII to AT-1. Other possibilities to block RAS system are the use of β-blockers or direct renin inhibitors162.
Epigenetics in cardiovascular disease
After publication of 3 billion base-pair sequence that makes up the human genome 163, genomic approaches cannot explain most of inheritance, cells function or their disruption during disease. In 1968 the term epigenetics has been defined as ‘the causal interaction between genes and their products’ 164. 30 years later, Holliday and Pugh identified DNA methylation165. With the identification of histone modifications166, the world had to move
from one dimension (linear DNA sequence) to 3 dimensions, with discovery of the importance of chromatin structure in the regulation of gene expression. Later on, in 1993 also non coding RNAs (ncRNAs) and miRNAs have been discovered 167,168.
For didactic purposes epigenetic modifications are classified in DNA methylation, histone
DNA methylation
The most studied DNA methylation is cytosine methylation. This leads to the formation of CpG islands. CpG islands are defined as sequences with more than 200 bases G+C and 60% of human gene promoters are associated with CpG islands. Usually CpG islands are unmethylated in normal cells and methylated in tissue specific manner in early development or in differentiated tissues.
Usually, CpG methylation is associated with gene silencing.
DNA methylation has also been described in humans at CHG and CHH (H may be A, C or T). It has been found in stem cells and is enriched in gene bodies directly correlated with gene expression and depleted in protein binding sites and enhancers. It decreases during differentiation and is restored in induced pluripotent stem cells (IPS) role in maintenance of pluripotent state.
DNA methylation can inhibit gene expression by various mechanisms. 1) methylated DNA can recruit methyl CpG binding domain (MBD). MBD in turn recruit histone modifying complexes to methylated sites; 2) DNA methylation can also directly inhibit transcription by inhibiting the binding of other proteins to their targets. In contrast non
Figure 3. Schematic view of the main epigenetic modifications. The most common is probably DNA methylation which is represented in panel A. It may be driven by maintenance or de novo DNA methyl transferase (DNMT). In panel B, histone modifications are classified in turn in histone acetylation, methylation and phosphorylation. Then, in panel C chromatin remodeling is represented. 169
methylated sites generate a chromatin structure which allows gene expression; 3) Sometimes DNA methylation may also direct DNA activation when methylation occurs at gene bodies. It is common in ubiquitously expressed genes.
DNA methylation is mediated by DNA methyltransferase (DNMT) enzymes that catalyze the transfer of methyl group from S-adenosyl methionine to DNA. In mammals 5 members of DNMT family have been identified but just 3 possess methyltransferase activity: DNMT1, DNMT3A e DNMT3B. They are classified in de novo and maintenance DNMT. De novo are thought to be responsible for establishing methylation during embryonic development. DNMT has preference for hemimethylated DNA. DNMT1 is the most abundant in the cell and is transcribed mostly during the S phase of the cell cycle. 169
Histone Modifications
Nucleosome is made of core histones H2A, H2B, H3 and H4. Histone H1 is called linker histone; it binds the linker DNA. All histones may be subjected to post-transcriptional modifications on histone tail. Common histone modifications are acetylation, methylation, phosphorylation, ubiquitination.
Histone modifications have important role in DNA repair, DNA replication, alternative splicing and chromosome condensation. Histone acetylation occurs on Lysine residues in H2B, H3 and H4 and this is driven by a balance in the activity of Histone acetyltransferase/Histone deacetylases (HAT/HDAC).
HDACs are classified in four groups according to their function and sequence similarity. § Class I HDAC include HDAC1-2-3-8; members of this group have nuclear localization. § Class II HDAC include class IIa and class IIb HDAC. Class IIa HDAC are HDAC4-5-7-9 and class IIb HDAC are HDAC6 and HDAC10. Class II HDAC may migrate from cytoplasm to the nucleus in different tissues including heart, skeletal muscle, brain tissue and may be linked to cardiac defects 170.
§ Class III HDAC comprise Sirtuin 1-7; members of this group are NAD+ dependent. They can be found in both nucleus and cytoplasm.
§ Class IV HDAC (HDAC11) is structurally distinct from the other HDACs and can be found in both nucleus and cytoplasm.
Histone methylation which is given by an equilibrium in the activity of Histone methyltransferase/Histone demethylases (HMT/HDM), is usually associated to
transcriptional activation/repression (depending on which residue is methylated and on the methylation degree).
All histones are subject to post-transcriptional modifications, which mainly occur in histone tails.
In relation to its transcriptional state, human genome can be divided into actively transcribed euchromatine and transcriptionally inactive heterochromatine. Euchromatine is characterized by high levels of acetylation and trimethylated H3K4, H3K36 and H3K79. On the other hand, heterochromatin is characterized by low levels of acetylation and high levels of H3K9, H3K27, H4K20 methylation.
Histones can be modified at different sites simultaneously. A very complex epigenetic landscape may be generated, with several epigenetic modifications, with a cross-talk among the different marks. Communication among histone modifications can occur within the same site, in the same histone tail and among different histone tails.
Of the enzymes that modify histones, HMTs, HDMs and kinases are the most specific to individual histone subunits and residues. Conversely, most of the HATs and HDACs are not highly specific and modify more than one residue. Many transcriptional co-activators (e.g., GCN5, PCAF, CBP, p300, Tip60 and MOF) have been reported to possess intrinsic HAT activity, whereas many transcriptional co-repressor complexes (e.g., mSin3a, NCoR/SMRT and Mi-2/NuRD) contain subunits with HDAC activity. HDACs in the human genome function to reset chromatin by removing acetylation at active genes, whereas HATs, by contrast, are mainly linked to transcriptional activation.
Nucleosomes are a barrier to transcription that block access to activators and transcription factors to their sites on DNA.
Position of nucleosome on transcription start sites (TSS) has influence on transcription initiation: loss of nucleosome is related to gene transcription while occlusion of TSS by a nucleosome is associated to gene repression. The nucleosome remodeling machinery is influenced by DNA methylation and has been linked with specific modifications. 169
Chromatin remodeling
Chromatin remodeling is regulated by two classes of enzymes. The first covalently modifies histone proteins, the second are specialized multi-protein complexes that use ATP to regulate gene expression by modulating the distribution of nucleosomes on DNA to make DNA-binding sites accessible to transcription factors, which can bind to DNA
only when this is free from the nucleosome core. 169 Epigenetics in the heart
Epigenetic mechanisms, including DNA methylation, histone alterations, and RNA-based transcriptional control, can alter the cellular gene expression profile and thus promote cardiovascular pathology. Few data are available about epigenetic regulation in the healthy myocardium; in 1985, Marion et al., reported that PKA induces relaxation of H3 associated DNA segments which condense the chromatin for transcription of the structural and functional proteins supporting normal cardiomyocyte contractility 171. As already mentioned in this thesis, HDAC9 and HDAC5 (class II HDAC) have anti-hypertrophic activity. Silencing of HDAC5 and HDAC9 in experimental mice model leads to major sensibility to the development of cardiac hypertrophy and failure in responding to pro-hypertrophic stimuli. This is because these enzymes bind and inhibit MEF2c a transcriptional factor that promotes gene expression of pro hypertrophic genes143.
Also, in response to hypertrophic stimuli two calcium calmodulin dependent protein kinases CaMK and protein kinase D phosphorylate HDAC5 e HDAC9. Once phosphorylated HDAC5 e HDAC9 bind to 14-3-3 protein, a chaperone protein that transport HDAC from nucleus to cytoplasm. This causes separation of MEF2c from HDAC5 e HDAC9 and they can interact with p300 an histone acetyl transferase that promotes transcription172.
HDAC4 is also involved in cardiac hypertrophy. HDAC4 is regulated by oxidation/reduction. When it is oxidized it moves from nucleus to cytoplasm inhibiting pro-hypertrophic genes. On the other hand when HDAC4 is reduced, it does not migrate into the cytoplasm, allowing pro-hypertrophic genes to be transcribed173.Also HDAC2 (class I HDAC) has a role in cardiac hypertrophy: down regulation of HDAC2 in a mouse model confers more resistance to pro-hypertrophic stimuli; by contrast, mice overexpressing HDAC2 are over sensitive to this stimulus 174. Class I HDAC was sufficient to block cardiac hypertrophy induced by AngII infusion and TAC 175,176. In general, class I HDAC are considered mediators of cardiac hypertrophy, while class II HDAC are anti-hypertrophic. Mainly, HDAC have been found to interact with MEF2 and other proteins implicated in the regulation of genes associated to cardiac hypertrophy and HF 175,177.
with co-activators and co-repressors. SWI/SNF includes Brahma (Brm) or Brg1 ATPase which has a critical role in the heart development 178. Recently, it was reported that the interaction between Brg1 and the DNA-binding protein Parp1 is responsible for the shift in the different MHC isoforms expression in pathological cardiac hypertrophy 179. As already mentioned, cardiac hypertrophy is associated to re-expression of fetal genes ANP, BNP, β-MHC and down regulation of α-MHC and SERCA2a (Fig. 9); this differential expression in diseased and healthy heart appears to be driven by interaction of Brm to the promoter of ANP and BNP leading to expression activation; binding of Brg1 to the promoter of α-MHC results in gene suppression 179–182 .
Figure 9. HDAC inhibition attenuates the expression of genes that encode proteins implicated in cardiac hypertrophy. The expression of ANP, BNP, and β-MHC genes is increased, whereas SERCA2 and α-MHC genes are decreased. HDAC inhibition attenuates cardiac hypertrophy by restoring the expression of these genes.183
HDAC inhibition is of course of great interest in the clinic. It was already shown that trichostatin A (TSA) blocks cardiac hypertrophy mediated by transverse aortic binding and calcium signaling as well as β-adrenergic agonists such as isoprotenerol 176,184,185. Unfortunately, it has been shown that HDAC inhibition not only affect the cardiomyocytes but also other non muscle cells in the heart, leading to inflammatory response activation 186.
Maspin, an inhibitor of histone deacetylase
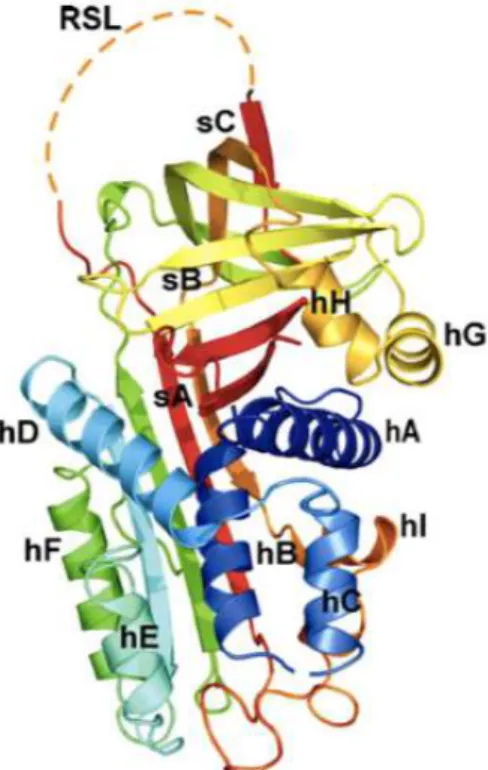
Maspin, also called SERPIN B5, is a member of serine proteases inhibitors superfamily (SerPIn). This family contains both inhibitory and non inhibitory members. Maspin is a non inhibitory serpin.
In general, serpin members share the same basic structure. They are characterized by reactive center loop called RCL (Reactive Site Loop), a flexible region found on the top