INDICE
RIASSUNTO... - 3 - INTRODUZIONE ... - 4 - 1. LA 3-IODOTIRONAMINA (T1AM)... - 4 - 1.1 SINTESI E METABOLISMO... - 5 - 1.2 RECETTORI DI T1AM... - 7 - 1.3 TRASPORTO INTRACELLULARE ... - 7 -1.4 EFFETTI DELLA SOMMINISTRAZIONE DI T1AM ... - 8 -
2. LA TECNOLOGIA MICROARRAY ... - 13 -
2.1 APPLICAZIONI DELLA TECNOLOGIA MICROARRAY... - 13 -
2.3 PRINCIPI DELLA TECNOLOGIA MICROARRAY DUAL-COLOR ... - 14 -
2.4 TIPOLOGIE DI SUPPORTI MICROARRAY ... - 16 -
Microarray a cDNA ... - 16 -
Microarray ad oligonucleotidi ... - 16 -
3. SCOPO DELLA TESI... - 17 -
MATERIALI E METODI ... - 18 -
1. ANIMALI E TRATTAMENTO CON T1AM... - 18 -
2. ESTRAZIONE DELL’RNA... - 18 -
3. QUANTIFICAZIONE, VERIFICA DELLA PUREZZA E DELL’INTEGRITA’ DELL’RNA ... - 19 -
4. DISEGNO SPERIMENTALE ADOTTATO PER L’ESPERIMENTO MICROARRAY - 22 - ... - 22 -
5. VETRINI UTILIZZATI ... - 22 -
6. AMPLIFICAZIONE E MARCATURA DELL’RNA... - 22 -
7. QUANTIFICAZIONE DELL’ aRNA E VERIFICA DELL’EFFICIENZA DI MARCATURA... - 25 -
8. FRAMMENTAZIONE E IBRIDIZZAZIONE ... - 26 -
9. LAVAGGI POST-IBRIDIZZAZIONE... - 27 -
10. ACQUISIZIONE DELL’IMMAGINE DEL VETRINO ED ANALISI DEI DATI. - 28 - 11. INTERPRETAZIONE BIOLOGICA DELLA LISTA DI GENI DIFFERENZIALMENTE ESPRESSI ... - 29 -
Valutazione della purezza e dell’integrità dell’RNA estratto... - 32 -
Valutazione dell’efficienza di amplificazione e di marcatura dell’RNA ... - 33 -
Analisi microarray ... - 34 -
DISCUSSIONE ... - 37 -
CONCLUSIONI ... - 44 -
BIBLIOGRAFIA ... - 45 -
RIASSUNTO
Gli ormoni tiroidei, tiroxina (T4) e il suo derivato triiodotironina (T3), giocano un ruolo chiave
nella regolazione del metabolismo e, dato che provocano una riduzione del peso corporeo aumentando la velocità metabolica, sono utilizzati per curare l’obesità. Gli effetti collaterali ad essi associati, quali tachicardia e aritmia, ne limitano però l’utilizzo solo ai casi di obesità dovuta ad ipotiroidismo.
La 3-iodotironamina (T1AM), un metabolita endogeno dell’ormone tiroideo che deriva dalla
decarbossilazione e successiva deiodinazione di T4, potrebbe rappresentare una valida
alternativa agli ormoni tiroidei nella cura dell’obesità, poiché incrementa il metabolismo dei lipidi senza effetti cardiotossici. Nel cuore isolato di ratto, infatti, T1AM induce una riduzione
della portata e della frequenza cardiaca. Nel criceto, la somministrazione di T1AM determina
una diminuzione del quoziente respiratorio, il che significa un aumento del catabolismo lipidico e una riduzione del catabolismo glucidico. L’utilizzo dei lipidi a scapito dei glucidi è confermato anche dalla comparsa di chetonuria. Inoltre, una significativa perdita della massa grassa corporea in seguito alla somministrazione di T1AM indica che i lipidi precedentemente
accumulati sono mobilizzati e utilizzati. Questi risultati suggeriscono che, in risposta a T1AM,
le richieste energetiche vengono soddisfatte dal metabolismo dei lipidi piuttosto che da quello dei carboidrati.
Obiettivo di questa tesi è stato identificare i meccanismi molecolari che possano spiegare il ruolo di T1AM nella modulazione del metabolismo dei lipidi. A tal fine, mediante la
tecnologia microarray è stata confrontata l’espressione genica del tessuto adiposo sottocutaneo di otto ratti a cui è stato somministrato cronicamente T1AM e otto ratti non
trattati di controllo.
Sulla base dei risultati ottenuti, T1AM sembra influenzare l’espressione di alcuni geni
associati con il re-uptake e il metabolismo dei lipidi, così come di geni implicati nel processo dell’adipogenesi, suggerendo che gli effetti fenotipici osservati nell’animale dopo somministrazione di T1AM possano essere dovuti, almeno in parte, ad un’azione diretta
INTRODUZIONE
1. LA 3-IODOTIRONAMINA (T1AM)
La 3-iodotironamina (T1AM) è una tironamina la cui sintesi e proprietà biologiche sono state descritte per la prima volta nel 2004 dal gruppo di Thomas S. Scanlan, dell’Oregon Health and Science University, in collaborazione con il gruppo del Prof. Riccardo Zucchi dell’Università di Pisa (Scanlan et al. 2004). Scanlan e collaboratori, utilizzando T1AM sintetico come standard in saggi di cromatografia liquida accoppiata a spettrometria di massa in tandem, dimostrarono che T1AM è un componente endogeno degli estratti di amine biogeniche provenienti dal cervello, dal fegato, dal cuore e dal sangue di roditore. Le tironamine (Figura 1) sono derivati della tiroxina (T4) e delle iodotironine.
Figura 1. Struttura e nomenclatura delle tironamine (TxAM). La x indica il numero di atomi di iodio presenti nella molecola.
La via biosintetica delle tironamine non è, ad oggi, ancora ben caratterizzata, ma poiché esse differiscono da T4, e dai suoi derivati per deiodinazione, solo per l’assenza del gruppo
carbossilico sulla catena laterale della β-alanina, si ipotizza che proprio la decarbossilazione della catena laterale della β-alanina sia un passaggio fondamentale nella loro biosintesi (Figura 2). L’enzima endogeno in grado di decarbossilare le iodotironine non è stato, però, ancora identificato (Wu et al. 2005).
1.
1.1 SINTESI E METABOLISMO
Ruolo delle deiodinasi nella biosintesi di T1AM
La deiodinazione per via enzimatica gioca un ruolo chiave nell’attivazione dell’ormone tiroideo dalla sua forma latente T4, alla sua forma attiva, T3 (Köhrle 2002, Bianco and Kim
2006). Il pathway di attivazione di T4 è mediato da due isoenzimi, la deiodinasi di tipo I
(Dio1) e la deiodinasi di tipo II (Dio2). T4, inoltre, è convertito in rT3 dalla deiodinasi di tipo
III (Dio3) (Huang 2005) secondo un putativo meccanismo di deattivazione.
Piehl e collaboratori, nel 2008, hanno dimostrato che le tironamine sono substrati efficienti delle tre deiodinasi Dio1, Dio2 e Dio3 (Piehl et al. 2008). In particolare questi autori hanno osservato che la reazione di deiodinazione che coinvolge le tironamine possiede un rapporto
Figura 2. Struttura chimica delle tironine e delle tironamine. T4 è convertito sia in T3 che in rT3 dalle deiodinasi indicate. T1AM ha lo stesso scheletro chimico di T4 e teoricamente può essere prodotto attraverso la
Vmax/Km comparabile a quello caratteristico della reazione di deiodinazione della tiroxina. Inoltre, è stato osservato che la selettività delle deiodinasi per le tironamine differisce da quella per le iodotironine. Per esempio, T4AM non è un substrato di Dio1 e Dio2 e non può
essere deiodinato in T3AM. Al contrario, T4AM è un ottimo substrato di Dio3 ed è quindi
rapidamente deiodinato in rT3AM. La deiodinazione sequenziale di rT3AM da parte degli
isoenzimi Dio1 e Dio2 è in grado di generare T1AM (Piehl et al. 2008). Il processo
isoenzima-selettivo appena descritto suggerisce l’esistenza di una specifica via per la biosintesi di T1AM
(Figura 2). Le molecole di ingresso di questa ipotetica via biosintetica sarebbero T4AM e
T3AM, a loro volta originati dalla rispettiva decarbossilazione di T4 e rT3. Tuttavia, sebbene la
logica che presuppone l’esistenza di questa via sia molto interessante, la relazione esatta che intercorre tra l’ormone tiroideo e T1AM non è stata ancora definita in maniera chiara. In
secondo luogo, benché T1AM sia chiaramente considerato un derivato chimico endogeno di
T4, non sono ancora state individuate prove certe in vivo di questa relazione.
La 3-iodotironamina (T1AM) come substrato delle sulfotransferasi (SULT)
Le tironamine sono substrati anche delle sulfotransferasi (SULT). Le sulfotransferasi sono enzimi in grado di catalizzare la solfatazione di numerosi composti endogeni, inclusi i neurotrasmettitori monoamminici e gli ormoni tiroidei (Pietsch, Scanlan and Anderson 2007). Il metabolismo delle tironamine via solfatazione è stato ipotizzato essere un possibile meccanismo per la loro deattivazione. Nel 2006, Pietsch e collaboratori, in uno studio riguardante le tironamine come substrati delle sulfotransferasi del fegato, hanno osservato che le sulfotransferasi SULT1A2, SULT1A3, e SULT1E1 esibivano la loro maggiore attività nei confronti di T1AM (Pietsch et al. 2007). Nell’uomo, nel topo e nel ratto, il tessuto cardiaco e
cerebrale sono bersagli noti dell’azione di T1AM (Saba et al. 2010). E' stato osservato che
questi tessuti, una volta trattati con T1AM, ne attivano la solfatazione (Pietsch et al. 2007). Il
significato potenziale dell’azione delle SULT sull’attività di T1AM potrebbe essere
l’attenuazione, e la conseguente regolazione, degli effetti da esso indotti.
Conversione di T1AM in acido 3-iodotiroacetico
Nel 2009 Wood e collaboratori hanno osservato che, dopo incubazione con cellule HepG2 o con omogenati di tessuto tiroideo umano, T1AM veniva convertito in acido 3-iodotiroacetico. Questa conversione risultava molto efficiente sia in vivo che in vitro (Wood et al. 2009). L’enzima responsabile di questa conversione è tuttora sconosciuto, ma due ragionevoli candidati potrebbero essere la monoamina ossidasi o l’ossidasi sensibile alla semicarbazide
(Wood et al. 2009). La conversione di T1AM nel corrispettivo acido tiroacetico iodinato potrebbe rappresentare un meccanismo di deattivazione, simile alla solfatazione.
1.2 RECETTORI DI T1AM
T1AM è stato dimostrato essere un potente agonista di TAAR1, un recettore delle ammine “in traccia” accoppiato a proteine G (Zucchi et al. 2006, Hart et al. 2006). T1AM, infatti, è in grado di attivare TAAR1 di ratto e di topo con valori di EC50 rispettivamente di 14 e 112 nM. Ulteriori studi focalizzati sul legame tra TAAR1 e T1AM hanno identificato l’esatto
farmacoforo di T1AM per il legame con TAAR1 e la sua attivazione (Hart et al. 2006,(Tan et al. 2007, Tan et al. 2008, Snead et al. 2008) Nelle cellule beta del pancreas, inoltre, è stato dimostrato che T1AM interagisce con i recettori α2 adrenergici producendo un effetto iperglicemico (Regard et al. 2007).
1.3 TRASPORTO INTRACELLULARE
Nel 2009, Ianculescu e collaboratori dimostrarono l’esistenza di un trasporto intracellulare specifico per T1AM. Con un nuovo metodo di screening tramite RNAi in cellule HeLa,
ponendo come bersaglio l’intera superfamiglia dei trasportatori di soluti (SLC) (composta da 403 geni), questi autori identificarono otto trasportatori, che una volta inattivati mediante knockout, causavano una riduzione del trasporto intracellulare di T1AM. E’ possibile che
questi trasportatori partecipino in maniera collettiva alla regolazione della concentrazione intracellulare di T1AM (Ianculescu, Giacomini and Scanlan 2009). Il trasporto di T1AM
potrebbe essere mediato anche da trasportatori non-SLC, soprattutto considerando che vengono scoperti continuamente nuovi geni che codificano per trasportatori. Alternativamente, potrebbe essere coinvolto un meccanismo di trasporto completamente indipendente. Per esempio, la megalina è stata identificata come recettore endocitico per l’assorbimento cellulare degli ormoni steroidei, inclusi la vitamina D, gli androgeni e gli estrogeni e si è visto che può agire come recettore per un’ampia varietà di ligandi (Lin and Scanlan 2005). Il meccanismo di trasporto di T1AM potrebbe quindi dipendere da
trasportatori di membrana specifici oppure da meccanismi di endocitosi mediata da recettore. E’ stato ipotizzato che i meccanismi di trasporto intracellulare possano servire alla regolazione dell’azione di T1AM seguendo schemi diversi. Ad esempio, l’azione di T1AM
attraverso i suoi ligandi extracellulari, come TAAR1, potrebbe essere terminata dal suo ingresso nella cellula, in maniera simile al meccanismo di re-uptake dei neurotrasmettitori
monoamminergici. D'altro canto, l'ingresso nella cellula potrebbe servire per fornire a T1AM
l’accesso ai suoi bersagli intracellulari, come il trasportatore monoamminico vescicolare VMAT2, inibito da T1AM, o, in alternativa, potrebbe consentire a T1AM di svolgere funzioni
intracellulari ancora sconosciute (Ianculescu et al. 2009).
1.4 EFFETTI DELLA SOMMINISTRAZIONE DI T1AM
Effetti sul cuore
Nel tessuto cardiaco di ratto, l’analisi quantitativa delle tironamine endogene ha rivelato che la concentrazione media di T1AM è di 68 pmol g-1, simile a quella di epinefrina, dopamina e
adenosina (Ganguly et al. 1986, Milanés, Fuente and Laorden 2000), e più alta, rispettivamente di 20 e 2 volte di quella di T4 e T3 (Schröder-van der Elst and van der Heide
1990).
Nel 2007 Chiellini e collaboratori caratterizzarono gli effetti di T1AM su preparati di cuore di ratto isolato. In tale modello fisiologico, l’aggiunta di concentrazioni micromolari di T1AM
provoca la riduzione della gettata e del battito cardiaci (Chiellini et al. 2007, Scanlan et al. 2004). Questi risultati sono in linea con quelli ottenuti somministrando T1AM direttamente
agli animali per via intraperitoneale (Scanlan et al. 2004). Gli stessi autori hanno osservato, inoltre, che, sempre in risposta a T1AM, anche la pressione aortica ed il flusso coronarico
diminuivano, seppure in maniera meno marcata rispetto alla gettata cardiaca. Si può parlare quindi di effetti inotropi e cronotropi negativi di T1AM sul cuore.
Chiellini e collaboratori hanno, inoltre, osservato che la risposta a T1AM si sviluppa entro
tempi brevissimi (30 secondi) ed in maniera prontamente reversibile. Questi due aspetti sono in accordo con un meccanismo non trascrizionale, mediato dalla stimolazione di un recettore GPCR. In generale, nel tessuto cardiaco, gli effetti di T1AM sono, quindi, opposti a quelli
indotti, in tempi più lunghi, dall’ormone tiroideo. Sempre nello stesso studio, Chiellini e collaboratori hanno osservato che gli effetti derivanti dall’aggiunta di T1AM erano evidenti
anche in cardiomiociti isolati suggerendo che questi ultimi possano essere i bersagli diretti dell’azione di questa tironamina.
Oltre a TAAR1, noto come target di T1AM, altri recettori della famiglia TAAR sono espressi nel cuore di ratto. Tra questi T1AM sembra essere in grado di legarsi a TAAR3, TAAR4 e TAAR8a in maniera specifica e saturabile, con una bassa costante di dissociazione (Chiellini et al. 2007). Tuttavia, non essendo disponibili antagonisti selettivi per i vari sottotipi TAAR,
non è possibile distinguere tra i diversi siti di legame potenziali presenti in questi recettori. In generale, gli esperimenti funzionali eseguiti da Chiellini e collaboratori suggeriscono l’esistenza di un nuovo sistema aminergico capace di modulare la funzione cardiaca. In particolare, nei preparati di cuore isolato di ratto non è stato osservato nessun cambiamento dei livelli di cAMP in risposta a T1AM. Gli effetti funzionali di T1AM, inoltre, sono risultati opposti a quelli indotti da agenti che sono noti essere in grado di aumentare le concentrazioni tissutali di cAMP. Tuttavia, pur considerando queste osservazioni, non è da escludere che l’aumento di cAMP tissutale possa avvenire lo stesso, attivando vie di segnale non ancora caratterizzate, capaci di avere effetti inotropi e cronotropi sul cuore. Un’altra ipotesi verosimile è che l’attivazione di TAAR1 possa associarsi ad altre vie del secondo messaggero che non portano a variazioni della concentrazione cellulare di cAMP. Infine, un’ulteriore ipotesi è che il recettore che media gli effetti di T1AM nel cuore non sia TAAR1, ma bensì un altro TAAR. Quest’ultima ipotesi è coerente con l’espressione preferenziale di TAAR8a osservata nei preparati di cuore isolato di ratto (Chiellini et al. 2007).
Effetti sul trasporto delle monoammine
T1AM è una molecola normalmente presente a livello cerebrale ed essendo strutturalmente
simile ai neurotrasmettitori monoamminici, è stato ipotizzato che possa interagire con i loro trasportatori (Ianculescu and Scanlan 2010).
Nel 2007 Snead e collaboratori osservarono che T1AM era in grado di inibire il trasporto
delle monoammine, sia a livello vescicolare che di membrana plasmatica. In particolare, osservarono che T1AM è un inibitore non competitivo sia dei trasportatori di membrana della
dopamina e della norepinefrina, che dei trasportatori vescicolari di monoamine. La capacità inibitoria di T1AM nei confronti di questi trasportatori è simile a quella dei normali substrati
endogeni, ma non si sa se T1AM sia presente, a livello delle sinapsi o nei neuroni del sistema
nervoso centrale, in quantità necessaria ad inibire il trasporto monoamminico. L’inibizione di T1AM è specifica per le catecolamine, poiché la stessa molecola non ha mostrato la medesima
attività inibitoria sui trasportatori della serotonina (Snead et al. 2007). Alla luce di queste considerazioni è possibile che la modulazione del trasporto delle monoammine possa contribuire agli effetti di ipotermia e riduzione della efficienza cardiaca osservati in vivo, così come, in alternativa, che T1AM, attraverso TAAR1, agisca in maniera sinergica con le monoammine nel determinare gli effetti suddetti.
Effetti iperglicemici
Nel 2007 Regard e collaboratori osservarono che T1AM può agire come regolatore del
rilascio di insulina (Regard et al. 2007). La somministrazione intraperitoneale di 50 mg/kg di T1AM, infatti, incrementava il livello ematico di glucosio e diminuiva quello di insulina sia in
topi wild type che Cre-negativi. In particolare, dopo 2 ore dalla somministrazione di T1AM, la
glicemia raggiungeva il 250% del valore basale, mentre, dopo 8 ore, tornava ai livelli di base. Al contrario, dopo 2 ore dall’iniezione di T1AM, l'insulinemia scendeva del 40% rispetto al
valore basale e la somministrazione di insulina esogena, a quel punto, portava ad una rapida discesa dei livelli di glucosio nel sangue. Quest’ultima osservazione, in particolare, dimostra che i tessuti periferici rimangono sensibili all’insulina dopo il trattamento con T1AM. La
somministrazione di T1AM causa anche l’aumento ematico del glucagone. Ciò è in accordo
con la riduzione T1AM-indotta del rilascio di insulina da parte delle isole pancreatiche,
mentre l’iperglicemia stessa probabilmente promuove la secrezione di glucagone da parte delle cellule α (Gromada et al. 2004).
Regard e collaboratori focalizzarono la loro attenzione sulla funzione di T1AM rispetto ai
membri della famiglia Gi e dei recettori ad essi accoppiati. Utilizzando una linea di topi
ingegnerizzati (RIP-PTX), è stata fatta esprimere nelle cellule pancreatiche delle isole β la tossina pertussis (PTX), che è in grado di disaccoppiare le proteine della famiglia Gi dai
GPCR ad esse accoppiati. L’espressione di PTX nelle cellule β genera topi iperinsulinemici con un elevato rilascio di insulina indotto dal glucosio. Nei topi ingegnerizzati, l’aumento della tolleranza al glucosio e della resistenza al diabete indotto dalla dieta suggerisce l’importanza, nella regolazione del metabolismo del glucosio, della via di segnale che coinvolge le proteine Gi e i GPCR delle cellule β. T1AM, infatti, non ha effetti sui livelli di
insulina nei topi RIP-PTX, mentre inibisce in vitro il rilascio di insulina glucosio-indotto in cellule provenienti da isole pancreatiche umane e di topo, effetto che viene bloccato dal pretrattamento delle stesse cellule con PTX (Regard et al. 2007). Questi risultati suggeriscono che T1AM sia in grado di inibire la secrezione di insulina attivando un recettore delle cellule β
accoppiato a Gi. Utilizzando oociti di Xenopous come sistema di espressione, non è stato
osservato accoppiamento di TAAR1 alla proteina Gi. (Regard et al. 2007). Quest’ultimo dato
ha portato gli autori ad esplorare l’ipotesi alternativa che T1AM potesse attivare Gi ed inibire
la secrezione di insulina attraverso un recettore delle cellule β diverso da TAAR1. Poiché T1AM possiede caratteristiche chimiche simili a quelle delle catecolamine, i ligandi classici
pancreatiche, Adra2a è stato considerato un possibile candidato per il ruolo di mediatore associato a Gi, degli effetti di T1AM sulle cellule β. Esperimenti di "binding" hanno
dimostrato che T1AM è in grado di legare Adra2a con un’affinità simile a quella
dell’epinefrina. A conferma di ciò, la co-somministrazione di yohimbina (antagonista di Adra2a) e di T1AM inibiva gli effetti iperglicemici indotti in condizioni normali dal solo
T1AM. Inoltre, T1AM non causava iperglicemia in topi knockout per il gene Adra2a. Questi
dati suggeriscono in maniera chiara che, nelle cellule β, gli effetti inibitori di T1AM sul
rilascio di insulina in vivo siano mediati da Adra2a in modo Gi dipendente (Regard et al.
2007).
Ulteriori studi sul ruolo di TAAR1 nella risposta della cellula β nei confronti di T1AM,
hanno utilizzato la linea cellulare MIN6 di insulinoma, in cui i livelli di espressione del recettore α2A adrenergico e di TAAR1 sono invertiti rispetto alle cellule β. L’esposizione delle
cellule MIN6 a T1AM provocava un aumento della secrezione di insulina, ulteriormente
incrementato dal trattamento con PTX o yohimbina. Ciò suggerisce che T1AM può stimolare
la secrezione di insulina attraverso la via di segnale che coinvolge TAAR1 e la proteina Gs e
inibire la secrezione di insulina attraverso il recettore α2A adrenergico e la proteina Gi. Gli
effetti derivanti dall’attivazione di TAAR1, quindi, sembrano dominare nelle cellule MIN6, nelle quali l’espressione di TAAR1 è maggiore di quella del recettore α2A, mentre nelle
cellule β in vivo, dove il recettore α2A è molto più espresso rispetto a TAAR1, gli effetti del
recettore α2A predominano (Regard et al. 2007). Topi knockout per il recettore α2A sviluppano
ipoglicemia dopo somministrazione di T1AM, a conferma che gli effetti dell’attivazione di
TAAR1 sulla secrezione di insulina sono visibili solo in assenza del recettore α2A (Regard et
al. 2007).
Effetti sulla quantità di cibo ingerito
Uno studio recente si è occupato degli effetti di T1AM sulla quantità di cibo ingerita dai
roditori (Dhillo et al. 2009). Poiché T1AM è presente nel cervello e TAAR1 è espresso nel
nucleo ipotalamico, è stato ipotizzato che T1AM possa giocare un ruolo importante nel
regolare l’omeostasi energetica. E’ noto, infatti, che alcuni nuclei ipotalamici, come il nucleo paraventricolare ed il nucleo arcuato, siano importanti nella regolazione dell’appetito e del bilancio energetico. Nello studio di Dhillo e collaboratori, la quantità di T1AM somministrata
era marcatamente inferiore rispetto a quella utilizzata nella maggior parte degli studi pubblicati precedentemente. Un’iniezione intraperitoneale di 1.42 µg Kg-1 di T1AM era,
animali, mentre non aveva alcun effetto sul tasso metabolico o sull’attività locomotoria. Anche la somministrazione intraventricolare diretta di 0.043-0.43 µg Kg-1 di T1AM nel
nucleo arcuato dei ratti incrementava in maniera significativa la quantità di cibo ingerita, portando gli autori a concludere che T1AM è un composto oressigeno, che può agire
attraverso il nucleo arcuato per indurre aumento del consumo di cibo nei roditori (Dhillo et al. 2009).
Effetti sul metabolismo
Negli ultimi anni, diversi studi si sono concentrati sugli effetti di T1AM sul metabolismo e sull’utilizzo delle riserve energetiche.
In uno studio effettuato da Braulke e collaboratori è stato visto che una singola dose (50 mg Kg-1) di T1AM era in grado di modificare il metabolismo energetico aumentando il catabolismo lipidico a scapito di quello glucidico (Braulke et al. 2008). In particolare, questi autori osservarono, sia nel criceto siberiano che nel topo, una modifica del quoziente respiratorio (QR) dai valori normali (0.90 per il criceto, 0.83 per il topo), derivanti da un uguale utilizzo di lipidi e carboidrati, a valori di circa 0.7, derivanti invece da un utilizzo quasi esclusivo delle riserve lipidiche. In linea con queste osservazioni, il trattamento dei due modelli animali con T1AM provocava chetonuria ed una significativa perdita di grasso
corporeo.
Lo spostamento dei valori di QR dopo trattamento con T1AM avviene più lentamente rispetto all’ipotermia, alla bradicardia e alla iperglicemia. Questi tre effetti si manifestano, al massimo, dopo un’ora dall’iniezione di T1AM, mentre il cambiamento dei valori di QR viene raggiunto 4.5 ore dopo l’iniezione. Nei criceti siberiani il contenuto di chetoni nelle urine raggiunge un picco massimo a 16 ore dall’iniezione di T1AM, mentre ipotermia, bradicardia e iperglicemia tornano ai valori basali dopo 4-5 ore (Braulke et al. 2008). La riduzione del QR, indice di un diminuito metabolismo glucidico, dura per diverse ore dopo il ripristino del normale tasso metabolico e della temperatura corporea ottimale, indicando che un po' di tempo è necessario per riconvertire il macchinario metabolico dall'utilizzo esclusivo dei lipidi a quello del glucosio come principale fonte energetica. Sembra, quindi, che T1AM sia un ormone endogeno che rallenta il metabolismo mediante un'interruzione rapida dell'utilizzo dei carboidrati, accompagnata da un aumento compensatorio dell'utilizzo dei lipidi.
2. LA TECNOLOGIA MICROARRAY
La tecnologia dei microarray è stata ideata ed utilizzata per la prima volta, in studi di espressione genica, intorno alla metà degli anni ’90 (Schena et al. 1995, Schena 1996). Da allora ha attratto l’interesse di molti ricercatori, si è diffusa rapidamente in vari laboratori di ricerca biologica e biomedica ed è stata perfezionata e adattata ad esigenze diverse (Patel et al. 2005). Questa enorme diffusione della metodica è dovuta al fatto che con i microarray, in un unico esperimento, ossia attraverso una singola analisi, è possibile ottenere un’istantanea dell’attività di tutti i geni che la cellula esprime in un determinato momento della propria vita o come risposta ad un determinato stimolo esterno. Su un supporto di materiale vario (es. vetro o plastica) sono fissate, in posizioni note ben precise, sequenze oligonucleotidiche a singolo filamento (le sonde o probe), capaci di riconoscere ed ibridizzarsi a sequenze complementari (i bersagli o target) preparate a partire dall’RNA messaggero dei campioni in esame. Le sequenze target sono marcate con una (tecnologia one–color) o più molecole (tecnologia dual-color) fluorescenti (fluorocromi), che segnalano l’avvenuta ibridazione sul vetrino e permettono di quantificare l’abbondanza del target ibridato e quindi il livello d’espressione del gene corrispondente (Duggan et al. 1999).
2.1 APPLICAZIONI DELLA TECNOLOGIA MICROARRAY
Data la loro enorme potenzialità, i microarray si sono rivelati molto utili nella ricerca di base e, da qualche anno, cominciano ad avere le prime applicazioni cliniche.
Nel settore oncologico, ad esempio, lo studio dell’espressione genica mediante microarray ha permesso di identificare classi-geniche tumore specifiche, evidenziando l’elevato livello di individualità dei vari sottogruppi tumorali (Alon et al. 1999, Golub et al. 1999, Alizadeh and Staudt 2000, Ross et al. 2000)). Ad esempio, ad oggi è possibile valutare la probabilità di ricorrenza del cancro al seno attraverso un test di laboratorio, il Mammaprint (http://www.agendia.com/pages/mammaprint/21.php). Questo test valuta, nel tessuto delle
pazienti asportato chirurgicamente, il profilo di espressione di 70 geni associati con i meccanismi di proliferazione, invasione, metastasi e angiogenesi ed è in grado di predire se le pazienti presentano un rischio alto o basso di ricorrenza della malattia.
In campo farmacologico, i microarray sono applicabili allo studio di variazioni dell’espressione genica che spesso accompagnano il trattamento con i farmaci. Lo studio dell’effetto dei farmaci sull’espressione dei geni consente di ottenere informazioni utili per individuare i target molecolari e quindi comprendere il meccanismo d’azione dei farmaci, i loro possibili effetti collaterali e la differente risposta al trattamento osservabile in pazienti diversi (Yun et al. 2010, Kemmel et al. 2010, Ju et al. 2009, Mei et al. 2006, McQuillin, Rizig and Gurling 2007).
Nel campo delle malattie infettive, i microarray sono utili nella comprensione del meccanismo molecolare di azione degli agenti patogeni, dal momento che consentono di identificare l’insieme dei geni che si attivano a seguito di un’infezione (Hossain and Chakraborty 2006).
2.3PRINCIPI DELLA TECNOLOGIA MICROARRAY DUAL-COLOR
Nei microarray di tipo “dual color” le sequenze target originate dai due campioni oggetto del confronto vengono marcate con due tipi distinti di fluorocromi, che emettono a lunghezza d’onda diversa, e ibridizzate, contemporaneamente, sullo stesso supporto contenente le sonde di interesse, l’array. Una volta avvenuta l’ibridazione, il segnale di fluorescenza, in corrispondenza di ciascuna posizione dell’array, viene indotto ed acquisito mediante uno scanner ad alta precisione. Lo scanner possiede un doppio laser, cioè produce due raggi di luce con lunghezze d’onda corrispondenti ai picchi d’eccitazione dei due fluorocromi. Vengono quindi eseguite due scansioni con la generazione di due immagini, una per ciascun fluorocromo: nella prima la fluorescenza di un fluorocromo (es. Cy3) è visualizzata in verde, nella seconda la fluorescenza dell’altro fluorocromo (es. Cy5) è visualizzata in rosso (Figura 3).
L’ immagine composita, data dalla sovrapposizione delle due immagini singole, viene processata in modo da “ripulire” il segnale dal fondo o background e normalizzare i dati ottenuti, eliminando l’influenza di variabili confondenti, quindi viene determinata l’intensità dei segnali su ciascuno spot e la prevalenza dell’uno o dell’altro colore. Questi dati vengono tradotti in un valore di espressione differenziale, espresso solitamente come logaritmo in base 2 del rapporto tra l’intensità registrata per il campione trattato e l’intensità registrata per il campione di controllo (valore M):
Il rapporto I trattato / I controllo è definito Fold Change (FC) e rappresenta la differenza
d’espressione del gene in esame nei due campioni a confronto. Il valore M sarà uguale a 0 per geni non differenzialmente espressi, maggiore o minore di 0 rispettivamente per geni sovraespressi o sottoespressi nel campione trattato.
Il passo successivo consiste nella determinazione della significatività del valore così calcolato. Questa fase avviene mediante l’utilizzo di diversi modelli statistici, come l’ANOVA (ANalysis Of VAriance, (Kerr and Churchill 2001b, Kerr and Churchill 2001a) o SAM (Significance Analysis of Microarray, (Tusher, Tibshirani and Chu 2001).
Uno dei problemi legati all’utilizzo di due fluorocromi diversi è rappresentato dal fatto che le due molecole possono funzionare con efficienza diversa. In questo caso è opportuno normalizzare il segnale pianificando a monte l’utilizzo di disegni sperimentali (Es. Balalnced Block Design o Dye-swap design) che prevedano l’inversione dei fluorocromi tra i due insiemi di sequenze target dei trattati e dei controlli (Dobbin et al. 2005, Dabney and Storey 2007)
Figura 3. Esempio di un esperimento di dual color microarray, dalla preparazione dell’ array, che sfrutta metodi automatizzati, alla rilevazione del segnale fluorescente dei due fluorocromi. Questo viene acquisito mediante uno scanner a doppio laser, che produce due immagini distinte per ciascun fluorocromo. Queste vengono sovrapposte per il calcolo dell’espressione relativa di ciascun gene (immagine da Duggan et al., 1999).
2.4 TIPOLOGIE DI SUPPORTI MICROARRAY
In base alla lunghezza delle probe utilizzate, i microarray possono essere suddivisi in: microarray a cDNA e microarray ad oligonucleotidi.
Microarray a cDNA
Le probe sono molecole di cDNA, di lunghezza compresa tra le 200 e le 2000 basi che corrispondono a sequenze espresse, ottenute da librerie di cDNA; sono amplificate mediante PCR e successivamente depositate su vetrino.
Questo tipo di microarray è detto a bassa densità perché il numero di geni che può contenere è compreso tra 100 e 500.
Microarray ad oligonucleotidi
Le probe sono sequenze oligonucleotidiche sintetizzate direttamente sulla superficie del vetrino. Appartengono a questa tipologia i microarray ad oligonucleotidi corti di Affymetrix e i microarray ad oligonucleotidi lunghi di Agilent.
Sia i microarray ad oligonucleotidi lunghi, sia quelli ad oligonucleotidi corti sono detti ad alta densità perché possono contenere fino a decine di migliaia di geni.
Microarray ad oligonucleotidi corti
Gli oligonucleotidi sono lunghi circa 25 basi e sono sintetizzati direttamente su vetrino. Per ciascun gene, come probe, sono utilizzati più di 40 oligonucleotidi disegnati in una regione che ha la minore omologia di sequenza possibile con altri geni. Venti di questi oligonucleotidi sono perfect matches (PM), cioè perfettamente complementari all’mRNA del gene, mentre gli altri venti sono mismatches (MM), cioè identici agli oligo PM eccetto che per la posizione centrale in cui un nucleotide è stato sostituito dal suo complementare. La presenza degli oligonucleotidi MM permette di riconoscere le ibridazioni aspecifiche e di distinguere dal rumore di fondo anche i segnali più deboli, aumentando così sia la specificità che la sensibilità del sistema. La lunghezza di 25 basi delle sequenze oligonucleotidiche le rende
facilmente accessibili per l’ibridazione. L’alta specificità dei risultati ottenuti, nonostante l’utilizzo di oligonucleotidi molto corti, è dovuta alla presenza degli MM probe, mentre l’elevata sensibilità deriva dal fatto che per ciascun gene sono presenti fino a 20 probe PM.
Microarray ad oligonucleotidi lunghi
Gli oligonucleotidi sono lunghi 60 basi e sono sintetizzati direttamente su vetrino. Per ciascun gene, come probe, sono utilizzati centinaia di oligonucleotidi identici disegnati in una regione con la minor omologia di sequenza possibile con altri geni. La lunghezza di 60 basi delle sequenze oligonucleotidiche, rende il processo di ibridizzazione tra probe e target altamente specifico senza inficiare la cinetica di ibridazione.
3. SCOPO DELLA TESI
La stretta relazione chimica tra T1AM e la forma attiva dell'ormone tiroideo T3, sia in termini di struttura che di processamento enzimatico, insieme al perdurarepiù a lungo nel tempo degli effetti di T1AM sul metabolismo, rispetto, ad esempio, agli effetti iperglicemici e bradicardici, fa supporre che l'azione di T1AM, come quella di T4/T3, possa determinare dei cambiamenti nei profili di espressione genica, responsabili dei fenotipi metabolici osservati.
Allo scopo di verificare questa ipotesi, oggetto di questa tesi è stato lo studio dell'espressione genica differenziale tra tessuto adiposo di ratti trattati con T1AM e quello di ratti non trattati.
Poiché un'azione in grado di modificare il trascrittoma difficilmente emerge con trattamenti in singola dose, i ratti utilizzati per l'esperimento microarray sono stati sottoposti a più iniezioni intraperitoneali di T1AM, secondo uno schema farmacologico adeguato.
Il lavoro di questa tesi rientra in un progetto di ricerca più ampio, che prevede lo studio dell'espressione genica differenziale anche nel tessuto cardiaco ed epatico degli stessi ratti trattati, allo scopo di comprendere meglio, da un punto di vista molecolare, i processi metabolico-funzionali che vedono coinvolto T1AM.
MATERIALI E METODI
1. ANIMALI E TRATTAMENTO CON T1AM
Gli animali usati in questo studio sono ratti Wistar di sesso maschile del peso di 100-125 grammi.
Otto ratti sono stati trattati con la molecola T1AM, alla dose di 10mg/Kg, per via
intraperitoneale due volte al giorno per cinque giorni consecutivi. Contemporaneamente, altrettanti ratti di controllo sono stati sottoposti allo stesso numero di iniezioni intraperitoneali senza T1AM.
I ratti sono stati, poi, decapitati e il tessuto adiposo è stato rapidamente prelevato dall’interno-coscia e immediatamente immerso in azoto liquido. I campioni di tessuto sono stati conservati a -80°C fino al loro utilizzo.
2. ESTRAZIONE DELL’RNA
L’RNA totale è stato estratto da tessuto adiposo di ratto utilizzando il kit RNeasy® Lipid Tissue (Qiagen, Valencia, CA, USA).
RNeasy® Lipid Tissue
I campioni di tessuto congelati, di peso pari a circa 500mg, sono stati frantumati in ghiaccio secco e, da ciascuno sono stati presi circa 30mg che sono stati immediatmente immersi in 1ml di QIAzol Lysis Reagent. Al fine di ridurre la viscosità del lisato, prodotta dalla rottura delle cellule, i campioni sono stati omogeneizzati con il TissueLyser (Qiagen, Valencia, CA, USA) ad una frequenza di 20Hz per 10 minuti.
Successivamente, in accordo con quanto suggerito dal protocollo del kit, a ciascun campione omogenato sono stati aggiunti 200µl di cloroformio e la miscela è stata centrifugata a 4°C per 15 minuti a 12.000 xg. In questo passaggio il campione si separa in tre fasi di cui quella superiore, contenente gli acidi nucleici, è stata trasferita in un nuovo tubo a cui è stato aggiunto 1 volume di EtOH al 70%. La miscela così ottenuta è stata caricata su una colonnina di purificazione e centrifugata per 15 secondi a 10.000 rpm per permettere il legame dell’RNA alla membrana della colonna e separarlo così dal DNA.
Successivamente, per eliminare eventuali residui di DNA, è stata eseguita una digestione su colonna con DNasi: dopo aver lavato la membrana con 350µl di Buffer RW1, 80 µl della miscela costituita da 10µl di DNasi e 70µl di Buffer RDD è stata caricata direttamente sulla membrana della colonnina, che è stata lasciata a temperatura ambiente per 15 minuti. La DNasi è stata eliminata aggiungendo alla colonna 350µl di Buffer RW1.
Infine, l’RNA è stato lavato due volte con 500µl di RPE a cui, precedentemente, erano stati aggiunti 4 volumi di EtOH assoluto. Al primo lavaggio il campione è stato centrifugato per 15 secondi a 10.000 rpm e al secondo lavaggio per 2 minuti a 10.000 rpm.
Dopo aver accuratamente gettato il liquido residuo, il campione è stato nuovamente centrifugato per 1 minuto a 13.000 rpm per eliminare tutti i residui di EtOH. L’RNA è stato eluito con 50µl di acqua nucleasi-free mediante centrifugazione per 1 minuto a 10.000 rpm.
3. QUANTIFICAZIONE, VERIFICA DELLA PUREZZA E DELL’INTEGRITA’ DELL’RNA
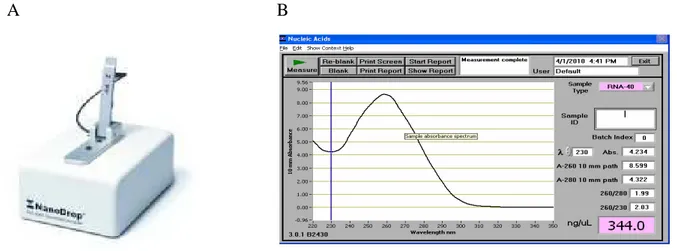
Per verificare la resa dell’estrazione e la purezza del campione ottenuto, 1µl di RNA estratto è stato misurato allo spettrofotometro Nanodrop ND-1000 (NanoDrop Technologies, Inc. Wilmington, Del, USA) (Figura 4A) utilizzando il programma “Nucleic Acid Measurements”. La quantità del campione è stata valutata mediante lettura dell’assorbanza a 260 nm; il grado di purificazione rispetto alle proteine, mediante il rapporto delle assorbanze 260/280 e quello rispetto ad altri contaminanti organici, mediante il rapporto delle assorbanze 260/230 (Figura 4.B). I rapporti di assorbanza 260/280 e assorbanza 260/230 maggiori di 2 sono indice di un RNA sufficientemente libero da proteine e da altre sostanze organiche.
A B
Figura 4 .A: Spettofotometro Nanodrop ND-100; B: Rappresentazione grafica delle lettura delle assorbanze a 260, 280 e 230.
Per verificare l’integrità dell’RNA è stato utilizzato il Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA, USA) (Figura 5.A) in combinazione con il kit RNA 6000 Nano LabChip (Agilent Technologies, Palo Alto, CA, USA). Un µl di RNA estratto da ciascun campione è stato caricato sull’RNA 6000 Nano chip (Agilent Technologies, Palo Alto, CA, USA) (Figura 5.B) insieme ad un RNA di controllo. In maniera del tutto simile alla classica corsa elettroforetica, nel chip i frammenti di RNA migrano all’interno di una matrice polimerica guidati da un gradiente elettrico e si separano tra loro in base al peso molecolare. Un fluoroforo contenuto nella matrice polimerica si intercala all’RNA e un raggio laser induce l’emissione di fluorescenza. Il software 2100 Expert (Agilent Technologies, Palo Alto, CA, USA ), associato allo strumento, quantifica l’informazione in formato digitale generando un elettroferogramma (Figura 6) in cui la fluorescenza emessa dai vari frammenti nucleotidici è messa in relazione con il loro tempo di migrazione. Dal confronto dell’elettroferogramma del campione in esame con quello dell’RNA di controllo, il software calcola un parametro numerico, il RIN (RNA Integrity Number) che è indicativo dell’integrità dell’RNA (Schroeder et al. 2006). Questo parametro varia da 1 a 10: un RIN = 1 è indice di RNA completamente degradato, mentre un RIN = 10 è indice di RNA perfettamente integro.
Figura 5.A: Bioanalyzer Agilent 2100; B: Chip utilizzato per la separazione dei frammenti di RNA. Con un solo chip è possibile verificare contemporaneamente l’integrità di 12 campioni di RNA: il chip contiene 16 pozzetti dei quali 3 sono usati per il caricamento del gel (G), per il caricamento del campione di controllo e i restanti 12 per il caricamento dei campioni da analizzare. Il chip è costituito da microcanali che creano una rete di connessione tra i 16 pozzetti. I microcanali, prima del caricamento dei campioni, sono riempiti con una matrice polimerica contenente una molecola fluorescente. Una volta che i canali ed i pozzetti sono stati riempiti il chip si trasforma in un circuito elettrico integrato. Caricati tutti i campioni, il chip è inserito all’interno del bioanalyzer e ogni pozzetto del chip collimerà perfettamente con i 16 elettrodi posti nella parte interna del coperchio del bioanalyzer, attraverso i quali avverrà il passaggio di corrente.
B
Figura 6. Elettroferogramma rappresentativo di un RNA con RIN=8.
4. DISEGNO SPERIMENTALE ADOTTATO PER L’ESPERIMENTO MICROARRAY
L’esperimento microarray è stato eseguito utilizzando un “Balanced Block Design”. Sia per la classe dei controlli che per quella dei trattati, metà dei campioni è stata marcata con Cy3 e l’altra metà con Cy5. Su ogni array è stato ibridizzato un campione marcato con Cy3 appartenente alla classe dei controlli e un campione marcato con Cy5 appartenente alla classe dei trattati o viceversa, per un totale di 8 array (Figura 7).
5. VETRINI UTILIZZATI
Sono stati utilizzati vetrini “Whole Rat Genome 4x44K” (AgilentTechnologies, Palo Alto, CA, USA ) sui quali sono depositate 44.000 sonde oligonucleotidiche lunghe 60 basi che rappresentano 41.015 trascritti di ratto.
6. AMPLIFICAZIONE E MARCATURA DELL’RNA
L’RNA estratto dal tessuto adiposo di ratto è stato amplificato e marcato utilizzando il kit Two-Color Microarray-Based Gene Expression Analysis (Agilent Technologies, Palo Alto, CA, USA).
Two-Color Microarray-Based Gene Expression Analysis kit
Il protocollo prevede i seguenti passaggi: - preparazione Spike-In A e B
- denaturazione dell’RNA e sintesi di cDNA a doppio filamento
- amplificazione e marcatura dell’RNA con Cianina 3 (Cy3) e Cianina 5 (Cy5)
- purificazione dell’RNA amplificato e marcato. Figura 7. Disegno sperimentale "Balanced Block Design".
Preparazione Spike- In A e B
Gli Spike-In A e B sono due controlli positivi costituiti da una miscela di trascritti di RNA virale: ogni Spike-In contiene quantità differenti di 10 trascritti di RNA poliadenilato, sintetizzati in vitro e derivanti dal trascrittoma di Adenovirus E1A ( Tabella 1). Questi controlli sono amplificati e marcati insieme all’RNA del campione di interesse: lo Spike-In A è unito al campione di RNA che sarà marcato con Cy3 mentre lo Spike- In B è unito al campione di RNA che sarà marcato con Cy5. Al momento dell’ibridizzazione, i due campioni di interesse, contenenti i rispettivi Spike-In, saranno mescolati insieme in uguale quantità e i due Spike-In si ibridizzeranno con le sonde ad essi complementari presenti sul microarray. Il rapporto logaritmico delle intensità dei segnali rosso e verde, per ogni trascritto degli Spike-In, sarà usato per monitorare il processo di amplificazione, di marcatura e di ibridizzazione dei campioni.
In accordo con quanto dettato dal protocollo, gli Spike-In sono stati mescolati vigorosamente con un vortex e sono stati denaturati a 37°C per 5 min.
Poiché per l’amplificazione-marcatura sono utilizzati 1000ng di RNA totale (vedi paragrafo successivo), ciascun Spike-In è stato diluito 3200 volte attraverso tre diluizioni scalari: 1:20-1:40 e 1:4 ( Tabella 2). Nell’esperimento è stata utilizzata la diluizione 1:4.
Denaturazione dell’RNA e sintesi di cDNA a doppio filamento
La quantità di RNA richiesta per l’amplificazione è compresa nell’intervallo 50-5000ng. In questo esperimento sono stati utilizzati 1000ng di RNA per ciascun campione. Dopo aver unito all’RNA (1000ng) 2µl della diluizione 1:4, precedentemente preparata, degli Spike-In (A o B a seconda del fluoroforo utilizzato nella successiva reazione di marcatura) e 1,2µl di primer oligo-dT, è stata aggiunta acqua nucleasi-free per portare il volume a 11,5µl. La miscela è stata incubata a 65°C per 10min e successivamente posta in ghiaccio per 5min per denaturare il campione e consentire l’appaiamento dei primer oligo-dT alla coda di polyA degli mRNA.
Successivamente, sono stati aggiunti 8,5µl di cDNA Master Mix contenente 4µl di 5x First Strand Buffer, 2µl di DTT 0,1M, 1µl dNTP mix (dATP, dCTP, dGTP e dTTP 10mM ciascuno), 1µl di MMLV-RT (300U/µl ) e 0,5µl di RNaseOut e la miscela è stata incubata a 40°C per 2 ore. In questa fase si forma cDNA a doppio filamento poiché l’enzima MMLV-RT è polifunzionale: è una DNA polimerasi-RNA dipendente che sintetizza il primo filamento di cDNA, complementare all’mRNA ed è anche una DNA polimerasi-DNA dipendente che sintetizza il secondo filamento di cDNA. Terminate le 2 ore a 40°C, il campione è stato incubato a 65°C per 15min, per inattivare l’enzima MMLV-RT.
Tabella 2. Diluizioni scalari degli Spike-In in base alla quantità iniziale di RNA totale utilizzata.
Amplificazione e marcatura dell’RNA con Cianina 3 (Cy3) e Cianina 5 (Cy5)
Per la reazione di amplificazione e marcatura, alla precedente miscela, sono stati aggiunti 60µl di Transcription Master Mix costituita da 20µl di 4x Transcription Buffer, 6µl di DTT 0,1M, 8µl di NTP mix, 6,4µl di PEG 50%, 0,5µl di RNaseOUT, 0,6µl di Inorganic pyrophosphatase, 0,8µl di T7 RNA polymerase e 2,4µl di CTP-Cy3 o CTP-Cy5. La miscela è stata incubata a 40°C per 2 ore. In questa fase la polimerasi T7 sintetizza e contemporaneamente marca con Cy3 o Cy5 numerose molecole di RNA antisenso (aRNA).
Purificazione dell’RNA amplificato e marcato
In accordo con quanto suggerito dal protocollo di Agilent, la purificazione dell’aRNA marcato è stata eseguita utilizzando le colonnine di purificazione e i buffer di lavaggio del kit RNeasy Mini (Qiagen, Valencia, CA, USA).
A ciascun campione sono stati aggiunti 20 µl di acqua nucleasi-free per portare il volume a 100 µl e, successivamente sono stati uniti 350 µl di Buffer RLT e 250 µl di etanolo al 100%. La miscela è stata trasferita nell’apposita colonnina di purificazione e centrifugata per 30 sec a 13.000 rpm.
La colonnina è stata lavata due volte con 500 µl di RPE a cui, precedentemente, erano stati aggiunti 4 volumi di EtOH assoluto. Al primo lavaggio il campione è stato centrifugato per 30 sec a 13.000 rpm e al secondo lavaggio per 1 min a 13.000 rpm. Dopo aver accuratamente gettato il liquido residuo, il campione è stato nuovamente centrifugato per 30 sec a 13.000 rpm per eliminare tutti i residui di EtOH. L’RNA è stato eluito con 30 µl di acqua nucleasi-free mediante centrifugazione a 13.000 rpm per 1 min. In questa fase sono rimossi i sali e i fluorofori non legati all’aRNA.
7. QUANTIFICAZIONE DELL’ aRNA E VERIFICA DELL’EFFICIENZA DI MARCATURA
Per verificare la resa dell’amplificazione e l’efficienza di marcatura, 1 µl di RNA amplificato e marcato è stato misurato allo spettrofotometro NanoDrop ND-1000 (NanoDrop Technologies, Inc. Wilmington, Del, USA) (Figura 4.A) utilizzando il programma “Microarray Measurements”.
La quantità di acido nucleico ottenuta è stata calcolata mediante lettura dell’assorbanza a 260nm e l’efficienza di marcatura mediante lettura dell’assorbanze a 555nm (lunghezza d’onda alla quale assorbe il Cy3) e a 647nm (lunghezza d’onda alla quale assorbe il Cy5). I valori di assorbanza ottenuti permettono di calcolare i nanogrammi di aRNA, le picomoli di nucleotidi totali e le picomoli di fluorocromo presenti nel campione. Le formule che si utilizzano sono le seguenti :
ng aRNA = (A260) x 37(mg/ml)-1 cm-1 (coefficiente di estinzione molare medio dei nucleotidi per l’RNA) x vol
pmoli nucleotidi TOT= (A260 x vol x 37(mg/ml)-1 cm-1 (coefficiente di estinzione molare medio dei nucleotidi
per l’RNA) x 1000 (fattore di conversione da nmol a pmol)/ 324 g/mol(peso molare di un nucleotide)
pmoli Cy3 = (A550) x vol / 0,15µl pmol-1 cm-1 (coefficiente di estinzione molare Cy3(ε)
pmoli Cy5 = (A650) x vol / 0,25ml pmol-1 cm-1 (coefficiente di estinzione molare Cy5(ε)
dove vol rappresenta il volume totale del campione espresso in µl. Tali formule si ricavano dalla legge di Lambert e Beer secondo la quale l’assorbanza (A) è proporzionale alla concentrazione (C) della sostanza assorbente, allo spessore dello strato attraversato (cammino
ottico, l) e al coefficiente di estinzione molare (ελ):
A = ελCl
I valori di assorbanza registrati dal Nanodrop devono essere moltiplicati per 10 per compensare la differenza di cammino ottico rispetto ad un comune spettrofotometro.
Il rapporto (R) tra le picomoli di nucleotidi totali e le picomoli di nucleotidi marcati è un indice dell’efficienza della marcatura.
8. FRAMMENTAZIONE E IBRIDIZZAZIONE
L’RNA amplificato è stato frammentato, prima dell’ibridizzazione sui microarray ad oligonucleotidi, per migliorare la cinetica della reazione.
La frammentazione è stata eseguita in un volume di 55µl così composto: 825ng di aRNA marcato con Cy3, 825ng di aRNA marcato con Cy5, 11µl di 10x Blocking Agent, 2,2µl di 25x Fragmentation Buffer e acqua nucleasi-free. Questa miscela è stata incubata a 60°C per 30 min esatti. Dopodichè la reazione è stata bloccata con 55 µl di 2x GEx Hybridization Buffer HI-RPM.
Il tempo di incubazione rappresenta un fattore critico per la dimensione dei frammenti che si ottengono.
Dal momento che sui vetrini utilizzati sono presenti quattro array, su ciascun vetrino sono state ibridizzate quattro coppie di campioni. Le miscele di ibridizzazione sono state centrifugate per 1 min a 13.000rpm e 100 µl di ognuna sono stati depositati, in successione, su una delle quattro aree, delimitate da una guarnizione, di un vetrino coprioggetto (Figura 8.A). Dopodichè, sul vetrino coprioggetto è stato posizionato il microarray con la superficie contenente le sonde a contatto con la soluzione di ibridizzazione. Il sandwich così formato è stato inserito in una cameretta di ibridizzazione (Figura 8.B) ed è stato messo a ruotare a 4rpm in una stufa a 65°C per 17h (Figura 8.C).
9. LAVAGGI POST-IBRIDIZZAZIONE
Dopo aver smontato la cameretta di ibridizzazione, i vetrini sono stati separati dai vetrini coprioggetto in una soluzione di SSPE 6X e Triton X-102 0.005% e sono stati lavati, in successione, con una soluzione di SSPE 6X e Triton X-102 0.005% per 1 min, quindi con una soluzione di SSPE 0.06X e Triton X-102 0.005% preriscaldata a 37°C , per 1 min; infine sono stati immersi 10 sec in Acetonitrile quindi, nella Stabilization and Drying Solution per 30 sec. L’immersione del vetrino nella Stabilization and Drying Solution ha lo scopo di proteggere i fluorofori, in particolare il Cy5, dal decadimento del segnale provocato dall’ozono, in quanto questa soluzione contiene un composto, dissolto in Acetonitrile, che
Figura 8 A: Vetrino coprioggetto; B: Cameretta di ibridizzazione ; C: Stufa di ibridizzazione.
cattura l’ozono. Appena tolti dalla Stabilization and Drying Solution i vetrini sono stati sottoposti a scansione laser.
10. ACQUISIZIONE DELL’IMMAGINE DEL VETRINO ED ANALISI DEI DATI
L’immagine del vetrino è stata acquisita mediante lo scanner a due laser Agilent G2565BA (Agilent Technologies, Palo Alto, CA, USA) ad una risoluzione di 5µm e con una modalità di scansione denominata XDR (eXtended Dynamic Range). L’XDR è un algoritmo di doppia scansione degli array a due differenti guadagni del fotomoltiplicatore. La prima scansione viene effettuata al 100% del guadagno mentre la seconda al 10% del guadagno; infine, l’algoritmo di quantizzazione dei dati produce un file unico nel quale convergono le informazioni provenienti da entrambe le scansioni.
L’immagine, in formato TIFF, è stata analizzata mediante il software Feature Extraction 10.5 (Agilent Technologies, Palo Alto, CA, USA), che consente di effettuare il controllo di qualità dell'esperimento. Successivamente, l’immagine è stata sottoposta a sottrazione del background e normalizzazione dei segnali.
La libreria di analisi LIMMA (“LInear Model of MicroArray data”) è stato utilizzato per produrre una lista di geni differenzialmente espressi statisticamente significativi.
Per ciascun gene è stato determinato un valore di M (log2(Irosso/Iverde) e una significatività
11. INTERPRETAZIONE BIOLOGICA DELLA LISTA DI GENI DIFFERENZIALMENTE ESPRESSI
Di seguito sono descritti gli strumenti informatici utilizzati per l’interpretazione biologica dei dati microarray.
Pathway Express (http://vortex.cs.wayne.edu/projects.html).
Pathway Express è stato concepito per eseguire pathway analysis su liste geni. (Khatri et al. 2007) Una volta sottomessa una lista di geni differenzialmente espressi, Pathway Express ,facendo riferimento al database KEGG (Kyoto Enciclopedia of Genes and Genomes), è in grado di creare un elenco di pathway associati a tali geni. In particolare, questo strumento esegue una classica analisi di arricchimento, basata su una distribuzione ipergeometrica, con l’obbiettivo di identificare una serie di pathway che contengono un numero di geni differenzialmente espressi che sia significativamente diverso da quello che ci si potrebbe aspettare per effetto del caso. Questo tipo di analisi produce un set di P-value che definiscono la significatività del pathway dal punto di vista statistico (un basso valore di p-value corrisponde ad un’alta significatività). In aggiunta, Pathway Express calcola un Fattore di
Perturbazione PF(g) relativo ad ogni gene all’interno del singolo pathway. Questo fattore di
perturbazione prende in considerazione sia il fold-change normalizzato del gene che il numero di geni che si trovano a monte di esso (ovvero la sua posizione nel pathway). In pratica il fattore di perturbazione riflette l’importanza relativa di ogni gene differenzialmente espresso nel pathway di appartenenza. Infine, Pathway Express calcola un Fattore di Impatto del singolo pathway, utilizzando un termine probabilistico che tiene in considerazione il numero e la somma dei fattori di perturbazione dei geni mappati al suo interno.
Figura 9. Tipica schermata in output di Pathway Express. La schermata è organizzata in quattro pannelli. Il Pannello A riassume i risultati con un grafico a barre. Il Pannello B fornisce ulteriori dettagli relativi ad ogni pathway: (i) numero di geni nel pathway, (ii) numero di geni nell’array utilizzato che sono presenti nel pathway, (iii) numero di geni presenti nella lista sottomessa che sono nel pathway, (iv) il fattore di impatto del singolo pathway, (v) il codice identificativo del pathway, ecc. Il Pannello C fornisce informazioni sui singoli geni appartenenti alla lista fornita come imput incluso l’ID del gene, il nome ed il simbolo del gene, il numero di pathway in cui il gene è stato mappato, ed il fold-change fornito dall’utente stesso. Il doppio click sul nome del pathway nel pannello B fornisce la lista di geni mappati in quel pathway (con il loro corrispondente fattore di perturbazione) tramite il pannelo D. (immagine da Purvesh Khatri et al. 2007)
GeneCards® (www.genecards.org)
GeneCards® (Rebhan et al. 1998, Safran et al. 2002, Safran et al. 2003) è un software di navigazione che permette di avere accesso in maniera veloce e comprensibile a tutte le informazioni (genomiche, proteomiche, trascrittomiche, associazione con malattie e dati funzionali) riguardanti i geni umani conosciuti e predetti. GeneCards® unisce le informazioni su tali geni che sono contenute nei grandi database pubblici disponibili in rete come HGNC
(http://www.genenames.org/), NCBI (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene), ENSEMBL (http://www.ensembl.org/index.html), UniProtKB (http://www.uniprot.org/).
Le informazioni riguardanti il singolo gene sono organizzate in sezioni distinte con l’utilizzo di un formato “a carte”.
PubGene (http://www.pubgene.org/)
Pubgene è un database che permette di ottenere informazioni su geni e proteine di diversi organismi. La filosofia che ne sta alla base permette di definire Pubgene come un database “gene-centrico”. Geni e proteine oltre ad essere referenziati tra di loro sono cross-referenziati anche rispetto a termini in grado di definire la loro funzione biologica, la loro importanza nelle malattie e la loro relazione con sostanze chimiche. Il risultato è un “network letterario” che è in grado di organizzare le informazioni in modo da renderle facili da consultare (Jenssen et al. 2001).
OntoExpress (http://vortex.cs.wayne.edu/ontoexpress/)
Onto-Express (OE) (Draghici et al. 2003, Khatri et al. 2002) è un database di annotazioni integrato negli Onto-tools. E’ uno strumento disegnato per elaborare i dati di annotazione funzionale dei geni, disponibili in rete, allo scopo di definire un profilo funzionale della condizione sperimentale studiata.. Le tipologie di dati da sottomettere ad Onto-Express possono essere: una lista di accession numbers di GenBank, gli ID dei probe Affimetrix, o gli ID dei cluster di UniGene.
Un gene può essere incluso in una data categoria funzionale sulla base di specifiche evidenze sperimentali ( indicato come “experimented”) o sulla base di inferenze teoriche, es. similarità con una proteina avente funzione nota (indicato come “inferred”). I geni per i quali non sono disponibili informazioni vengono indicati come “non-recorded”.
Onto-Express fornisce i risultati della propria analisi in forma grafica, costruendo un profilo funzionale per ognuna delle categorie di Gene Ontology (GO): tipo di componente cellulare, processo biologico e funzione molecolare, funzione biochimica e ruolo cellulare. Poichè l’espressione di un gene può essere regolata sulla base della sua posizione sul cromosoma (es. imprinting), OntoExpress costruisce anche un profilo addizionale di localizzazione cromosomica.
RISULTATI
I ratti trattati con T1AM non mostravano alterazioni evidenti del comportamento, né
cambiamenti nella quantità di cibo assunto.
Valutazione della purezza e dell’integrità dell’RNA estratto
Ogni campione di RNA estratto è risultato di ottima qualità sia come grado di purificazione che integrità, come indicato dai rapporti di assorbanza 260/280 e 260/230 e dai valori di RIN riportati in Tabella 3.
Campioni RIN Ass 260/280 Ass 260/230
C1 9.4 2.00 2.19 C2 9.2 2.00 2.18 C3 9.3 1.99 2.18 C4 9.1 2.03 2.09 C5 9.2 2.01 2.13 C6 9.5 1.99 2.18 C7 9.3 2.00 2.23 C8 9.4 2.00 2.08 T1 9.0 2.00 2.21 T2 9.1 2.00 2.21 T3 9.1 2.00 2.00 T4 8.9 2.02 2.00 T5 9.2 2.00 2.21 T6 9.4 2.00 2.19 T7 9.1 2.19 2.00 T8 9.5 2.00 2.02
Valutazione dell’efficienza di amplificazione e di marcatura dell’RNA
L’analisi spettrofotometrica dei campioni di RNA amplificato e marcato ha rivelato una resa di amplificazione compresa tra 9-14.7 µ g totali di aRNA e un’efficienza di marcatura compresa tra 10.8-12 pmol fluoroforo /µg aRNA per il Cy3 e 13-15 pmol fluoroforo /µg aRNA per il Cy5. I campioni di aRNA ottenuti non presentavano, inoltre, contaminazione da proteine o da altre sostanze organiche, dato che i rapporti delle assorbanze 260/280 e 260/230 erano tutti maggiori di 2 (Tabella 4).
µg TOT di RNA µg TOT di aRNA Efficienza di Marcatura Ass 260/280 Ass 260/230
1µg C1 11.0 µg 15.0 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg C2 9.4 µg 11.5 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg C3 11.0 µg 15.0 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg C4 14.6 µg 11.0 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg C5 11.0 µg 14.0 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg C6 12.0 µg 12.0 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg C7 13.0 µg 13.0 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg C8 11.5 µg 11.0 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg T1 11.8 µg 12.0 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg T2 11.0 µg 15.0 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg T3 10.6 µg 11.0 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg T4 10.0 µg 14.0 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg T5 12.5 µg 11.0 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg T6 11.6 µg 14.5 pmol Cy5/ µg aRNA ≥2 ≥2
1 µg T7 14.7 µg 10.8 pmol Cy3/ µg aRNA ≥2 ≥2
1 µg T8 9.00 µg 15.0 pmol Cy5/ µg aRNA ≥2 ≥2
Tabella 4. Resa dell’ aRNA ottenuta impiegando 1µg di RNA tot, efficienza di marcatura del Cy3 e del Cy5 e grado di purificazione dell’aRNA rispetto alle proteine e ad altri contaminanti organici.
Analisi microarray
T1AM, somministrato cronicamente, ha modificato l’espressione di 378 geni: 268
sovraespressi e 110 sottoespressi. L’analisi di pathway mediante Pathway Express, ha mappato 60 dei 378 geni differenzialmente espressi in 70 pathway tra i quali l’apoptosi, il pathway del segnale delle MAPK, del segnale di mTOR, del segnale delle VEGF, dei ritmi circadiani, del segnale delle adipocitochine e del segnale dell’insulina (Tabella 5).
L’utilizzo delle informazioni presenti nelle banche dati di GeneCards® (www.genecards.org) e di PubGene (http://www.pubgene.org/) e di quelle ontologiche di OntoExpress (http://vortex.cs.wayne.edu/ontoexpress/) ha permesso di identificare numerosi altri geni implicati nell’omeostatsi del colesterolo, nel metabolismo dei lipidi e nel processo di adipogenesi (Tabella 5).
ELABORAZIONE INFORMAZIONE GENETICA ELABORAZIONE INFORMAZIONE AMBIENTALE
SISTEMI DELL’ORGANISMO MALATTIE UMANE
Fattori di trascrizione
basali Pathway del segnale mediato dal calcio
Pathway del segnale delle adipocitochine
Leucemia mieloide acuta
Riparazione basi recise Interazione tra citochine e recettori delle citochine
Presentazione e processamento
degli antigeni Morbo di Alzheimer
Riparazione non-omologa Interazione ECM-recettori Orientamento degli assoni Carcinoma delle cellule basali
Proteasoma Pathway del segnale via ErbB Pathway del segnale del recettore delle cellule B
Carcinoma della vescica
Proteolisi mediata dall’ubiquitina
Pathway del segnale via
Hedgehog Ritmi circadiani Leucemia mieloide cronica
PROCESSI CELLULARI
Pathway del segnale delle
MAPK Cascate del complemento e della coagulazione Carcinoma colorettale Giunzioni aderenti Pathway del segnale di Notch Pathway del segnale di Fc
epsilon RI
Carcinoma endometriale
Apoptosi Sistema del segnale del
fosfatidilinositolo Pathway del segnale di GnRH Glioma Ciclo cellulare Pathway del segnale delle VEGF Pathway del segnale dell’
insulina
Malattia di Huntington
Adesione focale Pathway del segnale di Wnt Migrazione transendoteliale dei leucociti
Melanoma
Giunzioni gap Molecole di adesione cellulare
(CAMs) Depressione a lungo termine
Carcinoma polmonare non a piccole cellule
Regolazione del
citoscheletro di actina Pathway del segnale Jak-STAT Potenziamento a lungo termine Carcinoma pancreatico Regolazione dell’autofagia Pathway del segnale di mTOR Melanogenesi Morbo di Parkinson
Giunzioni strette Interazione neuroattiva
ligando-recettore Citotossicità mediata da cellule natural killer Pathway del cancro Pathway del segnale di
TGF-beta
Trasduzione olfattiva Pathway del cancro
Pathway del segnale di PPAR Immunodeficienza primaria
Sistema angiotensina-renina Carcinoma della prostata
Pathway del segnale del recettore delle cellule T
Carcinoma delle cellule renali
Trasduzione del gusto Carcinoma polmonare a piccole cellule
Pathway del segnale dei recettori Toll-like
Lupus eritematoso sistemico
Carcinoma della tiroide Contrazione del muscolo
cardiaco
Tabella 6. T1AM regola l’espressione di geni implicati nell’omeostasi del colesterolo, nel metabolismo dei lipidi e nell’adipogenesi.
Funzione biologica Gene Direzione
del Fold-Change
Ldlrap1 (LDL receptor adaptor protein 1) ↑
Lrp10 (low-density lipoprotein receptor-related
protein10) ↑
ApoD (Apolipoprotein D) ↑
Scarb1 (scavenger receptor class B, member 1) ↑
Hdlbp (high density lipoprotein binding protein) ↑
Sirt6 (sirtuin 6) ↑
Omeostasi colesterolo
Osbpl5 (oxysterol binding protein-like 5) ↑
Acsl5 (acyl-CoA synthetase long-chain family
member 5) ↑
Psap (prosaposina) ↑
Pex5 (peroxisomal biogenesis factor 5 ) ↑
Adra2C (adrenergic, alpha-2C-, receptor) ↓
Metabolismo dei lipidi
G0S2 (G(0)/G(1) switch gene 2) ↓
Cebpb (CCAAT/enhancer binding protein (C/EBP),
beta) ↓
Paqr3 (progestin and adipoQ receptor family
member III ) ↑
Igfbp2 (insulin-like growth factor binding protein 2) ↓
Pmp22 (peripheral myelin protein 22) ↑
Adipogenesi