UNIVERSITÁ DEGLI STUDI ROMA TRE
Facoltà di Scienze Matematiche, Fisiche e Naturali Dipartimento di Biologia
Scuola Dottorale in Biologia
Sezione di Biologia Applicata alla Salute
dell’Uomo (BASU) - XXII
MECHANISMS UNDERLYING THE PROTECTIVE
EFFECTS OF ESTROGEN IN DIFFERENT CELLULAR
CONTEXTS
MECCANISMI ALLA BASE DEGLI EFFETTI
PROTETTIVI DEGLI ORMONI ESTROGENI IN
DIFFERENTI CONTESTI CELLULARI
To my family for its understanding, endless patience and encouragement when it was most required To Alessandro always beside me, here and across the world With all my heart
INDEX SUMMARY ... I RIASSUNTO ... IV 1. BACKGROUND ... 1 1.1OVERVIEW ON ESTROGEN ... 1 1.2 ESTROGEN RECEPTORS ... 4 1.3ER ACTION MECHANISMS ... 5
1.3.1 Nuclear signals (NS) of estrogens 1.3.2 Membrane initiated signals (MIS) of estrogens 1.4ER LOCALIZATION ... 10
1.5CELL FUNCTIONS REGULATED BY MIS ... 11
1.6ERΒ RAPID ACTIONS ... 12
1.7ER TISSUTAL DISTRIBUTION ... 14
1.8SELECTIVE ESTROGEN MODULATORS (SERM) ... 14
2. AIMS ... 16
3. ERα-MEDIATED SIGNAL TRANSDUCTION PATHWAYS IN CANCER CELL PROLIFERATION ...17
3.1INTRODUCTION ... 17
3.2RESULTS ... 19
3.2.1 Effect of E2 and naringenin on cell proliferation 3.2.2 Mechanisms underlying the ERα activities modulation by naringenin Nar decreases ERα palmitoylation Nar rapidly impairs ERα-caveolin-1 association Nar rapidly stimulates p38/MAPK Nar prevents the association of ERα with signaling protein involved in proliferation Nar rapidly stimulates p38/MAPK 3.3DISCUSSION ... 31
4. ERβ-MEDIATED SIGNAL TRANSDUCTION PATHWAYS IN CANCER CELL PROLIFERATION ...34
4.1INTRODUCTION ... 34
4.2RESULTS ... 37 4.2.1 ERβ is a palmitoylable protein
4.2.2 ERβ palmitoylation is negatively modulated by E2 4.2.3 Role of ERβ palmitoylation
ERβ palmitoylation is necessary for receptor-protein association and plasma membrane localization
ERβ palmitoylation is necessary for E2-induced signaling and pro-apoptotic effects
ERβ palmitoylation is necessary for DLD-1 cell growth decrease ERβ palmitoylation is important for E2-inducecd ERβ up regulation ERβ palmitoylation is not necessary for E2-induced transcriptional activity
4.3.DISCUSSION ... 56
5. SIGNAL TRANSDUCTION PATHWAYS ACTIVATED BY ERα AND ERβ COEXPRESSION ...60
5.1INTRODUCTION ... 60
5.2RESULTS ... 62
5.2.1 ER in skeletal muscle cells 5.2.2 E2 effects on skeletal muscle cell proliferation 5.2.3 E2 effects skeletal muscle differentiation markers 5.2.4 Mechanisms underlying the E2 effects in L6 myoblasts 5.2.5 ERα and ERβ involvement in E2-induced L6 myoblast differentiation. 5.2.6 E2 effects on C2C12 myoblasts 5.2.7 ERα and ERβ involvement in E2 effect on ROS production in L6 myoblasts. 5.3DISCUSSION ... 86
6. CONCLUSION ... 92
7. REFERENCES ... 95
ACKNOWLEDGEMENTS ...118 APPENDIX ... Material and Methods available on CD-ROM
I
SUMMARY
Estrogens, in particular 17β-estradiol (E2) the most potent estrogen in humans, play key roles in development and maintenance of normal sexual and reproductive functions. In addition, in both men and women, they exert a vast range of biological effects in the cardiovascular, musculoskeletal, immune, and central nervous systems (Gustafsson, 2003; Deroo and Korach, 2006, Heldring et al., 2007) and they are now generally thought to play protective effects against several degenerative diseases (e.g., cardiovascular and neurodegenerative pathologies). This expanded view of estrogen action reflects the findings of a large number of clinical and epidemiological studies which gathered on the effects of exogenous estrogen administration on the post-menopausal women health (Deroo and Korach, 2006).The biological actions of E2 are mediated by two estrogen receptor isoforms (ERα and ERβ) which belong to the nuclear receptor super-family (NR3A1 and NR3A2, respectively) (Ascenzi et al., 2006). The mechanisms of action of ERs are complex and involve signal pathways starting from plasma membrane that lead to protein kinase activation (membrane-initiated steroid signals, MISS), and direct or indirect transcription of target genes (nuclear mechanism, NS). These pathways seem to synergize each other to determine the overall effects of E2 in target tissues. However, if the nuclear-initiated signals of E2 have been studied for long time, the membrane-started signals of ER need more investigation to be fully understood. The specific role of rapid signals of each ER isoform in estrogen regulating cell functions is still unclear and several controversies related to their physiological relevance are still unsolved.
The overall aim of this thesis is to investigate the molecular mechanisms underlying some E2-induced protective effects evaluating the role played by each ERα- and ERβ-mediated mechanism. For this purpose we choose different experimental models: ER-devoid human cervix epitheloid carcinoma cells (HeLa) transiently transfected with the expression vector of ERα or ERβ; human hepatocellular carcinoma cell line (HepG2) which contains endogenous ERα; human colon cancer cells (DLD-1) that express only ERβ and rat skeletal myoblasts (L6) where both estrogen receptor isoforms (ERα and ERβ) are expressed.
At the present, most of finding point to the concept that ERα membrane starting pathways is mainly involved in E2-induced cell proliferation that may contribute to malignant tumor growth. However, we recently demonstrated that ERα shows anti-proliferative activities in presence of different ligands (e.g., naringenin, Nar) as well (Totta et al., 2004; Virgili et al., 2004) and the mechanisms underlying are here investigated. Present data show that ERα plasma membrane localization and
II
the ERα-caveolin-1 interaction are impaired through the modulation of ERα post-transductional modification (i.e., palmitoylation) by the nutritional compound naringenin. As a consequence, the ERα association with adaptors and/or signaling proteins [e.g., c-Src and the modulator of non genomic action (MNAR)] is prevented such as the ERα ability to activate ERK and AKT phosphorylation important for cell proliferation. On the other hand, a pro-apoptotic cascade [caspase-3 activation and and poly(ADP)-ribose polymersase (PARP) cleavage] involving the p38 activation is induced by the ERα:Nar complex. The ERα-mediated transcriptional activity of an estrogen responsive element (ERE)-containing promoter is not affected by Nar (Galluzzo et al., 2008) which decouples ERα rapid pathways from its direct transcriptional activity. Beside the knowledge of the mechanisms underlying the (anti)estrogenicity of dietary compounds such as flavonoids these data bring to light the new important role played by ERα signals. Due to the allosteric properties of this molecule, different ligands induce ER to assume different conformation responsible for specific protein-protein interaction which, in turn, drive cell to different fate.
At present, inadequate information is available on the role played by ERβ, even if its role as dominant negative of ERα proliferative activities has been reported (Paruthiyil et al., 2004; Strom et al., 2004). Besides this role, these results show that ERβ is also able to mediate specific signal transduction pathways underlying the protective effects of estrogen against cancer cell proliferation. Present results demonstrate that in DLD-1 colon cancer cells in which only the ERβ1 isoform is expressed, like ERα, ERβ undergoes the palmitoylation which allows to a small ERβ pool to localize at the plasma membrane and associate with caveolin-1 and the p38. Upon E2 stimulation, ERβ undergoes de-palmitoylation increasing receptor association to caveolin-1 and p38. The ERβ-caveolin-1 and ERβ-p38 physical association are responsible for the ERβ level increase at the plasma membrane impairing the association of signaling proteins important for ERα-mediated cell survival and proliferation (i.e., Src, ERK and AKT) (Galluzzo et al, 2007). On the other hand, the ERβ-mediated E2-induced p38 activation deeply impact on DLD-1 colon cancer cells. P38 activation is fundamental for the downstream pro-apoptotic cascade which involves the caspase-3 activation and PARP cleavage, but also for both for the rapid increase of ERβ mRNA translation and for the slow ERβ (ESR2) gene transcription (Caiazza et al, 2007). The final consequence is an increased level of ERβ in DLD-1 cells which, in the presence of E2, further increase the hormone protective effect against tumor growth.
Overall these data allow to conclude that E2 in presence of ERα or ERβ activates very different rapid signals important for the final cellular
III
fate. In particular, ERα:E2 is able to activate signal cascades addressing to cell proliferation, even if ERα is also able to exert protective effects against cancer cells proliferation in dependence on the bound ligand (Galluzzo et al., 2008). On the other hand, beside its role as negative modulator of ERα activities previously reported (Matthews and Gustafsson, 2003; Paruthiyil et al, 2004; Strom et al, 2004), ERβ is able to activate specific rapid signals started from plasma membrane which trigger a pro-apoptotic cascade. The presence of both ER isoforms, for instance in skeletal muscle cells, lead us to investigate the presence and the impact of ERα- and ERβ-dependent pathways on the final effects of estrogen in this cellular context.
Present results demonstrate that in actively proliferative rat myoblasts (L6), E2 does not increase cell proliferation or apoptosis, whereas E2 stimulates the cell differentiation increasing the expression of myogenesis markers [i.e., myogenin, myosin heavy chain (MHC), and the glucose transporter type 4 (Glut-4) translocation]. In particular, the rapid extra-nuclear signals (i.e. AKT activation) and the presence of ERα on plasma membrane are necessary and sufficient for the first step of differentiation process (Glut-4 translocation). Although ERα is the isoform less expressed in L6 cells, its contribute has been demonstrated to be significant. In addition, the use of ER specific agonists and antagonist (i.e., PPT and THC) demonstrate that in absence of ERβ activities the E2 effect is more evident, suggesting that ERβ opposes the ERα functions. On the other hand, ERβ is the receptor isoform mainly expressed and even if its contribute seems to be negligible to E2-induced differentiation, ERβ exerts important functions to protect the skeletal muscle cell from H2O2-induced oxidative stress that could occur during muscle activity (Galluzzo et al., 2009a). These data lead us to conclude that the E2 effects in cells co-expressing ERα and ERβ could not only depend on the protein expression level of ERs, but also on the balance between the signals originated by each isoform.
All together reported data argue that both ERα and ERβ activities mediate the estrogen protective effect depending on cellular context and that a dominant protective ER player in the intricate interplay among ERα and ERβ dependent signaling does not exist. Furthermore, the complexity of the mechanism of ER action suggests a more finely tuned control exerted by E2-induced rapid signals on cellular molecular events. In particular, the extra-nuclear signals induced by E2 occur before the appearance of nuclear effects and the cell context in which the genomic events occur will be different depending on which signal pathway is activated. Thus, the integration between these molecular events is required to obtain the complete cellular response.
IV
RIASSUNTO
Gli estrogeni, ed in particolare il 17β-estradiolo (E2) il più potente estrogeno presente nell’uomo, svolgono un ruolo fondamentale nello sviluppo e nel mantenimento delle normali funzioni sessuali e riproduttive, ma la loro azione si esplica, sia nell’uomo che nella donna, anche in sistemi non riproduttivi come, ad esempio, l’apparato cardiovascolare, l’apparato muscolo scheletrico, il sistema immunitario (Gustafsson, 2003; Deroo and Korach, 2006, Heldring et al., 2007). Inoltre più di recente è stato attribuito agli estrogeni un importante ruolo protettivo contro alcune patologie degenerative (e.g., cardiovascolari e neurodegenerative). Questa più ampia conoscenza sulle funzioni degli estrogeni deriva da studi epidemiologi basati sugli effetti sulla salute di donne in post-menopausa sottoposte a terapie estrogeniche (Deroo e Korach, 2006).
Le azioni biologiche esercitate da E2 sono mediate da due isoforme del recettore degli estrogeni (ERα ed ERβ) appartenenti alla superfamiglia dei recettori nucleari (NR3A1 e NR3A2, rispettivamente) (Ascenzi et al., 2006). Il meccanismo d’azione di ERα e ERβ è complesso e coinvolge vie di trasduzione del segnale che partono dalla membrana plasmatica (membrane-initiated steroid signals, MISS) e portano all’attivazione di protein-chinasi, e la trascrizione diretta ed indiretta di geni target (nuclear mechanism, NS). Questi meccanismi sembrano sinergizzare l’un l’altro per determinare l’effetto complessivo di E2 nei tessuti bersaglio, tuttavia, se i meccanismi genomici sono ormai ben noti, i segnali che partono dalla membrana plasmatica richiedono ulteriori studi per essere chiariti. Il ruolo specifico di ERα ed ERβ nel regolare le funzioni cellulari dipendenti da E2 non è completamente noto e le numerose controversie sull’importanza fisiologica ricoperta dai segnali rapid di E2 non sono ancora risolte.
Scopo di questa tesi è proprio quello di investigare i meccanismi molecolari alla base di alcuni effetti protettivi esercitati dagli estrogeni valutando il coinvolgimento dei meccanismi specifici mediati da ciascuna isoforma di ER. Per questo studio differenti linee cellulari sono state scelte come modello sperimentale: cellule umane di carcinoma alla cervice uterina (HeLa) rese responsive ad E2 in seguito a transfezione transiente con i vettori di espressione per ERα o ERβ; cellule di epatocarcinoma umano che contengono endogenamente solo il recettore ERα (HepG2); cellule umane di adenocarcinoma al colon che esprimono solo l’isoforma β di ER e mioblasti scheletrici di ratto (L6) dove sono co-espressi ERα de ERβ.
Al presente, la maggior parte dei dati a nostra disposizione riporta che i segnali rapidi mediati da ERα sono prevalentemente coinvolti negli effetti proliferativi di E2 che possono contribuire a trasformazione e crescita tumorale. Tuttavia, dati ottenuti di recente nel nostro laboratorio, hanno
V
dimostrato che ERα svolge anche attività antiproliferative in presenza di differenti ligandi (e.g., naringenina, Nar) (Totta et al., 2004; Virgili et al., 2004) ed i meccanismi alla base sono oggetto di studio di questa tesi.
I dati qui presentati dimostrano che la localizzazione di ERα e la sua associazione con la caveolina-1 sono modificate dalla molecola di origine nutrizionale Nar attraverso la modulazione della modificazione post-traduzionale di ERα (i.e., palmitoilazione). Di conseguenza, l’associazione di ERα con proteine adattatrici e di seganle [e.g. c-Src e la proteina modulatrice delle attività non genomiche di ER (MNAR)], è prevenuta così come la fosforilazione e attivazione di ERK e AKT importanti per la proliferazione cellulare. D’altra parte il complesso ERα:Nar induce una cascata pro-apoptotica che coinvolge l’attivazione della p38/MAPK, l’attivazione della caspase-3 ed il taglio proteolitico poli(ADP)-ribosio polimerasi (PARP). L’attività trascrizionale mediatada ERα di gene contententi nel promotore l’elemento di risposta agli estrogeni (ERE) non è influenzata dall naringenina (Galluzzo et al., 2008) che disaccoppia, così, le attività rapide di ERα dalla sua capacità trascrizionale diretta. Oltre alla comprensione del meccanismo alla base dell anti(estrogenicità) di composti nutrizionali come i flavonoid, questi dati portano alla luce un nuovo importante ruolo esercitato dai segnali rapidi di ERα. Date le proprietà allosteriche di questa proteina, diversi ligandi possono assumere differenti conformazioni responsabili delle specifiche interazioni protein-proteina che, a loro volta, determinano diversi destini cellulari.
Al presente, le informazioni relative al ruolo rivestito da ERβ non sono sufficienti, anche se gli è stato attribuito un ruolo come regolatore negativo delle attività proliferative di ERα (Couse and Korach, 1999; Weihua et al., 2003; Paruthiyil et al., 2004; Strom et al., 2004). I risultati di questa tesi dimostrano che accanto a questo ruolo, ERβ attiva rapidamente vie specifiche di segnale coinvolte negli effetti protettivi di E2 contro la proliferazione cellulare. In particolare, in cellule di cancro al colon (DLD-1), così come ERα, anche ERβ viene modificato post-traduzionalmente mediante palmitoilazione che consente ad una piccola frazione di ERβ di localizzarsi a livello della membrana plasmatica, associarsi alla caveolina-1 e alla p38/MAPK. La stimolazione con E2 induce la de-palmitoilazione di ERβ e l’aumento della sua associazione con la caveolina-1 e la p38. Queste interazioni protein-proteina sono responsabili dell’aumento di ERβ al livello della membrana plasmatica e prevengono l’associazione (e.g., Src e MNAR) e l’attivazione di proteine di segnale (e.g., ERK e AKT) importanti per la proliferazione mediata da ERα (Galluzzo et al, 2007). D’altra parte, l’attivazione della p38 dipendente da E2 influenza fortemente il destino cellulare delle DLD-1 inducendo una cascata pro-apoptotica (attivazione
VI
della caspase-3 e taglio proteolitico della PARP) ed essendo responsabile contemporaneamente del rapido incremento della traduzione dell’mRNA di ERβ e della lenta trascrizione del gene ESR2 di ERβ (Caiazza et al, 2007). Il complessivo incremento dei livelli proteici di ERβ amplifica ulteriormente l’effetto protettivo dell’ormone contro la crescita tumorale.
L’insieme di questi dati ci permette di concludere che E2 attiva vie di segnale diverse in presenza di ERα o ERβ che portano a proliferazione o ad apoptosi, rispettivamente. Anche se ERα è anche in grado di indurre una cascata apoptica in presenza di differenti ligandi (e.g., Nar).
La contemporanea espressione di entrambe le isoforme di ER per esempio in cellule muscolari scheletriche ci ha spinto a studiare l’impatto delle vie rapide mediate da ERα ed ERβ sull’effetto finale di E2 in questo contesto cellulare. I dati ottenuti dimostrano che in cellule muscolari in attiva proliferazione (L6) E2 non induce un incremento della proliferazione né apoptosi, mentre stimola il differenziamento agendo sui marker del differenziamento [miogenina, catena pesante della miosina (MHC), e trasportatore del glucosio di tipo 4 (Glut-4)]. In particolare le attvitità rapide di ERα (i.e., attivazione AKT) sono necessarie e sufficienti per le prime fasi del differenziamento (traslocazione sulla membrana del Glut-4). Anche se meno rappresentato, l’isoforma α di ER contribuisce in modo significativo. Inoltre, l’eliminazione delle attività di ERβ, mediante l’uso di agonisti e antagonisti selettivi di ER (i.e., PPT and THC) rende l’effetto di E2 più evidente, suggerendo ancora il suo ruolo di regolatore negativo di ERα. Tuttavia, anche se ERβ, l’isoforma maggiormante espressa, non contribuisce all’incremento dei marker del differenziamento, esercita un ruolo fondamentale nella protezione delle cellule da stress ossidativo indotto da H2O2 (Galluzzo et al., 2009a) che protebbe verificarsi durante l’attività muscolare. Questi dati ci permettono di concludere che l’effetto finale di E2 in cellule che co-esprimono ERα e ERβ dipendono non solo dai livelli proteici ma dal bilancio dei segnali rapidi attivati da ogni isoforma.
L’insieme di questi dati ci permette di sostenere che entrambi ERα e ERβ mediano gli effetti protettivi di E2 in dipendenza dal contesto cellulare e che non esiste un attore pricipale nelle intricate azioni tra ERα e ERβ. Inoltre, i segnali extra-nucleari indotti da E2 avvengono prima della comparsa degli effetti nucleari ed il contesto cellulare nel quale gli eventi genomici avvengono saranno diversi in dipendenza dalle vie rapide attivate. Di conseguenza, per ottenere una completa risposta cellulare è richiesta l’integrazione di tutti gli eventi molecolari.
1
1. BACKGROUND
1.1 OVERVIEW ON ESTROGENS
Estrogens, like other steroid hormones, are naturally occurring cyclopentanophenanthrene compounds whose synthesis begins with cholesterol. Estrogens play key roles in development and maintenance of normal sexual and reproductive function in both men and women (Heldring et al., 2007). Although, their levels are significantly higher in women of reproductive age. They are mainly produced by the ovary and in part by the adrenal cortex, and three estrogens occur naturally in the female (Ruggiero et al., 2002; Ascenzi et al., 2006).In premenopausal women, 17β-estradiol (E2), produced by the ovary granulosa, is the estrogen produced in the largest quantity and is the most potent as it has the highest affinity for its receptors. In pre-menopausal women, circulating estradiol levels fluctuate from 40 to 200-400 pg/mL during the menstrual cycle (Ruggiero et al., 2002). After menopause, estradiol levels drop to less than 20 pg/mL. The second estrogen is estrone (E1), a less potent metabolite of estradiol. Estrone is produced from androstenedione, the immediate precursor of estrone, in the liver and adipose tissue. In post-menopausal women, the ovary ceases to produce estradiol while the adrenal gland continues to produce androstenedione. As the result, the level of estrone remains unchanged while the plasma level of estradiol falls significantly. The third endogenous estrogen is estriol (E3), also a metabolite of estradiol. Estriol is the principal estrogen produced by the placenta during pregnancy, and is found in smaller quantities than estradiol and estrone in non-pregnant women(Ruggiero et al., 2002; Ascenzi et al., 2006; Chen et al., 2008).
In the target tissues such as reproductive organs, estrogens exhibit important biological functions. Among these, the ovarian cycle, the development of both sexual primary and secondary characters are dependent on estrogen (Ikeda and Inoue, 2004). Besides regulation of physiological functions in reproductive tissues, estrogens exert a vast range of biological effects and influence many other physiological processes in mammals including cardiovascular health, bone integrity, cognition, and behaviour (Gustafsson, 2003; Heldring et al., 2007).
Estrogens play important roles in bone homeostasis, being involved in modeling of bone during adolescence. They initiate pubertal growth and later limit longitudinal bone growth by inducing closure of the epiphysial growth plate. Even in adult life, sex steroids appear to influence the remodeling of bone. E2, in particular, is crucial for the maintenance of bone mass in females (Migliaccio et al., 1996) as is evident from the rapid loss of trabecular bone and development of osteoporosis that occurs after
2
ovariectomy or at menopause (Turner, 1990, 1999). Several factors, known to be important in regulating differentiation and function of osteoblasts and osteoclasts, are regulated by E2. In osteoblasts, E2 stimulates synthesis and secretion of the anabolic growth factor IGF-I and inhibits that of the cytokines, IL-1, tumor necrosis factor (TNF), and IL-6 (Roodman, 1996) that are involved in bone resorption (Nilsson et al., 2001).
Estrogen appears to be protective in the cardiovascular system, in part due to favourable modulation of serum lipid profiles (Ettinger et al., 1996; Farish et al., 1996). The 30% of these effects are due to a reduction of total cholesterol by increasing the LDL receptor (LDLr) expression in the liver (Di Croce et al., 1996; Marino et al., 2001a) and inhibiting the LDLr oxidation at vascular level (Zhu et al., 2002). Recent evidence indicates that the 70% of estrogen effect is also due to its direct action on the vasculature, as demonstrated by gender differences in smooth muscle contractility, with a greater response to noradrenaline or phenylephrine in aortas isolated from male than female rats (Stallone et al., 1991; Kanashiro et al., 2001). E2 rapidly induces the activity of endothelial nitric oxide synthase (eNOS), which produces the vasodilator nitric oxide (NO) (Kim and Bender, 2005). Furthermore, estrogens are able to prevent the proliferation and the migration of vascular smooth muscular cells (VSMC) both directly (Razandi et al., 2000) and indirectly blocking the mitogenic action of growth factors (Mendelsohn and Karas, 1999; Incerpi et al., 2003). Prior to menopause, women have a lower incidence of coronary heart disease compared with age-matched men (Van der Schouw et al., 1996; Barrett-Connor, 1997; Phillips et al., 1997). Moreover, estrogen replacement therapy reduces mortality due to coronary heart disease in post-menopausal women (Stampfer et al., 1991; Ettinger et al., 1996). Deprivation of estrogens by ovariectomy results in enhanced vascular contraction in aortas of female rats (Kanashiro et al., 2001). In ovariectomized rats, chronic estrogen replacement suppresses endothelium-dependent contractions mediated by the cyclo-oxygenase/prostaglandin H synthase pathway (Davidge et al., 1998). Studies in humans also demonstrate a beneficial vascular action of estrogen, with endothelium-dependent vasodilatation being enhanced by the administration of 17β-estradiol at physiological levels (Herrington et al., 1994; Gilligan et al., 1994a,b; New et al., 1997; Leung et al. 2007).
In central nervous system, estrogen has been found to contribute to promote synapse formation and plasticity and to limit neuronal damage and death, besides its capability to protect from neurodegenerative process. Furthermore, the 17β-estradiol plays an important role as antioxidant reducing the oxygen reactive species (ROS) by increasing the protein level
3
of tioredoxin, an important protein that protect from lipid peroxidation after oxidative stress (McCullough and Hurn, 2003; Lee et al., 2003; Hammes and Levin, 2007).
Postmenopausal women are in an estrogen-deprived state and are at risk for stroke and other neurodegenerative diseases (Garcia-Segura et al., 2001). Epidemiological evidence suggests that postmenopausal estrogen therapy (ET) reduces the risk or delays the onset of Alzheimer disease (Henderson et al., 1994; Paganini-Hill et al., 1994). Estrogen loss from natural or surgical menopause has been associated with a decline in cognitive function (Sherwin, 1997, 1998) that is reversed by ET. ET has been shown to affect cognitive function during brain aging as well (Resnick et al., 1998; Maki et al., 2001; Resnick and Maki, 2001; Simpkins and Singh, 2008).
Given this widespread role for estrogen in human physiology, it is not surprising that estrogen is also implicated in the development or progression of numerous diseases, which include but are not limited to osteoporosis, neurodegenerative diseases, cardiovascular disease, insulin resistance, lupus erythematosus, endometriosis, obesity and various types of cancer (breast, ovarian, colorectal, prostate, and endometrial). Indeed, in addition to proliferative effects on normal cells, estrogen is considered as a stimulant for the initiation and promotion of tumors in these organs (Ikeda and Inoue, 2004). This expanded view of estrogen action reflects the findings of a large number of clinical and epidemiological studies which gathered on the effects of exogenous estrogen administration on the post-menopausal women health. Based on data from both clinical and animal studies, estrogen is implicated in the development of breast cancer stimulating proliferation of mammary cells by increasing cell division and DNA synthesis (Deroo and Korach, 2006). Risk factors associated with breast cancer reflect cumulative exposure of the breast epithelium to E2 (Henderson and Feigelson, 2000; McEwan, 2004; Ascenzi et al., 2006). There is also evidence that estrogens, together with gonadotropins, contribute to the etiology of ovarian cancer in humans (Chu et al., 2000; Deroo and Korach, 2006). Whereas, clinical studies indicate that the incidence of colon cancer is lower in women than in men (Jemal et al., 2004), and data from the Women’s Health Initiative indicate a significantly reduced incidence of colon cancer in postmenopausal women receiving combined ‘Hormone Replacement Therapy’(HRT; estrogen plus progestin) (Rossouw et al., 2002; Ascenzi et al., 2006).
4 1.2 ESTROGEN RECEPTORS
The biological actions of estrogens are traditionally mediated by binding to one of two specific estrogen receptors (ERs), ERα or ERβ, that are encoded by different genes located on different chromosomes (locus 6q25.1 and locus 14q23-24.1, respectively) (Gosden et al., 1986; Enmark et al., 1997; Luisi et al., 2006; Zhou et al., 2006). ERα and ERβ, like all the members of the nuclear receptor super-family, are modular proteins sharing common regions, named A/B, C, D, and E/F, as well as a high sequence homology (Fig. 1.1). These regions participate in the formation of independent but interacting functional domains. The N-terminal domain (A/B region) is involved in both inter-molecular and intra-molecular interactions as well as in the activation of gene transcription. The DNA binding domain (DBD, C region) allows ER to dimerize and to bind to the specific ERE sequence on DNA through its two “zinc finger” structures. The hinge domain (D region) has a role in receptor dimerization and in binding to chaperone heat-shock proteins (Hsp). The ligand binding domain (LBD, E/F region, C-terminal) comprises the E2-binding domain and works, synergistically with the N-terminal domain in the regulation of gene transcription (Mosselman et al., 1996; Nilsson et al., 2001; Claessens and Gewirth, 2004; Kumar et al., 2004).
Figure 1.1: A schematic structural comparison of human ERα and ERβ functional domains. Receptor domains are illustrated with different colored boxes, and the approximate size of each domain is indicated. The A/B domain contains the ligand-independent transcriptional-activation function AF-1, the C domain represents the DNA-binding-domain (DBD), the D domain corresponds to the hinge region, and the E domain contains the hormone-binding domain (LBD) and the hormone-dependent transcriptional-activation function AF-2. The number inside each box of ERβ refers to the percentage of amino acid identity.
5
ERs contain two regions called activation functions (AFs) important for ligand-dependent transcriptional activity (Fig. 1.1) (Mosselman et al., 1996; Nilsson et al., 2001; Claessens and Gewirth, 2004; Kumar et al., 2004). AF-1 and AF-2 regions of ERs, interacting with a number of trancription co-activators, can activate transcription independently but in most cases, they synergize with one another in a promoter- and cell-context specific manner (McEwan, 2004). AF-1 could be activated even in a ligand-independent manner, depending on the phosphorylation status of ER. In particular, the Ser118 residue in the AF-1 region of ERα, as well as residues Ser106 and Ser124 in the AF-1 region of ERβ, are the phosphorylation sites essential for the ligand-independent activation of ERs through the Ras-mitogen activated protein kinase (MAPK) signaling cascade (see Ortì et al., 1992; Lannigan, 2003).
Recent progress in studies on genomic and cDNA sequences has accelerated the identification of gene splice variants in the NR super-family. Numerous mRNA splice variants exist for both ERs and the best-characterized splice variants are ERα46 and ERβcx, which are frequently co-expressed with their wild-type counterparts. The exact function and potential role of these and other ERs splice variants in physiology and human disease remain to be elucidated (see Herynk and Fuqua, 2004; Marino et al., 2006a).
1.3 ER ACTION MECHANISMS
The mechanisms of ERα and ERβ action are complex pathways that involve two distinct types of signaling which lead to protein kinase activation (rapid membrane-initaited mechanism) and direct or indirect transcription of target genes (nuclear mechanism) (Fig. 1.2). All these pathways synergize each other to determine the overall effects of E2. 1.3.1 Nuclear signals (NS) of estrogens
In the “classical” mechanism of action, estrogens diffuse into the cell membrane and bind to ERs causing ERs to dissociate from heat shock proteins, to dimerize and to traslocate into the nucleus. The nuclear ERα- or ERβ-E2 complex directly binds DNA through the ERE (estrogen responsive element) sequences or indirectly through protein-protein interactions with activator protein-1 (AP-1) or stimulating protein (Sp-1), resulting in recruitment of coregulatory proteins (coactivators or corepressors) to the promoter, increased or decreased mRNA levels, protein synthesis, and physiological responses (Ascenzi et al, 2006; Deroo and Korach, 2006). A large subset of coregulatory proteins (e.g., steroid receptor coactivator-1, 2, and 3) helps the hormone-receptor complex to recruit histone
6
acetyltransferases and methyltransferases which, in turn, possess chromatin-remodeling ability and tether activated receptors to the basal transcriptional machinery (Smith and O’Malley, 2004). Both ERα and ERβ are capable of regulating gene transcription through this classical mechanism involving ERE. ERβ seems to be a weaker transactivator (Cowley and Parker, 1999). AF-1 activity of ERβ is weak compared with that of ERα on ERE, whereas their AF-2 activities are similar (Cowley and Parker, 1999). In turn, when both AF-1 and AF-2 functions are active in a particular cell and/or on a particular promoter, the activity of ERα greatly exceeds that of ERβ, whereas ERα and ERβ activities are similar when only AF-2 is required (McInerney et al, 1998; Cowley and Parker, 1999; Ascenzi et al, 2006). It has been postulated that differences in the ERα and ERβ activities are due to differences in the ability of the receptors to interact with coregulatory proteins, because of the low amino acid identity in A/B domain of ERs (Figure 1.1) (Smith and O’Malley, 2004; Ascenzi et al, 2006).
Only a fraction of the known mammalian EREs reflects the consensus palindromic element ERE (GGTCAnnnTGACC), initially described based on the ERE in the Xenopus laevis vitellogenin A2 promoter (Klein-Hitpass et al., 1986; Ponglikitmongkol et al., 1990). For instance, for the 38 estrogen-responsive genes reviewed by Klinge (Klinge, 2001), most of the functional EREs located within the promoters or 30-untranslated regions are not the traditional consensus sequence. Thus, many target genes contain response elements that bear little similarity to consensus EREs and affects the affinity that a given receptor isoform has for binding DNA (Loven et al., 2001).
Another category of gene promoters, lacking any ERE-like sequences, requires a second DNA-binding transcription factor (e.g., Sp-1 and AP-1) to mediate ER association with the DNA (O’Lone et al, 2004). Although ERα and ERβ have similar effects on ERE-mediated gene transcription, the receptors show opposite effects on promoters containing AP-1 (Paech et al, 1997). E2 activates AP-1-mediated gene transcription when bound to ERα but inhibits promoter activity when bound to ERβ (Paech et al, 1997). The converse is true for anti-estrogens, such as tamoxifen, raloxifene, and ICI 164384, which are AP-1 transcriptional suppressors via ERα and activators via ERβ (Paech et al, 1997; Weyant, et al, 2001; Loven et al., 2001). Similar to AP-1, E2 binding to ERα induces transcriptional activation when associated with Sp-1 in GC-rich regions. However, E2 binding to ERβ does not result in the formation of a transcriptionally active complex at a promoter containing Sp-1 elements (Saville et al, 2000). As an example ERα and ERβ, in the presence of E2, oppose each other’s function in the regulation of the cyclin D1 promoter (Liu et al, 2002). There is considerable
7
evidence that cyclin D1, important for the progression of cells through the G1 phase of the cell cycle, is a well defined target for ERα-E2 action in mammary carcinoma cells (Altucci et al, 1996; Foster and Wimalasena, 1996; Prall et al, 1997), although no detectable “perfect” or ERE-like sequence in the cyclin D1 gene promoter has been reported (Herber et al, 1994). Deletion of AP-1 and Sp-1 responsive element motifs in the cyclin D1 gene promoter resulted in attenuation of promoter responsiveness to E2 (Marino et al, 2002, 2003). Unlike ERα, E2-bound ERβ represses cyclin D1 expression (Acconcia et al, 2005a) and blocks ERα-E2-mediated induction when both receptor isoforms are present (Matthew and Gustafsson, 2003). Consequently, these differences in transcriptional activity between the ERα and ERβ may account for the major differences in their tissue specific biological actions.
Figure 1.2: Schematic model illustrating the action mechanisms of E2. In the first panel (a) is depicted the classical interaction of the activated receptor with ERE on DNA. In panels b and c are representations of the indirect effects of ERs on transcription interactions. This occurs through protein-protein interactions with the Sp1 (b), AP-1 (c). The panel d represents the E2-non-genomic mechanism. AP-1, activating factor-1; Sp-1, stimulating factor-1.
8
1.3.2 Membrane initiated signals (MIS) of estrogens
The nuclear initiated signals of steroid hormones occurs after a time-lag of at least 2 hours after E2 stimulation and explains some of hormone functions in physiological and pathological situations (Farach-Carson and Davis, 2003; Marino et al., 2005). This picture was challenged when a physiological dose of E2 was reported to increase the uterine cAMP level in ovariectomized rats within 15 seconds (Szego and Davis, 1998), an effect too rapid to be accounted for genomic action(s). This event was not abrogated by transcriptional inhibitors and was termed “rapid or non-genomic”. Actually the term “non-genomic” is not adequate when referring to rapid changes that may also initiate new gene transcription (Farach-Carson and Davis, 2003; Kampa and Castanas, 2006). Various signaling pathways are activated upon E2 binding to ERs. These rapid events may be classified into four main signaling cascade: phospholipase C (PLC)/protein kinase C (PKCs) (Morley et al., 1992; Marino et al., 1998, 2001a, 2001b; Picotto et al., 1999; Perret et al., 2001; Incerpi et al., 2003), Ras/Raf/MAPK (Marino et al., 2002; Watter et al., 1997; Russel et al., 2000; Dos Santos et al., 2002; Migliaccio et al., 2002; Tanaka et al., 2003; Klinge et al., 2005; Woo et al., 2005), phosphatidyl inositol 3 kinase (PI3K)/AKT (Castoria et al., 1999, 2001; Simoncini et al., 2000; Marino et al., 2003; Björnström and Sjöberg , 2005; Levin, 2005; Acconcia et al., 2005a; Marino et al., 2005; Chambliss et al., 2005), and cAMP/protein kinase A (PKA) (Gu and Moss, 1996; Farhat et al., 1996; Picotto et al., 1996; Chen et al., 1998; Malyala et al., 2005). These pathways present numerous interactions with several other pathways. The ERα:E2 complex interacts with the IGF-1 receptor, leading to IGF-1 receptor activation and hence to MAPK signaling pathway activation (Kahlert et al., 2000). In addition, the ERαE2 complex activates the EGF receptor by a mechanism that involves activation of guanine nucleotide exchange proteins (G-proteins), Src, and matrix metalloproteinases, leading to an increase in extracellular regulated kinases (ERK) and PI3K/AKT activities (Dos Santos et al., 2002; Driggers et al., 2002; Improta-Brears et al., 1999; Razandi et al., 2003; Zhang et al., 2004; Kupzig et al., 2005). In endothelial cells the Src/PI3K/AKT pathway mediates rapid E2-dependent activation of eNOS and the release of nitric oxide. AKT and PKC could also modulate the MAPK pathway through Raf phosphorylation (Chambliss et al., 2000, 2005; Marino et al., 2005; Kim and Bender, 2005). It is important to note that activation of signaling pathways by E2 is cell type-specific. Indeed, the effect of E2 on PKC activity has been observed in the preoptic area of female rat brain slices, but not in the hypothalamus or cortex (Ansonoff and Etgen, 1998). The activation of G-protein/Src/PI3K/MAPK pathway by E2 was evident in late,
9
but not early, differentiated rat pre-adipocytes (Dos Santos et al., 2002). The differential requirement of Src/PI3K or intracellular calcium for MAPK activation is also observed in diverse cell types (Dos Santos et al., 2002; Björnström and Sjöberg, 2005; Kupzig et al., 2005). Different PKC isoforms are rapidly activated by E2 in HepG2 and MCF7 cells (Marino et al., 2001b). As a whole, these studies indicate that the rapid actions of E2 depend on a number of conditions such as the set of signal transduction molecules and downstream targets present in the target cell, thus the responses are likely to be diverse. All these results point to the concept that ERαis the primary endogenous mediator of rapid E2 actions.
Figure 1.3: Schematic model illustrating the relationship between the mechanisms of action of E2. Palmitoylation (PA) allows the estrogen receptor (ER) localization at the plasma membrane. E2 binding induces ER association to signaling proteins, and triggers the activation of signaling cascades. The kinase activations phosphorylate ER, modulate transcriptional coactivators recruitment, and enhance AP-1 and Sp-1 activation. After dimerization ERs directly interact with ERE on DNA. ERs-DNA indirect association occurs through protein-protein interactions with the Sp-1 and AP-1 transcription factors.
10 1.4ER LOCALIZATION
The rapidity by which E2 induces rapid signals as well as the localization of signaling complex outside the nucleus raises the requirement of a plasma membrane ER. Debate continues over whether structural changes target nuclear ERs in separate pools localizing them to the membrane (Chambliss, et al., 2000; Acconcia and Kumar, 2005; Marino et al., 2005; Kampa and Castanas, 2006), or whether membrane ER represents a novel receptor (Ahola et al., 2002; Filardo et al., 2002; Ropero et al., 2002; Toran-Allerand et al., 2002; Thomas et al., 2005; Vivacqua et al., 2006). Besides these data, much evidence favors the idea that the membrane-localized ER is the same protein as the nuclear-localized receptor (Pappas et al., 1995; Norfleet et al., 1999; Razandi et al., 1999; Marino et al., 2002, 2003) and that ERα and ERβ must be considered a population of protein(s) which localization in the cell is able to dynamically change, shuttling from membrane to cytosol and to the nucleus, depending on ligand binding (Razandi et al., 1999; Dan et al., 2003; Marino et al., 2005; Leclercq et al., 2006). Current evidence indicates that a small population of ERα and ERβ localize at the plasma membrane exists within caveolar rafts. It is at the plasma membrane that E2-liganded ER associates with the scaffolding protein caveolin-1 and a variety of signal transduction cascade activation occurs [e.g., PLC, PKC, ERK, PI3K, and nitric oxide synthase (NOS)]. ERs do not contain a trans-membrane domain (Björnström and Sjöberg, 2005; Ascenzi et al., 2006), thus the ability of ERα and ERβ to associate with the plasma membrane could be due to its association with membrane proteins and/or by post-translational addition of lipids to ERα (Acconcia et al., 2005b; Levin, 2005).
Fatty acids and isoprenoids are two of the most common lipid moieties found on post-translational modified proteins bound to membranes. No consensus sequences for N-acylation (i.e., miristoylation) or S-prenylation have been found in ERα and ERβ (Acconcia et al., 2003). On the contrary, S-acylation (i.e., palmitoylation) does not require any consensus sequence, but just reactive Cys residues (Bijlmakers and Marsh, 2003). Cys residues present in the ERαand ERβLBD could undergo S-acylation. In particular, the amino acid sequence encompassing the Cys447 residue of ERαand Cys399 of ERβis highly homologous to that surrounding the S-palmitoylated Cys132 residue of human caveolin-1 (Acconcia et al., 2003). Based on this observation we demonstrated that ERαundergoes S-palmitoylation which represents the major determinant for its residence at the plasma membrane, for its association with caveolin-1 (Acconcia et al., 2003; 2005b; Marino et al., 2006b) and ERα-mediated activation of signal transduction pathways important for cell proliferation
11
(Acconcia et al., 2004; Acconcia et al., 2005b). The Cys 399 of ERβ is also palmitoylated as demonstrated in this thesis.
A physiological E2 concentration reduces by 50% the amount of palmitate incorporated in ERα within 60 min without any change in the protein level, while E2 induces a 90% reduction in the ability of ERα to form a complex with caveolin-1 (Acconcia et al., 2005b). E2-induced reversible S-palmitoylation of ERα could account for the coexistence of both membrane-bound and soluble isoforms of ERα (Marino and Ascenzi, 2006). S-palmitoylation is necessary for E2-induced rapid events. In fact, neither the ERα Cys447Ala mutant nor palmitoyl-acyl-transferase inhibition supports E2-induced ERK and PI3K/AKT pathway activation in human cancer cells (Acconcia et al., 2005b). These results have been recently confirmed by others authors (Pietras et al., 2005; Pedram et al., 2007).
Because ERα has no intrinsic kinase domains the localization of ERs at the plasma membrane facilitate the association between ER and signaling proteins allowing the activation of rapid events. Src, Shc, proline-, glutamic acid-, leucine- rich protein /modulator of non-genomic activity of estrogen receptor (PELP1/MNAR), the p85α subunit of PI3K, receptor tyrosine kinases (i.e., EGF and IGF-1 receptors), as well as G-protein isoforms (i.e., Gαs and Gαq) have all been reported to serve as components of large complexes of interacting proteins. Through the mediation of these molecules, E2 activates the MAPK and PI3K/AKT pathways (Acconcia and Kumar, 2005; Kennedy et al., 2005; Levin, 2005; Song et al., 2005; Greger et al., 2006). Protein-protein complex formation occurs only 5 to 15 min after E2 stimulation (Greger et al., 2006), thus, the conformational changes of the ER LBD domain, which follows E2 entry into the cell, seems to be important in allowing the ER:E2 complex to detach from the membrane and allocate with growth factor receptors or adapter proteins to activate downstream signals
1.5 CELL FUNCTIONS REGULATED BY MIS
The rapid activities of ERs are widely accepted and disagreement on the involvement of nuclear receptors is quite settled. However, other controversies in this field are still present and related to whether or not all of these rapid effects are of physiological relevance (Warner and Gustafsson, 2006). The main difficulties are linked to the experimental models used. In fact, the study of signaling pathways can be done mainly on isolated, often immortalized, cells and it is very complicated to obtain similar information on a whole organism in which the use of signaling inhibitors could have many side effects other than to inhibit just one kinase. Nevertheless, the physiological significance of rapid membrane-starting pathways has been
12
clarified at least for some E2 targets. In the nervous system, E2 affects neural functions (e.g., cognition, behavior, stress responses, and reproduction) in part by inducing such rapid responses (Farach-Carson and Davis, 2003). In the skeleton, ERα, present in caveolae of bone-forming osteoblasts, transmits survival signals through activation of the Src/Shc/ERK pathway and prolongs the life span of osteoblasts (Kousteni et al., 2003). At the same time, E2 delivers a pro-apoptotic signal to bone-resorbing osteoclasts, shortening their life span (Kousteni et al., 2003). Although these studies have been done mainly in cell-culture systems, their results suggest that ER rapid signaling actions have also a role in vivo. In the liver, rapid E2- induced signals (i.e., PLC/PKC) are deeply linked to the expression of the LDL receptor and to a decreased level of serum LDL-cholesterol (Marino et al., 2001a). Finally, vascular protection by E2 in ischemia/reperfusion injury in vivo requires E2-induced activation of endothelial NOS, as mediated by the PI3K/AKT pathway (Chambliss et al., 2000; Simoncini et al., 2000). The mechanism(s) by which E2 exerts proliferative effects is assumed to be exclusively mediated by rapid membrane-starting actions (Marino et al., 1998; Castoria et al., 1999, 2001; Marino et al., 2001b, 2002, 2003). E2 treatment of mammary-derived MCF-7 cells triggers the association of ERα with Src and p85α leading to DNA synthesis (Castoria et al., 2001). In HepG2 cells multiple and parallel membrane starting pathways are rapidly activated by the ERα-E2 complex (Marino et al., 1998, 2002, 2003) and the blockade of PLC/PKC, ERK, and PI3K/AKT pathways completely prevents the E2-induced DNA synthesis (Marino et al., 2002, 2003). ERK/MAPK and PI3K/AKT pathways, rapidly activated by the ERα-E2 complex, also have a critical role in E2 action as a survival agent. In fact, these pathways enhance the expression of the anti-apoptotic protein Bcl-2, block the activation of the p38/MAPK, reduce the pro-apoptotic caspase-3 activation, and promote G1-to-S phase transition via the enhancement of the cyclin D1 expression (Marino et al., 2002, 2003; Acconcia et al., 2005a).
1.6 ERβ RAPID ACTIONS
Less information is available on the role played by the ERβ:E2 complex to activate rapid non-genomic mechanisms and its contribution in E2 proliferative effects.
ERβ appears to act as a dominant regulator in E2 signaling, and when co-expressed with ERα it causes a concentration-dependent reduction of ERα-mediated transcriptional activation (Matthews and Gustafsson, 2003) and the repression of ERα-mediated effects including cell proliferation. Consistent with this notion, E2 increases cell proliferation and causes tumor
13
formation in MCF-7 cells expressing only ERα (Matthews and Gustafsson, 2003). On the other hand, ERβ inhibits the E2-induced proliferation of transfected MCF-7 cells and prevents tumor formation in a mouse xenograft model in response to E2 (Paruthiyil et al., 2004). This effect is linked to the ERβ repressive effect on ERα-induced gene transcription of cell cycle components (e.g., c-myc, cyclin D1, and cyclin E) which are associated with proliferation. These findings suggested a possible role for ERβ as tumor suppressor in breast cancer (Matthews and Gustafsson, 2003; Paruthiyil et al, 2004; Strom et al, 2004). Furthermore these studies support a functional antagonism between ERα and ERβ with respect to the E2-induced cell proliferation, suggesting without clarify the signal transduction pathways involved.
However, the ability of the ERβ:E2 complex to activate rapid non-genomic mechanisms has been reported, even if these data are limited and conflicting. A sub-population of ERβ transfected into Chinese Hamster ovary cells is capable of stimulating IP3 production, ERK/MAPK activation, and c-JNK phosphorylation (Razandi et al, 1999). Geraldes and coworkers reported that E2 reduces ERK activity through ERβ stimulation in porcine smooth muscle cells (Geraldes et al, 2003). Recently, ERβ has been reported to rapidly induce a persistent membrane-initiated activation of p38/MAPK without any interference on survival proliferative pathways, thus impairing the activation of cell cycle components (i.e., cyclin D1 expression) (Acconcia et al., 2005a). Although the scarce information does not allow a complete discussion on the contribution of ERβ in E2-induced rapid signals, these data indicate that also ERβ could originate cell-specific signal transduction cascade. Also for ERβ, the rapidity by which these cellular cascades are activated raises the need for a receptor localized at the plasma membrane. Although a subpopulation of ERβ localized within caveolar rafts, responsible for rapid endothelial nitric oxide synthase stimulation by E2 has been reported in the plasma membrane of endothelial cells (Chambliss et al. 2000), the mechanism allowing ERβ localization at the plasma is completely unknown.
As a whole, these evidence demonstrate the mechanism by which E2 exerts proliferative properties is to be exclusively mediated by ERα-induced rapid membrane-starting actions (Marino et al., 1998; Castoria et al., 1999; Lobenhofer et al., 2000; Marino et al., 2001b, 2002, 2003; Castoria et al., 2001; Acconcia et al., 2005a), whereas E2 induces cell death through ERβ non-genomic signaling (Acconcia et al., 2005a). All these results point to the concept that the ability of ER:E2 membrane starting pathways to signal through multiple cascades, undoubtedly has an impact on all aspects of cellular function, contributing to E2-induced cell proliferation and survival,
14
all essential features of cell physiology as well as of tumor biology (Levin, 2005). Thus the membrane-initiated signals of ERα and ERβ are both required to obtain the complete cellular response to E2.
1.7ER TISSUTAL DISTRIBUTION
Both ERs are widely distributed throughout the body, displaying distinct or overlapping expression patterns in a variety of tissues (Couse and Korach, 1999; Pettersson and Gustafsson, 2001). In particular, ERα mRNA is highly expressed in epididymis, testis, ovary, kidney, and adrenal. Moderate amounts of ERα are also present in the prostate gland, bladder, liver, and thymus. The highest amounts of ERβ mRNA were detected in the prostate gland, brain, ovary, gastrointestinal tract and bladder, hematopoietic and central nervous systems. ERα and β are, however, coexpressed in a number of tissues including the mammary gland, epididymis, thyroid, adrenal, bone, and certain regions of the brain. Although both ER subtypes may be expressed in the same tissue, they might not be expressed in the same cell type. In the rat ovary, ERβ is the predominant ER in the granulosa cells, whereas ERα is largely present in the thecal and interstitial cells (Hiroi et al., 1999; Sar and Welsch, 1999; Nilsson et al., 2001). The uterus and pituitary gland are special in that ERβ is expressed during development and ERα when the tissue matures (Brandenberger et al., 1997; Nishihara et al., 2000). Uterus, bladder, lung, and testis show intermediate levels of ERβ, whereas low but detectable levels of ERα are observed in the pituitary gland, thymus, various brain sections, and spinal cord (Deroo and Korach, 2006). Nonetheless, ERα and ERβ proteins have been simultaneously detected in many cell types including neurons and thymocytes (Greco et al., 2001; Mor et al., 2001), and these as well as other cell types that coexpress both ER subtypes are targets for potential interplay between the two receptors. As previously reported, when coexpressed with ERα, ERβ appears to act as a dominant regulator of estrogen signaling causing a concentration dependent reduction in ERα-mediated transcriptional and rapid activities (Pettersson et al., 2000; Liu et al., 2002; Matthews and Gustafsson, 2003). Form these data raises the hypothesis that the final effect of E2 of cell physiology could dependent on the subtle balance of ERα and ERβ expression level, even if other underlying mechanisms can not be excluded.
1.8 SELECTIVE ESTROGEN MODULATORS (SERM)
The central role of the ER signaling network in cancer, cardiovascular disease, osteoporosis, and neurological disease and an increasingly detailed understanding to the underlying cell biology have made ER an attractive
15
target for pharmacological intervention. Selective estrogen receptor modulators (SERMs) are ER ligands that can have varying agonist or antagonist activities given the cell context (Burger, 2000; Osborne et al., 2000). They have been developed and characterized to obtain more favorable tissue and receptor subtype selective effects (Veeneman, 2005; Heldring et al., 2007). These ligands that display tissue-selective pharmacology: as anti-estrogens (or antagonists) they oppose the action of estrogens in certain tissues, while mimicking the action of endogenous estrogens (agonists) in others. For instance, Tamoxifen is the prototypical SERM that, because of its agonist activity in the liver, reduces serum total cholesterol and LDL levels (Williams et al., 1997). Unfortunately, its strong agonist activity in the endometrium leads to endometrial hyperplasia and low-grade cancers.
Non steroidal plant-derived flavonoids present in the human diet show high degree of structural resemblance to certain selective estrogen modulators (SERMs) (Brzezinski and Debi, 1999). In fact, the flavonoids compete with E2 in binding to ERα and induce the classical genomic activity of ER through the transcription of ERE-containing genes (Kuiper et al., 1998). These observations raise the hypothesis that flavonoids may act as an estrogen ‘mimetic’ in different tissues and organs. However, flavonoids may have protective roles in several E2-related diseases (i.e., osteoporosis and cardiovascular diseases in post-menopausal women) (Lissin and Cooke, 2000; Ewies, 2002; Wuttke et al., 2003). Furthermore, epidemiological and in vitro studies indicate that this family of dietary components displays divergent effects from E2 in affecting cancer growth (Middleton et al., 2000; Wuttke et al., 2003). In particular, the ability of flavonoids (e.g., naringenin and quercetin) to hamper cell proliferation via their binding to ERα and to interfere with its rapid signaling has been showed (Totta et al., 2004; Virgili et al., 2004; Galluzzo et al., 2009b). From this evidence, these dietary compounds represent a very selective tool, which might be of great relevance for fully understanding the ER rapid signal transduction pathways and their impact on E2-dependent cell functions.
16
2. AIMS
The nuclear-initiated signals of E2 have been studied for long time. However, in addition to its well known role as a regulator of gene transcription sustained by the classical nuclear ERs (Nilsson et al., 2001; Acevedo and Kraus, 2004), E2 also shows rapid extra-nuclear actions (Levin, 2005; Marino et al., 2005; Ascenzi et al., 2006). At present it has been assumed that the rapid membrane initiated signals after E2-bound to ERα are mainly involved in E2-dependent proliferative effects (Marino et al., 1998; Castoria et al., 1999, 2001; Marino et al., 2001b, 2002, 2003; Lobenhofer et al., 2000; Fernando and Wimalasena, 2004). For instance, as previously reported, non-nuclear recruitment of MAPK signaling cascades by ERα has been demonstrated in cardiomyocytes (Nuedling et al., 1999) breast cancer (Castoria et al., 1999) and bone (Endoh et al., 1997; Jessop et al., 2001) leading to responses that include cell growth, cell cycle progression, and survival. Furthermore, recent data point to ERα palmitoylation as the mechanism responsible for the receptor localization to the cell membrane and the regulation of the E2-induced cell proliferation (Acconcia et al., 2005b). From these data, it is reasonable to assume that the stimulatory effect of estrogen on cell proliferation also contribute to malignant tumor growth in these tissue through ERα. At present if ERα and the downstream rapid signals are only involved in mediating these adverse E2 effects is still unclear.
On the other hand, less information are available on membrane-started action of ERβ and its physiological role. Contradictory evidence on the ability of ERβ to activate or inactivate Src and p38 kinases has also been reported (Castoria et al. 2001, Kousteni et al. 2001, Geraldes et al. 2003, Mori-Abe et al. 2003; Acconcia et al., 2005a). At present, the contribution of ERβ on E2-induced cell proliferation is due only to its action as dominant negative of ERα activities (Couse and Korach, 1999; Weihua et al., 2003; Paruthiyil e al., 2004; Strom et al., 2004). Thus, a full understanding of the existence of ERβ specific extranuclear signals and their impact on the final E2 effect remains to be determined.
Aim of present thesis is to investigate the presence and the impact of membrane-initiated signals mediated by ERα and ERβ on E2-dependent cell functions. In particular the role played by each ER rapid signal in protective effects of E2 versus proliferation and their integration when both ERs are co-expressed in the same cellular context will be studied.
17
3. ERα-MEDIATED SIGNAL TRANSDUCTION PATHWAYS IN CANCER CELL PROLIFERATION
3.1 INTRODUCTION
The mechanism by wich E2 exerts proliferative properties has been assumed to be exclusively mediated by ERα-dependent membrane-initiated signals (Marino et al., 1998; Castoria et al., 1999, 2001; Marino et al., 2001b, 2002, 2003; Lobenhofer et al., 2000; Fernando and Wimalasena, 2004) pointing to ERα as the isoform mainly involved in E2 adverse effects responsible for tumor growth. On the other hand, data from animal model and clinical studies support the involvement of ERα in the protective role for estrogens, for instance on cardiovascular system (Mendelsohn, 2000; Mendelsohn and Karas, 2005; Kim and Bender, 2005; Deroo and Korach, 2006). Indeed, the estrogens are able to directly prevent the proliferation and the migration of vascular smooth muscular cells (VSMC) (Razandi et al., 2000). In humans, both ERα and ERβ are expressed in vascular endothelial cells and vascular smooth muscle cells (Mendelsohn, 2000; Incerpi et al., 2003; Mendelsohn and Karas, 2005). Although it is possible that ERβ inhibits the vascular smooth muscle cell proliferation acting as dominant negative of ERα activities (Watanabe et al., 2003; Kim and Levin, 2006), we recently demonstrated that also ERα may activate rapid signals devoted to the block of cancer cell proliferation These signals started upon exogenous ligands bind to ERα (Totta et al., 2004; Virgili et al., 2004).
Naringenin (5,7,4’-trihydroxyflavanone, Nar; chemical structure Fig. 3.1), especially abundant in citrus fruits and in tomatoes, is reported to have anti-proliferative effects in different cancer cell lines (e.g., colon, breast, and uterus cancer cell lines) (So et al., 1996; Kawaii et al., 1999; Birt et al., 2001; Manthey et al., 2002; Frydoonfar et al., 2003; Harmon and Patel, 2004, Totta et al., 2004; Virgili et al., 2004). Among several mechanisms proposed for Nar induced anti-proliferative effects (i.e., antioxidant activities and kinase and glucose uptake inhibition) (Harmon and Patel, 2004; Moon et al., 2006), the ability of Nar to hamper cell proliferation by binding to ERs is particularly intriguing. Thus the study of the ER mediated anti-cancer action of nutritional flavonoids, could help to clarify the involvement of ER activities in regulating cell proliferation.
We previously showed that Nar concentrations, physiologically achieved in the plasma (1-10 µM) after the consumption of meals rich in Nar, enhanced ERβ-mediated signals important for cancer cell apoptosis and impaired ERα-mediated signaling important for E2-induced cancer cell proliferation (Totta et al., 2004; Virgili et al., 2004). In the presence of ERα, Nar prevented the activation of ERK/MAPK and PI3K/AKT signaling
18
without impairing the transcription of an ERE-containing gene construct. Moreover, Nar activated the rapid phosphorylation of p38/MAPK which, in turn, induced a pro-apoptotic cascade (e.g., caspase-3 activation) (Totta et al., 2004; Galluzzo and Marino, 2006). These data raised the possibility that Nar antagonistic effects on E2-related cancers were dependent on flavonoid ability to modulate ERα association to the plasma membrane and the protein-protein interaction important for ERα-mediate proliferative effects of E2.
We hypothesize that Nar binding to ERα could selectively modulate the receptor post-translational lipid modification palmitoylation. Here this possibility has been investigated comparing the effect of E2 and Nar on ERα palmitoylation and on ERα association to either membrane (i.e., caveolin-1) or adaptors (i.e., MNAR) or signaling proteins (i.e., c-Src). Finally, the influence of Nar-induced regulation of ERα palmitoylation on signaling cascade activation has been evaluated.
This study was conducted on human cervix epitheloid carcinoma cells (HeLa), devoid of any ERs and rendered E2-sensitive by transient transfection with a human ERα expression vector. This model allows analyzing the flavonoids effects on ERα activities without any interference of ERβ or ER splice variants. Furthermore, HeLa cell transfected with empty vector were used as control.
19 3.2 RESULTS
3.2.1 Effect of E2 and naringenin on cell proliferation
Our first target was to confirm the effects of naringenin in comparison with E2, on well known cell functions modulated by ERα:E2 complex such as promotion of cell growth. In a different way from E2 (Fig. 3.2a), the treatment of ERα-transfected HeLa cells with naringenin significantly decreased cell number (Fig. 3.2b). These effects of naringenin, in line with results reported for other flavonoids in several cell lines (Kuiper et al., 1998), were time and dose-dependent within the range utilized (0.01 µM-100 µM) (data not shown). However, none of flavonoid concentration utilized, as well as E2, influenced the growth of Hela cells transfected with the empty plasmid (Fig. 3.2a and 3.2b). The date confirms that Nar impairs cancer cell growth through an ERα-dependent mechanism.
Figure 3.2: Effect of E2 and Nar on cell growth. Time course analysis of cell growth in empty plasmid or ERα transfected-HeLa cells in the absence (Vehicle) or in the presence of E2 (10 nM) (a) or Nar (10 µM) (b). The data are the mean values ± S.D. of three independent experiments carried out in duplicate. *P<0.001, compared with un-stimulated cell values (Vehicle), was determined using Student’s t-test.
a
20
3.2.2 Mechanisms underlying the ERα activities modulation by naringenin
Nar decreases ERα palmitoylation
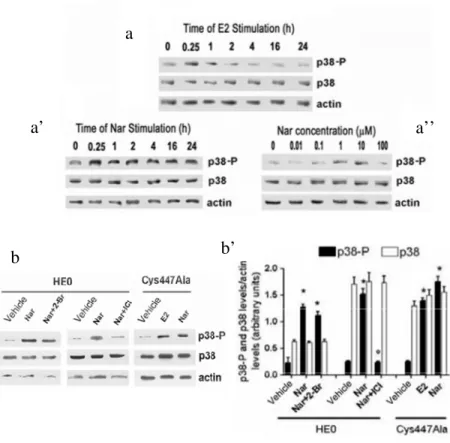
As previously demonstrated, ERα is a palmitoylated protein (Acconcia et al., 2004; Marino et al., 2006). In fact, [3H]-palmitate incorporated into ERα-transfected HeLa cells for up to 240 min reached a steady-state regimen after 10 min, the half-time of ERα palmitoylation was about 1.5 min (Fig. 3.3a). This is consistent with a rapid turnover of fatty acid on ERα supporting the idea that ERα-palmitoylation is a dynamic event involving cycles of acylation and deacylation (Linder and Deschenes, 2006). As previously reported (Acconcia et al., 2005b), 10 nM E2 stimulation induced the decrease of [3H]-palmitate incorporation with a half-time of about 30 min. Two hundred forty min after E2 stimulation 25 ± 0.5 % of ERα was still palmitoylated (Fig. 3.3b). A similar kinetic behavior was obtained pre-treating cells with the palmitoyl acyl transferase (PAT) inhibitor 2-Bromo palmitate (2-Br) 30 min before E2 stimulation (Fig. 3.3b). The Nar concentration (i.e., 1 to 10 µM) rapidly decreased the amount of [3 H]-palmitate incorporated in HeLa cells transfected with wild type ERα; the same result was obtained at higher Nar concentrations (i.e., 100 µM), whereas a lower Nar concentration (i.e., 0.01 and 0.1 µM) was ineffective (data not shown). The half-time of Nar-induced ERα-de-palmitoylation (Nar concentration = 10 μM) in cells containing transfected ERα (HeLa) (Fig. 3b) was about 8 min. In cells containing endogenous ERα (HepG2), the half-time of Nar-induced ERα-de-palmitoylation was about 1 min (Fig. 3.3c). Two hundred forty min after Nar stimulation 6.0 ± 0.5 % and 8.1 ± 1.2 % of ERα was still palmitoylated in HeLa and HepG2 cells, respectively (Fig. 3.3b and 3.3c). No change in the ERα protein level accompanied the E2- and Nar-induced decrease of [3H]-palmitate incorporation into ERα in both cell lines (Fig. 3.4).


![Figure 3.3: Effect of Nar on ERα palmitoylation. (a) Time course of [ 3 H]-palmitate incorporation in ERα transfected HeLa cells](https://thumb-eu.123doks.com/thumbv2/123dokorg/2844962.5515/31.630.96.565.95.548/figure-effect-era-palmitoylation-course-palmitate-incorporation-transfected.webp)