Review Article
Ischemia/Reperfusion Injury following
Acute Myocardial Infarction: A Critical Issue for
Clinicians and Forensic Pathologists
Margherita Neri,
1Irene Riezzo,
2Natascha Pascale,
2Cristoforo Pomara,
2and Emanuela Turillazzi
21Section of Forensic Pathology, Morphology, Surgery and Experimental Medicine Department, University of Ferrara,
Ospedale “Sant’Anna”, Via Fossato di Mortara 70, 44121 Ferrara, Italy
2Section of Forensic Pathology, Clinical and Experimental Medicine Department, University of Foggia, Ospedale Colonnello D’Avanzo,
Viale Degli Aviatori 1, 71100 Foggia, Italy
Correspondence should be addressed to Emanuela Turillazzi; emanuela [email protected]
Received 4 August 2016; Revised 26 October 2016; Accepted 30 November 2016; Published 13 February 2017 Academic Editor: Veronica Tisato
Copyright © 2017 Margherita Neri et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Acute myocardial infarction (AMI) is a leading cause of morbidity and mortality. Reperfusion strategies are the current standard therapy for AMI. However, they may result in paradoxical cardiomyocyte dysfunction, known as ischemic reperfusion injury (IRI). Different forms of IRI are recognized, of which only the first two are reversible: reperfusion-induced arrhythmias, myocardial stunning, microvascular obstruction, and lethal myocardial reperfusion injury. Sudden death is the most common pattern for ischemia-induced lethal ventricular arrhythmias during AMI. The exact mechanisms of IRI are not fully known. Molecular, cellular, and tissue alterations such as cell death, inflammation, neurohumoral activation, and oxidative stress are considered to be of paramount importance in IRI. However, comprehension of the exact pathophysiological mechanisms remains a challenge for clinicians. Furthermore, myocardial IRI is a critical issue also for forensic pathologists since sudden death may occur despite timely reperfusion following AMI, that is one of the most frequently litigated areas of cardiology practice. In this paper we explore the literature regarding the pathophysiology of myocardial IRI, focusing on the possible role of the calpain system, oxidative-nitrosative stress, and matrix metalloproteinases and aiming to foster knowledge of IRI pathophysiology also in terms of medicolegal understanding of sudden deaths following AMI.
1. Introduction
Acute myocardial infarction (AMI) is a leading cause of mor-bidity and mortality in the world [1]. Reperfusion strategies are the current standard therapy for AMI [2, 3]. They may, however, result in paradoxical cardiomyocyte dysfunction and worsen tissue damage, in a process known as “reperfu-sion injury” [4–9]. Ischemic reperfu“reperfu-sion injury (IRI) typi-cally arises in patients presenting with an acute ST-segment elevation myocardial infarction (STEMI), in whom the most effective therapeutic intervention is timely and effec-tive myocardial reperfusion [7, 10–14]. Reperfusion itself is known as a “double-edged sword” [4, 15] due to the spectrum of reperfusion-associated pathologies. Outcomes subsequent to IRI accrue in a time-dependent fashion [16],
beginning with oxidative stress, inflammation, intracellular
Ca2+ overload, and rapidly proceeding to irreversible cell
death by apoptosis and necrosis [13, 16]. Different forms of myocardial IRI are recognized, of which only the first two are reversible: reperfusion-induced arrhythmias, myocardial stunning, microvascular obstruction, and lethal myocardial reperfusion injury [13].
In particular, sudden death is the most common pattern for ischemia-induced lethal ventricular arrhythmias (VAs) during the acute phase of myocardial infarction [17], and it is well known that reperfusion itself can lead to life-threatening VAs [17] and, ultimately, induce sudden mortality.
The exact mechanisms of IRI are not fully known [18]. Molecular, cellular, and tissue alterations such as cell death,
Volume 2017, Article ID 7018393, 14 pages https://doi.org/10.1155/2017/7018393
inflammation, neurohumoral activation, and oxidative stress are considered to be of paramount importance for IRI development [10, 19]. However, comprehension of the exact pathophysiological mechanisms of IRI [20, 21] remains a challenge for clinicians [22, 23], and the existence of reper-fusion injury is still a matter of debate in the scientific com-munity, essentially due to a lack of a definitive clinical doc-umentation. Many gaps still exist between experimental ani-mal models and human clinical experience, with subsequent difficulties in translating experimental results on cardio-protection to clinical practice [22–24]. Despite the difficulties that still exist in fully comprehending myocardial IRI, early and aggressive reperfusion strategies remain the most impor-tant intervention and are strongly advocated. The develop-ment of ischemic conditioning strategies to limit the extent of infarcted tissue caused by ischemia/reperfusion injury markedly enhances the ability of the heart to withstand an ischemic insult [25].
Finally, myocardial IRI is a critical issue also for forensic pathologists since sudden death may occur despite timely reperfusion following AMI, that is one of the most frequently litigated areas of cardiology practice [26, 27].
In this paper we explore the literature regarding the pathophysiology of myocardial IRI, focusing on the possible role of the calpain system, oxidative-nitrosative stress, and matrix metalloproteinases. We discuss these mechanisms within the broad scenario of IRI, also discussing the medi-colegal issues related to sudden deaths occurring during the acute phase of myocardial infarct following reperfusion interventions.
2. The Calpain System
The process of IRI is not yet completely understood in its underlying pathophysiological mechanisms. Several path-ways have been proposed, including cytosolic and
mitochon-drial Ca2+overload, release of reactive oxygen species (ROS),
acute inflammatory response, and impaired metabolism [20, 21]. These alterations may collaboratively act and produce irreversible damage to ischemic reperfused cardiomyocytes.
The possibility that the calpain system could play a role in generating myocardial IRI has been experimentally investigated in the literature [28–32], and several studies have focused on the effects of calpain inhibitors in improving myocardial dysfunction in different animal models [33–37].
Calpains are a family of Ca2+-dependent nonlysosomal
cys-tein procys-teinase localized in the cytosol in their inactive form [38]. Calpain activation, which may occur under several con-ditions, is thought to be a key mechanism in activating a number of substrates such as growth factor receptors, cyto-skeletal proteins, microtubules associated proteins, and mito-chondria, so playing a crucial role in cell cycle, apoptosis, and differentiation [38–40].
The calpain superfamily is complex, and more than 25 calpains or calpain-like molecules have been discovered. Calpains 1 and 2 are biologically activated when they arrange as dimer with a 30 kDa subunit. Both biologically active
calpains are usually called𝜇-calpain (calpain 1 + 30 kDa
sub-unit) and m-calpain (calpain 2 + 30-kDa subsub-unit). The
terms 𝜇-calpain and m-calpain indicate, respectively, the
micromolar and millimolar Ca2+concentrations required for
their activation [19]. Calpains may appear in the form of both “ubiquitous” isoenzymes that are present in almost all cells
(such as𝜇-calpain, m-calpain, and calpains 5, 7, 10, 13, and 15)
and “tissue specific” calpains expressed only in special tissues and cells, such as calpains 3 and 6 and others [31].
In brief, it has been hypothesized that, under physio-logical conditions, inactive calpains are stored in cellular cytosol and bound in a substrate competitive manner to their endogenous inhibitor calpastatin. The elevation of intracel-lular calcium levels is the key to the calpain activation process. Calpain conformational changes permit its translocation into
cellular membrane, where phospholipids reduce the Ca2+
threshold for calpain activation or close the Ca2+ channels
leading up to protein activation [41]. Several pathological cardiac events are associated with an imbalance of calcium homeostasis related to myocardial ischemia/reperfusion injury [29–31]. Experimental studies on isolated perfused mammalian hearts demonstrated an increase in intracellular
Ca2+concentrations in response to ischemia/reperfusion [31,
41]. Myocardial ischemia favours intracellular ion accumula-tion (sodium, calcium) till dropping in pH and tissue acido-sis. Reperfusion evokes rapid alterations in ion flux and inter-acts with ischemia in altering the physiology of ion exchange [42]. Among others, a final result of the dangerous inter-play between ischemia and reperfusion is intracellular cal-cium overload.
The kinetics of calpain activation are not completely understood, and whether or not translocation to the sar-colemma is needed for calpain activation during IRI remains undetermined [43]. In their elegant experiment, Hernando et al. [37] suggested that calpain translocation to the cardiomy-ocytes membranes during ischemia is independent of its acti-vation since intracellular acidosis occurring during ischemia is likely to inhibit calpain activation. As intracellular pH nor-malizes following reperfusion, calpain activation occurs. Des-pite translocation, calpain seems to remain inactive even after 60 minutes of ischemia and only on reperfusion is it activated [37].
Activated calpain has a number of substrates such as growth factor receptors, cytoskeletal proteins, microtubules associated proteins, and mitochondria, thus playing a crucial role in the processes of cell cycle, apoptosis, and differentia-tion, negatively affecting cardiomyocyte function.
Firstly, the calpain system is part of the integrated prote-olytic system which is crucial to the maintenance of the struc-ture and function of the cardiac sarcomere. An imbalance of this system is the key to the sarcomeric dysfunction linked to several cardiovascular diseases, including hypoxia, IRI, myocardial infarction, and end-stage heart failure. Protein degradation (proteolysis) within cardiac sarcomere is regu-lated mainly by three systems: the ubiquitin proteasome sys-tem (UPS); autophagy/lysosomal degradation; and the cal-pain system [44]. Degradation of myofibrillar proteins involved in the contractile process is an effect of calpain acti-vation. The degradation process following IRI involves either structural or regulatory proteins of contractile apparatus. In vitro study [45] showed that many of these proteins are
Reperfusion Calpain system activation Degradation of structural proteins and increased
sarcolemmal fragility Apoptosis activation and mitochondria damage Proteolysis of myofibrillar proteins/ loss/disorganization of T-tubules structure Ischemia Membrane damage and
rupture Alteration of myofibrillarcontractility
Cell death Oxidative stress Heart failure Ca2+overload Ca2+overload ↓ pH pH↑ recovery
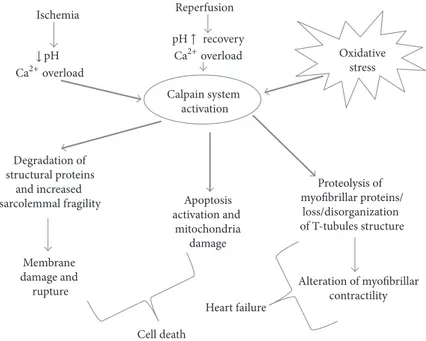
Figure 1: Schematic representation of calpain activation during myocardial IRI. Ca2+overload and pH recovery in reperfusion phase are
crucial in the activation of the calpain system. Increased sarcolemmal fragility may lead to membrane rupture and cell death. In addition, both the death-receptor and mitochondrial mediated apoptotic pathways seem to be affected by calpain activation. The degradation of myofibrillar proteins and the loss/disorganization of T-tubules structure are key factors in post-MI heart failure development.
potential targets of activated calpains, thus contributing to the development of postischemic injury in the human myo-cardium. Several experimental studies demonstrated that the loss/disorganization of T-tubules structure is a key factor in heart failure development [46–48]. Calpain-mediated disruption of T-tubules integrity through the proteolysis of junctophilin is demonstrated to be one of the major factors involved in an experimental model of cardiac muscle failure [49, 50].
Calpain deregulation is known to be an effective mech-anism of apoptosis induction in cardiac sarcomeres through different pathways [51–53], and apoptosis of myocardial cells is considered an important mechanism of IRI [54–56].
Conclusively, an uncontrolled activation of calpain has been found to be implicated in the pathophysiology of several cardiovascular disorders [57] including myocardial IRI [58], and the inhibition of calpains has been shown to attenuate myocardial stunning and reduce infarct size after ischemia reperfusion [59] (Figure 1). However, the exact role of calpain in acute myocardial IRI remains controversial [60].
3. Oxidative Stress and Mitochondria
An oxidant and antioxidant imbalance (oxidative stress) favours the accumulation of oxidants, from both increased ROS production and decreased ROS scavenging ability, thus leading to cellular damage in the cardiomyocytes [61]. Oxida-tive stress is often associated with elevated levels of ROS or reactive nitrogen species (RNS) in the cellular and subcel-lular levels [61], leading to proteins, lipids, and DNA dam-age [62]. Furthermore, in cardiomyocytes, increased ROS/ RNS levels can induce alterations of proteins involved in
excitation-contraction coupling with increased susceptibility to proteolysis [62–65].
In the first few minutes IRI, and especially myocardial reperfusion, induces a high production of ROS by a variety of sources [66–69]. Since Arroyo et al. provided direct evi-dence of ROS formation during myocardial ischemia and postischemic reperfusion by trapping these free radicals using nitrone DMPO [70], several preclinical and clinical studies [71–74] have demonstrated the potential cardiopro-tective value of antioxidants. While small amounts of ROS could result in cardioprotection via preconditioning [75], the excessive production of ROS during reperfusion seems espe-cially important in inducing injury.
Mechanisms leading up to the dysfunction and the initial sources of ROS during IRI are not completely clear [76]. Nitric oxide (NO) production is considered a key factor in IRI. NO is an important bioactive substance which plays an important role in the regulation of normal body function and disease occurrence, and it is recognized as an ubiquitous signalling molecule with a multitude of bio-logical actions and targets. Signalling may involve direct reactions between NO and a molecular target or can occur through indirect reactions of secondary ROS [77]. In fact, actions of NO are multifaceted, and its interactions with oxygen or oxygen-related reactive intermediates (e.g., super-oxide) yield numerous RNS and ROS. These account for most of the so-called indirect effects attributed to NO through oxidation, nitrosation, and nitrate reactions referred to as oxidative, nitrosative, and nitrative stress, respectively. The physiological production of NO in the heart maintains coronary vasodilator tone and inhibits platelet aggregation and neutrophil and platelet adhesion, so performing an active
role in cardioprotection [78–80]. Beyond its beneficial effects, it has been speculated that NO excess can induce cellular injury either due to direct toxicity [81, 82] and to the reaction
with superoxide (O2−) to form the potent oxidant
peroxynit-rite (ONOO2) [83] which in turn exerts cytotoxicity via its reaction with a variety of molecular targets [84, 85]. The formation of highly reactive species, such as peroxynitrite, is a possible mechanism by which NO elicits its dangerous effects [83].
Much about NO biological actions remains contradictory, especially with regard to pathophysiologic disturbances in NO signalling. There is an ongoing debate about the levels of NO involved and whether there is a clearly defined threshold at which NO shifts from being beneficial to being destructive. Some authors hypothesize that the biological function of NO depends mostly on concentration and time course of exposure to NO, supposing that cytotoxic events, such as arrest of the cell cycle, cell senescence, or apoptosis, can occur at high NO concentrations [86]. However, other authors suggest that the chemical and biological reactivity of NO that has been studied using very high NO concentrations is of doubtful physiological relevance [87].
Zhang and Cai have shown that exogenously applied netrin-1 exerts robust cardioprotective effects against IRI, via an increase in NO formation [88]; the same group have fur-ther demonstrated that endogenously increased NO produc-tion could mediate cardioprotecproduc-tion by modulating oxidative stress and mitochondrial function [76]. Under physiologi-cal oxidative stress, NO mediates S-nitrosylation of critiphysiologi-cal protein thiols and thus averts them from further oxidative modifications by ROS, thereby rendering cardioprotection [89]. It is argued that NO protects the heart against IRI [79, 90]; however, excessive NO formation is thought to con-tribute to contractile dysfunction [91, 92].
During reperfusion NO release may be stimulated through a number of mechanisms including the change in shear stress in the coronary vasculature during reperfusion,
increased intracellular Ca2+levels as a result of ischemia, and
the thermodynamically favoured production of NO from L-arginine and molecular oxygen due to reperfusion [93, 94]. NO is produced endogenously within the myocardium by three distinct isoforms of NO synthase (NOS) [95]. Neuronal NOS (NOS1) and endothelial NOS (NOS3) are constitu-tively expressed within cardiomyocytes while inducible NOS (NOS2) is only expressed within cardiomyocytes during inflammatory responses which occur during many patho-physiological conditions of the myocardium [96].
Mitochondria play a critical role in the pathogenesis of myocardial IRI. They occupy 30–50% of the cardiomyocyte cytoplasmic volume and are critical in cardiac energy balance since energy supply for cardiomyocytes is mostly derived from mitochondrial oxidative phosphorylation (OXPHOS). On the other hand, they are a favoured target of intracellular damage [97–99]. These cell organelles are the major con-tributors of ROS as well as the major target for ROS-caused damage [100–106]. Mitochondrial dysfunction, reflected in the structure, function, and number of mitochondria within the cardiomyocyte, leads to diminished energy production, loss of myocyte contractility, altered electrical properties,
and eventual cardiomyocyte cell death [100]. In this context, the mitochondrial permeability transition pore (MPTP) is thought to play a critical role in myocardial IRI (Figures 2 and 3)
MPTP refers to a mitochondrial channel which mediates the abrupt change, or transition, in inner mitochondrial membrane permeability which occurs under certain condi-tions [107]. The opening of the MPTP renders the inner mito-chondrial membrane nonselectively permeable to molecules less than 1.5 kDa and elicits mitochondrial membrane depo-larization and uncoupling of oxidative phosphorylation. It also favours collapsing the mitochondrial membrane poten-tial, and uncoupling oxidative phosphorylation, thus leading to impairment of energy and ATP metabolism and cell necro-sis [108–111]. MPTP opening also causes mitochondrial swel-ling, and outer mitochondrial membrane rupture, thus favouring the deposition of proapoptotic factors such as
cyto-chrome 𝑐 and SMAC/Diablo from the intermembranous
space into the cytosol, thereby initiating apoptotic cell death [107].
During ischemia/reperfusion, intertwined biochemical events occur leading to MPTP opening. In the ischemic
period, following factors such as Ca2+, long-chain fatty
acids, and ROS accumulation, the likelihood that MPTP will occur upon reperfusion gradually increases [112, 113]. During ischemia, due to increased glycolysis, an accumulation of lactic acid and reduction of pH occur. To restore the pH,
the Na+/H+ antiporter is activated, but it acts inefficiently
because Na+cannot be pumped out of the cell, as the Na+/K+
ATPase is inhibited by the absence of intracellular ATP.
Con-sequently, the cytosolic Ca2+concentration increases.
More-over, the existing decrease in the adenine nucleotide concen-tration, which is associated with an increased phosphate con-centration, is likely to sensitize MPTP opening in response
to Ca2+; however, low pH inhibits the opening. When
reperfusion occurs, the mitochondria recover their ability to respire and rescue the sustained mitochondrial membrane potential, which is required for ATP synthesis. In addition, strong production of ROS occurs when the inhibited res-piratory chain is reexposed to oxygen. Thus, the following resulting conditions are nearly optimal for MPTP opening:
high Ca2+levels within the mitochondrial matrix, increased
levels of phosphate and oxidative stress, depletion of adenine nucleotide concentration, and rapid restoration of physiolog-ical value of pH [113–115].
In their milestone paper, Griffiths and Halestrap [116] demonstrated that MPTP are closed during ischemia and open the first few (2-3) minutes of reperfusion. Subsequent data has confirmed that pore opening occurs during reper-fusion of the heart after ischemia, but not in the ischemic period [107]. Thus MPTP is an important new target for cardioprotection during reperfusion [114].
4. The Matrix Metalloproteinases
One group of enzymes that is important in mediat-ing IRI injury is the family of matrix metalloproteinases (MMPs). The MMPs are a large family of calcium-dependent,
Ischemia Hypoxia ROS Reperfusion MPTP opening Mitochondrion Cell death iNOS production Ion pump disturbance NADPH oxidases Xanthine oxidases NOS uncoupling Anaerobic glycolysis and Mitochondria swelling and membrane rupture Release of proapoptotic factors Impairment of
energy and ATP metabolism Direct cytotoxic effect Superoxide Peroxynitrite Cytotoxic effect ↓ pH ↓ ATP ↑ Ca2+ and Na+ ↑ O2 ↑ NO
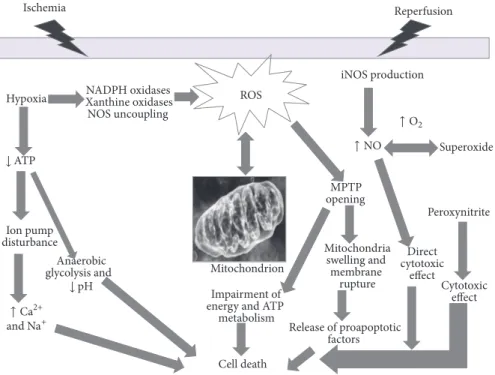
Figure 2: Schematic representation of oxidative stress contributing to tissue injury and cell death in IRI. Following ischemia, hypoxia results
in reduction of ATP production, ion pump function unbalance, leading to overload of Na+and Ca2+, activation of anaerobic glycolysis, and,
finally, reduction of pH. During the initial ischemic phase, the activation and upregulation of enzymes (such as NADPH oxidase, a superoxide-generating enzyme comprising a membrane-bound catalytic subunit) occurs, that are capable of producing ROS, when molecular oxygen is reintroduced in the reperfusion phase. ROS induces cell dysfunction and death via other mechanisms: activation of metalloproteinases and calpains, mitochondrial permeability transition pore (MPTP) opening which contributes to swelling and lysis of cells. This may elicit the release of proapoptotic factors in the cytosol, thus contributing to cell death. ROS indirectly interact with nitric oxide (NO) production, partly mediated by the inducible NOS (iNOS), the high-capacity NO-producing enzyme. Unlike the other two NOS isoforms, iNOS is not constitutively expressed in cells, and its production is elicited by several stimuli like IRI. NO cytotoxic effects are both direct and indirect mediated by NO reaction with superoxide to form the potent oxidant peroxynitrite which in turn exerts cytotoxicity.
(a) (b)
(c) (d)
Figure 3: Histomorphological pictures showing the phenotypic results of altered pathways in IRI. (a) Mild calpain 1 expression in the left ventricle cardiac tissue of a patient who died following early reperfused AMI (calpain 1, antibody anti-calpain 1, Santa Cruz, USA). (b) NOX2 expression in the left ventricle cardiac tissue of a patient who died following prompt fibrinolysis in acute STEMI. (c) Strong immunopositivity to anti-nitrotyrosine antibody (Abcam, Cambridge, UK). (d) Mild immunopositivity to anti-iNOS (inducible nitric oxide synthase) antibody (Santa Cruz, CA, USA) in the left ventricle sample of a patient who died following reperfusion therapy in STEMI.
zinc-containing endopeptidases that have the ability to remodel the extracellular matrix in both physiological and pathological processes. MMPs are regulated at different levels including transcriptional, posttranscriptional, and posttrans-lational levels. Moreover, they are controlled via their endoge-nous inhibitors, the tissue inhibitor of metalloproteinases (TIMPs), and by their intra- and extracellular localization [117]. Of all MMPs, MMP-2 (also known as gelatinase A or type IV collagenase) plays a critical role in cardiovas-cular diseases [117]. MMP-2 activity is also regulated via nonproteolytic posttranslational modifications of the
full-length zymogen form, by𝑆-glutathiolation, 𝑆-nitrosylation,
and phosphorylation [118–120]. The NO product, ONOO−,
directly activates MMP-s 2 [118] via a nonproteolytic
mech-anism involving the𝑆-glutathiolation of the propeptide
cys-teine sulfhydryl group in a reaction requiring only
micromo-lar concentrations of ONOO− in conjunction with normal
intracellular levels of glutathione [119]. In turn, it was
demon-strated that ONOO−inactivate TIMP-4 and TIMP-1, leading
to a net increase in MMP activity [118].
In IRI, the sudden availability of molecular oxygen during reperfusion reenergizes mitochondria and reactivates the electron transport chain, causing a significant increase in
the biosynthesis of ROS (including ONOO−) [94, 121] which
stimulates MMP-2 activity [118].
It has been demonstrated that MMP-2 exerts rapid effects in modulating different cellular functions independent of its action on the extracellular matrix (ECM). These include effects on platelet aggregation [122], vascular tone [123, 124], and acute mechanical dysfunction of the heart immediately after ischemia and reperfusion [125, 126]. In ischemia/reperfusion, injury may result in the partial proteol-ysis of the thin-filament regulatory protein troponin I (TnI) [60, 124, 125, 127–129], and studies on animal models have validated this observation, showing that MMP-2 degrade Tn I myofilaments [130].
MMP-2 has a proapoptotic role as demonstrated in adult rat cardiomyocyte by Menon et al. who show that
inhibition of MMP-2 inhibits 𝛽-AR-stimulated apoptosis
[131, 132]. Furthermore, MMP-2 is present in mitochondria [130], and cardiac-specific transgenic expression of active MMP-2 causes abnormalities in mitochondria ultrastructure, impaired respiration, increased lipid peroxidation, cell necro-sis, and reduced recovery of contractile performance during post-IRI [133].
Finally, a complex interplay exists between the calpain and MMP systems since there appears to be overlap in the substrates and/or biological actions of MMP-2 and calpains in various cellular pathways [117]. Kandasamy et al. have hypothesized that either MMP-2 targets a subset of proteins similar to calpain, or calpain has been incorrectly identified as the protease responsible for some intracellular proteolytic activities. Indeed, much of the evidence for calpain degrada-tion of substrates in cardiac cells rests on the use of calpain inhibitors such as calpastatin, which has been found to inhibit MMP-2 activity in vitro [117].
Other MMPs are thought to be involved in myocardial injury following AMI, such as MMP-9, first known as 92-kDa type IV collagenase or gelatinase B, a structurally complex
metalloproteinase that intervenes in the degradation of ECM in a large spectrum of physiology and pathophysiology processes involving tissue remodelling, including cardiac remodelling after AMI.
MMP-9 is expressed in the heart by endogenous cardiac cell types (e.g., cardiomyocytes, endothelial cells, and fibrob-lasts) and is also produced by nonresident cells that infiltrate the infarct in response to ischemic injury (e.g., leukocytes) [134–136].
Different and opposite functions have been hypothesized for MMP-9. Potential detrimental consequences of MMP-9 release and activation may include stimulating inappropriate extracellular matrix degradation, activating inflammatory mediators, and/or increasing capillary permeability [137, 138]. On the other hand, potential beneficial effects of early MMP-9 activation include removing matrix and necrotic myocytes, releasing growth factors and cell surface receptors, remodelling the extracellular matrix for scar formation, pro-cessing inflammatory mediators such as interleukin-1𝛽, and influencing angiogenesis [137]. MMP-9 has been correlated with an increase in infarct size and left ventricle fibrosis after experimental AMI [138–142].
Furthermore, increased myocardial MMP-9 expression or activity has been found in experimental myocardial injuries such as permanent coronary artery occlusion [143, 144] or reperfusion injury model in animals [145, 146], and the possible role of MMP-9 activation in myocardial IRI has been explored [147].
Following myocardial acute ischemia and reperfusion, neutrophil-derived MMP-9 is released in the myocardium and its levels increase as early as several minutes after AMI, remaining high for the first week in many animal models [137, 144, 148]. In the early phase of reperfusion, MMP-9 activation is likely to be localized in the perineutrophil area and might be initiated by neutrophils adhering to the ECM [137]; its tem-poral trend mirrors leukocyte infiltration [149]. The action of MMP-9 appears to be complex; it directly degrades ECM pro-teins and activates cytokines and chemokines to regulate tis-sue remodelling. MMP-9 deletion or inhibition has proven to be beneficial in a variety of animal models of cardiovascular disease. On the other hand, MMP-9 cell-specific overexpres-sion has also proven beneficial [137, 144, 146, 150] (Figure 4).
5. Ischemia/Reperfusion Injury and
Medicolegal Issues
Early reperfusion reduced mortality in AMI so that, in many countries, the hospital mortality has declined to about 5% [151]. There is no doubt that early reperfusion, both pharmacological and mechanical, is the only way to prevent progression to myocardial necrosis and thus to limit the size of the infarct. However, myocardial IRI has been described following reperfusion therapies including percutaneous coro-nary intervention (PCI), thrombolysis, and corocoro-nary bypass grafting [7, 13, 16].
VAs upon reperfusion have been recognized since the advent of recanalization techniques [152]; however their pathophysiological and prognostic significance is still contro-versial [153]. Several arrhythmogenic mechanisms have been
ECM breakdown Inactive pro-MMPs Ischemia/reperfusion Oxidative stress Different biological activities Transcription and activation Active MMPs
Degradation of TnI myofilaments Release of proapoptotic factors Mitochondria ultrastructure alteration
Increased lipid peroxidation Contractile dysfunction TIMPs
MMP-2
MMP-9
Post-AMI cardiac remodelling
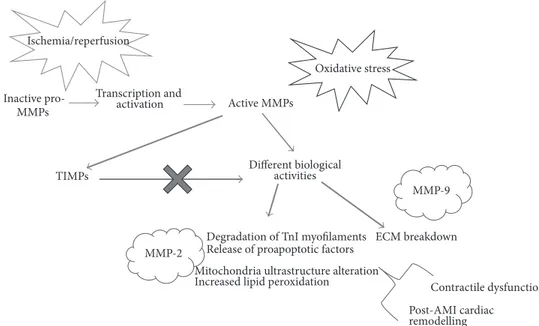
Figure 4: The matrix metalloproteinases system. MMPs activity results from different levels of regulation: transcription, activation, and inhibition by tissue inhibitors of metalloproteinases (TIMPs). During ischemia/reperfusion, oxidative stress stimulates the activity of MMPs, like MMP-2. Several biological activities of MMPs may contribute to myocardial contractile dysfunction and cell death. MMPs can both degrade extracellular matrix (ECM) and modulate different cellular mechanisms, thus leading to contractile dysfunction and modulation of cardiac remodelling and healing.
proposed to be involved in IRI-induced arrhythmias [154, 155]. Some of VAs that occur almost directly at the moment of reperfusion (namely, ventricular premature beats and accelerated idioventricular rhythms) are usually harmless and well tolerated [152]; however it has been reported that ventricular tachycardia and ventricular fibrillation occurring immediately after reperfusion remain the most important causes of sudden death following restoration of blood flow [156, 157].
Severe arrhythmias may not be common but the fact that they are life threatening makes them a relevant issue also for pathologists. In fact, the occurrence of such fatal events may represent a potential source of malpractice claims for cardiologists and it is noteworthy that AMI, in some manner, remains one of the most challenging areas with an associated high risk of alleged medical malpractice [158] and one of the clinical settings in which claims are most likely to arise [159, 160].
There is no doubt that prompt mechanical or/and phar-macological myocardial reperfusion represents the only real-istic strategy in STEMI and that it has greatly improved AMI outcome. However, patients may have increasing optimistic expectations about the benefits of the procedures, as well as in many other cardiological clinical settings [161–163], and especially in cases with fatal outcome litigations and malpractice claims may arise thus leading to medicolegal autopsies which are critical in proving or excluding medical malpractice. It is now recognized that there are a spectrum of responses of the myocardium to ischemia/reperfusion [16, 157], and knowledge on the biochemical and molecular substrates of myocardial IRI has considerably improved.
Reperfusion induces typical patterns of myocardial injury; contraction bands, calcium loading in the irreversibly injured myocytes, and hemorrhage in the region due to leakage of blood out of damaged blood vessels have been associated with IRI [16]. There is a growing appreciation that the pathobiologic response to ischemia/reperfusion injury is characterized by changes involving, among others, oxidative
stress, mitochondria, and Ca+homeostasis disturbance, with
each leading to unique histomorphological footprint. As the cellular and molecular processes of myocardial IRI are more and more unravelled, the histopathology of reperfused AMI has been revisited and deserves further studies [3, 164]. We believe that, in postmortem examination in cases of fatal outcome of reperfused AMI, forensic experts should be very careful as this type of postmortem examination requires a deep knowledge and investigation of the complex ionic and biochemical alterations which could result in an unsta-ble electrical substrate capaunsta-ble of initiating and sustain-ing arrhythmias. A sound knowledge of the pathophysi-ological changes underlying myocardial IRI and, namely, reperfusion arrhythmias is critical for forensic pathologists to make correct opinions concerning the real mechanism of death. Forensic pathologists, like clinicians, must think correlatively and move towards the explanation of the death on the basis of the underlying complex mechanisms. As a general concept, but mostly when deep pathophysio-logical derangements occur potentially leading to death, structural and anatomical knowledge obtained from autop-tic observation is not so useful and cannot provide sat-isfactory explanations, independently of functional know-ledge.
6. Therapeutic Challenges
Although there is no doubt that in AMI the reopening, as soon as possible, of occluded coronary arteries using either thrombolytic therapy or primary percutaneous coronary intervention is of vital importance for limiting the infarct size, thus representing an effective tool in AMI [162, 163], currently no similarly valid options exist in the treatment of myocardial IRI. Since the 1980s research has been focused on therapeutic agents that would render myocardial cells more resistant to the deleterious effects of ischemia and reperfusion [3, 164], the concept of “cardioprotection” encompasses the manipula-tion of the cellular events by different therapeutic tools during ischemia and reperfusion to reduce the amount of myocardial cells death [3, 164]. Ischemic conditioning strategies (ischemic preconditioning, IPC; remote ischemic precondi-tioning, RIPC; and ischemic postcondiprecondi-tioning, iPOST) have been widely investigated in laboratory settings. Nevertheless, there still exists some difficulty in translating experimental results and controlled animal models into a heterogeneous population of human patients [12, 13, 24, 165]. An incomplete understanding of how cardioprotective signalling may be initiated at the level of the cardiomyocytes may, in part, explain the lack of success [166]. A deeper knowledge of the cellular and molecular mechanisms underlying IRI has led to the development of cardioprotective strategies, focusing on epigenetic regulation, limitation of cell death (both necrosis and apoptosis), stem cell regenerative therapies, gene therapy, and the use of growth factors [167]. Among the mechanisms through which postischemic myocardial damage has been shown to occur, mitochondrial dysfunction and the opening of MPTP are key steps. This crucial role renders them very attractive targets for therapeutic intervention [168– 170]. In this context, a potential cardioprotective effect of intracoronary administration of 4-chlorodiazepam (4-CLD, a benzodiazepine derivative of diazepam) in animal models of IRI, has been recently demonstrated [171], thus suggesting a therapeutic role of intracoronary infusion of 4-CLD in AMI [171].
Furthermore, several lines of evidence support a potential role of platelet-rich plasma (PRP), an autologous product rich in growth factors obtained from a blood sample, in the healing of MI injury [172–175]. Platelets contain a wide amount of growth factors that are crucial in the reparative process following ischemic myocardial injury. In addition, they are rich in Factor XIII, a plasma transglutaminase, that has been shown to be critical in post-MI healing [176–178]. Factor XIII influences several steps of the reparative process, the formation of the three-dimensional fibrin meshwork, and the ECM components. Furthermore it is essential in adult stem cells recruitment, neoangiogenesis, and collagen deposit, thus playing a pivotal role at the intersection of several pathways involved in myocardial healing [179, 180].
7. Conclusions
AMI is a major cause of mortality worldwide. Early and successful myocardial reperfusion with either thrombolytic agents or primary percutaneous coronary intervention is the
most effective strategy to reduce infarct size and improve clin-ical outcome. However, the process of restoring blood flow to the ischemic myocardium can induce myocardial reperfusion injury, which can paradoxically reduce the beneficial effects of myocardial reperfusion. Thus reperfusion itself may lead to accelerated and additional myocardial injury beyond that generated by ischemia alone [181]. Different clinical mani-festations of this injury exist [13]; however, RAs remain the most important causes of sudden death following reperfusion therapy [182] even when the latter is technically successful. Thus myocardial IRI is both a critical clinical and medi-colegal problem. For clinicians a better understanding of the pathophysiology of myocardial IRI may open the way to new therapeutic strategies [25, 182, 183]. For forensic pathologists, the value of fostering a knowledge of IRI pathophysiology should be highlighted as this can lead to an increased aware-ness of this potentially fatal event related to myocardial IRI, even in the case of optimal and early treatment. The clear investigation and comprehension of IRI may be an additional value which may diminish the risk of exposure of physicians to malpractice claims.
Competing Interests
The authors declare that they have no competing interests.
References
[1] D. Mozaffarian, E. J. Benjamin, A. S. Go et al., “Heart disease and stroke statistics-2015 update: a report from the American Heart Association,” Circulation, vol. 131, no. 4, pp. e29–e322, 2015. [2] American College of Emergency Physicians, Society for
Car-diovascular Angiography and Interventions, P. T. O’Gara et al., “2013 ACCF/AHA guideline for the management of ST-eleva-tion myocardial infarcST-eleva-tion: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines,” Journal of the American College
of Cardiology, vol. 61, pp. e78–e140, 2013.
[3] P. Z. Gerczuk and R. A. Kloner, “An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials,” Journal of the American College
of Cardiology, vol. 59, no. 11, pp. 969–978, 2012.
[4] E. Braunwald and R. A. Kloner, “Myocardial reperfusion: a double-edged sword?” Journal of Clinical Investigation, vol. 76, no. 5, pp. 1713–1719, 1985.
[5] H. M. Piper, D. Garc´ıa-Dorado, and M. Ovize, “A fresh look at reperfusion injury,” Cardiovascular Research, vol. 38, no. 2, pp. 291–300, 1998.
[6] S. Verma, P. W. M. Fedak, R. D. Weisel et al., “Fundamentals of reperfusion injury for the clinical cardiologist,” Circulation, vol. 105, no. 20, pp. 2332–2336, 2002.
[7] D. M. Yellon and D. J. Hausenloy, “Myocardial reperfusion injury,” The New England Journal of Medicine, vol. 357, no. 11, pp. 1074–1135, 2007.
[8] T. Reffelmann and R. A. Kloner, “The no-reflow phenomenon: a basic mechanism of myocardial ischemia and reperfusion,”
Basic Research in Cardiology, vol. 101, no. 5, pp. 359–372, 2006.
[9] P. Pagliaro, F. Moro, F. Tullio, M.-G. Perrelli, and C. Penna, “Cardioprotective pathways during reperfusion: focus on redox signaling and other modalities of cell signaling,” Antioxidants &
[10] E. Braunwald, “The war against heart failure: the Lancet lecture,”
The Lancet, vol. 385, no. 9970, pp. 812–824, 2015.
[11] A. E. Moran, M. H. Forouzanfar, G. A. Roth et al., “The global burden of ischemic heart disease in 1990 and 2010: the global burden of disease 2010 study,” Circulation, vol. 129, no. 14, pp. 1493–1501, 2014.
[12] R. M. Bell, H. E. Bøtker, R. D. Carr et al., “9th Hatter Biannual Meeting: position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection,”
Basic Research in Cardiology, vol. 111, article 41, 2016.
[13] D. J. Hausenloy and D. M. Yellon, “Myocardial ischemia-reper-fusion injury: a neglected therapeutic target,” Journal of Clinical
Investigation, vol. 123, no. 1, pp. 92–100, 2013.
[14] C. Greco, S. Rosato, P. D’Errigo, G. F. Mureddu, E. Lacorte, and F. Seccareccia, “Trends in mortality and heart failure after acute myocardial infarction in Italy from 2001 to 2011,” International
Journal of Cardiology, vol. 184, no. 1, pp. 115–121, 2015.
[15] T. Kalogeris, Y. Bao, and R. J. Korthuis, “Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning,” Redox Biology, vol. 2, no. 1, pp. 702–714, 2014.
[16] L. M. Buja, “Myocardial ischemia and reperfusion injury,”
Car-diovascular Pathology, vol. 14, no. 4, pp. 170–175, 2005.
[17] H. Bonnemeier, U. K. H. Wiegand, E. Giannitsis et al., “Tempo-ral repolarization inhomogeneity and reperfusion arrhythmias in patients undergoing successful primary percutaneous coro-nary intervention for acute ST-segment elevation myocardial infarction: impact of admission troponin T,” American Heart
Journal, vol. 145, no. 3, pp. 484–492, 2003.
[18] C. P. Baines, “How and when do myocytes die during ischemia and reperfusion: the late phase,” Journal of Cardiovascular
Pharmacology and Therapeutics, vol. 16, no. 3-4, pp. 239–243,
2011.
[19] Z.-Q. Zhao, “Oxidative stress-elicited myocardial apoptosis during reperfusion,” Current Opinion in Pharmacology, vol. 4, no. 2, pp. 159–165, 2004.
[20] A. Prasad, G. W. Stone, D. R. Holmes, and B. Gersh, “Reperfu-sion injury, microvascular dysfunction, and cardioprotection: the ’dark side’ of reperfusion,” Circulation, vol. 120, no. 21, pp. 2105–2112, 2009.
[21] A. T. Turer and J. A. Hill, “Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy,” The American
Journal of Cardiology, vol. 106, no. 3, pp. 360–368, 2010.
[22] M. T. Dirksen, G. J. Laarman, M. L. Simoons, and D. J. G. M. Duncker, “Reperfusion injury in humans: a review of clini-cal trials on reperfusion injury inhibitory strategies,”
Cardiovas-cular Research, vol. 74, no. 3, pp. 343–355, 2007.
[23] K. McCafferty, S. Forbes, C. Thiemermann, and M. M. Yaqoob, “The challenge of translating ischemic conditioning from ani-mal models to humans: the role of comorbidities,” Disease
Models and Mechanisms, vol. 7, no. 12, pp. 1321–1333, 2014.
[24] R. S. V. Heide and C. Steenbergen, “Cardioprotection and myo-cardial reperfusion: pitfalls to clinical application,” Circulation
Research, vol. 113, no. 4, pp. 464–477, 2013.
[25] P. Ferdinandy, D. J. Hausenloy, G. Heusch, G. F. Baxter, and R. Schulz, “Interaction of risk factors, comorbidities, and comedi-cations with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote condition-ing,” Pharmacological Reviews, vol. 66, no. 4, pp. 1142–1174, 2014. [26] R. Abbott and M. Cohen, “Medico-legal issues in cardiology,”
Cardiology in Review, vol. 21, no. 5, pp. 222–228, 2013.
[27] S. Y. Tan, “Medical malpractice: a cardiovascular perspective,”
Cardiovascular Therapeutics, vol. 30, no. 3, pp. e140–e145, 2012.
[28] E. Letavernier, L. Zafrani, J. Perez, B. Letavernier, J.-P. Hay-mann, and L. Baud, “The role of calpains in myocardial remod-elling and heart failure,” Cardiovascular Research, vol. 96, no. 1, pp. 38–45, 2012.
[29] J. Inserte, V. Hernando, and D. Garcia-Dorado, “Contribution of calpains to myocardial ischaemia/reperfusion injury,”
Cardio-vascular Research, vol. 96, no. 1, pp. 23–31, 2012.
[30] D. Garcia-Dorado, M. Ruiz-Meana, J. Inserte, A. Rodriguez-Sinovas, and H. M. Piper, “Calcium-mediated cell death during myocardial reperfusion,” Cardiovascular Research, vol. 94, no. 2, pp. 168–180, 2012.
[31] C. Neuhof and H. Neuhof, “Calpain system and its involvement in myocardial ischemia and reperfusion injury,” World Journal
of Cardiology, vol. 6, no. 7, pp. 638–652, 2014.
[32] S. Parameswaran and R. K. Sharma, “Altered expression of cal-cineurin, calpain, calpastatin and HMWCaMBP in cardiac cells following ischemia and reperfusion,” Biochemical and
Bio-physical Research Communications, vol. 443, no. 2, pp. 604–609,
2014.
[33] C. Perrin, A. Ecarnot-Laubriet, C. Vergely, and L. Rochette, “Calpain and caspase-3 inhibitors reduce infarct size and post-ischemic apoptosis in rat heart without modifying contrac-tile recovery,” Cellular and molecular biology (Noisy-le-Grand,
France), vol. 49, pp. OL497–OL505, 2003.
[34] S. Tissier, S. Lancel, X. Marechal et al., “Calpain inhibitors improve myocardial dysfunction and inflammation induced by endotoxin in rats,” Shock (Augusta, Ga.), vol. 21, no. 4, pp. 352– 357, 2004.
[35] Y. Yoshikawa, H. Hagihara, Y. Ohga et al., “Calpain inhibitor-1 protects the rat heart from ischemia-reperfusion injury: anal-ysis by mechanical work and energetics,” American Journal of
Physiology—Heart and Circulatory Physiology, vol. 288, no. 4,
pp. H1690–H1698, 2005.
[36] C. Neuhof, V. Fabiunke, K. Deibele et al., “Reduction of myo-cardial infarction by calpain inhibitors A-705239 and A-705253 in isolated perfused rabbit hearts,” Biological Chemistry, vol. 385, no. 11, pp. 1077–1082, 2004.
[37] V. Hernando, J. Inserte, C. L. Sart´orio, V. M. Parra, M. Poncelas-Nozal, and D. Garcia-Dorado, “Calpain translocation and acti-vation as pharmacological targets during myocardial ischemia/ reperfusion,” Journal of Molecular and Cellular Cardiology, vol. 49, no. 2, pp. 271–279, 2010.
[38] D. E. Goll, V. F. Thompson, H. Li, W. Wei, and J. Cong, “The calpain system,” Physiological Reviews, vol. 83, no. 3, pp. 731–801, 2003.
[39] J. J´anossy, P. Ubezio, ´A. Ap´ati, M. Mag´ocsi, P. Tompa, and P.
Friedrich, “Calpain as a multi-site regulator of cell cycle,”
Bio-chemical Pharmacology, vol. 67, no. 8, pp. 1513–1521, 2004.
[40] Y. Ono and H. Sorimachi, “Calpains: an elaborate proteolytic system,” Biochimica et Biophysica Acta—Proteins and
Proteo-mics, vol. 1824, no. 1, pp. 224–236, 2012.
[41] T. Moldoveanu, C. M. Hosfield, D. Lim, J. S. Elce, Z. Jia, and P.
L. Davies, “A Ca2+switch aligns the active site of calpain,” Cell,
vol. 108, no. 5, pp. 649–660, 2002.
[42] S. Seki, K. Horikoshi, H. Takeda et al., “Effects of sustained low-flow ischemia and reperfusion on Ca2+ transients and con-tractility in perfused rat hearts,” Molecular and Cellular
[43] X. Liu, T. V. Vleet, and R. G. Schnellmann, “The role of calpain in oncotic cell death,” Annual Review of Pharmacology and
Toxi-cology, vol. 44, pp. 349–370, 2004.
[44] A. L. Portbury, M. S. Willis, and C. Patterson, “Tearin’ up my heart: proteolysis in the cardiac sarcomere,” Journal of Biological
Chemistry, vol. 286, no. 12, pp. 9929–9934, 2011.
[45] J. Barta, A. T´oth, I. ´Edes et al., “Calpain-1-sensitive myofibrillar proteins of the human myocardium,” Molecular and Cellular
Biochemistry, vol. 278, no. 1-2, pp. 1–8, 2005.
[46] F. B. Sachse, N. S. Torres, E. Savio-Galimberti et al., “Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy,”
Cir-culation Research, vol. 110, no. 4, pp. 588–597, 2012.
[47] R. C. Balijepalli and T. J. Kamp, “Cardiomyocyte transverse tubule loss leads the way to heart failure,” Future Cardiology, vol. 7, no. 1, pp. 39–42, 2011.
[48] D. J. Crossman, P. R. Ruygrok, C. Soeller, and M. B. Cannell, “Changes in the organization of excitation-contraction coupling structures in failing human heart,” PLoS ONE, vol. 6, no. 3, Article ID e17901, 2011.
[49] C.-Y. C. Wu, B. Chen, Y.-P. Jiang et al., “Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure,” Journal of the American Heart
Association, vol. 3, no. 3, Article ID e000527, 2014.
[50] R. M. Murphy, T. L. Dutka, D. Horvath, J. R. Bell, L. M.
Del-bridge, and G. D. Lamb, “Ca2+-dependent proteolysis of
juncto-philin-1 and junctophilin-2 in skeletal and cardiac muscle,”
Journal of Physiology, vol. 591, no. 3, pp. 719–729, 2013.
[51] G. Bajaj and R. K. Sharma, “TNF-𝛼-mediated cardiomyocyte apoptosis involves caspase-12 and calpain,” Biochemical and
Bio-physical Research Communications, vol. 345, no. 4, pp. 1558–
1564, 2006.
[52] Y. Li, J. M. O. Arnold, M. Pampillo, A. V. Babwah, and T. Peng, “Taurine prevents cardiomyocyte death by inhibiting NADPH oxidase-mediated calpain activation,” Free Radical Biology and
Medicine, vol. 46, no. 1, pp. 51–61, 2009.
[53] Y. Li, Y. Li, Q. Feng, M. Arnold, and T. Peng, “Calpain activa-tion contributes to hyperglycaemia-induced apoptosis in car-diomyocytes,” Cardiovascular Research, vol. 84, no. 1, pp. 100– 110, 2009.
[54] R. A. Gottlieb, K. O. Burleson, R. A. Kloner, B. M. Babior, and R. L. Engler, “Reperfusion injury induces apoptosis in rabbit cardiomyocytes,” Journal of Clinical Investigation, vol. 94, no. 4, pp. 1621–1628, 1994.
[55] A. Saraste, K. Pulkki, M. Kallajoki, K. Henriksen, M. Parvinen, and L.-M. Voipio-Pulkki, “Apoptosis in human acute myocar-dial infarction,” Circulation, vol. 95, no. 2, pp. 320–323, 1997. [56] B. Freude, T. N. Masters, F. Robicsek et al., “Apoptosis is initiated
by myocardial ischemia and executed during reperfusion,”
Jour-nal of Molecular and Cellular Cardiology, vol. 32, no. 2, pp. 197–
208, 2000.
[57] B. A. Potz, A. A. Sabe, M. R. Abid, and F. W. Sellke, “Calpains and coronary vascular disease,” Circulation Journal, vol. 80, no. 1, pp. 4–10, 2016.
[58] P. N. Khalil, C. Neuhof, R. Huss et al., “Calpain inhibition reduces infarct size and improves global hemodynamics and left ventricular contractility in a porcine myocardial ischemia/ reperfusion model,” European Journal of Pharmacology, vol. 528, no. 1–3, pp. 124–131, 2005.
[59] Y. Kudo-Sakamoto, H. Akazawa, K. Ito et al., “Calpain-depend ent cleavage of N-cadherin is involved in the progression of
post-myocardial infarction remodeling,” Journal of Biological
Chemistry, vol. 289, no. 28, pp. 19408–19419, 2014.
[60] R. Bolli and E. Marb´an, “Molecular and cellular mechanisms of myocardial stunning,” Physiological Reviews, vol. 79, no. 2, pp. 609–634, 1999.
[61] J. Navarro-Yepes, M. Burns, A. Anandhan et al., “Oxidative stress, redox signaling, and autophagy: cell death versus sur-vival,” Antioxidants and Redox Signaling, vol. 21, no. 1, pp. 66–85, 2014.
[62] H. Wiseman and B. Halliwell, “Damage to DNA by reactive oxy gen and nitrogen species: role in inflammatory disease and pro-gression to cancer,” Biochemical Journal, vol. 313, no. 1, pp. 17–29, 1996.
[63] M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur, and J. Telser, “Free radicals and antioxidants in normal physi-ological functions and human disease,” International Journal of
Biochemistry and Cell Biology, vol. 39, no. 1, pp. 44–84, 2007.
[64] G. Bartosz, “Reactive oxygen species: destroyers or messen-gers?” Biochemical Pharmacology, vol. 77, no. 8, pp. 1303–1315, 2009.
[65] R. L. Auten and J. M. Davis, “Oxygen toxicity and reactive oxygen species: the devil is in the details,” Pediatric Research, vol. 66, no. 2, pp. 121–127, 2009.
[66] S. L. Thompson-Gorman and J. L. Zweier, “Evaluation of the role of xanthine oxidase in myocardial reperfusion injury,” Journal of
Biological Chemistry, vol. 265, no. 12, pp. 6656–6663, 1990.
[67] Y. Xia and J. L. Zweier, “Substrate control of free radical genera-tion from xanthine oxidase in the postischemic heart,” Journal
of Biological Chemistry, vol. 270, no. 32, pp. 18797–18803, 1995.
[68] D. B. Zorov, M. Juhaszova, Y. Yaniv, H. B. Nuss, S. Wang, and S. J. Sollott, “Regulation and pharmacology of the mitochondrial permeability transition pore,” Cardiovascular Research, vol. 83, no. 2, pp. 213–225, 2009.
[69] P. Venditti, P. Masullo, and S. Di Meo, “Effects of myocar-dial ischemia and reperfusion on mitochondrial function and susceptibility to oxidative stress,” Cellular and Molecular Life
Sciences, vol. 58, no. 10, pp. 1528–1537, 2001.
[70] C. M. Arroyo, J. H. Kramer, B. F. Dickens, and W. B. Weglicki, “Identification of free radicals in myocardial ischemia/reper-fusion by spin trapping with nitrone DMPO,” FEBS Letters, vol. 221, no. 1, pp. 101–104, 1987.
[71] G. A. Kurian, R. Rajagopal, S. Vedantham, and M. Rajesh, “The role of oxidative stress in myocardial ischemia and reperfusion injury and remodeling: revisited,” Oxidative Medicine and
Cellular Longevity, vol. 2016, Article ID 1656450, 14 pages, 2016.
[72] N. Marczin, N. El-Habashi, G. S. Hoare, R. E. Bundy, and M. Yacoub, “Antioxidants in myocardial ischemia-reperfusion injury: therapeutic potential and basic mechanisms,” Archives of
Biochemistry and Biophysics, vol. 420, no. 2, pp. 222–236, 2003.
[73] N. S. Dhalla, A. B. Elmoselhi, T. Hata, and N. Makino, “Status of myocardial antioxidants in ischemia-reperfusion injury,”
Cardiovascular Research, vol. 47, no. 3, pp. 446–456, 2000.
[74] L. B. Becker, “New concepts in reactive oxygen species and car-diovascular reperfusion physiology,” Carcar-diovascular Research, vol. 61, no. 3, pp. 461–470, 2004.
[75] X.-L. Tang, H. Takano, A. Rizvi et al., “Oxidant species trigger late preconditioning against myocardial stunning in conscious rabbits,” American Journal of Physiology—Heart and Circulatory
Physiology, vol. 282, no. 1, pp. H281–H291, 2002.
[76] K. L. Siu, C. Lotz, P. Ping, and H. Cai, “Netrin-1 abrogates ischemia/reperfusion-induced cardiac mitochondrial dysfunc-tion via nitric oxide-dependent attenuadysfunc-tion of NOX4 activadysfunc-tion
and recoupling of NOS,” Journal of Molecular and Cellular
Cardiology, vol. 78, pp. 174–185, 2015.
[77] D. A. Wink, I. Hanbauer, M. B. Grisham et al., “Chemical bio-logy of nitric oxide: regulation and protective and toxic mech-anisms,” Current Topics in Cellular Regulation, vol. 34, pp. 159– 187, 1996.
[78] S. Moncada and A. Higgs, “The L-arginine–nitric oxide path-way,” The New England Journal of Medicine, vol. 329, no. 27, pp. 2002–2012, 1993.
[79] S. P. Jones and R. Bolli, “The ubiquitous role of nitric oxide in cardioprotection,” Journal of Molecular and Cellular Cardiology, vol. 40, no. 1, pp. 16–23, 2006.
[80] J. W. Elrod, J. J. M. Greer, N. S. Bryan et al., “Cardiomyocyte-specific overexpression of NO synthase-3 protects against myo-cardial ischemia-reperfusion injury,” Arteriosclerosis,
Thrombo-sis, and Vascular Biology, vol. 26, no. 7, pp. 1517–1523, 2006.
[81] J.-C. Drapier -, C. Pellat, and Y. Henry, “Generation of EPR-detectable nitrosyl-iron complexes in tumor target cells cocul-tured with activated macrophages,” Journal of Biological
Chem-istry, vol. 266, no. 16, pp. 10162–10167, 1991.
[82] J. R. Lancaster Jr. and J. B. Hibbs Jr., “EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macro-phages,” Proceedings of the National Academy of Sciences of the
United States of America, vol. 87, no. 3, pp. 1223–1227, 1990.
[83] J. S. Beckman, T. W. Beckman, J. Chen, P. A. Marshall, and B. A. Freeman, “Apparent hydroxyl radical production by peroxy-nitrite: implications for endothelial injury from nitric oxide and superoxide,” Proceedings of the National Academy of Sciences of
the United States of America, vol. 87, no. 4, pp. 1620–1624, 1990.
[84] R. Radi, J. S. Beckman, K. M. Bush, and B. A. Freeman, “Peroxy-nitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide,” Archives of
Biochem-istry and Biophysics, vol. 288, no. 2, pp. 481–487, 1991.
[85] A. van der Vliet, C. A. O’Neill, B. Halliwell, C. E. Cross, and H. Kaur, “Aromatic hydroxylation and nitration of phenylalanine and tyrosine by peroxynitrite. Evidence for hydroxyl radical production from peroxynitrite,” FEBS Letters, vol. 339, no. 1-2, pp. 89–92, 1994.
[86] D. D. Thomas, L. A. Ridnour, J. S. Isenberg et al., “The chemical biology of nitric oxide: implications in cellular signaling,” Free
Radical Biology and Medicine, vol. 45, no. 1, pp. 18–31, 2008.
[87] C. N. Hall and J. Garthwaite, “What is the real physiological NO concentration in vivo?” Nitric Oxide—Biology and Chemistry, vol. 21, no. 2, pp. 92–103, 2009.
[88] J. Zhang and H. Cai, “Netrin-1 prevents ischemia/reper-fusion-induced myocardial infarction via a DCC/ERK1/2/
eNOS𝑠1177/NO/DCC feed-forward mechanism,” Journal of
Molecular and Cellular Cardiology, vol. 48, no. 6, pp. 1060–1070,
2010.
[89] M. Y. Lee and K. K. Griendling, “Redox signaling, vascular function, and hypertension,” Antioxidants and Redox Signaling, vol. 10, no. 6, pp. 1045–1059, 2008.
[90] M. B. West, G. Rokosh, D. Obal et al., “Cardiac myocyte-specific expression of inducible nitric oxide synthase protects against ischemia/reperfusion injury by preventing mitochondrial per-meability transition,” Circulation, vol. 118, no. 19, pp. 1970–1978, 2008.
[91] P. Heusch, S. Aker, K. Boengler et al., “Increased inducible nitric oxide synthase and arginase II expression in heart failure: no net nitrite/nitrate production and protein S-nitrosylation,”
Amer-ican Journal of Physiology—Heart and Circulatory Physiology,
vol. 299, no. 2, pp. H446–H453, 2010.
[92] S. S. Soski´c, B. D. Dobutovi´c, E. M. Sudar et al., “Regulation of inducible Nitric Oxide synthase (iNOS) and its potential role in insulin resistance, diabetes and heart failure,” Open
Cardiovascular Medicine Journal, vol. 5, no. 1, pp. 153–163, 2011.
[93] J. L. Zweier, J. Fertmann, and G. Wei, “Nitric oxide and peroxy-nitrite in postischemic myocardium,” Antioxidants and Redox
Signaling, vol. 3, no. 1, pp. 11–22, 2001.
[94] W. Yasmin, K. D. Strynadka, and R. Schulz, “Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts,” Cardiovascular Research, vol. 33, no. 2, pp. 422–432, 1997.
[95] M. T. Ziolo and D. M. Bers, “The real estate of NOS signaling: location, location, location,” Circulation Research, vol. 92, no. 12, pp. 1279–1281, 2003.
[96] M. J. Kohr, S. R. Roof, J. L. Zweier, and M. T. Ziolo, “Modulation of myocardial contraction by peroxynitrite,” Frontiers in
Physi-ology, vol. 3, article 468, 2012.
[97] R. P. Laguens and C. L. G´omez-Dumm, “Fine structure of myo-cardial mitochondria in rats after exercise for one-half to two hours,” Circulation Research, vol. 21, no. 3, pp. 271–279, 1967. [98] J. J. Kane, M. L. Murphy, J. K. Bisset, N. deSoyza, J. E. Doherty,
and K. D. Straub, “Mitochondrial function, oxygen extrac-tion, epicardial S-T segment changes and tritiated digoxin dis-tribution after reperfusion of ischemic myocardium,” The
Amer-ican Journal of Cardiology, vol. 36, no. 2, pp. 218–224, 1975.
[99] R. B. Jennings and C. E. Ganote, “Mitochondrial structure and function in acute myocardial ischemic injury,” Circulation
Research, vol. 38, no. 5, pp. 80–91, 1976.
[100] Y. Capetanaki, “Desmin cytoskeleton: a potential regulator of muscle mitochondrial behavior and function,” Trends in
Cardio-vascular Medicine, vol. 12, no. 8, pp. 339–348, 2002.
[101] V. Adam-Vizi and C. Chinopoulos, “Bioenergetics and the formation of mitochondrial reactive oxygen species,” Trends in
Pharmacological Sciences, vol. 27, no. 12, pp. 639–645, 2006.
[102] A. Y. Andreyev, Y. E. Kushnareva, and A. A. Starkov, “Mito-chondrial metabolism of reactive oxygen species,” Biochemistry
(Moscow), vol. 70, no. 2, pp. 200–214, 2005.
[103] M. Inoue, E. F. Sato, M. Nishikawa et al., “Mitochondrial genera-tion of reactive oxygen species and its role in aerobic life,”
Cur-rent Medicinal Chemistry, vol. 10, no. 23, pp. 2495–2505, 2003.
[104] M. P. Murphy, “How mitochondria produce reactive oxygen species,” Biochemical Journal, vol. 417, no. 1, pp. 1–13, 2009. [105] A. A. Starkov, “The role of mitochondria in reactive oxygen
species metabolism and signaling,” Annals of the New York
Academy of Sciences, vol. 1147, pp. 37–52, 2008.
[106] H. Tsutsui, S. Kinugawa, and S. Matsushima, “Mitochondrial oxidative stress and dysfunction in myocardial remodelling,”
Cardiovascular Research, vol. 81, no. 3, pp. 449–456, 2009.
[107] S.-B. Ong, P. Samangouei, S. B. Kalkhoran, and D. J. Hausenloy, “The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury,” Journal of Molecular
and Cellular Cardiology, vol. 78, pp. 23–34, 2015.
[108] D. J. Hausenloy and D. M. Yellon, “The mitochondrial perme-ability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion,” Journal of Molecular
and Cellular Cardiology, vol. 35, no. 4, pp. 339–341, 2003.
[109] G. Heusch, K. Boengler, and R. Schulz, “Inhibition of mitochon-drial permeability transition pore opening: the Holy Grail of cardioprotection,” Basic Research in Cardiology, vol. 105, no. 2, pp. 151–154, 2010.
[110] A. P. Halestrap and A. P. Richardson, “The mitochondrial per-meability transition: a current perspective on its identity and role in ischaemia/reperfusion injury,” Journal of Molecular and
Cellular Cardiology, vol. 78, pp. 129–141, 2015.
[111] F. Di Lisa, A. Carpi, V. Giorgio, and P. Bernardi, “The mitochon-drial permeability transition pore and cyclophilin D in cardio-protection,” Biochimica et Biophysica Acta—Molecular Cell
Research, vol. 1813, no. 7, pp. 1316–1322, 2011.
[112] J. N. Weiss, P. Korge, H. M. Honda, and P. Ping, “Role of the mitochondrial permeability transition in myocardial disease,”
Circulation Research, vol. 93, no. 4, pp. 292–301, 2003.
[113] G. Morciano, C. Giorgi, M. Bonora et al., “Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury,” Journal of Molecular and Cellular
Cardiology, vol. 78, pp. 142–153, 2015.
[114] A. P. Halestrap, S. J. Clarke, and S. A. Javadov, “Mitochondrial permeability transition pore opening during myocardial reper-fusion—a target for cardioprotection,” Cardiovascular Research, vol. 61, no. 3, pp. 372–385, 2004.
[115] E. Murphy and C. Steenbergen, “What makes the mitochondria a killer? Can we condition them to be less destructive?”
Bio-chimica et Biophysica Acta—Molecular Cell Research, vol. 1813,
no. 7, pp. 1302–1308, 2011.
[116] E. J. Griffiths and A. P. Halestrap, “Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion,” Biochemical Journal, vol. 307, no. 1, pp. 93–98, 1995. [117] A. D. Kandasamy, A. K. Chow, M. A. M. Ali, and R. Schulz, “Matrix metalloproteinase-2 and myocardial oxidative stress injury: beyond the matrix,” Cardiovascular Research, vol. 85, no. 3, pp. 413–423, 2010.
[118] S. Viappiani, A. C. Nicolescu, A. Holt et al., “Activation and modulation of 72 kDa matrix metalloproteinase-2 by peroxyni-trite and glutathione,” Biochemical Pharmacology, vol. 77, no. 5, pp. 826–834, 2009.
[119] T. Okamoto, T. Akaike, T. Sawa, Y. Miyamoto, A. Van der Vliet, and H. Maeda, “Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation,” The Journal of Biological Chemistry, vol. 276, no. 31, pp. 29596–29602, 2001.
[120] Z. Gu, M. Kaul, B. Yan et al., “S-nitrosylation of matrix metal-loproteinases: signaling pathway to neuronal cell death,”
Sci-ence, vol. 297, no. 5584, pp. 1186–1190, 2002.
[121] M. Sariahmetoglu, B. D. Crawford, H. Leon et al., “Regulation of matrix metalloproteinase-2 (MMP-2) activity by phosphory-lation,” FASEB Journal, vol. 21, no. 10, pp. 2486–2495, 2007. [122] G. Sawicki, E. Salas, J. Murat, H. Miszta-Lane, and M. W.
Rado-mski, “Release of gelatinase A during platelet activation medi-ates aggregation,” Nature, vol. 386, no. 6625, pp. 616–619, 1997. [123] C. Fernandez-Patron, M. W. Radomski, and S. T. Davidge,
“Vascular matrix metalloproteinase-2 cleaves big endothelin-1 yielding a novel vasoconstrictor,” Circulation Research, vol. 85, no. 10, pp. 906–911, 1999.
[124] C. Fernandez-Patron, K. G. Stewart, Y. Zhang, E. Koivunen, M. W. Radomski, and S. T. Davidge, “Vascular matrix metal-loproteinase-2-dependent cleavage of calcitonin gene-related peptide promotes vasoconstriction,” Circulation Research, vol. 87, no. 8, pp. 670–676, 2000.
[125] P.-Y. Cheung, G. Sawicki, M. Wozniak, W. Wang, M. W. Radom-ski, and R. Schulz, “Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart,” Circulation, vol. 101, no. 15, pp. 1833–1839, 2000.
[126] Z.-Q. Zhao, M. Nakamura, N.-P. Wang et al., “Reperfu-sion induces myocardial apoptotic cell death,” Cardiovascular
Research, vol. 45, no. 3, pp. 651–660, 2000.
[127] W. D. Gao, D. Atar, Y. Liu, N. G. Perez, A. M. Murphy, and E. Marban, “Role of troponin I proteolysis in the pathogenesis of stunned myocardium,” Circulation Research, vol. 80, no. 3, pp. 393–399, 1997.
[128] J. L. McDonough, D. K. Arrell, and J. E. Van Eyk, “Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury,” Circulation Research, vol. 84, no. 1, pp. 9–20, 1999.
[129] R. J. Solaro and H. M. Rarick, “Troponin and tropomyosin: pro-teins that switch on and tune in the activity of cardiac myofila-ments,” Circulation Research, vol. 83, no. 5, pp. 471–480, 1998. [130] W. Wang, C. J. Schulze, W. L. Suarez-Pinzon, J. R. B. Dyck, G.
Sawicki, and R. Schulz, “Intracellular action of matrix metal-loproteinase-2 accounts for acute myocardial ischemia and reperfusion injury,” Circulation, vol. 106, no. 12, pp. 1543–1549, 2002.
[131] B. Menon, M. Singh, R. S. Ross, J. N. Johnson, and K. Singh, “𝛽-Adrenergic receptor-stimulated apoptosis in adult cardiac
myocytes involves MMP-2-mediated disruption of𝛽1integrin
signaling and mitochondrial pathway,” American Journal of
Physiology - Cell Physiology, vol. 290, no. 1, pp. C254–C261,
2006.
[132] B. Menon, M. Singh, and K. Singh, “Matrix metalloproteinases
mediate𝛽-adrenergic receptor-stimulated apoptosis in adult
rat ventricular myocytes,” American Journal of Physiology—Cell
Physiology, vol. 289, no. 1, pp. C168–C176, 2005.
[133] H.-Z. Zhou, X. Ma, M. O. Gray et al., “Transgenic MMP-2 expression induces latent cardiac mitochondrial dysfunction,”
Biochemical and Biophysical Research Communications, vol. 358,
no. 1, pp. 189–195, 2007.
[134] G. V. Halade, Y.-F. Jin, and M. L. Lindsey, “Matrix metallopro-teinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation,” Pharmacology and
Therapeutics, vol. 139, no. 1, pp. 32–40, 2013.
[135] N. A. Turner and K. E. Porter, “Regulation of myocardial matrix metalloproteinase expression and activity by cardiac fibro-blasts,” IUBMB Life, vol. 64, no. 2, pp. 143–150, 2012.
[136] Z. Xie, M. Singh, and K. Singh, “Differential regulation of matrix metalloproteinase-2 and -9 expression and activity in adult rat cardiac fibroblasts in response to interleukin-1𝛽,” Journal of
Biological Chemistry, vol. 279, no. 38, pp. 39513–39519, 2004.
[137] M. Lindsey, K. Wedin, M. D. Brown et al., “Matrix-depend-ent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reper-fusion,” Circulation, vol. 103, no. 17, pp. 2181–2187, 2001. [138] W. D. McMillan, N. A. Tamarina, M. Cipollone et al., “The
relationship between MMP-9 expression and aortic diameter,”
Circulation, vol. 96, pp. 2228–2232, 1997.
[139] S. Heymans, A. Luttun, D. Nuyens et al., “Inhibition of plasmin-ogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure,” Nature Medicine, vol. 5, no. 10, pp. 1135–1142, 1999. [140] L. E. Rohde, A. Ducharme, L. H. Arroyo et al., “Matrix
metallo-proteinase inhibition attenuates early left ventricular enlarge-ment after experienlarge-mental myocardial infarction in mice,”
Circu-lation, vol. 99, no. 23, pp. 3063–3070, 1999.
[141] A. Ducharme, S. Frantz, M. Aikawa et al., “Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlarge-ment and collagen accumulation after experienlarge-mental myocardial