INDEX
Introduction 5
Background 28
Aim of the work 38
Chemistry 45
Estrogen Receptor Binding Assays 61
Molecular Modeling 70
Transcriptional Assays 76
Conclusions 82
Serine Palmitoyl Transferase (SPT) Inhibitors as Antineurodegenerative Agents 84
Experimental Section 86
5 INTRODUCTION
Estrogens: general aspects and chemical structure
Estrogens, androgens and progestins are three classes of steroidal hormones to which belong the most important sexual hormones produced by male (testis) and female (ovary) gonads.
The main functions of goandal hormones are:1
− Stimulation of the development of oocytes and sperm from the respective germinal cells, through a local action (paracrine and autocrine function);
− Stimulation of the development of secondary sexual organs and their function, and regulation of secretion of hypothalamic-pituitary hormones, through an action target cells (endocrine function).
In male, the modest production of estrogens takes place in the interstitial Leydig cells of testis.
In female, estrogens are produced in the ovaric follicle during the first phase of menstrual cycle, whereas, after ovulation, are synthesized by the luteinized cells of granulosa and of curpus luteum, together with
6
progesterone. Minimal amounts of estrogens are also produced by skeletal muscles, adipose tissue and, during pregnancy, by the placenta and the fetal adrenal.2
Estrogens are involved in controlling the release of gonadotropins in the hypothalamic-pituitary axis.1,2 The ipotalamic factor of liberation of gonadotropins (GnRH) is a deca-peptide that stimulates the adenohypophysis (or anterior pituitary) to release follicle stimulating hormone (FSH) and luteinizing hormone (LH), which act on ovary in female, while act on the testis in male.3 The function of both hormones is to promote gametogenesis and follicle development in female, and to promote spermatogenesis in male.
FSH is the main hormone that controls the secretion of estrogens in female; infact it allows androgens of granulosa to be converted into estrogens. In male it acts on Sertoli cells and stimulates the production of a protein that binds androgens.
The LH allows the ovulation to take place in female has and is the main hormone that controls the subsequent secretion of progesterone by the corpus luteum. In male, it stimulates the secretion of testosterone, acting on the Leydig cells.
7
The products of the secretory activity of the gonads exert a negative feedback effect towards both the hypothalamus and pituitary, by inhibiting the release of GnRH and FSH/LH, respectively. Instead, when the blood concentrations of estrogens and androgens are low, the control on the hypothalamic-pituitary axis is a positive feedback (GnRH is released and, consequently, FSH and LH are released).
Estrogens have also beneficial effects on heart and bones. They maintain the mineral density and reduce the risk of bone fractures; moreover, they affect brain centres responsible for maintainment and regulation of the body temperature.2
With the insorgence of menopause, the lower levels of estrogens cause several disorders, such as hot-flushes, increment of LDL-cholesterol, with high risk of cardiovascular disease and decrease in bone mineral density, that can lead to osteoporosis.2 This series of events can be take under control with an hormone-replacement-therapy (HRT); However, this can cause the onset of hormone-dependent diseases like breast and uterine cancers. The development and progression of breast tumors (with the risk of developing metastases) are in close correlation with the exposure to estrogens (endogenous and/or exogenous). Infact, breast carcinoma was the first to be recognized as an hormone-dependent tumor
8
(the complete removal of the ovaries induced regression of mammary tumors in a group of pre-menopausal patients).4
Estrogens are clinically used not only for the hormone treatment of post-menopausal women, but also for the treatment of primary hypogonadism.2
The main female estrogens are estradiol (17β-estradiol), estrone and estriol (Figure 1).2 Figure 1 HO O H3C HO OH H3C HO OH H3C OH
Estradiol Estrone Estriol
Estradiol is the main product of the ovarian secretion, but it is also produced from the adrenal cortex. Although a certain amount of estrone is produced in the ovary, most of this hormone (and estriol) is synthesized in the liver from estradiol, or in peripheral tissues from androstenedione and other androgens.
9
Chemically, estrogens are characterizad by a four-fused rings system, named cyclopentane-perhydro-fenantrene (Figure 2).
Figure 2. 17 16 15 14 13 12 11 10 9 7 8 6 5 4 3 2 1 A B C D
In estradiol, ring A is hydroxylated at C-3 position and the hydroxyl at C-17 position confers the estrogenic activity of this hormone. The absence of this hydroxyl in estrone and the presence of another hydroxyl (at position C-16) in estriol, determines a low hormonal activity of these estrogens and thus a lower affinity for the estrogen receptor.
Estrogen receptors
Once released into the bloodstream, estradiol binds strongly to an α2-binding globulin called SHBG (sex hormone α2-binding globulin), and, with lower affinity, to albumin. Bound-estrogen is relatively unavailable for diffusion into the cells, so is the free fraction which is physiologically
10
active. Since the target receptor has been reached, the complex SHGB-estradiol dissociates and the hormone interacts with the receptor. There are two types of estrogen receptors: membrane receptors and nuclear receptors.
Membrane estrogen receptor
The membrane estrogen receptors have been recently discovered. In some cases they mediates rapid responses (from seconds to minutes), not involvig any transcription process.5 The membrane receptors are largely expressed in the CNS. They are coupled to G proteins and have seven trans-membrane domains, with an extracellular N-terminal domain and a C-terminal intracellular domain. The third cytoplasmic loop of the receptor represents the region with which G protein is coupled. The mechanism of signal transduction is showed in Figure 3.
11 Figure 3. !"##$!%"&&'() *+,-./'0&) ,%.12%.-0("/0.&) 3/%'-) 4'11'&5'-1)
G protein consists of three subunits α, β, γ, and when the agonist binds to the receptor, activates a conformational change, which involves the cytoplasmic domain. The α subunit dissociates and activates an effector, which in this case is the phospholipase C (PLC). The latter leads to the formation of IP3 (inositol 1,4,5 triphosphate) which increases the
concentration of intracellular Ca++, by stimulating its release from the sarcoplasmic reticulum, and DAG (diacylglycerol) which in turn activates a membrane protein kinase (PKC, protein kinase C); this protein facilitates the entry of calcium ions into the cell. The increase in the concentration of intracellular Ca++ promotes the activation of NOS
(NO-12
synthase), that release NO (nitric oxide) used in the CNS as a neurotransmitter.
Nuclear estrogen receptors
Two nuclear estrogen receptor subtypes have been actually identified: estrogen receptor α (ERα) and β (ERβ), which are encoded by genes on different chromosomes and that function as ligand-modulated transcription factors, up- and down-regulating gene expression in target tissues.
Both receptors possess six distinct domains (A–F), which are depicted in Figure 4.6
Figure 4.
!"#$ %$ &$ '$ ($
)!"#$%&'()*
+,%(&'* %!"#$%&'()*+,%(&'*
&)!!-&'.&'/*
+,%(&'* *+,-./+,%(&'*!-&'.&'/* 0&'/#1*
13
- The N-terminal region (A/B), higly variable and including the AF1 region, which is involved in the activation of transcription.
- A central highly conserved C domain, named DNA binding domain (DBD). The DBD consists of two functionally distinct zinc-finger motifs and is responsible for specific binding of the receptors to the estrogen response element (ERE) or ERE-like sequences in the promoter of target genes.7
- The "hinge" D region, which separates the DBD and the ligand-binding domain. The flexibility in the secondary structure of this region allows conformational changes when the ligand binds to the receptor.
- The E domain, which binds to the ligand (LBD) and that include a second transcriptional activation function domain (AF-2). This region consists of 12α-helices, which form a hydrophobic pocket responsible for estradiol (E2) or ligand binding.8 LBD is formed by three layers of
antiparallel α-helices: a central layer of three helices (H5/6, H9 and H10), placed between two additional layers of helices (H1-4 and H7, H8, H11), and finally has two sections with β-sheet structure (S1, S2) and an α-helical portion (H12);
14
- The C-terminal domain (F) which promotes the transcription by RNA polimerase.
Nuclear receptors are found in the cytoplasm in the form of oligomeric complexes with particular stabilizing proteins, called Hsp90 (heat shock protein 90).9 The free-hormone, coming from the plasma or interstitial tissues, passes into the cell and binds to the receptor (LBD) by inducing conformational changes that allow the receptor to dissociate itself from the Hsp90, thus forming a homo- or hetero-dimer (Figure 5). The ligand-receptor complex is then actively transported into the nucleus, where the C domain of the receptor interacts with particular DNA sequences, called EREs (Estrogen Response Elements). Once bound to DNA, the ligand-receptor complex recruits and binds coactivators and corepressors, which subsequently modulates transcriptional activity of estrogen-responsive genes (Figure 5).
15 Figure 5. !"# !"# !"# !$# %&'(# ((()*# %&'(# ((()*# +,-# !"!# ./0123(1242# -5367/36,4#
Differences between ERα and ERβ
ERα and ERβ are highly homologous in their DBDs (∼96%) and possess moderate (∼53%) sequence identity in the LBD.8 The A/B and D
domains presents an homology of 26% and 27%, respectively. The F domain presents the less homology (22%). The gene for ERα encodes for 595 aminoacids, whereas the gene for ERβ encodes for 530 aminoacids (Figure 6)
16 Figure 6.
!"#$
!"#$
%$
%$ &$
&$ '"($
'"($
!" !#$" %&'" '$%" ()(" !" !*)" %!*" %*#" ('$" )&'*+)+,-.$ /0$ 10$ /2$ 34$'5!"
'5#"
6789:;8<=-!><?7@A-7B-'5$
The two binding cavities of ERα and ERβ differ in two amino acid residues: ERβ has a Met336 residue in place of Leu384 of ERα and a residue Ile373 in place of Met421 of ERα.10
Furthermore, the ERβ-binding pocket has a smaller volume than that of ERα, and there are also slight differences in the shape of these cavities because of differences in the amino acid residues lining the cavity borders.
The major functional domain difference between ERα and ERβ is contained within the N-terminal region (AF-1): in ERα, this domain is very active in stimulating the transcription, whereas the activity of the
17
AF-1 domain of ERβ is negligible.11 This dissimilarity in the N-terminal regions of ERα and ERβ is suggested to be the reason for the difference between the two receptors in their response to various ligands.
In addition to their domain differences, these receptors also exhibit different expression patterns. ERα is predominantly expressed in breast, uterus and vagina, whereas, ERβ is expressed in tissues such as the central nervous system, cardiovascular system, immune system, gastrointestinal system, kidney, lungs, and bone, with marginal expression in the breast. These differences in tissue distribution between the two receptors may explain some of the selective actions of estrogens in different tissues.
ER-agonists and -antagonists
Through cristallographic studies of the estrogen receptors complexed with an agonist or an antagonist, it is possible to highlight the different conformations that the estrogen receptors assume (depending on the nature of the ligand).
In particular, in the case of an agonist (estradiol), helix 12 closes on helix 4, causing a "close" conformation, in which the ligand remains
18
"locked"; in this way the interaction with the coactivator is allowed and consequently the trascription process is activated.
On the contrary, when an antagonist binds to the receptor, helix 12 cannot close on helix 4 and the conformation remains in an "open" state; the interaction with the coactivator is not allowed and the transcription process is not activated.12,13 These conformation is caused by the steric hindrance of the side-chain that is present in molecules which can act as antagonists (in the case of tamoxifene, for example, it is a N,N-dimethyl-aminoethyl group), and is stabilized by an H-bond that forms between the side-chain and an Asp351 residue of the binding cavity.
Interactions of agonists and antagonists with the ER binding cavity12
Agonists:
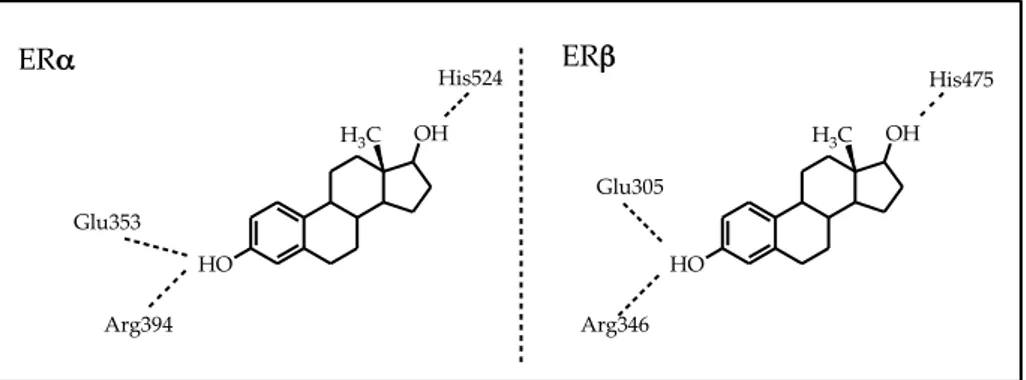
As shown in Figure 7 the hydroxyl group of the phenolic "A" ring of estradiol is engaged in a H-bond network with Glu353, Arg394 and a water molecule. The hydroxyl of the "D" ring forms an H-bond with His524. The other rings of the molecule are involved in hydrophobic interactions with the binding cavity.
19 Figure 7. HO OH H3C HO OH H3C Glu353 Arg394 Glu305 Arg346 His524 His475 ERα ERβ Antagonists:
Figure 8 shows the interactions of raloxifene with the ER binding cavity. Figure 8. S O HO OH O Raloxifene N Glu353 Arg394 His524 Asp351 ERα
20
The benzothiophene portion of the molecule mimics the phenolic "A" ring of estradiol. The hydroxy substituent on this portion forms H-bonds with Glu353 and Arg394. The phenolic ring of raloxifene binds to His524. Moreover, is present an additional interactions between the side-chain and Asp351, as mentioned above. The rest of the molecule is involved in hydrophobic interactions similar to that of estradiol.
SERMs (Selective Estrogen Receptor Modulators)
SERMs are molecules that display a particular and interesting tissue-selective pharmacology: they are agonists in some tissues, such as bone (maintaining the bone mineral density), cardiovascular system (LDL decrement and therefore reduced risk of cardiopathies) and CNS ("hot flushes" reduction) and antagonists in other tissues, such as brain and breast (antiproliferative effect, reducing the risk of developing tumors).
21 Figure 9. S HO OH O O N Raloxifene Tamoxifene O N Toremifene O N Cl
Due to their specific pharmacological properties, SERMs are useful in therapy for the prevention and treatment of diseases such as osteoporosis and breast cancer.9 However, some agents belonging to this class (as tamoxifene), if used for a prolonged therapy, may increase the risk of developing endometrial cancer, due to agonist action on this tissue.
To explain pharmacological responses induced by SERMs, different mecchanism of action are proposed:
1) Different conformation assumed by the receptor upon binding with SERMs;
2) Different ER-subtypes (α, β) with which the SERM interacts in various tissues and their different modes of activation of transcription;
3) Different corregulator proteins that bind to receptors. The point n. 1 has already been described in previous sections.
22
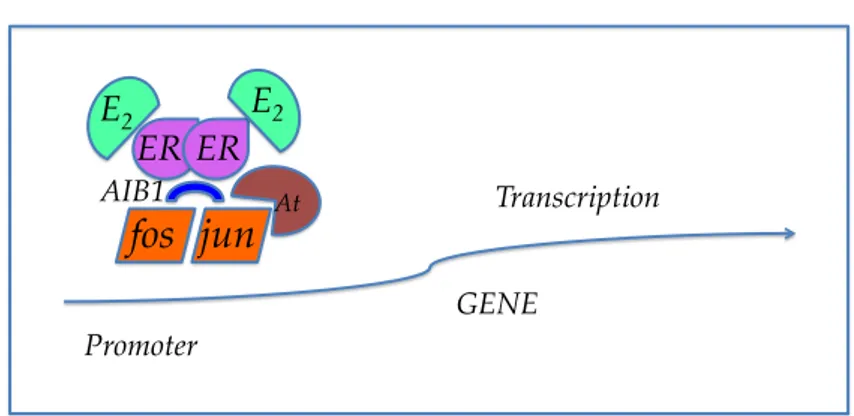
As regards the different modes of activation of transcription (point n. 2), it has been demonstrated that the agonist activity may have a mechanism by which the ligand-ER complex interacts with the DNA by means of particular proteins (such as jun and fos belonging to the AP-1 protein, Figure 10) and this leads to the activation of the transcription.14 With regard to the point n. 3, once the SERM-ER complex has formed, it can interact with a coactivator (CoA), which promotes the transcription (as is the case for the natural estrogens), or with a corepressore (CoR), which inhibits the mechanism of transcription, despite the receptor-ligand complex is attached to the ERE site in the DNA (Figure 11). The presence of certain coactivators or corepressors and the expression of transcription depends on the tissue and the receptor subtype of ER.
Figure 10. !"# !$# %&#
'()#
*+,#
%-./#!"# !$# 01+2+&31# 4!5!# 617),819:&9+)#23 Figure 11. !"# $%&# '!"()# *+,# -%)# !"# !"# ./01# '!"()# 23.$# .45# .41# !"!# 67897&7:;/<89=<>! "!/"!''?@A# .$6?B.6?@A# '!"()# Examples of SERMs Tamoxifene
It is a triphenylethylene derivative and is used to prevent and treat the early stages of tumor estrogen-dependent breast cancer, due to its antiestrogenic activity in this tissue. It is also characterized by a weak agonist activity on bone and cardiovascolar system.9 It has also been used
as a chemopreventive agent, approved by FDA in 1999, to reduce the risk of development of breast cancer in pre- and post-menopausal women.9 However, as mentioned above, the long-term treatment with tamoxifene may increase the risk of malignancy in the endometrium.15
24 Raloxifene
It is a benzothiophene derivative that proved to be an agonist on bone and adipose tissue: it preserves bone mineral density and produce a lipid-lowering effect. On the contrary, it has an antagonistic activity on the breast. This pharmacological profile produces an anti-osteoporotic effect and a cardioprotective effect, similar to that of estrogens, without the risk of breast cancer. Moreover, raloxifene does not have any proliferative effects on the uterus of postmenopausal women and it does not increase the risk of endometrial cancer.13
It is not effective against the symptoms of menopause, such as hot flushes.7
Toremifene
It is a triphenylethylene derivative. It has the same properties of tamoxifen and it seems not to increase the risk of developing uterine cancer. The FDA has restricted the use of this SERM for postmenopausal women with metastatic breast cancer. It also produce a modest reduction of LDL cholesterol.9
25
SERBAs (Selective Estrogen Receptor Beta Agonists)
SERBAs are ERβ-selective non-steroidal molecules. They may have those "ideals" features of SERMs, such as the ability to promote beneficial effects on the liver, bones, CNS, adipocytes (regulating fatty acid metabolism, lowering the blood levels of cholesterol) and the cardiovascular system (preventing myocardial hypertrophy and post-ischemic damage),16 and, at the same time, be free from the undesired proliferative effects on breast and uterus (mediated by the other receptor subtype, ERα).
ERβ-selective agonists may also be considered specific anti-inflammatory agents for the treatment of pathologies, such as some forms of asthma and inflammation involving T-helper lymphocytes).17
Furthermore, it has been demonstrated that ERβ-selective agonists are useful in therapeutic areas, such as prostate hyperplasia/cancer, bone demineralization, arthritis and intestinal inflammation.
26
Coumestrol and genistein (Figure 12) are examples of natural ERβ-selective agonists. Coumestrol is a benzofuran-condensed chromenone derivative present in many plants and foods (soybeans) and has a 7-fold ERβ-selectivity, with an ERβ-RBA* value of 140%. Genistein, an isoflavone derivative found in soybeans, is much more ERβ-selective (β/α ratio = 20).18 Figure 12. O O OH HO O O HO OH OH O HO CN OH Coumestrol Genistein DPN N O HO F OH ERB-‐‑041
* RBA values are calculated from IC50 values, determined in competitive radiometric or
fluorometric ligand-binding assays:
RBA(%) = (IC50estradiol / IC50ligand) x 100
Thus, an RBA of 100% represents an affinity equivalent to that of estradiolon either ERα or ERβ. In competitive radiometric ligand-binding assays, the tracer is generally [3H]estradiol, and K
d
values for estradiol are 0.2 nM for ERα and 0.5 nM for ERβ. Using Kd values of estradiol, ligand Kd values can be estimated from the RBA values:
27
Diarylpropionitrile (DPN) is a synthetic derivative characterized by a 70-fold ERβ-selectivity in binding assays (RBA of 0.25% for ERα and 18% for ERβ), and a 78-fold ERβ-selectivity in transcriptional assays (EC50 of
66 nM for ERα and 0.85 nM for ERβ).19
ERB-041 (Figure 12) is a synthetic benzoxazole derivative that proved to be one of the most effective ERβ-selective agonists, showing a 200-fold selectivity for ERβ (IC50 values of 1200 nM on ERα and 5.4 nM on
28 BACKGROUND
Estrogen receptor β as a therapeutic target
As described before (see the Introduction), the estrogen receptor (ER) exist in two isoforms: ERα and ERβ, which have different tissutal distributions and different functions. The first subtype is more expressed in the mammary gland, uterus, bone, brain, liver and cardiovascular system. The other subtype is found primarily in the prostate, immune system, digestive system, bone marrow, vascular endothelium and in some regions of the brain.
ERα mediates the "classical" estrogenic effects and, unfortunately, is also involved in the development of breast and endometrial estrogen-dependent cancer. For a long time, ERα has been considered the only target for estrogen-related therapies, using either agonists (as in hormone replacement therapy, in hot flushes and osteoporosis), or antagonists (as in hormone-sensitive breast cancer). The tissue-selectivity of certain molecules termed “selective estrogen receptor modulators” (SERMs) has offered, in many cases, great advantages in both types of therapeutic interventions.
29
The biological functions of ERβ have not been completely defined; the actually proposed roles include: antiproliferative function, regulation of apoptosis and modulation of the immune response. Furthermore, ERβ-selective agonists (SERBAs: ERβ-selective estrogen receptor beta agonists) are very promising in therapeutic areas such as prostate hyperplasia and cancer, bone loss, arthritis, and intestinal inflammation. Notably, the beneficial effects resulting from selective ERβ-stimulation would be expected to be free from the undesired ERα-mediated proliferative effects on breast and uterus, and this constitutes a great advantage for the prospective therapeutic use of such drugs.
For these reasons, the estrogen receptor β has emerged more and more, especially in recent years, as a therapeutic target and has inspired research efforts aimed at identifying and developing higly subtype-selective ERβ agonists.
Salicylaldoxime moiety as a bioisosteric replacement of the steroidal phenolic ring
In the research laboratory where I accomplished my PhD, two series of monoaryl-substituted salicylaldoximes (Figure 13) bearing a
para-30
hydroxylated aryl substituent at either position 4 (Structure A) or 5 (Structure B) of the central ring, were developed as agonists of estrogen receptor β.21,22 Figure 13. O H N OH HO STRUCTURE A A'ʹ NH O OH STRUCTURE B A'ʹ HO R R R R
These series were inspired by a consolidated pharmacophoric model (Figure 15) that has been derived from structural analysis of natural non-steroidal compounds such as genistein (isoflavone phytoestrogen, Figure 2), or synthetic non-steroidal compounds such as DPN and indazole derivatives (Figure 14). This pharmacophoric model is characterized by a "central core", represented by phenyl, aryl or an eterocycle, a phenolic ring and a para-hydroxy-phenolic function opposite to the phenolic ring.23
31 Figure 14. O OH HO O OH Genistein HO NN Cl OH Indazol Derivates CN HO OH DPN
A common structural feature typical of almost all synthetic ER-ligands possessing a good binding affinity is a phenolic ring that seems to mimic the steroid “A-ring” present in natural estrogens. This phenolic group is thought to be responsible for the strongest attractive polar interaction between ligand and receptor, through the formation of a hydrogen bond network that includes a bound water molecule and two amino acid residues of the ER ligand binding domain (Glu353 and Arg394).
In the two series of compounds developed in our research laboratory, the phenolic ring described above has been bioisosterically replaced by a salicylaldoxime moiety (Figure 15), characterized by a six-membered pseudo-cycle (A'-ring) that can form through an intra-molecular H-bond,
32
between the oximic nitrogen atom and the adiacent OH group. This A′-ring presents several features that indicate its similarity with the phenolic A-ring: (i) both rings have approximately same size and same planar π-conjugated (at least partially, in the case of the salicylaldoximes) hexagonal geometry; (ii) the OH of the oxime group is attached to an sp2 hybridized atom (nitrogen) that is intramolecularly hydrogen-bonded to the ortho phenol and has a pKa value around 10, within the pKa range of
typical phenolic OH groups; (iii) the position of the oxime OH group corresponds to the position 3 of the phenolic A ring, the position actually occupied by an OH in classical ER ligands.
Furthermore, accordingly to the pharmacophoric model, salicylaldoximes have an aromatic ring that can act as the “core structure” carrying additional substituents, at least one of which should be an aromatic group. Figure 15. HO OH central core substituent ERβ-‐‑Pharmacophoric model O H N R HO Mono-‐‑aryl Salicyl-‐‑aldoxime A'ʹ A
33
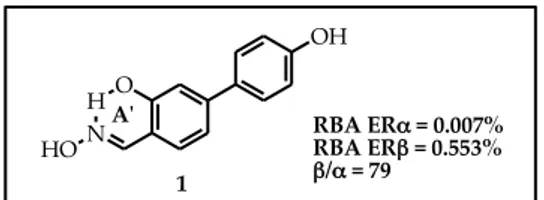
The simplest member of the STRUCTURE A series is represented by compound 1 (Figure 16). In biological binding assays (explained in the Introduction) it showed a good level of ERβ-selectivity (β/α ratio = 79), although its absolute affinity was not satisfactory (Table 1 pag. 62).21,22
Figure 16. O H N OH HO 1
A'ʹ RBA ERα = 0.007%
RBA ERβ = 0.553%
β/α = 79
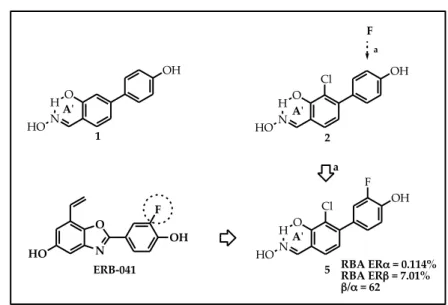
Starting from compound 1, the introduction of several small substituents in position 3 of the central ring were explored. In fact, this type of structural modification had led to good results in some indazole derivatives previously reported.23 In particular, the introduction of a halogen atom (Cl, Br, I), a small alkyl (methyl, ethyl) or a CN group, dramatically increased the ERβ affinities of the resulting molecules and also remarkably enhanced their ERβ-selectivities.23
Therefore, salicylaldoximes 2-4 (Figure 17, Table 1 pag 62) bearing a Cl, a CH3 and a CN respectively, were synthetized. In biological assays
34
compound 2 and 3 showed an ∼8-fold icrease of ERβ-affinity respect to 1. In terms of ERβ-selectivity, compound 2 showed a value similar to that of 1, while compound 3 showed a drastic decrement respect to 1. Compound 4 did not demonstrate significant levels of ERβaffinity and -selectivity. Figure 17. RBA ERα = 0.065% RBA ERβ = 4.21% β/α = 65 O H N OH HO b Cl O H N OH HO Cl a 1 2 a b A'ʹ A'ʹ O H N OH HO 3 A'ʹ O H N OH HO 4 A'ʹ CH3 CN CH3 CN c c HO NN Cl OH Indazol Derivates CN HO OH DPN RBA ERα = 0.249% RBA ERβ = 4.10% β/α = 16 RBA ERα = 0.004% RBA ERβ = 0.078% β/α = 20
Furthermore, on the basis of a very successful example reported in literature (ERB-041, a higly ERβ-selective benzoxazole derivative, possessing a β/α ratio = 200), a fluorine atom were inserted in position 3'
35
of the pheripheral substituent of 2, obtaining compound 5 (Figure 18). This last derivative improved the ERβ-affinity, respect to 2 and preserved the same value of ERβ-selectivity.
Figure 18. RBA ERα = 0.114% RBA ERβ = 7.01% β/α = 62 O H N OH HO a F O H N OH HO Cl O H N OH HO Cl F 1 2 5 a A'ʹ A'ʹ A'ʹ N O F OH HO ERB-‐‑041
The synthesized compounds were submitted to further biological testing to assess their pharmacological character. In particular, they were assayed for transcriptional activity through both receptor subtypes, together with estradiol for reference. In these assays, one of the best STRUCTURE A members, compound 2 (Table 1, pag. 62) reached only a partial activation of ERβ (EMAX = 60%); compound 5, bearing an
36
additional 3'-fluorine substituent, showed an improved potency (EC50 =
4.8 nM), maximal effect (EMAX = 85%), and subtype selectivity (β/α
EC50-selectivity ratio = 4.0).21,22 However, these STRUCTURE A
members can be considered only partial agonists, because the maximal activation of transcription (normalized to that resulting from 100 nM estradiol) was never reached. Besides, these compounds proved to be much less β-selective in transcriptional assays, than in binding assays, probably because ERα- and ERβ-ligand complexes interact differently with cellular coregulators, which act as ligand potency modulators (co-activators and co-repressors).
In the other series of salicylaldoximes (STRUCTURE B, Figure 1) the salicylaldoxime moiety was "inverted" (the relative position of the oxime and the phenol group was exchanged). The first and simplest member of this series (compound 6, Figure 19), showed, in estrogen receptor binding assay, an increase in ERβ-binding affinity respect to the "non inverted" analog 1, together with a notable level of ERβ-selectivity (β/α = 41). Docking studies highlighted that this compound was able to participate to an unprecedented H-bond with a threonine residue of the ERβ binding cavity (T299).
37
In transcriptional assays, compound 6 reached the maximal transcriptional activation (~100%), proving to be the first ERβ-full agonist among the synthesized oxime derivatives. However, even in the case of compound 6, the ERβ-‐selectivity in transcriptional assays was lower than that showed in binding assays, probably for the same reasons reported above. Figure 19. O H N OH HO 1 a OH N H O HO 6 a A'ʹ A'ʹ RBA ERα = 0.064% RBA ERβ = 2.64% β/α = 41
38 AIM OF THE WORK
Based on results described above and with the aim to develop highly ERβ-selective salicylaldoximes with better agonist features, we decided to make an optimization process to the STRUCTURE B series, starting from the simplest member (compound 6) and applying some of the same structural modifications that had given good results in the STRUCTURE A series, combined with other suggested by molecular modeling studies.
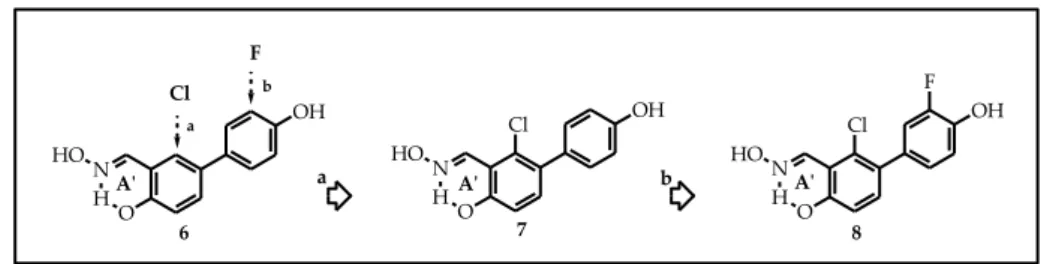
In particular, we first synthetized compounds 7 and 8 (Figure 20). Compound 7 is characterized by the "inverted" salicylaldoxime moiety and by the presence of a chlorine atom in position 6 of the central ring, adjacent to the oxime function; compound 8 differs from compound 7 for the presence of a fluorine atom in 3' position.
Figure 20. b Cl F a a OH N HO HO OH N H O HO OH N HO HO Cl F Cl 6 7 8 b
39
We also introduced relatively small substituents (such as methyl and chlorine) in position 3 of the central ring, a place for molecular variations that has been "unexplored" within these salicylaldoxime derivatives. We first synthesized compound 9 (Figure 21), resulting from the introduction of a methyl group in position 3 of the central scaffold of 7 (opposite to the chlorine atom) and then inverted the respective 3/6-positions of the methyl and chlorine groups (compound 10, Figure 21). The subsequent insertion of a meta-fluorine atom in the 4-aryl-substituent led to compound 11 (Figure 21). Figure 21. OH O N H CH3 OH O N H CH3 Cl Cl F 9 10 HO HO OH O N H Cl CH3 11 HO A'ʹ A'ʹ A'ʹ 6 OH O N H HO A'ʹ e b c a d Cl CH3 CH3 Cl F a,d b,c e
40
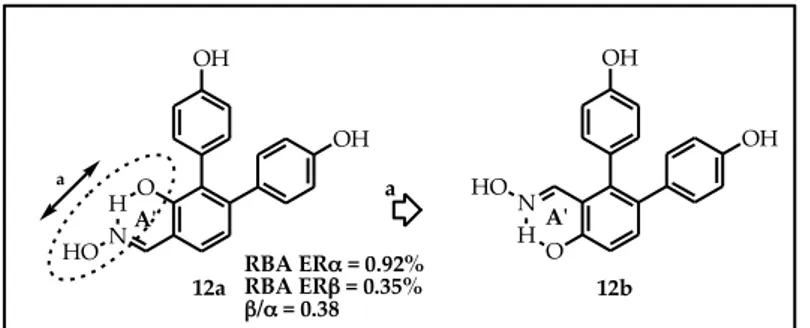
Furthermore, we decided to verify the effect due to a second para-hydroxyphenyl function, in position 6 of the central scaffold. Therefore we synthetized compound 12b (Figure 22) as an analogue of compound 12a, a salicylaldoxime of the STRUCTURE A series previously developed that in biological assays didn’t show good levels of ERβ- affinity and -selectivity.
Figure 22. N HO HO OH A'ʹ OH OH O H N a A'ʹ 12a 12b OH HO a RBA ERα = 0.92% RBA ERβ = 0.35% β/α = 0.38
As a further development of of STRUCTURE B salicylaldoximes, we explored other structural modification on this series of compounds.
We first inserted relative small groups (CH3, Cl) on the meta-position
41
Then we investigated the effect due to the shift, from para to meta position, of the hydroxyl group of the peripheral 4-aryl substituent (compound 13, Figure 23). Figure 23. OH N HO HO b c a 6 c a b Cl OH N H O HO 14 CH3 N HO HO 13 OH CH3 OH N H O HO 15 Cl A'ʹ
A'ʹ A'ʹ A'ʹ
OH N HO HO 16 Cl A'ʹ Cl Cl d d
Moreover, on the basis of the structure of genistein (a natural compound that is demonstrated to be a good SERBA), possessing a resorcinol moiety, we decided to insert a second hydroxyl function on the pheripheral substituent (compounds 17 and 18, Figure 24).
42 Figure 24. O OH HO O OH Genistein O OH N HO O OH N HO 17 18 H A'ʹ H A'ʹ Cl OH OH OH N H O HO 6 A'ʹ
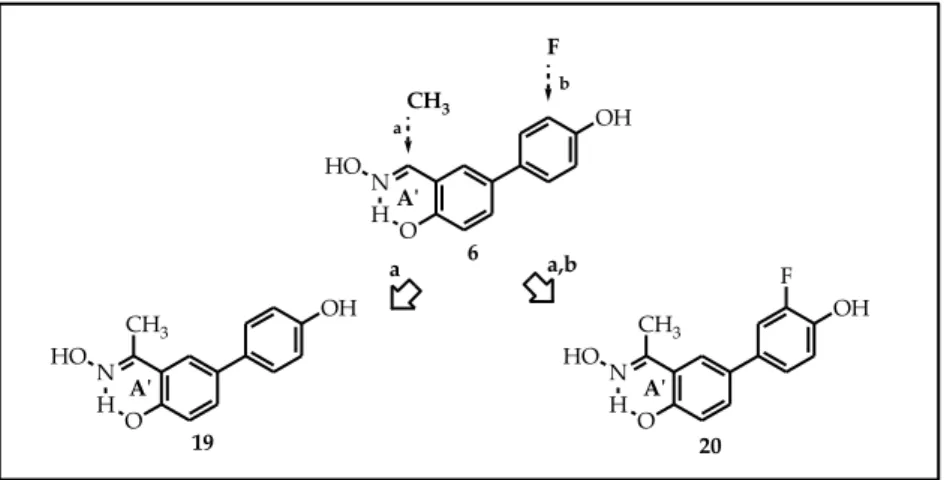
To investigate the steric effect due to the presence of a methyl group on the oximic carbon, we synthetized new keto-oxime derivatives 19 and 20 (Figure 25). Compound 19 is the keto-oxime analog of 6, while compound 20 differs from compound 19 for the presence of a fluorine atom in meta-position on the peripheral 4-aryl substituent.
43 Figure 25. O N HO O OH N HO 20 19 H H A'ʹ A'ʹ OH F CH3 CH3 CH3 F b a,b OH N H O HO 6 a a A'ʹ
To evaluate the importance of the presence of the chlorine atom on the central scaffold on “inverted” salycilaldoxime (STRUCTURE B), we decided to replace it with a methyl or a trifluoromethyl group (compounds 21 and 22, Figure 26).
Figure 26. OH N HO HO 6 A'ʹ 22 CF3 O OH N HO H CH3 O OH N HO H 21 b a CF3 CH3 a b
44
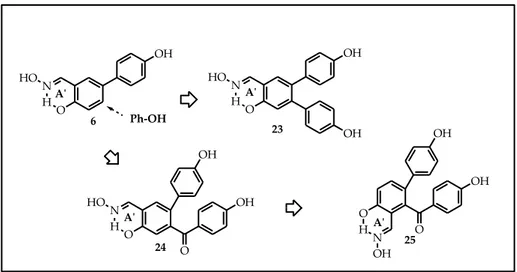
As highlighted by docking studies, compound 6, the simplest member of the “inverted” salicylaldoxime series, is able to form a particular interaction with a threonine 299 in the ERβ binding cavity; at the same time, it’s not involved in the interaction with the histidine 475, that is generally found in the “non inverted” salicylaldoximes. In order to possibly restore the interaction with histidine 475, we envisaged the possibility to attach a second para-hydroxy phenolic function directly, or through a carbonyl linker, to the central scaffold. Therefore, compounds 23-25 (Figure 27) were designed.
Figure 27. O N O OH OH HO H O OH OH 24 25 O HN OH A'ʹ A'ʹ N H O HO OH OH 23 A'ʹ OH N H O HO 6 A'ʹ Ph-‐‑OH
45
Finally, we decided to replace the salycilaldoxime moiety with an other bioisosteric replacement as the anthranyl function. This type of structural modification was previously experimented in some diaryl-substituted oximes, obtaining good results.24 So we designed new derivatives 26 and 27 (Figure 28), that are anthranyl analogues of 6 and 7, respectively.25 Figure 28. OH N HO HO 6 A'ʹ OH N HO HO Cl 7 A'ʹ 27 Cl N OH N HO H N OH N HO H 26 H H A'ʹ A'ʹ CHEMISTRY
The synthesis of compounds 7 and 8 started from 3-bromo-2-chloro-6-methoxybenzaldehyde 28, as shown in Scheme 1.
46 Scheme 1. 28 Cl CHO OCH3 Br 29. R=H 30. R=F Cl OHC H3CO OCH3 R 31. R=H 32. R=F Cl OHC HO OH R 7. R=H 8. R=F Cl O OH R N HO H a,b c d
a) Pd(PPh3)4, p-methoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOHass., 100 °C; b) Pd(PPh3)4, 3-fluoro-p-methoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOHass., 100 °C;c) BBr3, CH2Cl2, -78 → 0 °C; d)
NH2OH·HCl, EtOH, H2O, 50 °C.
Compound 28 was submitted to a Pd-catalyzed cross-coupling reaction, under "classical" Suzuki conditions, with the appropriate arylboronic acid, (phenylboronic, 4-methoxyphenylboronic, 3-fluoro-4-methoxyphenylboronic), forming the corresponding monoaryl-substituted adducts 29 and 30. The resulting adducts were treated with boron tribromide to obtain O-demethylated salicylaldehydes 31 and 32, which were then condensed with hydroxylamine hydrochloride, yielding final salicylaldoximes 7 and 8.
47 Scheme 2. a b c e 28 Cl CH3 OCH3 Cl OCH3 Br Cl OCH3 OAc Cl OCH3 OH Cl CHO OCH3 Cl CHO OCH3 Br 33 35 36 37 34 d
a) NBS, Bz2O2, 95 → 100 °C; b) AcONa/AcOH, 100 °C; c) NaOH 20%, MeOH, THF, 100 °C; d) PCC,
CH2Cl2, t.a.; e) Br2, AcOH, r. t..
Synthesis of 3-bromo-2-chloro-6-methoxybenzaldehyde 28 (Scheme 2) started from 3-chloro-2-methylanisole 33 that was submitted to a radical bromination with NBS in presence of Bz2O2, to obtain
benzhylbromide 34. This intermediate was first treated with AcONa in acetic acid and then with NaOH, methanol and THF, to obtain benzhyl alcohol 35. This was oxidized with MnO2 in toluene, to get aldehyde 37
that was brominated with Br2 in AcOH, affording derivative 28. This
synthetic approach was optimized by oxidizing the benzhyl alcohol with PCC, improving the yield of the reaction from 20% to 80%.26
The synthesis of methyl/chloro-substituted oximes 9-11 was slightly longer, as shown in Scheme 3 and 4. Commercially available phenols 38
48
and 47 underwent O-allylation upon treatment with allyl bromide. Claisen rearrangement of the resulting ethers 39 and 48, at 210 °C, yielded O-allyl-phenols 40 and 49. The terminal double bond of 40 and 49 was "shifted" to the internal position by alkaline isomerisation with potassium tert-butoxide in DMSO at 55 °C, to give β-methylstyrene derivatives 41 and 50 as E-isomers. Oxidative cleavage of the styrene-type double bond with sodium periodate in the presence of catalytic amounts of osmium tetroxide yielded aldehydes 42 and 51, which were then treated with bromine in glacial acetic acid, affording mono-brominated salicyaldehyde derivatives 43 and 52.
At this point, we initially tried to carry out a cross-coupling reaction of 43, 52 with the appropriate boronic acid, to promote the replacement of the bromine atom with a suitable aryl substituent. Unfortunately, we were not able to perform this reaction efficiently, probably because of the bidentate chelating effect of the free OH/CHO vicinal groups of the salicylaldehyde portion towards both the boron atom of the boronic acid and the palladium catalyst, thus diverting the reactants from the correct reaction pathway during the Suzuki step. Therefore, we first protected the phenol hydroxyl with a methyl group, by reaction with iodomethane and
49
potassium carbonate in acetone, and then submitted the resulting anisole derivatives 44, 53 to the Suzuki coupling reaction, obtaining monoaryl adducts 45, 54 and 55. Final steps included O-demethylation with BBr3,
to give intermediates 46, 56 and 57, and subsequent condensation with hydroxylamine hydrochloride, affording final oximes 9-11.
Scheme 3. Cl CH3 HO Cl CH3 O Cl CH3 HO Cl CH3 HO Cl CH3 HO OHC Cl CH3 HO OHC Br Cl CH3 H3CO OHC OCH3 Cl CH3 H3CO OHC Br Cl CH3 HO OHC OH Cl CH3 OH N H O HO a b c d e g f i h 38 39 40 41 42 43 44 45 46 9
a) K2CO3, Allyl bromide, CH3CN, 80 °C, b)210 °C; c) t-BuOK, DMSO 55 °C, d) OsO4, NaIO4, dioxane/H2O;
e) Br2, AcOH; f) MeI, K2CO3; e) Br2, AcOH; g) Pd(PPh3)4, p-metoxy-phenylboronic acid, Na2CO3 2M,
50 Scheme 4. CH3 Cl HO CH3 Cl O CH3 Cl HO CH3 Cl HO CH3 Cl HO OHC CH3 Cl HO OHC Br CH3 Cl H3CO OHC OCH3 CH3 Cl H3CO OHC Br CH3 Cl HO OHC OH R R CH3 Cl OH R N HO HO a b c d e g,h f i l 47 48 49 50 51 52 53 54. R=H 55. R=F 56. R=H 57. R=F 10. R=H11. R=F
a) K2CO3, Allyl bromide, CH3CN, 80 °C, b) 210 °C; c) t-BuOK, DMSO 55 °C, d) OsO4, NaIO4, dioxane/H2O;
e) Br2, AcOH; f) MeI, K2CO3; e) Br2, AcOH; g) Pd(PPh3)4, p-metoxy-phenylboronic acid, Na2CO3 2M,
toluene, EtOH ass., 100 °C; h) Pd(PPh3)4, 3-fluoro-p-metoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH
51
To prepare compound 12b (Scheme 5), derivative 29 was submitted to two reaction cycles under the same Suzuki conditions, using a total of 3.2 equivalents of boronic acid, afforded mostly the diaryl-substituted intermediate 84. The resulting adduct was treated with boron tribromide to obtain O-demethylated salicylaldehyde 85, which were then condensed with hydroxylamine hydrochloride, yielding final salicylaldoxime 12.
Scheme 5. a OHC H3CO OCH3 29 Cl OHC H3CO OCH3 58 OCH3 OHC HO OH OH OH OH N H O HO b c 59 12b
a) Pd(PPh3)4, p-metoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass., 100 °C; b) BBr3, CH2Cl2, -78 à
52
The synthesis of compound 16 started from 3-bromo-2-chloro-6-methoxybenzaldehyde 28, as shown in Scheme 6.
Scheme 6. 28 Cl CHO OCH3 Br 67 Cl OHC H3CO OCH3 Cl 68 Cl OHC HO OH Cl 16 Cl O OH Cl N HO H a b c a) Pd(PPh3)4, 3-chloro-p-methoxy-phenylboronic acid, Na2CO3, THF, 100 °C;b) BBr3, CH2Cl2, -78 → 0 °C; c) NH2OH·HCl, EtOH, H2O, 50 °C.
Compound 28 was submitted to a Pd-catalyzed cross-coupling reaction, with the 3-chloro-4-methoxyphenylboronic, Na2CO3, THF
100°C, forming the corresponding monoaryl-substituted adduct 67. The resulting adduct was treated with boron tribromide to obtain O-demethylated salicylaldehyde 68, which was then condensed with hydroxylamine hydrochloride, yielding final salicylaldoxime 16.
53
The synthesis of compounds 13-15 started from 5-bromosalycilaldehyde 58, as shown in Scheme 7 and 8.
Scheme 7. OHC HO Br OHC HO OHC HO a b c 60 61 62 13 N HO HO OH OCH3 OH
a) Pd(PPh3)4, m-methoxy-phenylboronic acid, Na2CO3 2M, toluene,EtOH ass., 100 °C;b) BBr3, CH2Cl2, -78 →
0 °C; c) NH2OH·HCl, EtOH, H2O, 50 °C. Scheme 8. OHC HO Br OHC HO OCH3 R OHC HO OH R HO OH R N HO a,b c d 60 63. R=CH3 64. R=Cl 65. R=CH3 66. R=Cl 14. R=CH3 15. R=Cl
a) Pd(PPh3)4, 3-methyl-p-metoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass., 100 °C; b) Pd(PPh3)4,
3-cloro-p-metoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass., 100 °C; c) BBr3, CH2Cl2, -78 à 0 °C;
54
5-Bromosalicylaldehyde 60 was submitted to a cross-coupling reaction in the presence of 3-methoxyphenylboronic or 3-methyl-4-methoxyphenylboronic or 3-chloro-4-3-methyl-4-methoxyphenylboronic acid, affording the monoaryl-substituted adducts 61, 63 and 64, respectively. The resulting adducts were treated with boron tribromide to obtain O-demethylated salicylaldehydes 62, 65 and 66, which were then condensed with hydroxylamine hydrochloride, yielding final salicylaldoximes 13-15.
The synthesis of compounds 18, 19 started from 3-bromo-2-chloro-6-methoxybenzaldehyde 28, and 5-bromosalycilaldehyde 60 as shown in Scheme 9. Scheme 9. OHC R2O Br OHC R2O OCH3 OH a 28. R1=Cl; R2=CH3 60. R1=H; R2=H 17. R=Cl 18. R=H R1 R1 R N H O HO OH OCH3 69. R1=Cl; R2=CH3 70. R1=H; R2=H OHC HO OH R1 OH 71. R1=Cl 72. R1=H b
a) Pd(PPh3)4, 2,4-dimetoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass, 100 °C; b) BBr3, CH2Cl2, -78
55
Aldhehydes 28 and 60 were submitted to Suzuki coupling reaction with 2,4-dimethoxy-phenylboronic acid. The resulting adducts 69, 70 were treated with boron tribromide; unfortunately, O-demethylated salycilaldehydes 70, 71 were not obtained, because of purification problems.
The synthesis of keto-oximes 20 and 21 started from 5-bromo-2-hydroxy-acetophenone 73, as shown in Scheme 10.
Scheme 10. HO Br H3CO OCH3 HO OH OH a d 73 N HO HO H3CO Br 74 O H3C O H3C O H3C O H3C CH3 b,c e 75. R=H 76. R=F 77. R=H 78. R=F 19. R=H 20. R=F R R R
a) MeI, K2CO3, b) Pd(PPh3)4, p-metoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass, 100 °C; c)
Pd(PPh3)4, 3-F-p-methoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass., 100 °C, d) BBr3, CH2Cl2, -78
à 0 °C; e) NH2OH·HCl, EtOH, H2O, 50 °C.
This ketone was protected with a methyl group, by reaction with iodomethane and potassium carbonate in acetone, and the resulting
56
anisole derivative 74 was submitted to a Suzuki coupling reaction with 4-methoxyphenylboronic or 3-fluoro-4-4-methoxyphenylboronic acid. The resulting adducts 75, 76 were treated with boron tribromide to obtain O-demethylated ketones 77, 78, which were then condensed with hydroxylamine hydrochloride, yielding final keto-oximes 19 and 20.
To synthesize compound 21, we tried different synthetic strategies (Scheme 11 and 12). In particular, we tried to directly formylate m-cresole 79 with sodium hydroxide in CHCl3, or with TiCl4 and
Cl2CHOCH3, but these attempts did not lead to salicylaldehyde 80
(Scheme 11). Scheme 11. OH CH3 OH CH3 CHO OH CH3 Br 79 80 81 a b
57
Therefore, we decided to brominate m-cresole 79 with Br2 in acetic
acid or in CHCl3, but intermediate 81 was not obtained (Scheme 11).
Then, we tried to replace the chlorine atom of 28 with a methyl group, through a cross-coupling reaction with methyl boronic acid, K3PO4, dry
toluene and S-Phos, but derivative 82 was not obtained (Scheme 12).
Scheme 12. OHC H3CO OCH3 OH a b 82 21 CH3 CH3 N H O HO OHC H3CO OCH3 29 Cl
a) Methylboronic acid, S-Phos, K3PO4, toluene, 100 °C.
The synthetic strategy to prepare compound 22 is described in Scheme 13. Scheme 13. a b c d e CF3 CH3 NO2 CF3 NO2 Br CF3 NO2 OAc CF3 NO2 OH CF3 CHO NO2 83 85 86 87 84 CF3 CHO NH2 22 CF3 O OH N HO H 88
a) NBS, Bz2O2, 95 → 100 °C; b) AcONa/AcOH, 100 °C; c) NaOH 20%, MeOH, THF, 100 °C; d) PCC,
58
3-chloro-2-methyl-nitrobenzene 83 was submitted to a radical bromination with NBS in presence of Bz2O2, affording benzhylbromide 84. This was first treated with AcONa in acetic acid, and then with NaOH, methanol and THF to obtain benzhyl alcohol 86. This alcohol was oxidized with PCC to get nitro-aldehyde 87. The nitro group was reduced with H2 in presence of Pd/C, to obtain amino-aldehyde 88.
Unfortunately, the subsequent bromination of this intermediate in para position respect to the amino group, or the conversion of the amino group in hydroxyl function was not successful.
To prepare compound 23 (Scheme 14), we submitted commercial 3,4-dichloro-salicylaldehyde 89 to two subsequent Suzuki reactions, but we did not obtain the diaryl derivative 90.
Scheme 14. OHC HO Cl Cl OHC HO OHC H3CO OHC H3CO Cl Cl N HO HO OCH3 OCH3 OCH3 OCH3 OH OH 89 90 91 92 c a,b,c d 23
a) Pd(PPh3)4, p-metoxy-phenylboronic acid, Na2CO3 2M, toluene, EtOH ass, 100 °C; b)
p-metoxy-phenylboronic acid, Pd2(DBA)3, CsCO3, dioxane 100 °C, c) p-metoxy-phenylboronic acid Pd(OAc)2, TBAB,
59
Therefore, we decided to use Fu conditions (Pd2(dba)3, CsCO3,
dioxane), but also in this case we failed in our pourpose. To avoid the bidentate chelating effect that the free OH/CHO vicinal groups of the salicylaldehyde portion have on both the boron atom of the boronic acid and the palladium catalyst, we protected the phenol hydroxyl with a methyl group, by reaction with iodomethane and potassium carbonate in acetone, and then submitted the resulting anisole derivatives 91 to the cross-coupling in water, in the presence of Pd(OAc)2 and
tetrabuthylammonium bromide, but also in this case we did not obtain the diaryl derivate 92.
The synthesis of the diaryl-oximes 24 and 25, charactherized by the presence of a carbonylic linker, was slightly longer, as shown in Scheme 15.
60 Scheme 15. H3CO Br COOH HO Br COOH a HO Br OCH3 O b O Br OCH3 O c HO Br OCH3 O HO Br OCH3 O d 93 94 95 96 97 98 O N O OH OH HO H O OH OH 24 25 O H N OH
a) BBr3, CH2Cl2, -78 → 0 °C; b) Graphite, MeSO3H, anisole, 80 °C; c) K2CO3, allyl-bromide, CH3CN, 80 °C;
d) N-Methyl-aniline, 180 °C.
Commercially available anisole 93 was treated with boron tribromide to obtain the phenol 94, that was submitted to Friedel-Craft acylation in presence of graphite and MeSO3H, affording the diaryl-phenol 95. This
phenol was converted to O-allyl derivative 96 upon treatment with allyl bromide. Claisen rearrangement of the resulting ether at 180 °C in PhNHMe, afforded O-allyl-phenols 97 and 98; but this rearrangement did not go to completion, and the amounts of O-allyl-phenols were not enough to prosecute the synthetic pathway.
61
The synthesis of antranilaldoximes 26 and 27 started from 3-chloro-2-methyl-ammine 96 (Scheme 16). Scheme 16. Cl CH3 NH2 Cl CH3 NH Cl NH2 CF3 O Cl NH CF3 O Cl CH3 N CF3 O Cl N CF3 O Me Me Br Br Br 99 100 101 103 102 104 a b b b
a) Trifluoro-acetammide, K2CO3, acetone, b) NBS, Bz2O2, 95 → 100 °C; c) MeI, acetone, K2CO3.
Aniline 99 was submitted to a radicalic bromination with NBS and Bz2O2, but the reactivity of the amino group did not allow the reaction to
happen. For this reason, we protected the amino group with trifluoroacetamide, to give 101, and we treated derivative 101 with iodomethane and potassium carbonate in acetone, affording derivative 102. Both 101 and 102 were submitted to a radicalic bromination with NBS, but these attempts were not successful.
62
All the synthesized oximes were obtained as single E-diastereoisomeric forms, presumably because the intramolecular hydrogen bond, which can only form in the E-isomer, contributes to the oxime stability. This selectivity had already been demonstrated for other oxime analogues previously reported and it was here confirmed by the chemical shift values of the oxime protons of all the new products, which were always found downfield from 8 ppm (δ ≥ 8, see the experimental section).21,22,24,25
63
ESTROGEN RECEPTOR BINDING ASSAYS
ERα- and ERβ-binding affinities of synthesized oximes were determined by a radiometric competitive binding assay with [3H]estradiol. Preparations of purified, full-length human ERα and ERβ (PanVera/Invitrogen) were used. Experimental methods have been previously described.27,28 These assays were performed by J. A. Katzenellenbogen, J. R. Gunther and K. E. Carlson, (Dept. of Chemistry, University of Illinois, 600 S. Mathews Avenue, Urbana, IL 61801, USA) who collaborate for several years with the research laboratory in which I accomplished my PhD. The relative binding affinity (RBA) values for compounds 7-16 are summarized in Table 1. RBA values are reported as percentages (%) of that of estradiol, which is set at 100%.
The Kd for estradiol for ERα is 0.2 nM, and for ERβ it is 0.5 nM. Ki
values for the new compounds can be readily calculated by using the formula:
64
Table 1.
Compounds Structure hERa
(%) hERβ (%) ratio β/α
Estradiol
HO OH H3C100 100 1
1
O H N OH HO A'ʹ 0.007 0.553 792
O H N OH HO Cl A'ʹ 0.065 4.21 653
O H N OH HO CH3 A'ʹ 0.249 4.10 164
O H N OH HO CN A'ʹ 0.004 0.078 205
H O N OH HO Cl F A'ʹ 0.114 7.01 626
OH N H O HO A'ʹ 0.064 2.64 4165
7
OH N H O HO Cl A'ʹ 4.46 130.3 308
OH N H O HO Cl F A'ʹ 1.88 87.11 469
OH O N H Cl CH3 HO A'ʹ 0.07 0.576 1010
OH O N H CH3 Cl HO A'ʹ 1.50 18.27 1011
OH O N H CH3 Cl F HO A'ʹ 0.392 7.90 2012b
N H O HO OH A'ʹ OH 88.4 101 1.166
Analyzing some important results obtained with the previously synthesized STRUCTURE A salicylaldoximes,21,22,29 (Table 1) we can see that the simplest member of this class (compound 1) was a very ERβ-selective ligand (β/α ratio = 79) but its ERβ-binding affinity was modest (0.55%). The introduction of substituents in the central ring had
12a
OH O H NA'ʹ OH HO 0.92 0.35 0.3813
N H O HO OH A'ʹ 0.003 0.101 2914
OH N H O HO CH 3 A'ʹ 0.035 0.763 2515
OH N H O HO Cl A'ʹ 0.012 0.906 6716
OH N H O HO Cl A'ʹ Cl 1.43 7.19 6.967
significant effects on binding affinities. Infact, the introduction of a chlorine in the central scaffold, as in 2, led to a 8-fold increase in the affinity (RBA = 4.21%), together with a remarkable level of ERβ-selectivity (β/α ratio = 65). The methyl-substituted derivative 3 showed a similar level of ERβ-affinity (RBA = 4.10%), respect to 4, but its subtype selectivity was not very satisfactory. On the other hand, the introduction of a cyano group, as in derivative 4, caused a loss of affinity for both receptor subtypes. Oxime 5 showed an increase in ERβ binding affinity, reaching the best value among the STRUCTURE A compounds (ERβ-RBA value of 7.01%, which corresponds to a Ki of 7.1 nM). Fortunately,
a high level of subtype-selectivity was also preserved (β/α = 64).
Evaluating the results obtained with salicylaldoximes of the
STRUCTURE B series,29 we can point that the simplest member of this
series, 6, showed a 5-fold higher ERβ-binding affinity (2.64%) compared to 1 (Table 1), together with a low value of ERα-binding affinity (0.064%), so that this compound maintained a significant level of ERβ-selectivity (β/α ratio = 41).
Among the new salicylaldoximes, compound 7, possessing a chlorine atom in the central scaffold, showed the best results, with an ERβ-affinity
68
higher than that of estradiol (RBA = 130%, corresponding to a Ki of 0.38
nM), and a noticeable ERβ-selectivity (β/α ratio = 29).
The introduction of a meta-fluorine in the peripheral aryl substituent, as in compound 8, led to an ERβ-selectivity (β/α ratio = 46) higher than that of 7, together with an ERβ-binding affinity remarkably high (RBA = 87%, corresponding to a Ki of 0.57 nM).
The introduction of a methyl group in the central ring of 7 led to compound 9, which showed a great drop in affinity for both receptor subtypes.
The inversion of the relative positions of the methyl and chlorine substituents (compound 10: RBA = 1.47% for ERα and 15.8% for ERβ) caused an improvement of the binding properties, respect to 9. However, the ERβ-selectivity of 10 was not high (β/α ratio = 11).
Compound 11, which differs from 10 by the presence of a meta-fluorine atom in the peripheral aryl substituent, (RBA = 0.392% for ERα and 7.90% for ERβ), showed a marked increase in ERβ-selectivity (β/α ratio = 20).
The presence of two para-hydroxyphenyl substituents, as in compound 12b, led to high ER-affinities (RBA = 88.4% for ERα and 101% for
69
ERβ), respect to that of the corresponding STRUCTURE A derivative 12a, without any appreciable selectivity (β/α ratio = 1.1).
Compound 13, in which the hydroxy group of the aryl substituent is shifted from para to meta position, and compound 14, bearing a methyl substituent adiacent to the 4'-hydroxy group, showed very low levels of ER-affinities and similar appreciable levels of ERβ-selectivity (β/α ratio = 29 for 13; β/α ratio = 25 for 14). The replacement of the methyl group of 14 with a chlorine atom, as in 15, caused a small increment in ERβ- affinity (RBA = 0.906), together with a decrement in ERα-affinity (RBA = 0.012), thus leading to a good level of ERβ-selectivity (β/α ratio = 67).
Finally, the addition of a chlorine atom in position 6 of the central scaffold of 15, as in compound 16, led to an increased level of ERβ- affinity (RBA = 7.19%), but, at the same time, an increment in ERα binding affinity (1.43%) was also found, so its level of ERβ-selectivity was quite low (β/α ratio = 6.9).
70 MOLECULAR MODELING
With the aim to rationalize the binding properties of the newly synthesized salicylaldoximes, an automated computational analysis was performed. AUTODOCK 4.0 software was used for the docking of the ligands into ERα and ERβ (PDB codes 2I0J and 2I0G, respectively).30 Docking studies were performed by the research group of Prof. A. Martinelli (Dept. of Pharmaceutical Science, University of Pisa, Via Bonanno 6, 56126, Pisa, Italy). Docking of the diarylsubstituted compound 12b into ERα and ERβ is shown in Figures 16A and B, respectively, and highlights strong interactions that confirm its high binding affinities for both subtypes. The interactions are the same in both ligand binding cavities, and this is in agreement with its similar affinity for ERα and ERβ resulted from binding assays. The pseudocycle/oxime system forms an H-bond with H475 (H524 in Erα). The para-hydroxyl of the 6-aryl substituent interacts with T299 (T347 in ERα). The para-hydroxyl group on the 5-aryl substituent is engaged in an H-bond network, including E305 (E353 in ERα), R346 (R394 in ERα), and a water molecule.
71
Figure. 17B and 16D show the docking of monoaryl-substituted salicylaldoximes 7 and 8, respectively, into ERβ. The pseudocycle/oxime system is involved in the H-bond network of the E305-R346-water system; the OH of the p-hydroxyphenyl ring forms a H-bond with T299. These interactions are similar to those previously found with their non-halogenated analog 6.22 Besides, the chlorine atom fits well into a lipophilic pocket delimited by A302, W335, M336, and L339. It is important to underline that in the case of STRUCTURE A derivatives such as 2 and 5, a completely different orientation was found, in which the p-hydroxyphenyl was engaged in the H-bond network of the E305-R346-water system and the pseudocycle/oxime system formed a H-bond with H475.22
Docking of the same compounds (7 and 8) into ERα, shows a completely different disposition (Figure 30A and 29C): these compounds are turned upside-down, such that their phenol 4’-OH group is involved in the H-bond network with E353-R394-water system, and their pseudocycle/oxime system forms a H-bond with H524. In the binding cavity of this receptor, the presence of a leucine (L384), instead of a less bulky methionine (M336 in ERβ), does not leave enough space for the
72
4’-hydroxyl to reach the OH of T347 (T299 in ERβ). So, the strong interaction between the 4’-hydroxyl and the threonine residue is possible only in the ERβ-LBD cavity.29
Docking of 9 into ERβ (Figure 29E), shows that this compound become rotated by 180°, respect to 7 and 8, resulting in a less stable complex. This is because the binding region in the proximity of M340 is not large enough to tolerate the presence of a CH3 in the para position
respect to the chloro-substituent. Infact, as previously seen in binding assays, compound 9 has a reduced affinity for ERβ.29
An orientation similar the one found for compounds 7 and 8 is restored if the position of the CH3/Cl is inverted, as in compounds 10
(Figure. 16F) and 11 (Figure 31B); the methyl substituent can now be accommodated in the pocket delimited by A302, W335, M336, and L339, while the chlorine atom now fits well in the pocket close to M340, being smaller than the methyl group present in an analogous position of 9.29
It is important to point that in this study we did not computationally evaluate the possible fit-induced effects (requiring the use of a flexible receptor); the analyzed ligands were docked into a rigid version of the